

Abstract
Background and Purpose
The concept of opioid ligands biased towards the G protein pathway with minimal recruitment of β‐arrestin‐2 is a promising approach for the development of novel, efficient, and potentially nonaddictive opioid therapeutics. A recently discovered biased μ‐opioid receptor agonist, PZM21, showed analgesic effects with reduced side effects. Here, we aimed to further investigate the behavioural and biochemical properties of PZM21.
Experiment Approach
We evaluated antinociceptive effects of systemic and intrathecal PZM21 administration. Its addiction‐like properties were determined using several behavioural approaches: conditioned place preference, locomotor sensitization, precipitated withdrawal, and self‐administration. Also, effects of PZM21 on morphine‐induced antinociception, tolerance, and reward were assessed. Effects of PZM21 on striatal release of monoamines were evaluated using brain microdialysis.
Key Results
PZM21 caused long‐lasting dose‐dependent antinociception. It did not induce reward‐ and reinforcement‐related behaviour; however, its repeated administration led to antinociceptive tolerance and naloxone‐precipitated withdrawal symptoms. Pretreatment with PZM21 enhanced morphine‐induced antinociception and attenuated the expression of morphine reward. In comparison to morphine, PZM21 administration induced a moderate release of dopamine and a robust release of 5‐HT in the striatum.
Conclusions and Implications
PZM21 exhibited antinociceptive efficacy, without rewarding or reinforcing properties. However, its clinical application may be restricted, as it induces tolerance and withdrawal symptoms. Notably, its ability to diminish morphine reward implies that PZM21 may be useful in treatment of opioid use disorders.
What is already known
PZM21 is a new opioid analgesic efficacious for the affective component of pain.
PZM21 exhibited reduced side effects and diminished rewarding activity.
What this study adds
PZM21 induces reflexive analgesia also at the spinal level and causes tolerance and withdrawal symptoms.
PZM21 does not induce reward and craving upon drug abstinence but suppresses morphine reward.
What is the clinical significance
Our data suggest that PZM21 may be a promising treatment for opioid use disorder.
Abbreviations
- CPP
conditioned place preference
- DAMGO
[d‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin
- MPE
maximum possible effect
1. INTRODUCTION
Although opioid analgesics are usually the first choice and most effective treatments for pain, numerous side effects, including a strong addictive potential, severely limit their clinical effectiveness (Webster et al., 2011). Substantial evidence indicates that opioid‐induced analgesia and adverse effects are processed by distinct cell signalling pathways. Analgesia is promoted by G protein signalling, whereas a range of undesirable effects are mediated through the regulatory protein β‐arrestin‐2 (Bohn et al., 1999; Raehal, Walker, & Bohn, 2005). Therefore, there is a growing interest in pharmacological approaches that allow separation of analgesia from opioid side effects by developing biased (functionally selective) ligands that preferentially activate G protein signalling with minimal engagement of the β‐arrestin‐2 signalling pathway (Brust et al., 2016; Chen et al., 2013; DeWire et al., 2013; Groer et al., 2007; Maillet et al., 2015; Manglik et al., 2016). The majority of the research has addressed the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 as a target for action of biased ligands, since μ‐receptor agonists are the most effective analgesics (Madariaga‐Mazón et al., 2017). However, the μ receptors also represent a key molecular trigger for reward processing and contribute to the development of addictive behaviour (Contet, Kieffer, & Befort, 2004). Thus, in addition to the exclusion of somatic side effects associated with opioid use, the main challenge for μ receptor ligands, biased towards G protein signalling, is reduction of opioid‐induced reinforcement. At present, there is no μ‐receptor agonist devoid of rewarding potential.
A novel, recently discovered G protein‐biased opioid analgesic, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9286, was described as a potent, selective μ receptor agonist and was reported to inhibit the emotional reaction to thermal nociceptive stimuli (named “affective analgesia”) with reduced http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627‐like side effects and addictive potential in mice (Manglik et al., 2016). In the present study, we aimed to further investigate behavioural and biochemical properties of PZM21 as well as possible mechanisms underlying its action and distinct effects on antinociception and addiction‐like behaviour. We show that acute treatment with PZM21 results in long‐lasting dose‐dependent antinociception that is mediated by the μ‐receptor, but repeated administration of this compound causes the development of antinociceptive tolerance and expression of withdrawal symptoms upon naloxone administration. Furthermore, PZM21 is devoid of opioid‐like reinforcing properties. However, its action is accompanied by slight, but dose‐dependent release of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=940 in the striatum. Interestingly, PZM21 induces a robust increase in striatal extracellular levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5. Furthermore, we demonstrated that pretreatment with PZM21 may influence behavioural responses to morphine in mice and notably, is able to diminish opioid reward.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the European Union regulations and the Directive 2010/63/EU and were approved by the II Local Bioethics Commission (permit numbers: 1213/2015, 1305/2016, 66/2017, 84/2018; Krakow, Poland). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The total number of animals as well as their suffering was minimized, according to the 3R principle. C57BL/6J mice and Sprague–Dawley rats were acquired from the Maj Institute of Pharmacology PAS breeding facility (Krakow, Poland); Wistar rats were obtained from Charles River (Hamburg, Germany). All animals were group housed, mice eight to 10 per cage (265 × 180 × 420 mm, Ehret Labor‐ und Pharmatechnik GMBH & Co.KG, Germany) and rats five per cage (380 × 200 × 590 mm, Ehret Labor‐ und Pharmatechnik GMBH & Co.KG, Germany) with aspen litter (MIDI LTE E‐002 Abedd, AnimaLab, Poland), under standard room temperature 22 ± 2°C, humidity 50 ± 5%, and 12/12 hr light–dark cycle, with free access to food and water (standard diet, Special Diets Services, England) and environmental enrichment. All behavioural tests were performed on adult male C57BL/6J mice (25–30 g, 8 weeks old at the beginning of the experiments, https://scicrunch.org/resources/Any/search?q=IMSR_JAX%3A000664&l=IMSR_JAX%3A000664), apart from experiments with intrathecal drug delivery and intravenous self‐administration, which were performed on Wistar https://scicrunch.org/resources/Any/search?q=RGD_13508588&l=RGD_13508588 and male Sprague–Dawley https://scicrunch.org/resources/Any/search?q=RGD_70508&l=RGD_70508 rats (280–350 g, 10 weeks old at the beginning of the experiments), respectively. Experiments were carried out on male rodents to compare our results with previously published data (Hill et al., 2018; Manglik et al., 2016) and to avoid the possible influence of the oestrous cycle and reproductive status of female rodents on the results obtained.
Animals were randomly assigned to treatment groups, and the experimenter was blinded to drug treatment until after data analysis has been performed. The n values in the experiments were chosen based on our previous experience with similar experimental protocols. The exact numbers of animals in each group used in the experiments are listed in Table S5 (n in each group >5). The criteria for excluding animals from experiments and statistical analysis in the present study included abnormal basal response in the tail flick test (higher than 6 s indicating attenuated pain sensitivity) and technical issues (catheter obstruction and equipment malfunction).
2.2. Intrathecal catheter implantation and drug delivery
The intrathecal drug administrations were achieved through implanting catheters according to the method described by Yaksh and Rudy (1976) under pentobarbital (60 mg·kg−1) anaesthesia, as reported previously (Rojewska et al., 2014). Briefly, a polyethylene catheter (PE 10, Intramedic, Clay Adams, Becton Dickinson and Company, Rutherford, USA) was sterilized by flushing with 70% ethanol and then sterile water prior to insertion. Rats were placed on a on a stereotaxic table (David Kopf Instruments, USA), and an incision was made in the atlantooccipital membrane. Then the catheter was carefully introduced through the atlantooccipital membrane to the subarachnoid space at the rostral level of the spinal cord lumbar enlargement (L4–L6), flushed with 10 μl of sterile water, and tightened with the tip. After implantation, the animals were allowed to recover for a minimum of 7 days and received enrofloxacin (KRKA, Slovenia) 0.1 ml s.c. per rat once daily for 2 days. The intrathecal injections were performed using a 50‐μl Hamilton syringe with a 30 1/2‐gauge needle; 5 μl was injected per animal, followed by 10 μl of saline.
2.3. Antinociception assessment: Tail flick
Tail flick was performed using a tail flick apparatus (Ugo Basile, Italy), and a light beam was used as a thermal nociceptive stimulus. The light beam was applied to the dorsal side of animals' tail, and the time latency to tail withdrawal or shaking was recorded. To avoid tissue damage, a cut‐off latency was set at 9 s. Responses were expressed as a percentage of the maximum possible effect (% MPE), calculated according to the formula: [(T 1 − T 0)/(T 2 − T 0)] × 100, where T 0 and T 1 are the tail flick latencies before and after drug injection, respectively, and T2 is the cut‐off time. To study for the influence of PZM21 on morphine‐induced antinociception, the compound was administered 30 min prior to morphine, and tail flick test was performed as described above.
2.4. Hot plate
The hot plate test was conducted using a hot plate analgesia meter (COTM, Poland). The mice were placed on the plate, which was preheated to 52.5°C, and the time latency to the first sign of spinally mediated withdrawal reflexes (later described as a paw flinching) was measured. Moreover, we have measured the time latency to the first sign of complex behaviour (including licking/biting of the paw and/or jumping). A maximum exposure time was set at 30 s (cut‐off) to avoid tissue damage. Both types of responses were expressed as % MPE.
2.5. Tolerance to thermal antinociception
To assess the development of tolerance to thermal antinociception induced by treatment with morphine or PZM21, the animals received injections with the drugs for seven consecutive days and were tested in the tail flick assay 1 hr after drug administration. To study for the influence of PZM21 on the development of tolerance to morphine antinociception, mice were pretreated with PZM21 30 min prior to morphine and then tail flick test was performed.
2.6. Conditioned place preference (CPP) test
The CPP procedure was conducted as previously described (Szklarczyk et al., 2012). Briefly, a CPP apparatus (Med Associates, USA) consisted of three different compartments. The CPP procedure began on Day 0 with 5 min of acclimatization to the apparatus. On Day 1 (preconditioning test), mice were allowed to freely explore the whole apparatus for 20 min, and time spent in each compartment was measured. The procedure was unbiased, so that no significant differences in compartment preference were found within each group during preconditioning test. During the conditioning days (Days 2–11), mice were injected with morphine or PZM21 (Days 3, 5, 7, 9, and 11) or saline (Days 2, 4, 6, 8, and 10) and immediately placed in the respective compartment for 60 min. To study the effects of PZM21 on morphine‐induced CPP, separate groups of mice were subjected to the procedure, and 30 min prior to the morphine injection, they received a pretreatment with PZM21. The postconditioning test was performed on Day 12 and was the same as the preconditioning one. The difference between the times spent in the drug‐ and vehicle‐paired compartments during the postconditioning session was considered to be a measure of CPP (CPP score).
2.7. Locomotor sensitization
The measurement of locomotor sensitization lasted for 6 days and was performed using custom‐made activity chambers. Each day, all animals were first injected with saline, placed in the chambers for 2 hr, received the appropriate injection (saline, morphine or PZM21), and were placed back in the boxes for an additional 2 hr. To study for the influence of PZM21 on sensitization to morphine‐induced hyperlocomotion, separate groups of mice received PZM21 injections 30 min prior to morphine. The expression of sensitization was tested after 8 days‐incubation period. All animals were habituated to the chambers for 2 hr, 1 day before the onset of the experiments.
2.8. Naloxone‐precipitated withdrawal
For https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638‐precipitated withdrawal mice were chronically (at 8:00 and 16:00 for five consecutive days) treated with saline, morphine or PZM21, as described by Abdel‐Zaher, Hamdy, Aly, Abdel‐Hady, and Abdel‐Rahman (2006). On the sixth day, 3 hr after the final saline or drug administration, all animals received 4 mg·kg−1 naloxone and immediately after the injection each animal was placed in a transparent acryl cylinder (20 cm in diameter, 50 cm in height) for 15 min to observe jumps, which were considered as a manifestation of withdrawal.
2.9. Intravenous drug self‐administration
A self‐administration procedure was performed as previously described (Solecki et al., 2013). Briefly, rats were anaesthetized with ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1) and implanted with a silastic catheter in the external jugular vein. For catheter implantation, a guide cannula (C313G, Plastics One Inc., USA), attached to silastic tubing (0.025 inner diameter, Bio‐Sil, Bio‐Rad, USA) and Marlex mesh via dental cement, was inserted subcutaneously between the shoulder blades and exited the skin via a dermal biopsy hole (3 mm). The other end of the tubing was threaded under the skin, inserted 3 cm into the right jugular vein, and then sutured securely to the underlying muscle tissue. After the catheter implantation, rats were allowed to recover for 7 days, during which the catheters were flushed with 0.3 ml of saline and 0.2 ml of heparin solution (Braun, Germany) in order to prevent occlusion. All animals were given an anti‐inflammatory and analgesic drug (tolfedine 4%, 1 ml·kg−1, i.p. ;Vetoquinol Biowet, Poland) and 5 ml of glucose to prevent dehydration during post‐surgery recovery. For the first 3 days after the operations, animals were treated with antibiotics added to the drinking water (Sul‐Tridin 24%, Biowet‐Pulawy, Poland). Self‐administration training was preceded by 2–3 days of food restriction to ~90% of free feeding levels. Rats were trained under a fixed ratio 1 schedule of reinforcement during which each active lever press led to intravenous drug infusion and conditioned stimulus (CS) cue presentation (tone + stimulus light for 6 s) in standard operant chambers (Med Associates, USA). Each active lever response was followed by a 20‐s time out during which lever pressing had no programmed consequences. Similarly, inactive lever presses had no programmed consequences. Each rat underwent 2 hr daily training sessions for 10 consecutive days.
2.10. Drug seeking under extinction conditions
Drug seeking under extinction conditions was performed as previously described (Solecki et al., 2013, 2018). After drug self‐administration training, rats underwent 3 days of forced abstinence in their home cages without access to drug or drug‐associated contextual and discrete cues. This forced withdrawal period did not include extinction to better model the medical detoxification experienced by many people with substance use disorders that occurs without behavioural extinction training. On Withdrawal Day 3, animals were placed in operant chambers for 2 hr, and active lever presses led to the discrete CS presentation (i.e., 6 s tone and light) with no drug delivery. A 20‐s timeout followed the CS termination, during which time responses were recorded but had no programmed consequences. Inactive lever presses had no programmed consequences. In such testing settings, drug seeking (i.e., active lever responding) was driven by both contextual cues and discrete CS presentation contingent upon active lever presses.
2.11. Brain microdialysis and analytical procedure
Mice were anaesthetized with ketamine (7.5 mg·kg−1) and xylazine (1 mg·kg−1), and a vertical microdialysis probe was implanted into the striatum using the coordinates (from Bregma): AP +1.0, L +1.8, and V −3.8. On the next day, the probe inlets were connected to a syringe pump (BAS, IN, USA), which delivered aCSF composed of the following (mM): NaCl 147, KCl 2.7, MgCl2 1.0, CaCl2 1.2; pH 7.4 at a flow rate of 1.5 μl·min−1. After 1 hr of the washout period, three basal dialysate samples were collected every 20 min, the animals were injected with the appropriate drugs as indicated in figure captions, and fraction collection continued for 240 min. At the end of the experiment, the mice were killed by decapitation under isoflurane anaesthesia; brains were removed and histologically examined to validate the probe placement. The dopamine and 5‐HT content of the dialysate fractions was analysed by HPLC with coulochemical detection. Chromatography was performed using an Ultimate 3000 System (Dionex, USA) and a coulochemical detector, Coulochem III (model 5300, ESA, USA), with a 5020 guard cell, 5014B microdialysis cell, and Hypersil Gold‐C18 analytical column (3 × 100 mm). The mobile phase was composed of 0.1‐M potassium phosphate buffer adjusted to pH 3.6, 0.5‐mM EDTA, 16 mg·L−1 1‐octanesulfonic acid sodium salt, and 2% methanol. The flow rate during analysis was set at 0.7 ml·min−1. The applied potential of the guard cell was +600 mV, while those of the microdialysis cells were as follows: E1 = −50 mV and E2 = +300 mV with a sensitivity set at 50 nA·V−1. The chromatographic data were processed by Chromeleon v. 6.80 (Dionex, USA) software and run on a personal computer. All obtained microdialysis data were presented as a percent of the basal level assumed to be 100% to allow comparison of the magnitude of dopamine and 5‐HT release.
2.12. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). For statistical analysis, GraphPad Prism 7.0 (GraphPad Prism Software Inc., USA, https://scicrunch.org/resources/Any/search?q=SCR_002798&l=SCR_002798 and Statistica (12.5, Stat‐Soft, Poland, https://scicrunch.org/resources/Any/source/nlx_144509-1/search?q=SCR_014213&l=SCR_014213 were used. Group sizes listed in the Table S5 present the number of independent samples/animals. Statistical analyses of behavioural data were performed using unpaired Student's t test (μ receptor antagonism in hot plate test), one‐way ANOVA (CPP experiments, antinociception in hot plate test, and naloxone‐precipitated withdrawal), two‐way ANOVA (drug seeking in self‐administration experiments), two‐way repeated measures ANOVA (antinociception and μ receptor antagonism in tail flick test, antinociceptive tolerance, locomotor sensitization test, and microdialysis experiments), or three‐way repeated measures ANOVA (drug self‐administration procedure) followed by Bonferroni post hoc tests where appropriate (performed only when F achieved P < .05 and there was no significant variance inhomogeneity). Data are presented on graphs as the mean ± SEM. Statistical significance was set at P < 0.05.
2.13. Materials
PZM21 was synthesized according to the previously published procedure (Manglik et al., 2016). A detailed synthesis procedure as well as data confirming high enantiomeric purity of the synthesised compound are included in Figures S1 and S2 and Tables S1 and S2. PZM21 displayed high affinity for μ‐receptors, similar to that of the prototypic μ‐receptor ligand https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1647 (Figure S3, Table S3), which is in accordance with previously described results regarding the μ‐ receptor‐mediated G protein activity of the compound (Manglik et al., 2016). The δ‐opioid receptor affinity of PZM21 was significantly lower than its affinity for the μ‐receptor (Figure S3, Table S3).
PZM21 was administered to mice at doses of 20, 40, or 80 mg·kg−1, depending on the experimental schedule. For intrathecal drug delivery, PZM21 was administered at doses of 2.5, 5, and 7.5 μg, and for intravenous drug self‐administration, PZM21 was used at doses of 0.05 and 0.5 mg·kg−1 (per infusion). Morphine (Pharma Cosmetic, Poland; 5, 10, and 20 mg·kg−1) or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7093 (Mundipharma, Poland; 0.06 mg·kg−1 [per infusion]) was used as positive control treatments. As a negative control, physiological saline was used. Cyprodime (Tocris, USA; 10 mg·kg−1) and naloxone (Merck, Poland; 4 mg·kg−1) were used as a selective μ‐receptor antagonist and non‐selective opioid antagonist, respectively. All the drugs were dissolved in saline and administered intraperitoneally in a volume of 10 ml·kg−1, intrathecally in a volume of 5 μl per administration or intravenously in a volume of 0.0125 ml per infusion. Drugs used for anaesthesia (ketamine [7.5 mg·kg−1 in mice and 100 mg·kg−1 in rats], xylazine [1 mg·kg−1 in mice and 10 mg·kg−1 in rats], and pentobarbital [60 mg·kg−1 in rats]) were supplied by Biowet‐Pulawy (Poland). The chemicals used for HPLC were purchased from Merck (Poland).
2.14. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. Antinociceptive effects of PZM21
Antinociceptive efficacy of PZM21 was assessed using two thermal antinociceptive tests: tail flick (mice and rats) and hot plate (mice). Administration of 10 mg·kg−1 morphine (i.p.), used as a positive control, resulted in antinociception (Figure 1a). Treatment with PZM21 (intraperitoneal) exerted an antinociceptive effect in the tail flick test at all of the tested doses: 20, 40, and 80 mg·kg−1 (Figure 1b), compared to saline. PZM21‐induced antinociception was dose‐dependent and lasted up to 8 hr. Pretreatment with a selective μ receptor antagonist, cyprodime (10 mg·kg−1, i.p.), prevented the antinociception induced by 40 mg·kg−1 PZM21 in the tail flick test (Figure 1c). In the hot plate test, we have distinguished two types of reaction: paw flinching and licking of the paw or jumping. The obtained data indicate that only morphine (10 mg·kg−1) induced a significant effect on the paw flinching reflex when compared to saline. PZM21 did not affect this reaction at any of the tested doses. At the same time, both PZM21 (at a dose of 80 mg·kg−1) and morphine (10 mg·kg−1) increased the latency to the second type of reaction (paw licking/jumping) in the hot plate test (Figure 1d). Pretreatment with cyprodime attenuated the effects induced by 40 mg·kg−1 PZM21 in hot plate test, for two types of reactions (Figure 1e). Therefore, we demonstrated that antinociceptive effects of PZM21 are mediated by μ‐receptors. What is more, intrathecal administration of PZM21 (2.5, 5, and 7.5 μg) caused dose‐dependent antinociception in tail flick test in rats (Figure 1f), which shows that this compound is effective for the reflexive, spinally mediated component of pain reaction.
Figure 1.
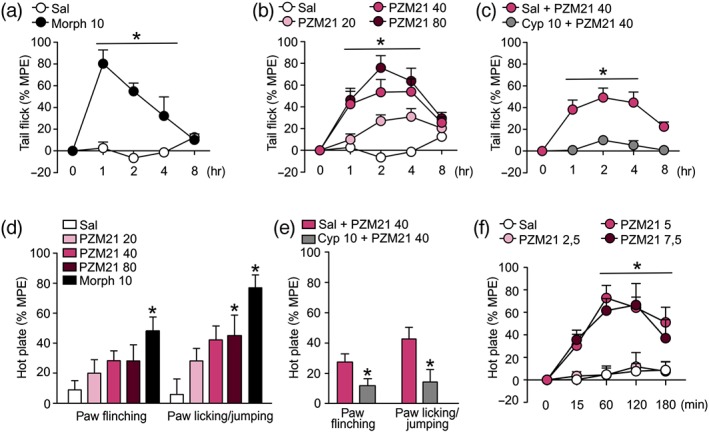
Effects of PZM21 on acute thermal antinociception. (a) Administration of morphine (10 mg·kg−1, i.p.), used as a positive control, resulted in an attenuated sensitivity to painful stimulus in the tail flick test (b) Treatment with PZM21 (20, 40, and 80 mg·kg−1, i.p.) caused dose‐dependent antinociceptive effect measured in the tail flick test. When compared to saline, the antinociceptive effect of 20 mg·kg−1 of PZM21 was statistically significant 2 and 4 hr after the drug administration, while treatment with doses of 40 and 80 mg·kg−1 of the compound induced antinociception that lasted from 1 to 4 hr after the treatment. Morphine and PZM21 groups are compared to the same saline controls. (c) A selective μ‐receptor antagonist, cyprodime (10 mg·kg−1, i.p.), administered 15 min prior to PZM21 (40 mg·kg−1), prevented antinociception in the tail flick test. (d) PZM21 had no effect on the paw flinching reaction in the hot plate test. However, at a dose of 80 mg·kg−1, it increased the latency to paw licking/jumping behaviour. Treatment with morphine significantly attenuated both types of reactions. Both responses were measured 90 min after drug administration. (e) Pretreatment with cyprodime attenuated the effects of 40 mg·kg−1 of PZM21 on both types of reaction in the hot plate test. * P < .05, significant effect of cyprodime. (f) Intrathecal administration of PZM21 (at doses of 5 and 7.5 μg) caused antinociceptive effects in the tail flick test in rats. Data are presented as the mean ± SEM. * P < .05, significant effect of treatment compared with appropriate controls. Numbers of animals used in experiments presented in Table S5. Cyp, cyprodime; MPE, maximum possible effect; Sal, saline
3.2. Influence of PZM21 on addiction‐like behaviour in mice
The influence of PZM21 on addiction‐like behaviour in mice was investigated in CPP and locomotor sensitization tests. Moreover, we measured the development of antinociceptive tolerance and naloxone‐precipitated withdrawal symptoms after chronic PZM21 administration in order to assess its potential to cause physical dependence. Treatment with PZM21 did not induce CPP at any of the tested doses (20, 40, and 80 mg·kg−1, i.p.), whereas morphine‐treated animals (10 mg·kg−1, i.p.) developed a significant preference to the drug‐associated compartment (Figure 2a). Interestingly, the obtained data suggest that treatment with PZM21 at a dose of 80 mg·kg−1 led to a drug‐induced aversion, compared with the saline group (Figure 2a). Repeated 6‐day administration of PZM21 did not influence animals' locomotor activity at any of the doses, while treatment with morphine induced locomotor sensitization (Figure 2b). Morphine‐treated animals showed increased expression of sensitization measured after 8‐day incubation period, while only a slight increase in locomotor activity was observed in the group treated with 80 mg·kg−1 PZM21 (Figure 2b). On the other hand, chronic administration of 80 mg·kg−1 of PZM21 as well as 10 mg·kg−1 of morphine resulted in the occurrence of naloxone‐induced jumps, considered as physical signs of withdrawal (Figure 2c). Moreover, repeated treatment with both 40 mg·kg−1 and 80 mg·kg−1 PZM21 resulted in the development of antinociceptive tolerance measured by the tail flick test (Figure 2d). Therefore, our results indicate that PZM21 is devoid of opioid‐like rewarding properties, although it induces physical dependence.
Figure 2.
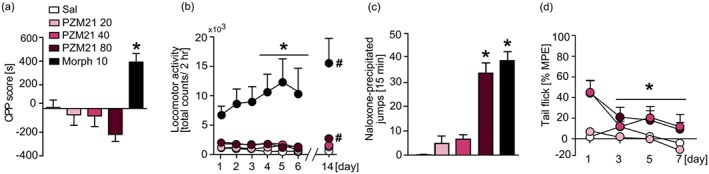
Influence of PZM21 on addiction‐like behaviour in mice. (a) In contrast to morphine (10 mg·kg−1, i.p.), PZM21 (20, 40, and 80 mg·kg−1, i.p.) did not induce a preference towards drug‐associated compartment in a CPP test at any of the tested doses. (b) Repeated treatment with morphine induced locomotor sensitization and expression, whereas that effect was not observed after PZM21 administration. Mice treated with 80 mg·kg−1 PZM21 presented a slight expression of sensitization after an 8‐day incubation period. (c) Chronic administration of PZM21 (80 mg·kg−1, but not 20 or 40 mg·kg−1) as well as morphine induced naloxone‐precipitated jumps, considered as a physical sign of withdrawal. (d) Repeated treatment with 40 and 80 mg·kg−1 PZM21 resulted in a decrease of antinociceptive efficacy of the compound. Tolerance was assessed using tail flick test performed on each experimental day, 1 hr after the drug administration. Data are presented as the mean ± SEM. In (a, c), * P<.05, PZM21‐ and morphine‐treated groups significantly different from saline controls; in (b, d) within group effects significantly different from the first day of experiment. # P < .05, expression of locomotor sensitization within groups significantly different from the last day of sensitization development. Numbers of animals used in experiments presented in Table S5. CPP, conditioned place preference; Morph, morphine; MPE, maximum possible effect; Sal, saline
3.3. Assessment of reinforcing properties of PZM21 in intravenous self‐administration paradigm in rats
PZM21, similar to saline, did not induce intravenous self‐administration during 10‐day training sessions in rats (Figure 3a–c), in contrast to robust oxycodone self‐administration, measured as a number of drug infusions (Figure 3a). Essentially, rats in the saline and PZM21 groups did not differentiate between active and inactive levers throughout training, whereas oxycodone self‐administering rats presented more responses on active in comparison to inactive lever, starting from Day 5 of training (Figure 3b,c). Finally, only rats that self‐administered oxycodone, but not saline or PZM21, demonstrated drug seeking behaviour, expressed as presses on previously active lever after 3 days of abstinence (Figure 3d). Thus, our data indicate that PZM21 does not act as a reinforcer and it does not induce craving upon drug abstinence.
Figure 3.
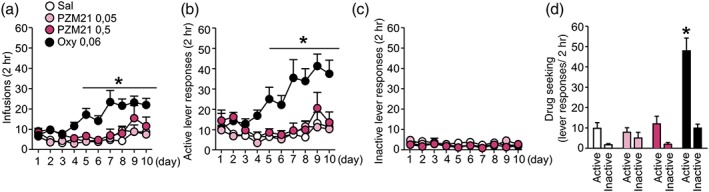
Evaluation of PZM21 effects on intravenous self‐administration in rats. (a) Rats that self‐administered oxycodone (0.06 mg·kg−1 per infusion, i.v.), but not PZM21 (0.05 and 0.5 mg·kg−1 per infusion, i.v.), presented an increasing number of infusions over time. (b) Only rats from the oxycodone group presented an increasing number of active lever responses. (c) No differences between groups were observed in inactive lever presses during self‐administration training. (d) Unlike the oxycodone group, rats in the saline and PZM21 groups did not present drug‐seeking behaviour after abstinence period, as they did not discriminate between active and inactive levers and made a similar number of responses on both levers. Data are presented as the mean ± SEM. In (a), * P < .05, within group effects significantly different from the first day of experiment; in (b–d), active and inactive lever responses significantly different within experimental groups. Numbers of animals used in experiments presented in Table S5. Oxy, oxycodone; Sal, saline
3.4. Increased monoamine release in the striatum in response to PZM21
Striatal dopamine and 5‐HT levels following drug administration were measured for 4 hr in freely moving mice. Both 40 and 80 mg·kg−1 (i.p.) PZM21 as well as 10 and 20 mg·kg−1 morphine (i.p.) markedly increased extracellular dopamine release (Figure 4a). The increase in dopamine release induced by 10 mg·kg−1 of morphine and 40 mg·kg−1 of PZM21 was comparable, whereas the effect of higher dose of PZM21 (80 mg·kg−1) was similar in magnitude to 20 mg·kg−1 of morphine. The action of both drugs on dopamine release is also presented as the total effect expressed as the AUC (Figure 4a). Moreover, administration of PZM21 at doses of 40 and 80 mg·kg−1 as well as morphine (10 and 20 mg·kg−1) caused an increase in 5‐HT extracellular levels. Treatment with 40 mg·kg−1 of PZM21 resulted in the extracellular elevation of striatal 5‐HT that was similar to 20 mg·kg−1 of morphine. However, higher dose of PZM21 (80 mg·kg−1) produced a robust release of 5‐HT, reaching above 350% of the basal level at the peak. PZM21 and morphine action on 5‐HT is also presented as the total effect expressed as the AUC in Figure 4b. An additional figure presenting microdialysis probe placement is provided in Figure S5.
Figure 4.
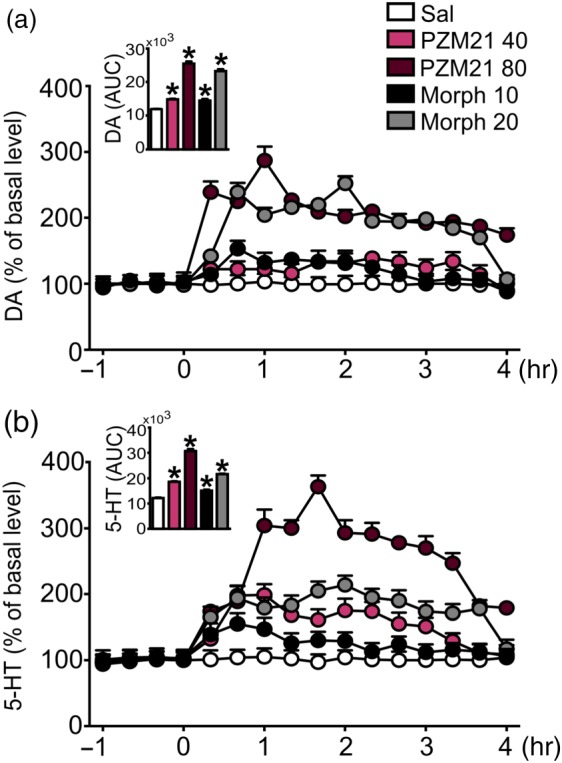
Effects of PZM21 on striatal dopamine and 5‐HT levels. (a) Administration of 40 and 80 mg·kg−1 PZM21 (i.p.) as well as 10 and 20 mg·kg−1 of morphine (i.p.) increased extracellular level of dopamine in the striatum. Basal extracellular levels were 3.71 ± 0.51 pg in a volume of 10 μl (n = 30). (b) All doses of PZM21 and morphine potentiated striatal 5‐HT release when compared to saline. Basal extracellular levels were 0.40 ± 0.06 pg in a volume of 10 μl (n = 30). Data are presented as the mean ± SEM. Bar graphs presenting cumulative data are expressed as AUC. * P < .05, significantly different from saline. Numbers of animals used in experiments presented in Table S5. DA, dopamine; Morph, morphine; Sal, saline
3.5. Consequences of pretreatment with PZM21 on morphine‐induced behaviour
We then investigated whether PZM21 may influence antinociceptive and addictive effects of morphine. For these experiments, we chose doses of 20 and 40 mg·kg−1 PZM21 (i.p.), as our previous data suggested a ceiling effect of PZM21 above the dose of 40 mg·kg−1 and these doses did not induce physical dependence or aversion. First, we determined the dose of morphine (5 mg·kg−1) that induced approximately 50% of MPE in the tail flick test and assessed how pretreatment with PZM21 will influence morphine‐induced antinociception. Our results show that the dose of 40 mg·kg−1 of PZM21 enhanced and prolonged antinociception evoked by morphine (Figure 5a). Next, we used a model of tolerance to morphine‐induced antinociception and assessed the effect of PZM21 under these conditions. The obtained data showed that pretreatment with PZM21 did not influence tolerance development during repeated administration of 10 mg·kg−1 of morphine (Figure 5b). To investigate whether PZM21 modulates addictive properties of morphine, we performed CPP and locomotor sensitization tests. The results obtained show that pre‐administration of 40 mg·kg−1, but not 20 mg·kg−1 of PZM21, prevented the formation of morphine‐induced CPP (Figure 5c). Pretreatment with PZM21 did not affect the development of morphine‐induced locomotor sensitization (Figure 5d). However, a slight tendency towards the reduction of morphine effects was observed. Moreover, PZM21 did not affect the expression of morphine sensitization after the 8‐day incubation period (Figure 5d). Taken together, our results show that PZM21 enhances antinociceptive effects of morphine and suppresses its rewarding properties.
Figure 5.
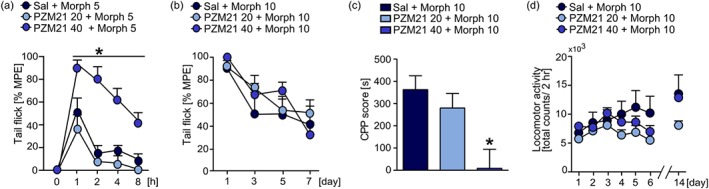
Influence of PZM21 on behavioural effects of morphine. (a) PZM21 (at dose of 40 mg·kg−1, i.p.), administered 30 min prior to morphine, enhanced antinociception evoked by 5 mg·kg−1 of morphine (i.p.) in the tail flick test. (b) Pretreatment with PZM21 had no effect on the development of tolerance to antinociception induced by 10 mg·kg−1 of morphine. Tolerance was assessed using tail flick test performed on each experimental day, 1 hr after the drug administration. (c) Pre‐administration of PZM21 at a dose of 40 mg·kg−1, but not 20 mg·kg−1, prevented the formation of conditioned response to morphine (10 mg·kg−1). (d) Pretreatment with PZM21 resulted in a tendency towards reduced development, but not expression, of locomotor sensitization induced by repeated administration of morphine (10 mg·kg−1). Data are presented as the mean ± SEM. * P < .05, significantly different from morphine controls. Numbers of animals used in experiments presented in Table S5. CPP, conditioned place preference; Morph, morphine; MPE, maximum possible effect; Sal, saline
4. DISCUSSION AND CONCLUSIONS
In the present study, we examined a novel G protein‐biased μ receptor ligand, PZM21, as a potential nonaddictive analgesic and assessed its ability to modulate morphine‐related behaviour in mice. We demonstrated that PZM21 (administered both intraperitoneal and intrathecal) efficiently exerts dose‐dependent, long‐lasting antinociception and that cyprodime, a μ‐receptor antagonist, blocks this effect. Further, we confirmed that PZM21 is a compound selective for μ‐receptors, while weakly interacting with δ‐opioid receptors. Therefore, our results suggest that PZM21 induces antinociception by acting selectively via μ‐receptor signalling. Interestingly, PZM21 did not elicit MPE in the tail flick test at any of the tested doses, suggesting that increasing the dosage beyond a certain level would not enhance antinociception, known as a ceiling effect (Trescot, Datta, Lee, & Hansen, 2008). PZM21 increased the latency to paw licking/jumping reaction in the hot plate test. Therefore, our study confirmed the effectiveness of PZM21 in the “supraspinal” component of pain processing, as was suggested by Manglik et al. (2016). However, our results clearly demonstrate that PZM21 action is not restricted to supraspinal CNS areas, because we observed antinociceptive effects after intrathecal administration of the compound in the tail flick test that is known to be a measure of spinal reflex (Deuis, Dvorakova, & Vetter, 2017). Taken together, the data from our study indicated that PZM21 is a compound with antinociceptive efficacy. Notably, G protein‐biased opioid analgesics were reported to have a broader therapeutic window than conventional opioids, which allows for antinociception in the absence of respiratory depression (Schmid et al., 2017). However, Hill et al. (2018) have shown that PZM21 depresses respiration; therefore, a question why PZM21 induces suppression of respiration regardless of its bias towards the G protein remains to be addressed in further studies.
Opioid drugs possess rewarding properties, which is one of the undesirable effects of their administration (Fields & Margolis, 2015). In our study, all of the tested doses of PZM21 failed to induce CPP, indicating that this compound is devoid of rewarding effects, and these results are consistent with the previous report (Manglik et al., 2016). However, it is possible that we did not capture PZM21‐induced reward‐related behaviour due to possible differences in the duration of action between morphine and the compound. Interestingly, at the highest dose (80 mg·kg−1), we observed a drug‐induced aversion, indicating that under certain conditions the compound may act as an antagonist. We also showed that in contrast to morphine, PZM21 did not induce locomotor sensitization, considered as a sign of drug‐induced plasticity (Marie, Canestrelli, & Noble, 2018). Furthermore, we demonstrated that PZM21 was not readily self‐administered by rats and did not induce drug seeking behaviour after an abstinence period, strongly suggesting that PZM21 did not present reinforcing properties at the tested doses. To our knowledge, this is the first report to assess PZM21 as a possible reinforcer. Therefore, these results show that PZM21 is devoid of opioid‐like rewarding and reinforcing activity, which is unique among opioid drugs, and also in biased agonists (Altarifi et al., 2017; Austin Zamarripa et al., 2018; Soergel et al., 2014).
In the present study, we observed that naloxone administration precipitated a withdrawal syndrome after chronic administration of PZM21. To date, studies regarding the potential of biased opioids to induce withdrawal are limited. Kliewer et al. (2019) suggested that reducing recruitment of β‐arrestin‐2 to μ‐receptors might not improve the safety profile of opioids, as genetically modified mice with receptors unable to recruit β‐arrestin‐2 displayed typical signs of withdrawal after chronic opioid treatment. What is more, our results showed that after repeated daily administration, PZM21 caused rapid development of antinociceptive tolerance at doses of 40 and 80 mg·kg−1, which is in line with the previous report (Hill et al., 2018). The β‐arrestin‐2‐mediated desensitization of μ receptors was previously suggested as a possible cause of tolerance to opioids (Kliewer et al., 2019; Mori et al., 2017; Przewlocka et al., 2002; Raehal et al., 2005; Yang et al., 2011). However, the currently accepted idea that G protein‐biased ligands should not produce tolerance has little evidence to support it and our data suggest that even opioids biased towards G protein might cause tolerance, limiting their utility as analgesics. Previous studies suggest that the development of tolerance not only may depend on long‐term adaptations connected with β‐arrestin‐2 function but also should be considered as an attribute of a particular ligand (Koch & Höllt, 2008). For example, according to some reports, tolerance to antinociceptive properties of morphine is mediated by the kinase https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518 rather than by the action of β‐arrestin‐2 (Kuhar et al., 2015; Marcus et al., 2015; Yuill, Zee, Marcus, & Morgan, 2016). Thus, it seems that the development of opioid tolerance may be ligand‐specific and involve pathways both dependent and independent of β‐arrestin‐2, leading to differential mechanisms of tolerance observed in vivo. Interestingly, high efficacy μ receptor agonists require lower receptor reserves to maintain an analgesic effect and in turn cause lower tolerance (Stevens & Yaksh, 1989). Our results showed that PZM21 displayed high μ receptor affinity but, in terms of antinociception, presented relatively low efficacy. This observation is consistent with our data revealing that PZM21 activates G protein signalling moderately, as the maximal stimulation of receptor‐mediated G protein activity is low and corresponds to a weak partial agonist activity. Therefore, a possible mechanism associated with PZM21‐induced tolerance might be connected with its agonist efficacy. However, it should be further investigated why PZM21 produces such a rapid development of tolerance to antinociception, regardless of its biased agonism. Taken together, our results indicate that PZM21 does not exhibit rewarding and reinforcing properties. However, chronic treatment with this compound leads to rapid development of tolerance and causes signs of physical dependence. Thus, our report demonstrates that PZM21 differentially influences motivational and physical aspects of addictive behaviour.
In a search for possible mechanisms underlying the lack of reward‐associated behaviour in PZM21‐treated animals, we measured extracellular monoamine levels in the striatum using brain microdialysis in mice. A single systemic administration of PZM21 slightly but dose‐dependently enhanced the extracellular release of dopamine. Our data show that administration of 40 mg·kg−1 of PZM21 induces similar release of dopamine to 10 mg·kg−1 of morphine and the effect of 80 mg·kg−1 of PZM21 is comparable to that evoked by treatment with 20 mg·kg−1 of morphine. Thus, the doses of both drugs that produced similar release of striatal dopamine had different effects on reward‐related memory when measured in the CPP paradigm. Namely, 10 mg·kg−1 of morphine induced strong preference towards the drug‐associated compartment, whereas 40 mg·kg−1 of PZM21 did not present rewarding properties. The enhancement of dopamine‐dependent neurotransmission within the striatum after opioid administration is commonly associated with their addictive properties (Barik et al., 2010; Di Chiara & Imperato, 1988; Spanagel, Herz, & Shippenberg, 1990). However, it was also proposed that dopamine is not necessary for morphine‐induced reward measured in the CPP paradigm and for heroin self‐administration (Borgkvist, Usiello, Greengard, & Fisone, 2007; Hnasko, Sotak, & Palmiter, 2005; Pettit, Ettenberg, Bloom, & Koob, 1984). Thus, the hedonic properties of opioids may only partly depend on dopamine release within the striatal circuity. On the other hand, an enhancement of striatal dopamine release is strongly related to opioid‐induced hyperlocomotion and locomotor sensitization (Murphy, Lam, & Maidment, 2001; Saito, 1990). We showed that PZM21 treatment does not influence locomotor activity. In agreement with our results, a previous study in mice revealed that β‐arrestin‐2 knockout may reduce the expression of some dopamine‐dependent behaviours such as locomotor activity (Bohn et al., 2003). Interestingly, we demonstrated a robust, dose‐dependent increase in 5‐HT striatal release following PZM21 treatment. The ratio of brain 5‐HT to dopamine has been reported to underlie the analgesic effectiveness of opioids (Major & Pleuvry, 1971) as well as the abuse potential of drugs, as 5‐HT neurons were shown to exert an inhibitory influence over mesolimbic dopamine neurons (Navailles & De Deurwaerdère, 2011; Rothman, Blough, & Baumann, 2008). Therefore, we propose that PZM21, through the modulation of 5‐HT release, might inhibit some dopamine‐related behaviours such as locomotor sensitization. Furthermore, 5‐HT‐dependent neurotransmission may enhance and prolong PZM21‐induced antinociception; however, this hypothesis needs to be examined in further studies.
Lastly, we investigated whether PZM21 may influence some outcomes of morphine administration. Pretreatment with PZM21 had no effect on the development of antinociceptive tolerance to morphine, but that it enhanced morphine‐induced antinociception. This observation suggests that PZM21 might be used in pain relief alone or in combination with other opioid drugs. On the other hand, PZM21 prevented the formation of morphine‐induced CPP. One possible explanation for PZM21‐mediated suppression of morphine reward is that it directly antagonized the morphine effects at the μ receptor due to its high affinity for this receptor. Interestingly, our studies on PZM21 suggest that it shows partial agonist characteristics. The use of partial agonists of μ receptors, such as buprenorphine or nalbuphine, appears to be a successful strategy for the attenuation of opioid‐induced reward and treatment of opioid use disorder (Abdel‐Ghany, Nabil, Abdel‐Aal, & Barakat, 2015; Nielsen et al., 2016; Robinson, Erickson, Browne, & Lucki, 2017; Tao, Liang, Sung, Wu, & Huang, 2006). Hence, the anti‐rewarding properties of PZM21 may be dependent on its pharmacological profile.
PZM21 was firstly described as a potent and selective μ receptor agonist (Manglik et al., 2016). However, our behavioural results suggest that it acts as a partial agonist/antagonist of μ receptors, as it displays a ceiling effect in antinociceptive tests, presents a tendency towards inducing aversion at high doses, and interacts with morphine, modulating some effects of its administration. Notably, a recently published Ca2+ imaging study has shown that PZM21 actually is a partial μ receptor agonist, because, when compared to DAMGO, it induces less activation of G protein‐coupled inwardly rectifying potassium https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=74 channels and less inhibition of the nociceptive https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=78 cation channels (Yudin & Rohacs, 2019). Therefore, our study provides a novel insight into pharmacological properties of this compound that differ from these described in the original publication (Manglik et al., 2016) and in many aspects are more consistent with reports by Hill et al. (2018) and Yudin and Rohacs (2019).
Taken together, the present study revealed new characteristics of PZM21. The drug does not induce rewarding and reinforcing effects, indicating that biased signalling could be an attractive direction for future pharmacological studies of novel opioid‐based therapeutics. However, our results point out that PZM21 does not evoke a very potent antinociception when compared to morphine and produces antinociceptive tolerance, suggesting that it may not be sufficient in clinical pain management, especially under chronic pain conditions. Further studies are required in order to assess the effects of PZM21 under different pain conditions and effects of its co‐administration with other opioid therapeutics. It is especially worth considering PZM21 as a pharmacological tool for the modulation of morphine effects, especially for diminishing reward‐related behaviour, and therefore, PZM21 may be considered a potential treatment for opioid use disorder.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
L.K. and R.P. designed the study and wrote the manuscript. R.B., A.B., and Sz.B. performed the synthesis and analysed enantiomeric purity of PZM21. L.K., U.S., L.W., W.S., W.M., and B.P. planned, performed, and analysed behavioural experiments. K.G. and A.W. conducted brain microdialysis and analysed the obtained data. F.Z. and S.B. performed and analysed binding experiments. All authors read and accepted the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1.
HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((R/S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea. HPLC (Chiralpak® AS‐H; 0.1% diethyl amine in isopropanol/hexane 35:65, 0.8 ml·min‐1 T = 21°C); tR = 11.0 min (S,R); tR = 13.7 min (S,S). Specific values are indicated in Table S1.
Figure S2. 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea – PZM21. HPLC (Chiralpak® AS‐H; 0.1% diethyl amine in isopropanol/hexane 35:65, 0.8 ml·min‐1 T = 21°C); tR = 13.8 min (S,S). Specific values are indicated in Table S2.
Figure S3. The μ‐receptors and δ‐receptors competitive binding of PZM21 in rat brain membrane homogenates. Data represent the specific binding of the radioligands to μ‐ORs and δ‐ORs in the presence of [3H]DAMGO, [3H]IleDelt II or PZM21. ‘Total’ on the x‐axis indicates the total specific binding of the radioligands in the absence of the compounds. The figure also indicates the level of 50% radioligand specific and nonspecific binding (0%), highlighted with dotted lines. Points represent means ± SEM for at least three experiments performed in duplicates. The affinity values are indicated in Table S3.
Figure S4. The G protein activity of PZM21 compared to DAMGO in [35S]GTPγS binding assays performed in rat brain membrane homogenates. [A] Data represents the specific binding of [35S]GTPγS binding in percentage over basal activity (100%) in the presence of increasing concentrations of PZM21 or DAMGO for control. [B] Data represents the fractional maximal response (settled as 1) of PZM21 normalized to DAMGO in the presence of increasing concentrations of PZM21 or DAMGO. The figure indicates the level of half maximal response (0.5) with a dotted line. In both figures ‘basal activity’ on the x‐axis indicates the total specific binding of the [35S]GTPγS in the absence of the compounds, which was settled as 100% or 0 (indicated with a dotted line) and also represents the monitored G protein basal activity. Points represent means ± SEM for at least three experiments were performed in duplicate. The curve parameters are indicated in Table S4.
Figure S5. Presentation of the microdialysis probe placement in the mouse striatum.
Table S1. HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((R/S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea.
Table S2. HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea – PZM21
Table S3. The μ receptor and δ‐ receptor affinity values of PZM21.
Table S4. The G protein activity of PZM21 compared to that of DAMGO.
Table S5. Numbers of animals used in behavioural experiments.
ACKNOWLEDGEMENTS
Funding for this study was provided by the Polish National Science Centre (Grants 2013/08/A/NZ3/00848, 2018/29/B/NZ7/00082, and 2018/31/B/NZ7/03954) as well as the National Research Development and Innovation Office (NKFIH, Hungary, Grant OTKA 108518).
Kudla L, Bugno R, Skupio U, et al. Functional characterization of a novel opioid, PZM21, and its effects on the behavioural responses to morphine. Br J Pharmacol. 2019;176:4434–4445. 10.1111/bph.14805
REFERENCES
- Abdel‐Ghany, R. , Nabil, M. , Abdel‐Aal, M. , & Barakat, W. (2015). Nalbuphine could decrease the rewarding effect induced by tramadol in mice while enhancing its antinociceptive activity. European Journal of Pharmacology, 758, 11–15. 10.1016/j.ejphar.2015.03.062 [DOI] [PubMed] [Google Scholar]
- Abdel‐Zaher, A. O. , Hamdy, M. M. , Aly, S. A. , Abdel‐Hady, R. H. , & Abdel‐Rahman, S. (2006). Attenuation of morphine tolerance and dependence by aminoguanidine in mice. European Journal of Pharmacology, 540, 60–66. 10.1016/j.ejphar.2006.03.059 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP collaborators (2017). The concise guide to pharmacology 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarifi, A. A. , David, B. , Muchhala, K. H. , Blough, B. E. , Akbarali, H. , & Negus, S. S. (2017). Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. Journal of Psychopharmacology (Oxford), 31, 730–739. 10.1177/0269881116689257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin Zamarripa, C. , Edwards, S. R. , Qureshi, H. N. , Yi, J. N. , Blough, B. E. , & Freeman, K. B. (2018). The G‐protein biased mu‐opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug and Alcohol Dependence, 192, 158–162. 10.1016/j.drugalcdep.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik, J. , Parnaudeau, S. , Saint Amaux, A. L. , Guiard, B. P. , Golib Dzib, J. F. , Bocquet, O. , … Tronche, F. (2010). Glucocorticoid receptors in dopaminoceptive neurons, key for cocaine, are dispensable for molecular and behavioral morphine responses. Biological Psychiatry, 68, 231–239. 10.1016/j.biopsych.2010.03.037 [DOI] [PubMed] [Google Scholar]
- Bohn, L. M. , Gainetdinov, R. R. , Sotnikova, T. D. , Medvedev, I. O. , Lefkowitz, R. J. , Dykstra, L. A. , & Caron, M. G. (2003). Enhanced rewarding properties of morphine, but not cocaine, in βarrestin‐2 knock‐out mice. The Journal of Neuroscience, 23, 10265–10273. 10.1523/JNEUROSCI.23-32-10265.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn, L. M. , Lefkowitz, R. J. , Gainetdinov, R. R. , Peppel, K. , Caron, M. G. , & Lin, F. T. (1999). Enhanced morphine analgesia in mice lacking beta‐arrestin 2. Science, 286, 2495–2498. 10.1126/science.286.5449.2495 [DOI] [PubMed] [Google Scholar]
- Borgkvist, A. , Usiello, A. , Greengard, P. , & Fisone, G. (2007). Activation of the cAMP/PKA/DARPP‐32 signaling pathway is required for morphine psychomotor stimulation but not for morphine reward. Neuropsychopharmacology, 32, 1995–2003. 10.1038/sj.npp.1301321 [DOI] [PubMed] [Google Scholar]
- Brust, T. F. , Morgenweck, J. , Kim, S. A. , Rose, J. H. , Locke, J. L. , Schmid, C. L. , … Bohn, L. M. (2016). Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Science Signaling, 9, ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X.‐T. , Pitis, P. , Liu, G. , Yuan, C. , Gotchev, D. , Cowan, C. L. , … Yamashita, D. S. (2013). Structure‐activity relationships and discovery of a G protein biased μ opioid receptor ligand, [(3‐methoxythiophen‐2‐yl)methyl]({2‐[(9R)‐9‐(pyridin‐2‐yl)‐6‐oxaspiro‐[4.5]decan‐9‐yl]ethyl})amine (TRV130), for the treatment of acute severe pain. Journal of Medicinal Chemistry, 56, 8019–8031. 10.1021/jm4010829 [DOI] [PubMed] [Google Scholar]
- Contet, C. , Kieffer, B. L. , & Befort, K. (2004). Mu opioid receptor: A gateway to drug addiction. Current Opinion in Neurobiology, 14, 370–378. 10.1016/j.conb.2004.05.005 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis, J. R. , Dvorakova, L. S. , & Vetter, I. (2017). Methods used to evaluate pain behaviors in rodents. Frontiers in Molecular Neuroscience, 10, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire, S. M. , Yamashita, D. S. , Rominger, D. H. , Liu, G. , Cowan, C. L. , Graczyk, T. M. , … Violin, J. D. (2013). A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. The Journal of Pharmacology and Experimental Therapeutics, 344, 708–717. 10.1124/jpet.112.201616 [DOI] [PubMed] [Google Scholar]
- Di Chiara, G. , & Imperato, A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences of the United States of America, 85, 5274–5278. 10.1073/pnas.85.14.5274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, H. L. , & Margolis, E. B. (2015). Understanding opioid reward. Trends in Neurosciences, 38, 217–225. 10.1016/j.tins.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer, C. E. , Tidgewell, K. , Moyer, R. A. , Harding, W. W. , Rothman, R. B. , Prisinzano, T. E. , & Bohn, L. M. (2007). An opioid agonist that does not induce μ‐opioid receptor—Arrestin interactions or receptor internalization. Molecular Pharmacology, 71, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R. , Disney, A. , Conibear, A. , Sutcliffe, K. , Dewey, W. , Husbands, S. , … Henderson, G. (2018). The novel μ‐opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. British Journal of Pharmacology, 175, 2653–2661. 10.1111/bph.14224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko, T. S. , Sotak, B. N. , & Palmiter, R. D. (2005). Morphine reward in dopamine‐deficient mice. Nature, 438, 854–857. 10.1038/nature04172 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer, A. , Schmiedel, F. , Sianati, S. , Bailey, A. , Bateman, J. T. , Levitt, E. S. , … Schulz, S. (2019). Phosphorylation‐deficient G‐protein‐biased μ‐opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nature Communications, 10, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, T. , & Höllt, V. (2008). Role of receptor internalization in opioid tolerance and dependence. Pharmacology & Therapeutics, 117, 199–206. 10.1016/j.pharmthera.2007.10.003 [DOI] [PubMed] [Google Scholar]
- Kuhar, J. R. , Bedini, A. , Melief, E. J. , Chiu, Y.‐C. , Striegel, H. N. , & Chavkin, C. (2015). Mu opioid receptor stimulation activates c‐Jun N‐terminal kinase 2 by distinct arrestin‐dependent and independent mechanisms. Cellular Signalling, 27, 1799–1806. 10.1016/j.cellsig.2015.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madariaga‐Mazón, A. , Marmolejo‐Valencia, A. F. , Li, Y. , Toll, L. , Houghten, R. A. , & Martinez‐Mayorga, K. (2017). Mu‐opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discovery Today, 22, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet, E. L. , Milon, N. , Heghinian, M. D. , Fishback, J. , Schürer, S. C. , Garamszegi, N. , & Mash, D. C. (2015). Noribogaine is a G‐protein biased κ‐opioid receptor agonist. Neuropharmacology, 99, 675–688. 10.1016/j.neuropharm.2015.08.032 [DOI] [PubMed] [Google Scholar]
- Major, C. T. , & Pleuvry, B. J. (1971). Effects of α‐methyl‐p‐tyrosine, p‐chlorophenylalanine, l‐β‐(3,4‐dihydroxyphenyl)alanine, 5‐hydroxytryptophan and diethyldithiocarbamate on the analgesic activity of morphine and methylamphetamine in the mouse. British Journal of Pharmacology, 42, 512–521. 10.1111/j.1476-5381.1971.tb07137.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik, A. , Lin, H. , Aryal, D. K. , McCorvy, J. D. , Dengler, D. , Corder, G. , … Shoichet, B. K. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature, 537, 185–190. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus, D. J. , Zee, M. , Hughes, A. , Yuill, M. B. , Hohmann, A. G. , Mackie, K. , … Morgan, D. J. (2015). Tolerance to the antinociceptive effects of chronic morphine requires c‐Jun N‐terminal kinase. Molecular Pain, 11, 34 10.1186/s12990-015-0031-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie, N. , Canestrelli, C. , & Noble, F. (2018). Role of pharmacokinetic and pharmacodynamic parameters in neuroadaptations induced by drugs of abuse, with a focus on opioids and psychostimulants. Neuroscience & Biobehavioral Reviews.. 10.1016/j.neubiorev.2018.06.006 [DOI] [PubMed] [Google Scholar]
- Mori, T. , Kuzumaki, N. , Arima, T. , Narita, M. , Tateishi, R. , Kondo, T. , … Narita, M. (2017). Usefulness for the combination of G‐protein‐ and β‐arrestin‐biased ligands of μ‐opioid receptors: Prevention of antinociceptive tolerance. Molecular Pain, 13, 1744806917740030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, N. P. , Lam, H. A. , & Maidment, N. T. (2001). A comparison of morphine‐induced locomotor activity and mesolimbic dopamine release in C57BL6, 129Sv and DBA2 mice. Journal of Neurochemistry, 79, 626–635. [DOI] [PubMed] [Google Scholar]
- Navailles, S. , & De Deurwaerdère, P. (2011). Presynaptic control of serotonin on striatal dopamine function. Psychopharmacology, 213, 213–242. 10.1007/s00213-010-2029-y [DOI] [PubMed] [Google Scholar]
- Nielsen, S. , Larance, B. , Degenhardt, L. , Gowing, L. , Kehler, C. , & Lintzeris, N. (2016). Opioid agonist treatment for pharmaceutical opioid dependent people. The Cochrane Database of Systematic Reviews, (5), CD011117. [DOI] [PubMed] [Google Scholar]
- Pettit, H. O. , Ettenberg, A. , Bloom, F. E. , & Koob, G. F. (1984). Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self‐administration in rats. Psychopharmacology, 84, 167–173. 10.1007/BF00427441 [DOI] [PubMed] [Google Scholar]
- Przewlocka, B. , Sieja, A. , Starowicz, K. , Maj, M. , Bilecki, W. , & Przewlocki, R. (2002). Knockdown of spinal opioid receptors by antisense targeting beta‐arrestin reduces morphine tolerance and allodynia in rat. Neuroscience Letters, 325, 107–110. 10.1016/S0304-3940(02)00246-X [DOI] [PubMed] [Google Scholar]
- Raehal, K. M. , Walker, J. K. L. , & Bohn, L. M. (2005). Morphine side effects in beta‐arrestin 2 knockout mice. The Journal of Pharmacology and Experimental Therapeutics, 314, 1195–1201. 10.1124/jpet.105.087254 [DOI] [PubMed] [Google Scholar]
- Robinson, S. A. , Erickson, R. L. , Browne, C. A. , & Lucki, I. (2017). A role for the mu opioid receptor in the antidepressant effects of buprenorphine. Behavioural Brain Research, 319, 96–103. 10.1016/j.bbr.2016.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojewska, E. , Popiolek‐Barczyk, K. , Jurga, A. M. , Makuch, W. , Przewlocka, B. , & Mika, J. (2014). Involvement of pro‐ and antinociceptive factors in minocycline analgesia in rat neuropathic pain model. Journal of Neuroimmunology, 277, 57–66. 10.1016/j.jneuroim.2014.09.020 [DOI] [PubMed] [Google Scholar]
- Rothman, R. B. , Blough, B. E. , & Baumann, M. H. (2008). Dual dopamine/serotonin releasers: Potential treatment agents for stimulant addiction. Experimental and Clinical Psychopharmacology, 16, 458–474. 10.1037/a0014103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, H. (1990). Inhibitory and stimulatory effects of morphine on locomotor activity in mice: Biochemical and behavioral studies. Pharmacology, Biochemistry, and Behavior, 35, 231–235. 10.1016/0091-3057(90)90231-6 [DOI] [PubMed] [Google Scholar]
- Schmid, C. L. , Kennedy, N. M. , Ross, N. C. , Lovell, K. M. , Yue, Z. , Morgenweck, J. , … Bohn, L. M. (2017). Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell, 171, 1165–1175.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel, D. G. , Subach, R. A. , Burnham, N. , Lark, M. W. , James, I. E. , Sadler, B. M. , … Webster, L. R. (2014). Biased agonism of the μ‐opioid receptor by TRV130 increases analgesia and reduces on‐target adverse effects versus morphine: A randomized, double‐blind, placebo‐controlled, crossover study in healthy volunteers. Pain, 155, 1829–1835. 10.1016/j.pain.2014.06.011 [DOI] [PubMed] [Google Scholar]
- Solecki, W. , Wickham, R. J. , Behrens, S. , Wang, J. , Zwerling, B. , Mason, G. F. , & Addy, N. A. (2013). Differential role of ventral tegmental area acetylcholine and N‐methyl‐d‐aspartate receptors in cocaine‐seeking. Neuropharmacology, 75, 9–18. 10.1016/j.neuropharm.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki, W. B. , Szklarczyk, K. , Pradel, K. , Kwiatkowska, K. , Dobrzański, G. , & Przewłocki, R. (2018). Noradrenergic signaling in the VTA modulates cocaine craving. Addiction Biology, 23, 596–609. 10.1111/adb.12514 [DOI] [PubMed] [Google Scholar]
- Spanagel, R. , Herz, A. , & Shippenberg, T. S. (1990). The effects of opioid peptides on dopamine release in the nucleus accumbens: An in vivo microdialysis study. Journal of Neurochemistry, 55, 1734–1740. 10.1111/j.1471-4159.1990.tb04963.x [DOI] [PubMed] [Google Scholar]
- Stevens, C. W. , & Yaksh, T. L. (1989). Potency of infused spinal antinociceptive agents is inversely related to magnitude of tolerance after continuous infusion. The Journal of Pharmacology and Experimental Therapeutics, 250, 1–8. [PubMed] [Google Scholar]
- Szklarczyk, K. , Korostynski, M. , Golda, S. , Solecki, W. , & Przewlocki, R. (2012). Genotype‐dependent consequences of traumatic stress in four inbred mouse strains.Genes, Brain and Behavior, 11, 977‐985. 10.1111/j.1601-183X.2012.00850.x [DOI] [PubMed] [Google Scholar]
- Tao, P.‐L. , Liang, K.‐W. , Sung, W.‐Y. , Wu, Y.‐T. , & Huang, E. Y.‐K. (2006). Nalbuphine is effective in decreasing the rewarding effect induced by morphine in rats. Drug and Alcohol Dependence, 84, 175–181. 10.1016/j.drugalcdep.2006.01.013 [DOI] [PubMed] [Google Scholar]
- Trescot, A. M. , Datta, S. , Lee, M. , & Hansen, H. (2008). Opioid pharmacology. Pain Physician, 11, S133–S153. [PubMed] [Google Scholar]
- Webster, L. , St Marie, B. , McCarberg, B. , Passik, S. D. , Panchal, S. J. , & Voth, E. (2011). Current status and evolving role of abuse‐deterrent opioids in managing patients with chronic pain. Journal of Opioid Management, 7, 235–245. 10.5055/jom.2011.0066 [DOI] [PubMed] [Google Scholar]
- Yaksh, T. L. , & Rudy, T. A. (1976). Chronic catheterization of the spinal subarachnoid space. Physiology & Behavior, 17, 1031–1036. 10.1016/0031-9384(76)90029-9 [DOI] [PubMed] [Google Scholar]
- Yang, C.‐H. , Huang, H.‐W. , Chen, K.‐H. , Chen, Y.‐S. , Sheen‐Chen, S.‐M. , & Lin, C.‐R. (2011). Antinociceptive potentiation and attenuation of tolerance by intrathecal β‐arrestin 2 small interfering RNA in rats. British Journal of Anaesthesia, 107, 774–781. 10.1093/bja/aer291 [DOI] [PubMed] [Google Scholar]
- Yudin, Y. , & Rohacs, T. (2019). The G protein‐biased agents PZM21 and TRV130 are partial agonists of μ‐opioid receptor‐mediated signaling to ion channels. British Journal of Pharmacology. 10.1111/bph.14702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuill, M. B. , Zee, M. L. , Marcus, D. , & Morgan, D. J. (2016). Tolerance to the antinociceptive and hypothermic effects of morphine is mediated by multiple isoforms of c‐Jun N‐terminal kinase. Neuroreport, 27, 392–396. 10.1097/WNR.0000000000000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((R/S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea. HPLC (Chiralpak® AS‐H; 0.1% diethyl amine in isopropanol/hexane 35:65, 0.8 ml·min‐1 T = 21°C); tR = 11.0 min (S,R); tR = 13.7 min (S,S). Specific values are indicated in Table S1.
Figure S2. 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea – PZM21. HPLC (Chiralpak® AS‐H; 0.1% diethyl amine in isopropanol/hexane 35:65, 0.8 ml·min‐1 T = 21°C); tR = 13.8 min (S,S). Specific values are indicated in Table S2.
Figure S3. The μ‐receptors and δ‐receptors competitive binding of PZM21 in rat brain membrane homogenates. Data represent the specific binding of the radioligands to μ‐ORs and δ‐ORs in the presence of [3H]DAMGO, [3H]IleDelt II or PZM21. ‘Total’ on the x‐axis indicates the total specific binding of the radioligands in the absence of the compounds. The figure also indicates the level of 50% radioligand specific and nonspecific binding (0%), highlighted with dotted lines. Points represent means ± SEM for at least three experiments performed in duplicates. The affinity values are indicated in Table S3.
Figure S4. The G protein activity of PZM21 compared to DAMGO in [35S]GTPγS binding assays performed in rat brain membrane homogenates. [A] Data represents the specific binding of [35S]GTPγS binding in percentage over basal activity (100%) in the presence of increasing concentrations of PZM21 or DAMGO for control. [B] Data represents the fractional maximal response (settled as 1) of PZM21 normalized to DAMGO in the presence of increasing concentrations of PZM21 or DAMGO. The figure indicates the level of half maximal response (0.5) with a dotted line. In both figures ‘basal activity’ on the x‐axis indicates the total specific binding of the [35S]GTPγS in the absence of the compounds, which was settled as 100% or 0 (indicated with a dotted line) and also represents the monitored G protein basal activity. Points represent means ± SEM for at least three experiments were performed in duplicate. The curve parameters are indicated in Table S4.
Figure S5. Presentation of the microdialysis probe placement in the mouse striatum.
Table S1. HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((R/S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea.
Table S2. HPLC data of 1‐[(S)‐2‐(Dimethylamino)‐3‐(4‐hydroxyphenyl)propyl]‐3‐((S)‐1‐(thiophen‐3‐yl)propan‐2‐yl)urea – PZM21
Table S3. The μ receptor and δ‐ receptor affinity values of PZM21.
Table S4. The G protein activity of PZM21 compared to that of DAMGO.
Table S5. Numbers of animals used in behavioural experiments.