

Abstract
Background and Purpose
Pulmonary arterial hypertension (PAH) is a progressive disease associated with high morbidity and mortality, despite advances in medical therapy. We compared the effects of infigratinib (NVP‐BGJ398), a new FGF receptor‐1 inhibitor, with or without the PDE‐5 inhibitor sildenafil, on vascular function and remodelling as well as on gene expression of signal transducers for receptors of TGF‐β (Smads‐1/2/4) and transcription factor of endothelial–mesenchymal transition (Twist‐1) in established experimental PAH. Types I and III pro‐collagen and TGF‐β expressions in lung fibroblasts were analysed in vitro after the different treatments.
Experimental Approach
PAH was induced in male Wistar rats with monocrotaline. 14 days later, treatments [sildenafil (SIL), infigratinib (INF) or their combination (SIL+INF)] were given for another 14 days. On Day 28, echocardiography and haemodynamic assays were performed, and lungs and pulmonary vessels were removed for analysis by histology, immunohistochemistry and RT‐PCR. Fibroblasts prepared from PAH lungs were also analysed for TGF‐β and pro‐collagen.
Key Results
Only the combination of infigratinib and sildenafil significantly improved right ventricular systolic pressure and vascular remodelling parameters (right ventricular hypertrophy, smooth muscle α‐actin, vessel wall thickness, and vascular collagen content). Infigratinib may act by reducing gene expression of Smads‐1/4 and Twist‐1 in lung tissue, as well as TGF‐β and types I and III pro‐collagen in lung fibroblasts.
Conclusions and Implications
In this model of monocrotaline‐induced PAH, the combination of the new inhibitor of FGF receptor‐1, infigratinib, and sildenafil effectively improved haemodynamics and decreased vascular remodelling.
What is already known
FGF is involved in the pathogenesis and vascular remodelling of pulmonary arterial hypertension (PAH).
Infigratinib is a new FGF receptor‐1 inhibitor used in cancer clinical studies with manageable toxicity.
What this study adds
Combination of infigratinib and sildenafil improved haemodynamics and decreased vascular remodelling in monocrotaline‐induced PAH.
Our results, obtained in monocrotaline‐induced PAH, may not apply to other models of PAH.
What is the clinical significance
Infigratinib, already used for human cancer treatment, could be combined with sildenafil for PAH.
Abbreviations
- 36B4
acidic ribosomal phosphoprotein P0
- Aext
external elastic lamina
- Aint
internal elastic lamina
- ALK
activin receptor‐like kinase
- bFGF
basic FGF
- FGFRs
FGF receptors
- INF
infigratinib
- PAH
pulmonary arterial hypertension
- PAT
pulmonary artery acceleration time
- PC
pro‐collagen
- PET
pulmonary artery ejection time
- PWT
per cent wall thickness
- RV
right ventricular
- RVSP
RV systolic pressure
- Smad
small mothers against decapentaplegic homolog
- SuHx
Sugen 5416/hypoxia
- Twist‐1
transcription factor of endothelial–mesenchymal transition
1. INTRODUCTION
http://www.guidetopharmacology.org/GRAC/DiseaseDisplayForward?diseaseId=804 (PAH) is a progressive disease of various aetiologies that carries a poor prognosis and results in right ventricular (RV) dysfunction (Klinger et al., 2019). PAH is characterized by haemodynamic alterations of the pulmonary circulation in which pulmonary artery pressures exceed 25 mmHg (Tuder, 2017) due to anatomical loss and obstructive remodelling of the pulmonary vascular bed, which result in right heart failure and functional decline (Humbert & Weatherald, 2018). Many drug therapies have been approved to improve quality of life and delay clinical deterioration; however, PAH remains an incurable disease. Pharmacological agents targeting the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989‐1, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509, or http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=270 pathways have shown benefits for patients with PAH. Nevertheless, these therapies failed to improve long‐term survival (Gall et al., 2017), perhaps due to their reduced effect on vascular remodelling (Hemnes & Humbert, 2017). Thus, new therapeutic approaches are required to attenuate pulmonary vascular remodelling process and to improve vascular function in PAH.
The fibroblast growth factors are involved in PAH pathogenesis and vascular remodelling. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924 is a ubiquitously expressed member of the FGF family and patients with PAH have increased plasma and urine bFGF levels (Benisty et al., 2004). Izikki et al. (2009) were able to prevent experimental pulmonary vascular remodelling by administering small interference RNA (siRNA) against rat FGF‐2. Additionally, pharmacological inhibition of FGF receptors (FGFRs) through intraperitoneal administration of the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1808 inhibitor http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5037 was shown to reduce pulmonary vascular remodelling (Zheng et al., 2015). However, PD173074 exhibits poor bioavailability and inhibitory activity at the cellular level and is not specific against this FGF receptor, as it also acts on http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=324 (Mohammadi et al., 1998). Guagnano et al. (2011) modified the molecular structure of PD173074 and developed http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7877, a compound with oral bioavailability in rodents, reduced toxicity, and increased specificity against FGFRs. This compound was recently tested in clinical trials for cancer treatment (Javle et al., 2018; Nogova et al., 2017), with promising results, and has been assigned the International Nonproprietary Name, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7877.
Considering the properties of infigratinib, we hypothesized that, when combined with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4743, these two agents would improve RV haemodynamics and mitigate the vascular remodelling process in an established model of monocrotaline‐induced PAH. In order to understand how this FGFRi may act on vascular remodelling, the gene expression of signal transducers for receptors of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1788 (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=303) and transcription factor of endothelial–mesenchymal transition (Twist‐1) were assessed in lung tissue. An in vitro analysis was also conducted to evaluate mRNA expression of types I and III pro‐collagen (PC) and TGF‐β in lung fibroblasts after different treatments.
2. METHODS
All animal care and experimental procedures complied with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the U.S. National Academy of Sciences Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Health Sciences Centre, Federal University of Rio de Janeiro (CEUA 063/18) and registered with the Brazilian National Council for Animal Experimentation Control. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Animals were housed at a controlled temperature (23°C) and controlled light–dark cycle (12–12 hr), with free access to water and food.
2.1. Animal preparation and experimental protocol
On Day 0, echocardiography was performed in 40 male Wistar rats (8 weeks old, weight 228 ± 37 g), which were then randomly assigned into two groups: (a) monocrotaline‐induced PAH (PAH, n = 32), in which animals received a single intraperitoneal injection of https://www.sigmaaldrich.com/catalog/product/sigma/c2401?lang=pt®ion=BR (60 mg·kg−1; C2401, Sigma Chemical Co., St Louis, MO, USA) and (b) control (CTRL, n = 8), in which animals received a similar volume of saline solution intraperitoneally. On Day 14, echocardiography was repeated to detect alterations indicative of PAH. Animals with confirmed PAH were randomized, by sealed envelopes, to receive oral saline (SAL), sildenafil citrate (EMS, Rio de Janeiro, RJ, Brazil; SIL) 20 mg·kg−1·day−1, infigratinib (Selleck Chemicals LLC, Houston, TX, USA) 5 mg·kg−1·day−1, or sildenafil plus infigratinib (SIL + INF), dosed as above, once daily for 14 days (Figure 1a). On Day 28, echocardiography was repeated, haemodynamic parameters were measured, and animals were killed for postmortem analyses (Figure 1b).
Figure 1.
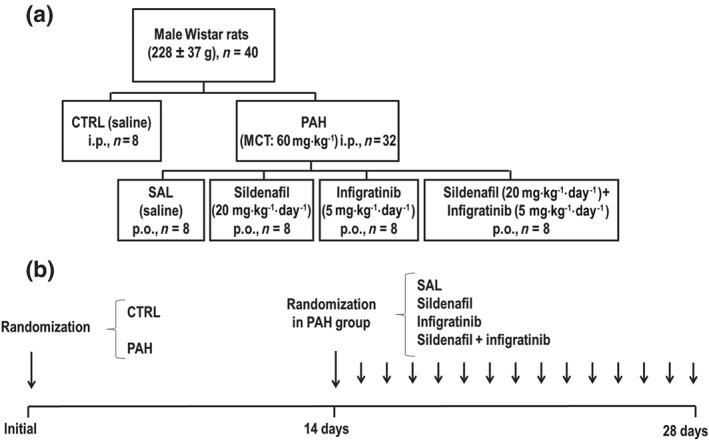
(a) Experimental design. (b) Experimental timeline. At the initial point, animals were randomized to CTRL and PAH groups. At 14 days, the PAH groups were further randomized to receive SAL (saline, PAH‐SAL, n = 8), sildenafil (20 mg·kg−1·day−1, PAH‐SIL, n = 8), infigratinib (5 mg·kg−1·day−1, PAH‐INF, n = 8), or a combination of sildenafil and infigratinib (PAH‐SIL + INF, n = 8). CTRL, control animals; MCT, monocrotaline; PAH, pulmonary arterial hypertension; SAL, saline
2.2. Echocardiography
Animals were anaesthetized with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2505 (Isoforine® 2.5 vol.%; Cristália, São Paulo, SP, Brazil) and shaved over the precordial region. Cardiovascular function was evaluated using an UGEO HM70A ultrasound system (Samsung, São Paulo, Brazil) coupled to a 30‐MHz transducer. Images were obtained from long and short parasternal axis views. Pulsed‐wave Doppler was used to measure pulmonary artery acceleration time (PAT) and pulmonary artery ejection time (PET), and the PAT/PET ratio was used as an indirect index of PAH. All parameters followed the American Society of Echocardiography and European Association of Cardiovascular Imaging guidelines, as validated in previous work on noninvasive assessment of murine pulmonary arterial pressure (Lang et al., 2015; Thibault et al., 2010).
2.3. Right ventricular hypertrophy
The heart was removed from the chest, weighed, and the ratio of the right ventricle to left ventricle plus septum weight (RV/LV + S) was calculated as the Fulton index of RV hypertrophy (RVH; Xiao et al., 2017).
2.4. Haemodynamic measurements
Rats were anaesthetized with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1626 100 μg·kg−1 i.p. (Unifental®, União Química, São Paulo, SP, Brazil) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3342 5 mg·kg−1 i.p. (Dormire®, União Química, São Paulo, SP, Brazil). If required, additional anaesthesia was provided by inhaled isoflurane through a nasal device. Animals were placed in dorsal recumbency, and the skin and muscle tissue of the anterior chest wall were removed until the ribs were visible. The thorax was then opened, and a heparinized 19G winged infusion set (Embramac, Itapira, SP, Brazil) was inserted into the RV. The RV systolic pressure (RVSP) was assessed with a physiological data acquisition system (LabChart 7.0, ADInstruments, Sydney, Australia, RRID:SCR_001620). Blood samples were collected through inferior vena cava puncture into a heparinized syringe. These samples were then centrifuged at 1,500× g for 5 min at 4°C for plasma retrieval for elisa (see below). At the end of the experiment, animals were killed by complete exsanguination through transection of the abdominal aorta.
2.5. Determination of pulmonary artery function
After haemodynamic measurements, the heart and lungs were removed. The main pulmonary artery was carefully dissected and cleaned of connective tissue. Arteries were attached to a force transducer and placed in vertical chambers filled with modified Tyrode's solution (123‐mM NaCl, 4.7‐mM KCl, 1.2‐mM MgCl2, 1.2‐mM KH2PO4, 15.5‐mM NaHCO3, 1.2‐mM CaCl2, and 11.5‐mM dextrose), which were oxygenated and kept at 37°C. After a stabilization period of 2 hr in 1.5× g tension, the solutions were exposed to increasing doses of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=485 (1 nM–10 μM). After the contraction plateau, the arteries were exposed to increasing doses of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 (1 nM–10 μM) to evaluate endothelial function by maximum relaxation capacity.
2.6. Quantification of lung collagen
To avoid parenchymal distortion, a positive end‐expiratory pressure (3 cmH2O) was applied to all animals through a mechanical ventilator before lung tissue removal (Flexivent; SCIREQ, Canada). The trachea was then clamped at end‐expiration, and the lungs removed from the thorax. The left lung was isolated, frozen in liquid nitrogen, and placed in Carnoy's solution. The lungs were dehydrated in a graded ethanol series and embedded in paraffin. Slices (4 μm thick) were cut, deparaffinized, and stained by Masson's trichrome. The slides were scanned in a Pannoramic MIDI II system (3DHISTECH, Budapest, Hungary, RRID:SCR_008674). The collagen fibres were quantified through a digital analysis system and specific software (Image‐Pro® Plus 6.3 for Windows®, RRID:SCR_007369) by an independent research (J. B. V.) blinded to slides allocation.
2.7. Quantification of α‐smooth muscle actin
Sections were deparaffinized and hydrated, and slides incubated with 10‐mM sodium citrate. Endogenous peroxidase activity was blocked with 3% http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2448. Slides were washed in TBS with 0.05% Tween‐20 (Sigma), blocked with a serum‐free protein block (Dako, Carpinteria, CA, USA), and immunostained with Vectastain ABC (Vector Laboratories, Inc., Burlingame, CA, USA, RRID:AB_2336827). Antibodies against α‐smooth muscle actin (Abcam, USA, Cat# ab7817, RRID:AB_262054) were incubated in TBS/Tween buffer overnight at 4°C. The slides were then scanned in Pannoramic MIDI II. Quantification of α‐actin was done by digital analysis in Image‐Pro® Plus 6.3 for Windows by an independent investigator (J. B. V.) blinded to group allocation. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.8. Smooth muscle wall thickness
Assessment of smooth muscle wall thickness was carried out using sections stained for α‐smooth muscle actin. Ten random fields per slide containing terminal arterioles (diameter < 150 μm) were captured (magnification ×40) with a digital camera, and the areas bounded by the external elastic lamina (Aext) and internal elastic lamina (Aint) of each vessel were measured with ImageJ (NIH, Bethesda, USA) by a previously calibrated examiner. The diameter (Dext) and per cent wall thickness (PWT) of the arterioles were calculated by the following formulas: Dext = 2 × sqrt (Aext/π), where “sqrt” denotes “square root” and percentage (PWT, %) = 100 × (1 − Aint/Aext). For each slide, the mean PWT of each animal was used as an individual data point in the respective experimental group.
2.9. Percentage of muscular arteries
In each rat, 50 peripheral pulmonary arteries (<150 μm) were identified as one of three structural types: muscular (M; with a complete medial coat of muscle), partially muscular (PM; with an incomplete coat, only a crescent of muscle being present), or non‐muscular (NM; no muscle apparent). The percentage of each type of pulmonary artery was determined as described elsewhere (Schermuly et al., 2004).
2.10. Plasma and lung tissue elisa
Plasma and lung tissue levels of vascular bFGF (Recombinant Murine FGF‐basic, PeproTech, NJ, USA, RRID:AB_147888) and plasma levels of TGF‐β (Recombinant Mouse Latent TGF‐β, Biolegend, CA, USA, RRID:AB_2563029) were quantified on Day 28. The results were normalized by total protein content by Bradford's technique and expressed as pg·mg−1. All measurements were performed in accordance with manufacturer guidelines by an independent researcher (C. L. B.) blinded to group allocation.
2.11. Expression of Smad‐1, Smad‐2, Smad‐4, and Twist‐1 in lung tissue
Quantitative RT‐PCR was performed to assess Smad 1, 2, and 4 and markers of endothelial–mesenchymal transition (Twist‐1) in lung tissue.
Central slices of the lungs and proximal sections of the pulmonary artery were cut, collected in cryotubes, flash‐frozen in liquid nitrogen, and stored at −80°C. Total RNA was extracted (RNeasy Plus Mini Kit; Qiagen, Hilden, Germany) and the RNA concentration measured by spectrophotometry (Nanodrop ND‐1000 system; Thermo Scientific, Wilmington, DE, USA). First‐strand cDNA was synthesized from total RNA (Quantitec Reverse Transcription Kit; Qiagen). Relative mRNA levels were measured with a SYBR green detection system in an ABI 7500 real‐time PCR analyzer (Applied Biosystems, Foster City, CA, USA). Samples were run in triplicate. For each sample, the expression of each gene was normalized to the acidic ribosomal phosphoprotein P0 (36B4) housekeeping gene and expressed as fold change relative to CTRL, using the 2–ΔΔCt method (Schmittgen & Livak, 2008), where ΔCt = Ct (reference gene) − Ct (target gene).
The following primers were used: Smad‐1 (5′‐AGAACCGATTCTGCCTTGGG‐3′ [forward] and 5′‐ACTCCGCATACACCTCTCCT‐3′ [reverse]), Smad‐2 (5′‐CGTCGGAAGAGGAAGGAACAA‐3′ [forward] and 5′‐GCGGAGTGAATGGCAAGATG‐3′ [reverse]), Smad‐4 (5′‐AAGCAATGACGCCTGTCTGA‐3′ [forward] and 5′‐CTGGGGTGAGCTCGATTTGT‐3′ [reverse]), and Twist‐1 (5′‐TCACAAGAATCAGGGCGTGG‐3′ [forward] and 5′‐AATGCAGAGGTGTGAGGGTG‐3′ [reverse]).
2.12. Expression of TGF‐β and types I and III PC in lung fibroblasts
Lung fibroblasts were isolated as previously described (Seluanov, Vaidya, & Gorbunova, 2010). Briefly, the right upper lung was removed, cut into small fragments, enzymatically digested in Liberase solution at 37°C for 30 min, rinsed three times with warm DMEM/F12 media with 10% FBS and 1× penicillin/streptomycin, centrifuged at 524× g, resuspended in complete DMEM/F12 media, transferred to a tissue culture dish, and, finally, placed in a humidified tissue culture incubator at 37°C and 5% CO2. After the cells reached 80–90% confluence, they were lysed, and total RNA was extracted from frozen tissues using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany), following the manufacturer's recommendations. The RT‐PCR analysis followed the description above. The following primers were used: TGF‐β (5′‐ATACGCCTGAGTGGCTGTC‐3′ [forward] and 5′‐GCCCTGTATTCCGTCTCCT‐3′ [reverse]), PC‐I (αI) (5′‐AGAAGTCTCAAGATGGTGGCCG‐3′ [forward] and 5′‐GGTCACGAACCACGTTAGCATC‐3′ [reverse]), PC‐III (αI) (5′‐CAGCTATGGCCCTCCTGATCTT‐3′ [forward] and 5′‐GTAATGTTCTGGGAGGCCCG‐3′ [reverse]), and 36B4 (5′‐CAACCCAGCTCTGGAGAAAC‐3′ [forward] and 5′‐CAACCCAGCTCTGGAGAAAC‐3′ [reverse]).
2.13. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). The sample size was calculated to allow detection of differences in RVSP between CTRL and PAH animals based on previous work from our group (de Mendonca et al., 2017). A sample size of n = 8 animals per group would provide the appropriate power (1 − β = 0.8) to identify statistically significant differences in RVSP, taking into account an effect size d = 1.57, a two‐sided t test, and a sample size ratio = 1 (G*Power 3.1.9.2, University of Düsseldorf, Düsseldorf, Germany, RRID:SCR_013726).
Each variable was tested for normality and variance using the Kolmogorov–Smirnov and Levene tests, respectively. Data are presented as mean ± SD unless otherwise specified. On Day 14, PAT/PET data were compared by Student's t test (P < .05). On Day 28, the CTRL group was compared to PAH groups by Bonferroni's correction for four comparisons, adjusted P < .0125. One‐way ANOVA followed by Holm–Šidák multiple comparisons or the Kruskal–Wallis test followed by Dunn's multiple comparisons (P < .05) were used to compare PAH groups. All tests were performed in GraphPad Prism version 6.01 (GraphPad Software, La Jolla, CA, USA, RRID:SCR_002798).
2.14. Materials
ACh, monocrotaline and phenylephrine were supplied by Sigma Chemical Co., (St Louis, MO, USA); sildenafil citrate by EMS (Rio de Janeiro, RJ, Brazil) and https://www.selleckchem.com/products/bgj398-nvp-bgj398.html by Selleck Chemicals LLC (Houston, TX, USA).
2.15. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017a, b).
3. RESULTS
3.1. Echocardiography and haemodynamics
On Day 14, the PAT/PET ratio was lower in PAH group than in CTRL group, suggesting pulmonary hypertension (Figure 2). On Day 28 (Figure 3a), the PAT/PET ratio was lower in PAH‐SAL and PAH‐INF groups compared to the CTRL group. On the other hand, it was higher in the PAH‐SIL and PAH‐SIL + INF groups than in the PAH‐SAL group.
Figure 2.
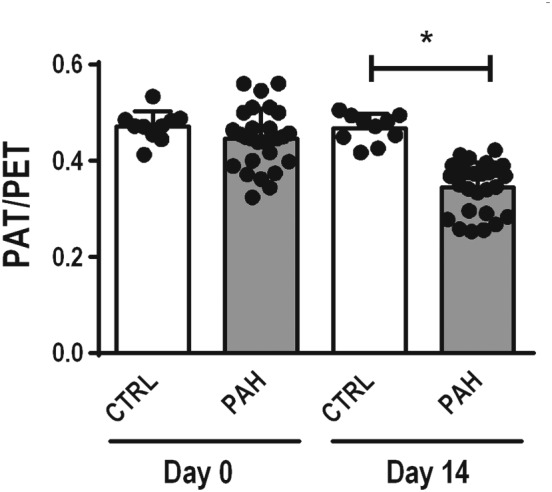
On Day 0, echocardiography was performed in 40 male Wistar rats, which were then randomly assigned into two groups: (a) monocrotaline‐induced PAH (PAH, n = 32), in which animals received 60 mg·kg−1 of monocrotaline intraperitoneally; and (b) control (CTRL, n = 8), in which animals received a similar volume of saline solution intraperitoneally. On Day 14, echocardiography was repeated to detect alterations indicative of PAH. On Day 14, the PAT/PET ratio was lower in PAH than CTRL groups, suggesting pulmonary hypertension. Data shown are individual values with means± SD. *P<.05, significantly different as indicated; Student's t‐test. PAT/PET, pulmonary artery acceleration time/pulmonary artery ejection time
Figure 3.
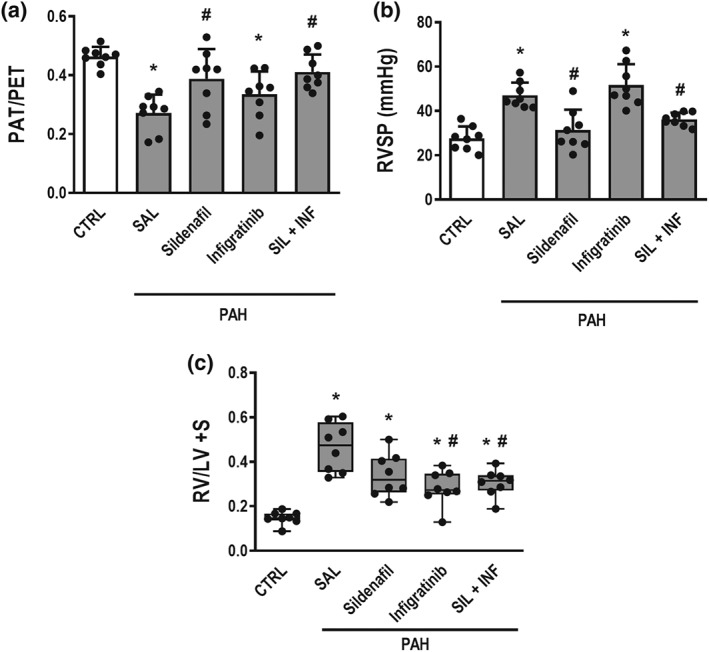
Data shown are individual values with means± SD. (a) Pulmonary artery acceleration time/pulmonary artery ejection time (PAT/PET) ratio. * P<0.05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P<0.05, significantly different from PAH‐SAL; one‐way ANOVA followed by Holm‐Šidák multiple comparisons.
(b) Right ventricle systolic pressure (RVSP). * P<0.05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P<0.05, significantly different from PAH‐SAL; one‐way ANOVA followed by Holm‐Šidák multiple comparisons. (c) Right ventricular hypertrophy (RV [right ventricle]/LV [left ventricle] + S [septum. *P<0.05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P<0.05, significantly different from PAH‐SAL; Kruskal–Wallis test followed by Dunn's multiple comparisons
As shown in Figure 3b, RVSP was higher in PAH‐SAL than in CTRL animals. After the 14 days of treatment RVSP was reduced in the PAH‐SIL and PAH‐SIL + INF groups, but not in PAH‐INF, when compared to values in the PAH‐SAL group. Heart rate was higher in all PAH groups when compared to CTRL. No differences in heart rate were observed among PAH groups.
3.2. Right ventricular hypertrophy
The Fulton index (RV/LV+S) was higher in PAH‐SAL when compared to CTRL animals. Treatment with infigratinib (PAH‐INF) or its combination with sildenafil (PAH‐SIL + INF) reduced this index (Figure 3c).
3.3. Endothelial dysfunction
The concentration‐response curve to phenylephrine was similar in all groups but the relaxation of the precontracted arterial tissue by ACh did differ between the experimental groups (Figure 4). The PAH‐SAL animals showed less relaxation than CTRL animals and the PAH‐SIL and PAH‐SIL + INF groups showed greater artery relaxation in response to increased dosages of ACh. No such behaviour was found in the PAH‐INF group.
Figure 4.
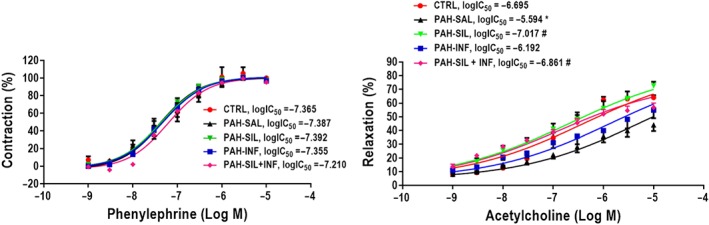
Pulmonary arterial rings were exposed to increasing doses of phenylephrine (Phe, 1 nM–10 μM, left panel). After the contraction plateau, the arteries were exposed to increasing doses of ACh (1 nM–10 μM, right panel) to evaluate endothelial function by maximum relaxation capacity. PAH‐SAL animals showed less relaxation than CTRL animals. The PAH‐SIL and PAH‐SIL + INF groups showed greater artery relaxation in response to increasing dosages of ACh; no such behaviour was found in the PAH‐INF group. The phenylephrine curve was similar in all groups. Data shown are means± SD. * P<0.05, significantly different from CTRL group; one way ANOVA with Bonferroni's correction. # P<0.05, significantly different from PAH‐SAL; one‐way ANOVA followed by Holm‐Šidák multiple comparisons
3.4. Vascular remodelling parameters
PAH‐SAL animals showed higher collagen (Figure 5a,b), vascular α‐actin, and wall thickness than CTRL animals (Figure 6a–c). Collagen content (Figure 5a,b) and vascular α‐actin (Figure 6a,c) only decreased in the presence of infigratinib (PAH‐INF and PAH‐SIL + INF groups). Vessel wall thickness was lower in all treated groups compared to PAH‐SAL (Figure 6a,c). The percentage of non‐muscular pulmonary arteries decreased, while that of partly and fully muscular pulmonary arteries increased, in PAH‐SAL compared to CTRL animals (Figure 7). PAH‐SIL animals showed a reduction in fully muscular pulmonary arteries compared to PAH‐SAL. The percentage of non‐muscular pulmonary arteries increased, whereas that of fully muscular pulmonary arteries decreased, in the PAH‐INF and PAH‐SIL + INF groups compared to PAH‐SAL. Furthermore, the combination of sildenafil and infigratinib reduced the percentage of partially and fully muscular pulmonary arteries in relation to sildenafil alone (Figure 7).
Figure 5.
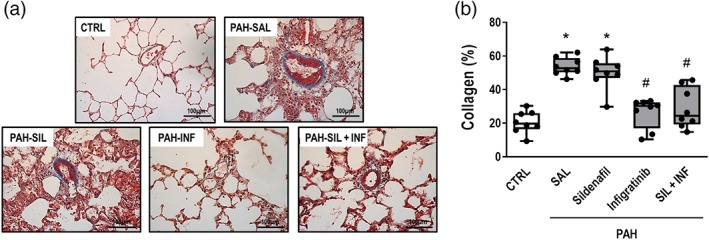
(a) Representative photomicrographs of pulmonary vessel collagen stained with Masson's trichrome (original magnification ×400). Note the intense collagen deposition in the pulmonary artery (black triangles) in the PAH‐SAL and PAH‐SIL groups. (b) Data are shown as box (interquartile, 25–75%, range) and whiskers (total range) plots with horizontal lines representing median values of eight animals per group. * P < .05, significantly different from CTRL group; one way ANOVA with Bonferroni's correction. # P < .05, significantly different from PAH‐SAL; Kruskal–Wallis test followed by Dunn's multiple comparisons test
Figure 6.
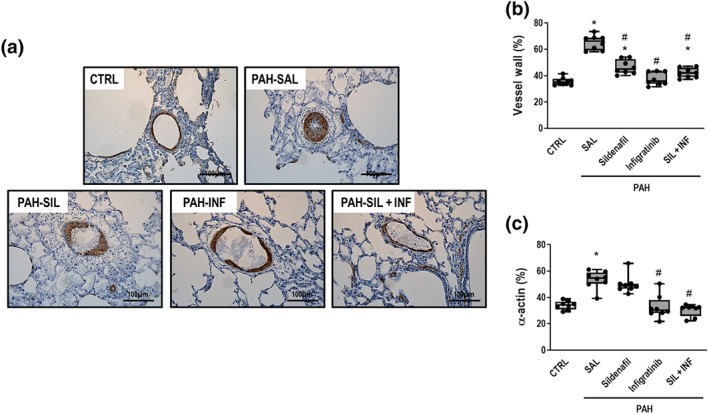
(a) Representative photomicrographs immunostained for α‐smooth muscle actin (original magnification ×400). Note intense smooth muscle α‐actin staining (black triangles) in PAH‐SAL and the PAH‐SIL groups. (b) and (c) Percentage of vessel wall thickness and smooth muscle α‐actin, respectively. Data are shown as box (interquartile, 25–75%, range) and whiskers (total range) plots with horizontal lines representing median values of eight animals per group. * P < .05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P < .05, significantly different from PAH‐SAL; Kruskal–Wallis test followed by Dunn's multiple comparisons test
Figure 7.
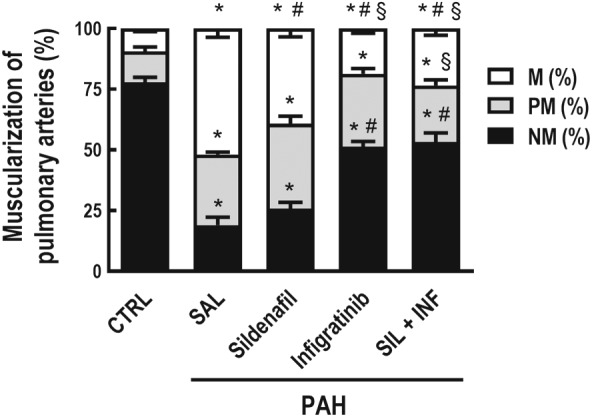
Percentage of non‐muscular (NM), partially muscular (PM), or fully (M) muscular pulmonary arteries relative to the total number of pulmonary arteries. A total of 50 intra‐acinar vessels were analysed in each lung from rats exposed to monocrotaline and the following treatments. Values are means and SD of eight animals per group. NM, PM, and M were compared among groups. Data shown are means± SD. * P<0.05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P<0.05, significantly different from PAH‐SAL; one‐way ANOVA followed by Holm‐Šidák multiple comparisons test
3.5. bFGF and TGF‐β protein levels in plasma and lung tissue
PAH‐SAL animals showed higher plasma and lung tissue bFGF protein levels compared to CTRL animals. PAH‐INF and PAH‐SIL + INF animals showed lower bFGF protein levels in plasma and lung tissue compared to the PAH‐SAL and PAH‐SIL groups. No major differences were observed in TGF‐β protein levels in plasma and lung tissue (Table 1).
Table 1.
Plasma and lung tissue protein markers
| Plasma | PAH | ||||
|---|---|---|---|---|---|
| CTRL | SAL | SIL | INF | SIL + INF | |
| bFGF (pg·mg−1) | 0.51 ± 0.08 | 1.16 ± 0.11* | 1.39 ± 0.58* | 0.62 ± 0.18#, † | 0.66 ± 0.28#, † |
| Lung tissue | PAH | ||||
|---|---|---|---|---|---|
| CTRL | SAL | SIL | INF | SIL + INF | |
| bFGF (pg·mg−1) | 0.55 ± 0.29 | 0.89 ± 0.04* | 1.31 ± 0.07* | 0.67 ± 0.09#, † | 0.59 ± 0.23#, † |
| TGF‐β (pg·mg−1) | 0.02 ± 0.01 | 0.02 ± 0.02 | 0.03 ± 0.03 | 0.03 ± 0.03 | 0.04 ± 0.02 |
Note. Plasma protein levels of basic FGF (bFGF) and lung tissue protein levels of bFGF and TGF‐β.
Values presented are mean ± SD of 8 animals per group. P<.05, significantly different from CTRL group; one‐way ANOVA followed by Holm‐Šidák multiple comparisons.
P<.05, significantly different from PAH‐SAL,
P<.05, significantly different from PAH‐SIL; one‐way ANOVA followed by Holm‐Šidák multiple comparisons. INF, infigratinib; PAH, pulmonary arterial hypertension; SAL, saline; SIL, sildenafil
3.6. Expression of mRNA for Smad‐1, Smad‐2, Smad‐4, and Twist‐1
Smad‐2 mRNA expression was higher in all PAH groups (Figure 8b). On the other hand, Smad‐1 mRNA expression was higher in PAH‐SAL than in CTRL animals. Treatment with infigratinib, whether alone or combined with SIL, decreased Smad‐1 mRNA expression (Figure 8a). Smad‐4 mRNA expression was higher in PAH‐SAL animals than in CTRL animals. Infigratinib alone or combined with sildenafil, also decreased Smad‐4 mRNA expression (Figure 8c). Twist‐1 mRNA expression was higher in PAH‐SAL rats than in CTRL rats. Again, infigratinib, whether alone or with sildenafil, decreased expression of Twist‐1 mRNA (Figure 8d).
Figure 8.
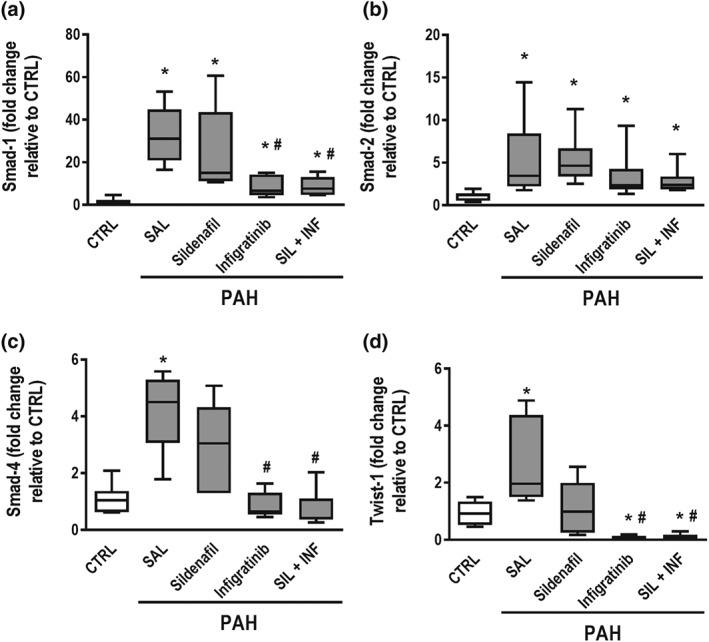
Real‐time PCR analysis of biological markers. (a) Smad‐1, (b) Smad‐2, (c) Smad‐4, and (d) endothelial–mesenchymal transition marker (Twist‐1) in lung tissue. Relative gene expression was calculated as a ratio of the average gene expression levels compared with the reference gene 36B4 and expressed as fold change relative to CTRL. Data are shown as box (interquartile, 25–75%, range) and whiskers (total range) plots with horizontal lines representing median values of eight animals per group. * P < .05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P < .05, significantly different from PAH‐SAL; Kruskal–Wallis test followed by Dunn's multiple comparisons test
3.7. Expression of mRNA for TGF‐β and types I and III PC in lung fibroblasts
Lung fibroblasts obtained from PAH‐SAL animals showed higher PC‐I, PC‐III, and TGF‐β mRNA expression compared to CTRL fibroblasts (Figure 9a–c). All treatment protocols were able to reduce PC‐I, PC‐III, and TGF‐β mRNA expressions in lung fibroblasts.
Figure 9.
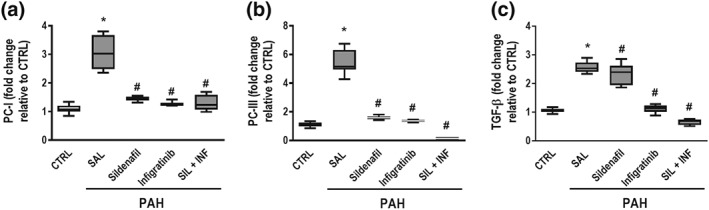
RT‐PCR from isolated lung fibroblast cells after 28 days. Lung fibroblasts obtained from PAH‐SAL animals showed higher PC‐I, PC‐III, and TGF‐β mRNA expressions compared to CTRL fibroblasts (a–c). All treatment protocols were able to reduce PC‐I, PC‐III, and TGF‐β mRNA expression in lung fibroblasts. Data are shown as box (interquartile, 25–75%, range) and whiskers (total range) plots with horizontal lines representing median values of eight animals per group. * P < .05, significantly different from CTRL group; one‐way ANOVA with Bonferroni's correction. # P < .05, significantly different from PAH‐SAL; Kruskal–Wallis test followed by Dunn's multiple comparisons test
4. DISCUSSION
In the rat model of monocrotaline‐induced PAH we have used, the combination of infigratinib and sildenafil (a) reduced RVSP and RVH and increased PAT/PET ratio, leading to lower RV overload; (b) decreased collagen fibre content and α‐actin deposition in pulmonary arterioles; (c) reduced vessel wall muscularization and the percentage of partially and fully muscular small pulmonary arteries (internal diameter < 150 μm); (d) decreased mRNA expression of Smads‐1/4 and Twist‐1, but not Smad‐2, in lung tissue; and (e) decreased gene expression of types I and III PC and TGF‐β in lung fibroblasts. As sildenafil, at this dosage, leads to vasodilation and the FGFRi infigratinib halts pulmonary vascular remodelling, the combination of both therapies may result in better overall effects (see Table S1), which would justify their use in PAH. We may infer from our findings that infigratinib acts through modulation of Smads‐1/4, but not Smad‐2, to reduce vascular remodelling in PAH.
Several models have been reported to induce PAH. More recently, the Sugen 5416/hypoxia (SuHx) model has been used because of its ability to reproduce relevant pathological features of human PAH, such as plexiform lesions. However, when these models are used to test new treatments for PAH patients, the expected benefits are not observed experimentally. In this context, iloprost treatment, which is approved for clinical use in PAH patients (Klinger et al., 2019), presented beneficial effects when tested in monocrotaline models (Yigitaslan & Sirmagul, 2012), but not in the SuHx model (Gomez‐Arroyo et al., 2012). Recently, similar behaviour was observed with treprostinil in the SuHx model, with decreased vascular resistance, but no direct effects on vascular remodelling (Chaudhary et al., 2018). The monocrotaline‐induced PAH model is simple, reliable, and economically suitable, despite some limitations regarding human PAH pathophysiology and translation of results from preclinical to clinical studies. Research using the monocrotaline model has identified important signalling pathways involved in human disease (Hill, Gillespie, & McMurtry, 2017).
In the present study, infigratinib, a new FGFR1 inhibitor, was used because of its better oral bioavailability and reduced toxicity (Guagnano et al., 2011). Infigratinib is known to act on FGFRs 1, 2, and 3, but mainly on FGFR1, specifically within the tyrosine‐protein kinase https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1805 (Guagnano et al., 2011), a receptor TK protein, also known as the c‐Kit ligand. During PAH development, this receptor can be linked to high circulating bFGF levels (Izikki et al., 2009). Once the TK is active, it in turn phosphorylates and activates signal transduction molecules that promote cell survival, proliferation, and differentiation. The choice of infigratinib dose (5 mg·kg−1) was based on preclinical (Guagnano et al., 2011) and clinical studies (Javle et al., 2018; Nogova et al., 2017) which reported minimal toxicity.
Infigratinib is believed to hinder vascular remodelling in experimental PAH, nevertheless, a minimal effect on vascular reactivity was expected. Therefore, we chose to combine infigratinib with sildenafil, one of nine FDA‐approved pharmacological therapies for PAH, which is known to result in vasodilation and have positive effects on exercise ability (Galie et al., 2005; Klinger et al., 2019). Sildenafil, but not infigratinib, reduced RVSP through its well‐known inhibitory effect on PDE‐5, increasing cGMP levels and thus inducing vasodilation. The increased RVSP that is observed during PAH may result from several alterations, such as endothelial dysfunction, vasoconstriction, smooth muscle hypertrophy, intimal proliferation, uncontrolled cell proliferation, and in situ thrombosis (Rabinovitch, Guignabert, Humbert, & Nicolls, 2014). Nevertheless, removing one of these components may not suffice to have a positive effect on RVSP. For instance, we observed that sildenafil acted on vascular relaxation, as observed by exposure of pulmonary artery rings to increasing dosages of ACh in vitro, and ultimately reduced RVSP with minimal effects on pulmonary vascular remodelling. Previous experimental studies (Merabet et al., 2019; Schermuly et al., 2004; Zhang et al., 2019) have shown positive effects of sildenafil on vascular remodelling in PAH models. However, these studies used higher sildenafil doses (30 and 25 mg·kg−1, respectively) than that used in the present study (20 mg·kg−1). This is further corroborated by the findings of Li et al. (2019), who used 8 mg·kg−1 and did not observe beneficial effects on vascular remodelling. We may infer that the association between reduced pulmonary vascular remodelling and reduced RVSP may become more apparent when the sildenafil dose is increased. Nevertheless, we do not believe that an increase in sildenafil dose would preclude the use of infigratinib, since the latter had a greater effect on vascular remodelling. In short, we believe that the combination of both drugs would be a good therapeutic strategy, because (a) sildenafil could be used at a lower dose, with beneficial effects on haemodynamics and probably fewer side effects, and (b) infigratinib demonstrated an important effect by reducing vascular remodelling. Interestingly, sildenafil and infigratinib act through different pathways in PAH.
Vascular relaxation through NO metabolism plays an important role as a determinant of RVSP, which would justify the combination of sildenafil and infigratinib (Ramani & Park, 2010). Furthermore, RVH and the degree of muscularization of pulmonary arteries only decreased after infigratinib treatment. It may be inferred that decreased pulmonary vascular remodelling is mutually associated with reduced RVH. On the other hand, decreased pulmonary vascular remodelling and reduced RVSP are not necessarily linked by causality, as vasoreactivity may be a confounding factor (Li et al., 2019).
We may infer from our data that infigratinib had a significant inhibitory effect on fibroblast activity in extracellular matrix synthesis, as shown by reduced vascular collagen, as well as activation of fibroblast cells towards myofibroblasts, demonstrated by reduced α‐actin staining (Shinde, Humeres, & Frangogiannis, 2017). There is evidence that c‐Kit activation and bone marrow‐derived progenitor cell crosstalk may be involved in vascular changes during PAH (Davie et al., 2004; Gambaryan et al., 2010; Gambaryan et al., 2011). By reducing the number of c‐Kit‐positive cells, infigratinib may hinder recruitment of new bone marrow cells into the already remodelled pulmonary arteries (Godinas et al., 2013). As the bFGF signalling pathway is associated not only with bFGF overproduction but also with increased FGFR expression (Tu et al., 2011), we evaluated the efficacy of FGFR1 inhibition by assessing protein levels of bFGF in plasma and lung tissue and found them to be reduced only in the presence of infigratinib.
Infigratinib did not modify TGF‐β protein levels in lung tissue. Therefore, the decrease in remodelling induced by this compound cannot be explained by TGF‐β levels. The bone morphogenetic protein receptors (BMPRs) are members of the TGF‐β superfamily. Once the BMPRs are activated, phosphorylation occurs through Smad proteins, allowing translocation of a signalling complex into the nucleus and shifting gene transcription towards extracellular matrix remodelling, proliferation, migration, and development (Humbert et al., 2004). The complexes are formed by the common ligand Smad‐4 with Smads‐1/5/8 or Smads‐2/3. The https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1839 which was previously activated, will determine which group of Smad (1/5/8 or 2/3) forms the complex with Smad‐4. Smad‐2 and Smad‐3 are phosphorylated directly by https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1788&familyId=318&familyType=CATALYTICRECEPTOR, whereas Smads‐1/5/8 are activated by https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1784&familyId=318&familyType=CATALYTICRECEPTOR (Biernacka, Dobaczewski, & Frangogiannis, 2011). Infigratinib reduced mRNA expression of Smad‐1, thus interfering with the ALK5 pathway. FGF inhibitors have been shown to suppress ALK1 activity in vitro (Tsai, Lee, & Wu, 2003). Inhibition of FGF may lead to lower levels of Smad‐1 and, thus, reduced formation of complexes with Smad‐4, thus reducing the gene transcription factor associated with organization of ECM components.
4.1. Limitations
The present study has some limitations. First, in vitro analyses to detect additive and synergic interactions between sildenafil and infigratinib were not performed. A practical approach was chosen instead, by combining two drugs used in human clinical trials. Second, the infigratinib dose was based on experimental and clinical studies conducted in cancer. Additionally, increases in infigratinib dose may result in more beneficial effects. Further studies are needed to evaluate the dose–response curve of infigratinib in this model of PAH, taking into account any adverse effects. Third, from a functional point of view, dual therapy did not lead to additive effects and this failure might be associated with the narrow time frame of observation (28 days). Additional experimental studies are needed to determine whether infigratinib is able to improve haemodynamics over a longer time. Fourth, the present results were obtained in monocrotaline‐induced PAH and should not be directly extrapolated to other PAH models, such as SuHx and schistosomiasis‐associated pulmonary hypertension, the latter being one of the most common causes of PAH in developing countries (Papamatheakis, Mocumbi, Kim, & Mandel, 2014). Fifth, fibroblasts were extracted at the end of the protocol, which means we cannot speculate on the effects of infigratinib treatments on other respiratory tract cell types.
In conclusion, we have found that, in a rat model of monocrotaline‐induced PAH, sildenafil improved RV systolic pressure, but without affecting vascular remodelling. Monotherapy with infigratinib blunted the vascular remodelling process. The combination of sildenafil and infigratinib effectively improved haemodynamics and decreased vascular remodelling, in this model of PAH.
AUTHOR CONTRIBUTIONS
N.S.F., P.R.M.R., and P.L.S. planned the study. N.S.F., L.M., and P.L.S. designed the experiments. N.S.F., L.M., C.L.B., and C.S.S. performed the experiments. N.S.F., C.L.B., J.S.S., J.B.V., F.F.C., and N.N.R. analysed the data. N.S.F. and P.L.S. wrote the manuscript with critical input provided by G.Z.S. and P.R.M.R.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1.
Percent change in parameters of interest compared to PAH‐SAL group
ACKNOWLEDGEMENTS
The authors express our gratitude to Andre Benedito da Silva, B.Sc., Laboratory of Pulmonary Investigation, Carlos Chagas Filho Institute of Biophysics, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil, for animal care; Arlete Fernandes, B.Sc., Laboratory of Pulmonary Investigation, Carlos Chagas Filho Biophysics Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil, for her help with microscopy; Maíra Rezende Lima, M.Sc., Laboratory of Pulmonary Investigation, Carlos Chagas Filho Biophysics Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil, for her assistance in molecular biology analysis; and Moira Elizabeth Schottler, B.A. and Filippe Vasconcellos, B.A., for their assistance in editing the manuscript.
Felix NS, de Mendonça L, Braga CL, et al. Effects of the FGF receptor‐1 inhibitor, infigratinib, with or without sildenafil, in experimental pulmonary arterial hypertension. Br J Pharmacol. 2019;176:4462–4473. 10.1111/bph.14807
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benisty, J. I. , McLaughlin, V. V. , Landzberg, M. J. , Rich, J. D. , Newburger, J. W. , Rich, S. , & Folkman, J. (2004). Elevated basic fibroblast growth factor levels in patients with pulmonary arterial hypertension. Chest, 126(4), 1255–1261. 10.1378/chest.126.4.1255 [DOI] [PubMed] [Google Scholar]
- Biernacka, A. , Dobaczewski, M. , & Frangogiannis, N. G. (2011). TGF‐β signaling in fibrosis. Growth Factors, 29(5), 196–202. 10.3109/08977194.2011.595714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary, K. R. , Deng, Y. , Suen, C. M. , Taha, M. , Petersen, T. H. , Mei, S. H. J. , & Stewart, D. J. (2018). Efficacy of treprostinil in the SU5416‐hypoxia model of severe pulmonary arterial hypertension: Haemodynamic benefits are not associated with improvements in arterial remodelling. British Journal of Pharmacology, 175(20), 3976–3989. 10.1111/bph.14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie, N. J. , Crossno, J. T. Jr. , Frid, M. G. , Hofmeister, S. E. , Reeves, J. T. , Hyde, D. M. , … Stenmark, K. R. (2004). Hypoxia‐induced pulmonary artery adventitial remodeling and neovascularization: Contribution of progenitor cells. American journal of physiology. Lung Cellular and Molecular Physiology, 286(4), L668–L678. 10.1152/ajplung.00108.2003 [DOI] [PubMed] [Google Scholar]
- Galie, N. , Ghofrani, H. A. , Torbicki, A. , Barst, R. J. , Rubin, L. J. , Badesch, D. , … for the Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group (2005). Sildenafil citrate therapy for pulmonary arterial hypertension. The New England Journal of Medicine, 353(20), 2148–2157. 10.1056/NEJMoa050010 [DOI] [PubMed] [Google Scholar]
- Gall, H. , Felix, J. F. , Schneck, F. K. , Milger, K. , Sommer, N. , Voswinckel, R. , … Ghofrani, H. A. (2017). The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. The Journal of Heart and Lung Transplantation: The Official Publication of the International Society for Heart Transplantation, 36(9), 957–967. 10.1016/j.healun.2017.02.016 [DOI] [PubMed] [Google Scholar]
- Gambaryan, N. , Perros, F. , Montani, D. , Cohen‐Kaminsky, S. , Mazmanian, G. M. , & Humbert, M. (2010). Imatinib inhibits bone marrow‐derived c‐Kit+ cell mobilisation in hypoxic pulmonary hypertension. The European Respiratory Journal, 36(5), 1209–1211. 10.1183/09031936.00052210 [DOI] [PubMed] [Google Scholar]
- Gambaryan, N. , Perros, F. , Montani, D. , Cohen‐Kaminsky, S. , Mazmanian, M. , Renaud, J. F. , … Humbert, M. (2011). Targeting of c‐Kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. The European Respiratory Journal, 37(6), 1392–1399. 10.1183/09031936.00045710 [DOI] [PubMed] [Google Scholar]
- Godinas, L. , Guignabert, C. , Seferian, A. , Perros, F. , Bergot, E. , Sibille, Y. , … Montani, D. (2013). Tyrosine kinase inhibitors in pulmonary arterial hypertension: A double‐edge sword? Seminars in Respiratory and Critical Care Medicine, 34(5), 714–724. 10.1055/s-0033-1356494 [DOI] [PubMed] [Google Scholar]
- Gomez‐Arroyo, J. , Saleem, S. J. , Mizuno, S. , Syed, A. A. , Bogaard, H. J. , Abbate, A. , … Voelkel, N. F. (2012). A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. American journal of physiology. Lung Cellular and Molecular Physiology, 302(10), L977–L991. 10.1152/ajplung.00362.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guagnano, V. , Furet, P. , Spanka, C. , Bordas, V. , Le Douget, M. , Stamm, C. , … Graus Porta, D. (2011). Discovery of 3‐(2,6‐dichloro‐3,5‐dimethoxy‐phenyl)‐1‐{6‐[4‐(4‐ethyl‐piperazin‐1‐yl)‐phenylamin o]‐pyrimidin‐4‐yl}‐1‐methyl‐urea (NVP‐BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. Journal of Medicinal Chemistry, 54(20), 7066–7083. 10.1021/jm2006222 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1091), D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemnes, A. R. , & Humbert, M. (2017). Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. European Respiratory Review: An Official Journal of the European Respiratory Society, 26(146), pii: 170093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, N. S. , Gillespie, M. N. , & McMurtry, I. F. (2017). Fifty years of monocrotaline‐induced pulmonary hypertension: What has it meant to the field? Chest, 152(6), 1106–1108. 10.1016/j.chest.2017.10.007 [DOI] [PubMed] [Google Scholar]
- Humbert, M. , Morrell, N. W. , Archer, S. L. , Stenmark, K. R. , MacLean, M. R. , Lang, I. M. , … Rabinovitch, M. (2004). Cellular and molecular pathobiology of pulmonary arterial hypertension. Journal of the American College of Cardiology, 43(12 Suppl S), 13S–24S. [DOI] [PubMed] [Google Scholar]
- Humbert, M. , & Weatherald, J. (2018). Right heart catheterisation is still a fundamental part of the follow‐up assessment of pulmonary arterial hypertension. The European Respiratory Journal, 52(1), pii: 1800738. https://doi.org/10.1183/13993003.00738‐2018 [DOI] [PubMed] [Google Scholar]
- Izikki, M. , Guignabert, C. , Fadel, E. , Humbert, M. , Tu, L. , Zadigue, P. , … Eddahibi, S. (2009). Endothelial‐derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. The Journal of Clinical Investigation, 119(3), 512–523. 10.1172/JCI35070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javle, M. , Lowery, M. , Shroff, R. T. , Weiss, K. H. , Springfeld, C. , Borad, M. J. , … Bekaii‐Saab, T. (2018). Phase II study of BGJ398 in patients with FGFR‐altered advanced cholangiocarcinoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 36(3), 276–282. 10.1200/JCO.2017.75.5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. J. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biology, 8(6), e1000412 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger, J. R. , Elliott, G. , Levine, D. J. , Bossone, E. , Duvall, L. , Fagan, K. , … Badesch, D. B. (2019). Therapy for pulmonary arterial hypertension in adults 2018: Update of the CHEST guideline and expert panel report. Chest, 155, 565–586. 10.1016/j.chest.2018.11.030 [DOI] [PubMed] [Google Scholar]
- Lang, R. M. , Badano, L. P. , Mor‐Avi, V. , Afilalo, J. , Armstrong, A. , Ernande, L. , … Voigt, J. U. (2015). Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography: Official Publication of the American Society of Echocardiography, 28(1), 1–39 e14. 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- Li, B. , He, W. , Ye, L. , Zhu, Y. , Tian, Y. , Chen, L. , … Hao, K. (2019). Targeted delivery of sildenafil for inhibiting pulmonary vascular remodeling. Hypertension, 73(3), 703–711. 10.1161/HYPERTENSIONAHA.118.11932 [DOI] [PubMed] [Google Scholar]
- de Mendonca, L. , Felix, N. S. , Blanco, N. G. , Da Silva, J. S. , Ferreira, T. P. , Abreu, S. C. , … Silva, P. L. (2017). Mesenchymal stromal cell therapy reduces lung inflammation and vascular remodeling and improves hemodynamics in experimental pulmonary arterial hypertension. Stem Cell Research & Therapy, 8(1), 220 10.1186/s13287-017-0669-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet, N. , Nsaibia, M. J. , Nguyen, Q. T. , Shi, Y. F. , Letourneau, M. , Fournier, A. , … Dupuis, J. (2019). PulmoBind imaging measures reduction of vascular adrenomedullin receptor activity with lack of effect of sildenafil in pulmonary hypertension. Scientific Reports, 9(1), 6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi, M. , Froum, S. , Hamby, J. M. , Schroeder, M. C. , Panek, R. L. , Lu, G. H. , … Hubbard, S. R. (1998). Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. The EMBO Journal, 17(20), 5896–5904. 10.1093/emboj/17.20.5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogova, L. , Sequist, L. V. , Perez Garcia, J. M. , Andre, F. , Delord, J. P. , Hidalgo, M. , … Wolf, J. (2017). Evaluation of BGJ398, a fibroblast growth factor receptor 1‐3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase I, dose‐escalation and dose‐expansion study. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 35(2), 157–165. 10.1200/JCO.2016.67.2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamatheakis, D. G. , Mocumbi, A. O. , Kim, N. H. , & Mandel, J. (2014). Schistosomiasis‐associated pulmonary hypertension. Pulmonary Circulation, 4(4), 596–611. 10.1086/678507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch, M. , Guignabert, C. , Humbert, M. , & Nicolls, M. R. (2014). Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circulation Research, 115(1), 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani, G. V. , & Park, M. H. (2010). Update on the clinical utility of sildenafil in the treatment of pulmonary arterial hypertension. Drug Design, Development and Therapy, 4, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermuly, R. T. , Kreisselmeier, K. P. , Ghofrani, H. A. , Yilmaz, H. , Butrous, G. , Ermert, L. , … Grimminger, F. (2004). Chronic sildenafil treatment inhibits monocrotaline‐induced pulmonary hypertension in rats. American Journal of Respiratory and Critical Care Medicine, 169(1), 39–45. [DOI] [PubMed] [Google Scholar]
- Schmittgen, T. D. , & Livak, K. J. (2008). Analyzing real‐time PCR data by the comparative C(T) method. Nature Protocols, 3(6), 1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Seluanov, A. , Vaidya, A. , & Gorbunova, V. (2010). Establishing primary adult fibroblast cultures from rodents. Journal of Visualized Experiments: JoVE, (44). 10.3791/2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde, A. V. , Humeres, C. , & Frangogiannis, N. G. (2017). The role of α‐smooth muscle actin in fibroblast‐mediated matrix contraction and remodeling. Biochimica et biophysica acta. Molecular Basis of Disease, 1863(1), 298–309. 10.1016/j.bbadis.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault, H. B. , Kurtz, B. , Raher, M. J. , Shaik, R. S. , Waxman, A. , Derumeaux, G. , … Scherrer‐Crosbie, M. (2010). Noninvasive assessment of murine pulmonary arterial pressure: Validation and application to models of pulmonary hypertension. Circulation. Cardiovascular Imaging, 3(2), 157–163. 10.1161/CIRCIMAGING.109.887109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, P. T. , Lee, R. A. , & Wu, H. (2003). BMP4 acts upstream of FGF in modulating thymic stroma and regulating thymopoiesis. Blood, 102(12), 3947–3953. 10.1182/blood-2003-05-1657 [DOI] [PubMed] [Google Scholar]
- Tu, L. , Dewachter, L. , Gore, B. , Fadel, E. , Dartevelle, P. , Simonneau, G. , … Guignabert, C. (2011). Autocrine fibroblast growth factor‐2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. American Journal of Respiratory Cell and Molecular Biology, 45(2), 311–322. 10.1165/rcmb.2010-0317OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder, R. M. (2017). Pulmonary vascular remodeling in pulmonary hypertension. Cell and Tissue Research, 367(3), 643–649. 10.1007/s00441-016-2539-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, R. , Su, Y. , Feng, T. , Sun, M. , Liu, B. , Zhang, J. , … Hu, Q. (2017). Monocrotaline induces endothelial injury and pulmonary hypertension by targeting the extracellular calcium‐sensing receptor. Journal of the American Heart Association, 6(4), pii: e004865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigitaslan, S. , & Sirmagul, B. (2012). Relation of bosentan, iloprost, and sildenafil with growth factor levels in monocrotaline‐induced pulmonary hypertension. Clinical and Experimental Hypertension, 34(3), 222–229. 10.3109/10641963.2011.631652 [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Chang, Z. , Zhao, F. , Zhang, P. , Hao, Y. J. , Yan, L. , … Zhou, R. (2019). Protective effects of 18β‐glycyrrhetinic acid on monocrotaline‐induced pulmonary arterial hypertension in rats. Frontiers in Pharmacology, 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Y. , Ma, H. , Hu, E. , Huang, Z. , Cheng, X. , & Xiong, C. (2015). Inhibition of FGFR signaling with PD173074 ameliorates monocrotaline‐induced pulmonary arterial hypertension and rescues BMPR‐II expression. Journal of Cardiovascular Pharmacology, 66(5), 504–514. 10.1097/FJC.0000000000000302 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Percent change in parameters of interest compared to PAH‐SAL group