

Abstract
(S)-2-hydroxypropylphosphonate [(S)-2-HPP, 1] epoxidase (HppE) reduces H2O2 at its non-heme-iron cofactor to install the oxirane “warhead” of the antibiotic fosfomycin. The net replacement of the C1 pro-R hydrogen of 1 by its C2 oxygen, with inversion of configuration at C1, yields the cis epoxide of the drug [(1R,2S)-epoxypropylphosphonic acid (cis-Fos, 2)]. Here we show that HppE achieves ~ 95% selectivity for C1 inversion and cis-epoxide formation via steric guidance of a radical-coupling mechanism. Published structures of the HppE•FeII•1 and HppE•ZnII•2 complexes reveal distinct pockets for C3 of the substrate and product and identify four hydrophobic residues – Leu120, Leu144, Phe182, and Leu193 – close to C3 in one of the complexes. Replacement of Leu193 in the substrate C3 pocket with the bulkier Phe enhances stereoselectivity (cis:trans ~ 99:1), whereas the Leu120Phe substitution in the product C3 pocket diminishes it (~ 82:18). Retention of C1 configuration and trans-epoxide formation become predominant with the bulk-reducing Phe182Ala substitution in the substrate C3 pocket (~ 13:87), trifluorination of C3 (~ 23:77), or both (~ 1:99). The effect of C3 trifluorination is counteracted by the more constrained substrate C3 pockets in the Leu193Phe (~ 56:44) and Leu144Phe/Leu193Phe (~ 90:10) variants. The ability of HppE to epoxidize substrate analogues bearing halogens at C3, C1, or both is inconsistent with a published hypothesis of polar cyclization via a C1 carbocation. Rather, specific enzyme-substrate contacts drive inversion of the C1 radical – as proposed in a recent computational study – to direct formation of the more potently antibacterial cis epoxide by radicaloid C–O coupling.
Keywords: iron, peroxidase, stereochemistry, oxirane, antibacterial
Graphical Abstract
INTRODUCTION
The strained oxirane (epoxide) moiety readily undergoes ring-opening by nucleophilic attack and can thus serve as the basis for covalent modification of drug targets.1 The naturally occurring antibiotic, fosfomycin [(1R,2S)-epoxypropylphosphonic acid; cis-Fos, 2], provides a classic example.2 Widely used to treat urinary tract infections, fosfomycin targets UDP-N-acetylglucosamine enolpyruvyl transferase (MurA), a key enzyme in bacterial peptidoglycan biosynthesis.3 Attack of a cysteine residue in the active site of MurA opens the epoxide, resulting in covalent coupling and enzyme inactivation.3,4
Enzymatic installation of an epoxide proceeds by one of two main routes: cyclic coupling of an oxygen-atom equivalent to both carbons of an olefin or cyclization of an alcohol.5 The epoxide warhead of fosfomycin is installed by the latter route.6 The enzyme S-2-hydroxypropylphosphonate [(S)-2-HPP, 1] epoxidase (HppE) removes a hydrogen atom (H•) from C1 of 1 and couples this carbon to the alcohol oxygen on C2 (Scheme 1A).7 For many years, HppE was thought to activate dioxygen at its non-heme Fe(II) cofactor to generate the H•-abstracting intermediate.6 However, more recent work showed that the oxidizing co-substrate of HppE is actually hydrogen peroxide (Scheme 1A).7 This same study speculated that O–O-bond heterolysis following addition of the peroxide to the Fe(II) cofactor would produce an Fe(IV)-oxo (ferryl) intermediate (Scheme S1, route A),7 known in other systems to abstract H• from unactivated aliphatic carbon centers.8 However, a more recent computational study questioned the feasibility of such a pathway and suggested, instead, cofactor-promoted homolysis of the peroxide bond to form a hydroxyl radical, oriented for abstraction of H• from C1 by hydrogen bonding to the Fe(III)-coordinated hydroxide derived from the other peroxide oxygen (Scheme S1, route B).9 To date, no experimental data to distinguish between these two possibilities have been reported.
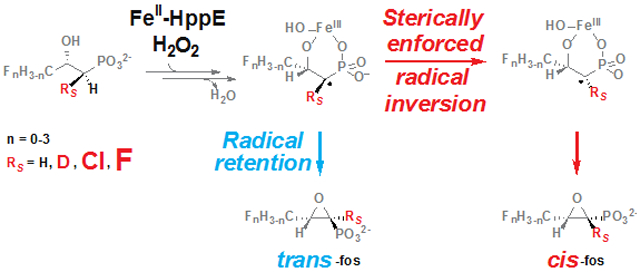
Scheme 1.

(A) Predominant transformation of (S)-2-HPP by HppE and (B) possible mechanisms for this outcome.
Multiple mechanisms have also been considered for the ensuing C1 ↔ O–C2 coupling step (Scheme 1B),6, 9-10 and no consensus has yet been reached. In the reactions of several other non-heme-iron enzymes that cleave strong C–H bonds to install heteroatoms (e.g., isopenicillin N synthase, the aliphatic halogenases, and several radical-SAM enzymes), carbon-centered radicals attack the Fe(III)-coordinated heteroatoms (X) to form the new C–X bonds, in the process reducing the Fe(III) to Fe(II).11-19 A recent study of an inorganic model complex demonstrated ether C–O-bond formation by this mechanism.20 Given that the iron cofactor of HppE also coordinates the (C2) oxygen of 1 that couples to C1 (Figure 1A), an analogous radical-coupling mechanism would seem likely for its cyclization step (Scheme 1B, blue arrows). However, two experimental observations – the unexpected stereochemical course of the native epoxidation reaction21 and the nature of a distinct oxidation of a substrate analog22-23 – have led to consideration of more complex C–O-coupling mechanisms. HppE removes the pro-R hydrogen from C1 of 1,24 consistent with the orientation of the substrate in published x-ray crystal structures of the complex (Figure 1A).25-26 Coupling of the C2 oxygen to C1 in the site vacated by the abstracted H• would yield the 1S configuration and trans epoxide (trans-Fos, 3), rather than the 1R configuration and cis epoxide of the actual antibiotic, 2. In other words, C1 ↔ O–C2 coupling proceeds with inversion of configuration at C1.7 By contrast, in the simplest radical-coupling mechanism, retention of configuration at C1 would be anticipated (Scheme 1B, blue arrows). Consequently, a mechanism involving an initial radical coupling of the coordinated phosphonate oxygen (with retention at C1) and subsequent nucleophilic attack (SN2) of the C2 oxygen upon the carbon of the unstable C-P-O heterocycle (with inversion), was proposed (purple arrows).7
Figure 1.

Structural models of the HppE substrate and product complexes from x-ray crystallographic studies. (A) Model of the HppE•FeII•(S)-2-HPP complex (PDB ID: 1ZZ8). (B) Model of the HppE•ZnII•fosfomycin complex (PDB ID: 2BNN).
HppE is capable of a number of alternative outcomes when challenged with structural isomers of its substrate (Scheme S2). In addition to its native epoxidation of 1 (Scheme S2A), it can dehydrogenate the alcohol groups of (R)-2-HPP and (S)-1-HPP to carbonyls (Schemes S2B-C).22-23 The former outcome implies that the enzyme can also remove H• from C2, when differences in substrate structure position this carbon appropriately. More interestingly, (R)-1-HPP undergoes an oxidative 1,2-phosphonate migration, yielding 2-phosphonopropanal (Scheme S2D).22 This outcome was rationalized in terms of H• abstraction from C2, electron transfer from this radical to an oxidized form of the iron cofactor, and migration of the phosphonate to quench the resulting C2 carbocation.10, 22 An analogous pathway – H• abstraction from C1 of 1, electron transfer from the C1 radical to the cofactor, and polar C1+ ↔ −O–C2 coupling – was suggested for the native epoxidation reaction.22 At that time, O2 was still thought to be the oxidizing co-substrate, leading to the proposal of H• abstraction by a superoxo-Fe(III) complex and electron transfer to a ferryl form of the cofactor, but an analogous mechanism via C1 carbocation can be formulated with H2O2 as the oxidant (Scheme 1B, orange arrows).7 The hypothesis of a planar C1 carbocation intermediate would, in principle, explain why the expectation of strict retention in the ring-closure step is not met, but it would not, a priori, lead one to expect high selectivity for inversion. The aforementioned computational study put forth a different explanation – facile inversion of the C1 radical9 – for the stereochemical course of the ring-closure step (Scheme 1B, red arrows), thus potentially rationalizing this crucial aspect of the reaction within the framework of the chemical mechanism that would, in light of precedent, seem most probable.
In this study, we sought to clarify the mechanism of the C–O-coupling step in the HppE reaction by assessing (i) whether substrate analogs harboring electron withdrawing groups, which would be expected to disfavor C1-carbocation formation, can be epoxidized and (ii) whether sterically consequential modifications to the substrate or enzyme can change the stereochemical course of the reaction. The ability of the enzyme to epoxidize analogs bearing halogens at C3 (1-3 fluorine atoms), C1 (fluorine or chlorine), or both weighs heavily against the carbocation mechanism (Scheme 1B, orange arrows), and the demonstration that modifications to the substrate or enzyme, chosen rationally on the basis of published x-ray crystal structures, can completely reverse the ring-closure stereochemistry provides argument against a chemically enforced C1 inversion, as in the mechanism involving intermediary radical coupling and SN2 heterocycle interconversion (Scheme 1B, purple arrows). The data thus drastically limit the mechanistic possibilities, decisively favoring a sterically enforced inversion of the C1 radical and subsequent radicaloid C1 ↔ O–C2 coupling (Scheme 1B, red arrows). The observation of diminished antibiotic activities of the trans-epoxide compounds generated either from the 3-F3-substituted analog of (S)-2-HPP by the wild-type enzyme or from the native substrate by site-directed variants implies that the ability of HppE to direct C1 inversion and cis-epoxide formation in the ring-closure step is essential to its biological role, which is, presumably, to produce a competition-suppressing, antimicrobial secondary metabolite.
RESULTS AND DISCUSSION
Mixed stereochemistry in the C–O-coupling (cyclization) step of the HppE reaction.
Although published work has established that the predominant outcome in the oxidation of 1 by HppE is, effectively, replacement of the C1 pro-R hydrogen by the C2 oxygen with inversion of configuration at C1 (producing cis-Fos, 2; Scheme 1A and Figure 2A),6-7 one study examining production of Fos in vivo reported observation of a small quantity of the trans epoxide, [(1S,2S)-epoxypropylphosphonic acid; trans-Fos, 3],27 potentially arising from competing retention at C1 in the HppE-catalyzed cyclization (Scheme 1B, blue arrows, and Figure 2A). Because even a small degree of stereo-ambiguity in this step would rule out the mechanism enforcing inversion (Scheme 1B, purple arrows), we deemed it important to test directly for in vitro production of 3 by HppE. Accordingly, we synthesized the racemic trans isomer (rac-3, composed of 3 and its 1R,2R enantiomer; see Supplemental Methods in the Supporting Information for details of the synthetic procedure) in order to compare its 1H- and 31P-NMR spectra to those of the commercially available cis compound, 2). Rac-3 is readily distinguished from 2 by (i) the diminished downfield shifts of its C1-1H and C2-1H resonances and ~ 2-fold weaker vicinal coupling21 (3JH-H; Figure 2B-C, top and middle spectra) and (ii) the increased downfield shift of its 31P resonance (Figure 2D, top and middle spectra) and stronger 31P-1H geminal coupling (2JP-H; Figure 2C, top and middle spectra). These distinctions afforded the tools to determine relative yields of the cis and trans products in the reactions of all combinations of modified substrates and variant enzymes examined in this work.
Figure 2.

Re-examination of the stereochemical selectivity in Fos production by HppE. (A) Cis-Fos (2) and trans-Fos (3) products of ring-closure with inversion and retention, respectively, of C1 configuration. The atoms are color-coded to facilitate interpretation of the 1H-NMR spectra in the lower panels. (B) 1H NMR spectra of commercial cis-Fos (top), synthetic standard trans-Fos (middle), and the isolated epoxide products from the HppE reaction (bottom). (C) Blow-up of the peaks arising from the C1-1H showing that the vicinal coupling constant, 3JH-H, to the C2-1H in cis-Fos (3JH-H = ~ 5.1 Hz) is almost twice that in trans-Fos (3JH-H = ~ 2.7 Hz). For ease of comparison, minor adjustments in peak positions (≤ 0.08 ppm) have been made to account for vari-ations in δ with differences in pH, counter ion composition, and concentration. (D) Corresponding 31P-NMR spectra measured in 100 mM NaOD in D2O. The dd feature (2JP-H = ~ 21.7, 3JP-H = ~ 5.1 Hz) at ~ 10.4 ppm is from trans-Fos, and the dd feature (2JP-H = ~ 18.7, 3JP-H = ~ 5.3 Hz) at ~ 9.9 ppm is from cis-Fos. The singlet at ~ 4.8 ppm is from the internal standard (PO43−).
1H-NMR (Figure 2B-C, bottom spectra) and 31P-NMR (Figure 2D, bottom spectrum) spectra of the products from the HppE reaction with 1 (see SI, Sec 2.2 for detailed procedures) confirm that, indeed, the enzyme does produce a small quantity (3-5 % of the total epoxide) of trans-Fos, 3, suggesting that ring closure with retention competes to a minor extent with the predominant C1-inverting pathway. To verify that formation of 3 still involves removal of the pro-R hydrogen,6-7,24 we synthesized both 1-[2H1]-(S)-2-HPP diastereomers (SI Sec 2.2 and 6.1; Figure S2) and subjected them to oxidation by HppE. Analysis by 31P-NMR spectroscopy (Figure 3; Figure S3) showed that, as expected, the (1S,2S) diastereomer, 1d, yields the monodeuterated cis- and trans-epoxide products, 2d and 3d, in the characteristic ~ 95:5 ratio (panel B, top), whereas the (1R,2S) diastereomer, 1e, yields the same ratio of cis and trans epoxides (2 and 3) with protium at C1 (panel B, bottom). These results confirm that the cis and trans products form by initial removal of the same (pro-R) hydrogen and subsequent ring-closure with opposite stereochemistry, weighing against the proposed mechanism involving an intermediary coupling step via the C–P–O heterocycle (Scheme 1B, purple arrows).
Figure 3.

Stereochemical courses of the transformations of stereospecifically 1-d1 labeled 2-HPP diasteromers by wild-type (wt) HppE. (A) Summary of reaction outcomes and (B) 31P NMR analysis of the products in reactions of wt HppE with the (1S,2S)-1-d1-2-HPP (1d) and (1R,2S)-1-d1-2-HPP (1e). Each trans:cis ratio is the mean value from three 31P-NMR assays, and the standard deviations given in parentheses are errors on each percentage value [i.e., 5:95 (0.2) implies a trans:cis ratio in the range of 4.8:95.2 – 5.2:94.8].
Structure-guided mutagenesis to alter cyclization stereochemistry.
The above evidence against the sole mechanism that could enforce net inversion in the ring-closure step implies that the enzyme controls the reaction stereochemistry sterically, by favoring either (i) radicaloid ring closure from the inverted configuration of the C1 radical (Scheme 1B, red arrows) or (ii) the “back-side” trajectory for attack of the C2 oxygen on the planar C1 carbocation (Scheme 1B, orange arrows). We examined the published HppE structures25, 28 (Figure 1) for active-site residues and enzyme-substrate/product interactions that might contribute to this steric control. From the best available structures of the HppE•1 (A) and HppE•2 (B) complexes, we identified four hydrophobic residues in the active site that are within 5 Å of the C3 methyl group in one of the two complexes and ≥ 0.5 Å closer in one complex than in the other. Leu193 (L193) and Phe182 (F182) are markedly (~ 1.5 Å) closer to C3 in the model of the substrate complex than in that of the product complex, and L144 appears to be slightly (~ 0.5 Å) closer. These three residues make up the substrate-C3 binding pocket. Conversely, L120 is nearly 3 Å closer in the model of the product complex and largely defines the product-C3 pocket. It appears – although there is no available structure to confirm – that the trans-epoxide product, 3, generated by ring closure with retention of configuration at C1, would project its C3 group toward the substrate-C3 pocket, and so we reasoned that amino acid substitutions contracting this pocket (by increasing the volume of the side chains lining it) might enhance selectivity for C1 inversion, whereas expansion of the pocket (by bulk-reducing substitutions to the residues defining it) might diminish the stereoselectivity. Conversely, we expected that bulk-enhancing substitutions of L120 would constrict the product-C3 pocket and diminish selectivity for C1 inversion and cis-epoxide (2) formation. We prepared the L193F and L144F/L193F variant proteins to evaluate the effect of constriction, and the F182A variant to evaluate the effect of expansion, of the substrate-C3 pocket. Similarly, we prepared the L120F variant to assess the impact of a constricted product-C3 pocket. All four proteins were sufficiently soluble and stable to be characterized after production in and purification from Escherichia coli, although the F182A protein was seen to lose activity upon freeze-thaw cycles. All four proteins proved capable of multiple turnovers of (S)-2-HPP with H2O2 as oxidizing co-substrate, although the total turnover numbers (TTNs) achieved with the variant proteins were diminished from that obtained with the wild-type enzyme (~ 40) to a minimum of ~ 3 for the L144F/L193F double variant (Figure S4). For the two variant proteins that exhibited the most striking effects on the reaction stereochemistry (F182A and L193F; vide infra), we determined the H2O2:product stoichiometries to compare to that of wild-type HppE, which we previously reported7 and verified here (Figure S5A-C) to be indistinguishable from the theoretical value of unity. Both variant proteins retain the ability to fully couple H2O2 reduction to (S)-2-HPP oxidation (Figure S5D-I).
Characterization of the reaction products by 31P-NMR spectroscopy (Figures 4B and S6) revealed that, as predicted from analysis of the published structures, expansion of the substrate-C3 pocket by replacement of the phenyl group of F182 with hydrogen (i.e., the F182A substitution) diminishes selectivity for C1 inversion in the ring-closure step. The F182A variant produces predominantly the trans-Fos isomer, 3 (Figure 4B, bottom, Figure S6). A cis:trans (2:3) ratio of 12:88 was obtained by averaging 3 replicates of the 31P-NMR analysis; standard deviations are given in parentheses. By use of the stereospecifically 1-[2H1]-labeled substrates, 1d and 1e, we confirmed that the variant protein still abstracts the pro-R hydrogen (Figure S7A): the former substrate yields the 12:88 mixture of monodeuterated epoxide products (2d:3d), whereas the latter substrate yields the equivalent mixture of epoxides lacking deuterium (2:3; Figure S7A, middle). Increased bulk in the product-C3 pocket, imparted by the L120F substitution, was also seen to diminish selectivity (Figure 4B, second from bottom, Figure S6), albeit to a more modest extent (2:3 ~ 82:18) than the F182A substitution. We saw the opposite effect on ring-closure stereoselectivity upon introducing the L193F substitution (Figure 4B, top; Figure S6), predicted by the structural analysis to constrict the substrate-C3 pocket. This variant protein is, remarkably, more stereoselective than the wild-type enzyme, producing a fraction of the trans epoxide, 3, so small (~ 1%) as to be challenging to detect. We again verified that this variant removes the pro-R hydrogen in the cyclization reaction (Figure S7B). The accuracy of these several predictions regarding ring-closure stereochemistry confirms the hypothesis on which they were founded – that HppE controls the outcome by a steric mechanism rather than by leveraging a multi-step pathway (Scheme 1B, purple arrows) that enforces inversion.
Figure 4.

Analysis by 31P-NMR spectroscopy of the product profiles in reactions of HppE and its variants with the 3-Fn-substituted (n = 0-3) (S)-2-HPP substrates (1, 1a-c). (A) Reaction scheme and explanation of product nomenclature. (B-E) 31P-NMR spectra from the reactions with the n = 0-3 compounds and the HppE variants indicated to the right of each spectrum. The trans:cis ratios shown at the center of the spectra were determined by comparison of the integrated intensities of the more down-field dd features of the trans-epoxide products and the more upfield dd features of the cis-epoxide compounds. Each value is the mean of three trials, with standard deviations shown in parentheses. Details of the analytical procedures are provided in the Supporting Information. For compounds with at least one fluorine (C-E), trans:cis ratios were also calculated by comparison of the intensities of the appropriate features in the 19F-NMR spectra and were found to be quantitatively consistent.
Use of halogen substitution to distinguish between polar and radicaloid C–O-coupling mechanisms.
We next sought to distinguish whether the enzyme directs C1 inversion (i) by tuning the relative stabilities within the active site of the two configurations of the C1 radical (Scheme 1B, red arrows) or (ii) by impacting the relative barrier heights of the two possible trajectories for attack of the C2 oxygen on the C1 carbocation (Scheme 1B, orange arrows and green arrows). We reasoned that carbocation formation would be disfavored by the presence of strongly electron-withdrawing groups on the substrate, whereas the radicaloid mechanism would be more robust to such modifications.10 We therefore synthetically incorporated halogen atoms at C3, C1, or both and evaluated the ability of HppE to cyclize these analogues. Comparison of the 1H-NMR spectra of the products generated by the wild-type enzyme from the 3-F3-substituted (S)-2-HPP analog (1c) to the spectra of the corresponding synthetic cis- and trans-epoxide standards (2c and 3c, respectively) showed that replacement of even all three C3 hydrogens by fluorine does not prevent HppE-catalyzed epoxidation (Figure S8). Intriguingly, trifluorination of C3 does markedly impact the stereochemistry: 1H-NMR (Figure S8), 31P-NMR (Figure 4C, middle spectrum; Figure S10), and 19F-NMR (Figure S11) spectra were all consistent with a cis:trans (2c:3c) product ratio of ~ 23:77, a selectivity reversed from that seen with the native substrate. All three fluorine atoms are required for this effect on the stereoschemistry: we found the 3-F- (1a) and 3-F2- (1b) substrates to yield cis:trans ratios (2a,b:3a,b) similar to those obtained with the native substrate, 1 (Figure 4D-E; Figures S12-16). With the C3-trifluorinated analogue (1c), the more cis-stereoselective L193F variant produces more of the cis-epoxide product (2c:3c ~ 56:44; Figure 4C, second from top; Figures S10-11) than does the wild-type enzyme, and the L144F/L193F double variant is even more selective for this product (Figure 4C, top; Figures S10-11), giving a cis:trans (2c:3c) ratio (~ 90:10) that approaches that for the wild-type enzyme with the native substrate. Conversely, the less cis-selective F182A variant produces almost exclusively (~ 1:99) trans epoxide 3c (Figure 4C, bottom; Figures S10-11). Thus, the trans-favoring effect of 3-F3 substitution is both counteracted by constriction of the substrate-C3 pocket and potentiated by expansion of the pocket. The parallel responses of the 3-F3-substituted and native substrates to these active-site amino acid substitutions implies that cyclization of the two compounds proceeds by the same mechanism. Most likely, the energetic cost of eclipsing the trifluoromethyl and phosphonate groups in the trajectory29 for C1 inversion/cis-epoxide formation is greater than that incurred with the unsubstituted C3 methyl group (Figure S9), resulting in more C1 retention and trans-epoxide production with the analog.
Fluorine substitution markedly destabilizes organic cations,10, 30-31 even when the carbon atom bearing the halogen is separated from the cationic site by an “insulating” sp3-hybridized carbon, as would be the case here for a polar cyclization mechanism. For example, the pKa of the ethylammonium cation is 10.8, whereas that of the 2,2,2-trifluoroethylammonium cation is 5.6.32-33 In this case, trifluorination diminishes the pKa of the β nitrogen substituent by ~ 5 units, corresponding to a relative destabilization of the cation by almost 30 kJ/mol (at 20 °C).34 Extrapolated to the proposed C1 carbocation intermediate in the HppE reaction, this effect would correspond to an increase in its reduction potential of ~ 300 mV. It seems unlikely that carbocation formation would remain robust to such a severe perturbation, implying that a radicaloid mechanism for C2–O ↔ C1 coupling is more likely. To provide additional evidence for this conclusion, we tested the (S)-2-HPP analogs containing either (i) halogens at both C3 (F3) and C1 (chlorine) or (ii) fluorine at C1 for the ability to serve as HppE substrates. In the case of the 1-Cl-3-F3 analogue, we were unable to obtain the single 1S,2S diastereomer that would be expected to project its C1-H toward the iron cofactor appropriately for H• abstraction, but we did verify by 19F-NMR analysis that the pure 1R,2S compound (1f) – expected to direct the chlorine toward the cofactor – is not a substrate for wild-type HppE (Figure 5, top; Figure S17B-C). Moreover, we found (also by 19F-NMR) that the racemic anti compound (rac-1g, consisting of a mixture of 1S,2S-1g and 1R,2R-1g) could be converted by HppE and its F182A variant to a mixture of the 2-ketone (4g), cis-epoxide (2g) and trans-epoxide (3g) products (Figure 5, bottom). Presumably, 1R,2R-1g is, by analogy to the reaction of (R)-2-HPP,23 dehydrogenated to the 2-ketone (4g), and 1S,2S-1g is successfully cyclized to the mixture of epoxides (Figure 5B, second from bottom). As for the native and 3-Fn-substituted substrates (1, 1a-c), we found that the F182A variant makes less of the cis epoxide (2g) from the rac-1g than does the wild-type enzyme (Figure 5B, bottom; Figure S17E-F).
Figure 5.

Summary of outcomes and 19F NMR analysis in the reactions of the wild-type (wt) and F182A HppE proteins with (1R,2S)-1-Cl-3-F3-HPP [(1R,2S)-1f] and rac-1-Cl-3-F3-2-HPP [(1S,2S)- and (1R,2R)-1g] compounds. (A) Reaction schemes depicting C1 pro-R hydrogen abstraction and formation of the cis and trans epoxides (2g and 3g, respectively) by the two proteins. aThe 2g:3g ratio given is the mean value from three 19F -NMR assays with the standard deviations given in parentheses. (B) 19F-NMR spectra of the reactions of (1R,2S)-1f with HppE (second from top), rac-1g with HppE and the F182A variant (second from bottom and bottom). Assignment of structures of products 2g, 3g, and 4g refers 19F NMR analyses of cis-, trans-F3-Fos and 3-F3-2-keto compounds (2c, 3c and 4c). Single peaks of 4g (second from bottom) and 4c (table S11) at ~−81-~−87 ppm correspond to C2 dehydrogenation. The doublets at −64.7 and −66.6 ppm correspond to cis- and trans-1-Cl-3-F3-Fos (2g and 3g), respectively, in analogy with the doublets at −64.2, −74.1 ppm of cis and trans-3-F3-Fos (2c and 3c). The single peak at −78.7 is from the internal standard sodium triflate.
We did succeed in synthesizing the desired single diastereomer of the 1-fluoro analogue, (1R,2S)-1-F-HPP (1h; note that the substituent priority order changes in the 1-F analog relative to the 1-Cl compound, Figure 6A). High-resolution mass spectra (HRMS; Figure 6B) of 1h (top), its reactions with wild-type HppE (second from top) or the trans-epoxide-favoring F182A variant (third from top), and the synthetic 2-ketone derivative, 4h (bottom), revealed that both HppE proteins effect a net dehydrogenation (removal of two hydrogens) of the 1-F-substituted analog. The corresponding 31P-NMR spectra (Figure 6C; Figure S18B-E) reveal a pair of major oxidation products with 31P resonances at δ ~ 2.5 ppm and ~ 3.5 ppm. These resonances are 6-7 ppm upfield from that of 1h (Figure 6C), similar to the 8-11 ppm shifts seen upon epoxidation of the 3-Fn-substituted compounds (1, 1a-c). The 31P spectra of both products reveal large geminal hyperfine couplings to 19F (2JP-F ~ 98 and 102 Hz) – as also seen for 1h and 4h – but neither product-associated resonance exhibits an obvious geminal coupling to 1H, unlike in 1h and 4h (Figure 6C; Figure S18C-E). This latter observation implies that one of the two hydrogens removed in the transformation of 1h to the pair of major products is from C1, as occurs in epoxidation of the other substrates. The NMR data thus show that HppE can cyclize 1h, despite the expectation that a hypothetical C1-carbocation intermediate would be severely destabilized by the fluorine substituent.
Figure 6.

Summary of stereochemistry, mass-spectrometric and NMR-spectroscopic characterization of the products generated by wild-type HppE and its F182A variant from (1R,2S)-1-F-HPP (1h). (A) Reaction schemes depicting C1 pro-R hydrogen abstraction and formation of the cis and trans epoxides (2h and 3h, respectively) by the two proteins. aEach cis:trans ratio given is the mean value from three 31P -NMR assays; bValues given were determined by 31F-NMR analysis, and the standard deviations given in parentheses are errors associated with each percentage value. (B) HRMS of synthetic 1h (top), the reactions of 1h with wild-type HppE (second from top) and its F182A variant (third from top), and synthetic 1-fluoro-2-oxo-propylphosphonate, 4h (bottom), another possible product of HppE-mediated oxidation (from C2 dehydrogenation) of 1h. (C) 31P NMR spectra showing production of primarily cis epoxide, 2h, in the reaction of the wild-type enzyme (second from top) and trans epoxide, 3h, in the reaction of the F182A variant (third from top). The flanking spectra are – as in panel B – of the 1h (top) and 4h (bottom) standards. Note that both 1h and 4h give rise to doublet-of-doublets (dd) spectra as a result of large geminal couplings of the 31P nucleus to the 19F (larger coupling) and 1H (smaller coupling) nuclei that are also bonded to C1, whereas both 2h and 3h give rise to doublet (d) features lacking the smaller geminal coupling to the C1 1H, which is removed in their formation from 1h. The multiplet at 22.9 ppm is from the internal standard, sodium propylphosphonate. (D) 19F-NMR spectra of the same samples as in panel B. The triplet at −217 ppm is from the internal standard, sodium fluoroacetate. (E) Blow-up of the 19F-NMR spectra of 2h and 3h, showing geminal couplings (2JF-P) consistent with those seen in the 31P-NMR spectra.
For each of the four pairs of 3-Fn-substituted (n = 0-3) cis and trans epoxides produced by HppE from the corresponding 3-Fn-(S)-2-HPP analog, the cis epoxide (2, 2a-c) has the lesser 31P δ, and the trans epoxide has the greater 31P δ. For the cases of 2/3 and 2c/3c, this trend was verified directly, by characterization of synthetic or commercially available standards, whereas for 2a/3a and 2b/3b, the rank order of the vicinal 1H-1H couplings (3JH-H) in each pair confirmed that the trend holds throughout the 3-Fn (n = 0-3) series. This trend would imply that the product from 1h with δ ~ 2.5 ppm is the cis epoxide, 2h, and the product with δ ~ 3.5 ppm is the trans epoxide, 3h. The absence of a 1H nucleus on C1 in these products precludes use of the rank order of 3JH-H values for confirmation of the assignment, but the 19F spectra of the products (Figure 6D-E) provide further verification. The two major products, tentatively assigned above as 2h and 3h, exhibit 19F resonances at δ ~ −137 and ~ −159 ppm, respectively. Blow-ups of their 19F spectra (Figure 6E) reveal, in addition to the major doublet splitting from the geminal 31P nucleus (2JF-P), smaller hyperfine couplings on each line of the doublet arising from 1H nuclei on C2 (3JF-H, doublet) and C3 (4JF-H, quartet). Deeper (derivative) analysis of the spectra revealed that the resonance with 19F δ ~ −137 ppm has the larger of the two vicinal F-H couplings (3JF-H 4.2 Hz compared to 1.5 Hz), suggesting that the associated product is the cis epoxide, 2h. With this assignment confirmed, it can be seen that, as in epoxidation of 1, 1a-c, and 1g, wild-type HppE markedly favors the cis product (2h:3h ~ 93:7), whereas the F182 variant markedly favors the trans product (2h:3h ~ 10:90) (Figure 6C-D; Figure S18). The consistent, pronounced shift to C1 retention and cis-epoxide formation caused by the F182A substitution implies that all of the analogs undergo epoxidation by a common mechanism. The combined results thus decisively favor a radicaloid ring-closure mechanism (Scheme 1B, red) – as supported by the computational analysis of Shaik and co-workers9 – over the previously proposed polar coupling via the C1 carbocation (Scheme 1B, orange and green).
Antimicrobial potencies of the (halogenated) cis and trans epoxide products.
The accumulated sets of substrate analogues and HppE variants afforded a panel of cis- and trans-epoxide compounds, which we evaluated for antimicrobial potency in comparison to the clinical antibiotic, Fos. We measured the areas of the zones of growth inhibition created by spotting reaction solutions on a lawn of Escherichia coli K12.2, 28 We first established a linear correlation between the area of inhibition and the concentration of commercial Fos within the range of 0.15 mM < [Fos] < 2 mM (Figure S19). We then validated use of raw reaction solutions for testing potency by confirming that the solution with wild-type HppE, 1.5 mM of the native substrate, and excess H2O2 (producing ~ 1.42 mM 2 and 0.08 mM 3) gave a zone of inhibition indistinguishable from that of 1.5 mM of pure commerical Fos. We next measured inhibition areas created by reactions that were, by choice of substrate (each at 1.5 mM with excess H2O2) and HppE variant, enriched in either 3 or the cis- or trans-epoxide product of 3-F- (2a or 3a), 3-F2- (2b or 3b), or 3-F3-(S)-2-HPP (2c or 3c) (Figure S20, 21). The reaction solution enriched in trans epoxide, 3 [F182A variant with native (S)-2-HPP], was ~ 50% less potent than that enriched in 2, and reactions enriched in any of the C3-fluorinated epoxides (either cis or trans) were at least 50% less potent than those enriched in 3. The uniformly deleterious impact of C3 fluorination suggests that increased electrophilicity of the epoxide warhead does not, at least in this bioassay, potentiate its activity. The fluorinated compounds may have diminished affinity for MurA or may suffer from increased decomposition (e.g., by hydrolysis) prior to reaching their target. More importantly, the implication of the diminished potency of the trans epoxide, 3, is that the steric enforcement of C1 inversion and cis-epoxide formation by HppE is important for its biological function. For the case of this enzyme, it appears that necessity was indeed the mother of invention.
Possible implications for mechanism of H•-abstraction step.
The mechanism deduced here for the HppE reaction – H• abstraction, radical inversion, and radicaloid ring closure – shares aspects of that previously elucidated for the first of two reactions catalyzed by the iron(II)- and 2-oxoglutarate-dependent (Fe/2OG) oxygenase, carbapenem synthase (CarC). CarC promotes the stereoinversion (epimerization) of C5 of (3S,5S)-carbapenam-3-carboxylate, yielding the 3S,5R compound, before desaturating between C2 and C3 to generate its (5R)-carbapenem final product. In the stereoinversion, H• is removed from C5 by a ferryl intermediate, and Tyr165 then donates H• to the opposite side of the bicyclic system.35 Calculations have suggested that the structure of the intermediate itself drives the facile inversion of the initial 5S radical to the 5R radical in a butterfly-like motion of the bicyclic system that is predicted to have a relatively low activation barrier (~ 24 kJ/mol) and a negative free-energy change (~ – 7 kJ/mol). It was proposed that this motion, which would be expected to move C5 away from the iron cofactor, is crucial to the avoidance of C5 hydroxylation via the generally facile coupling of the initial 5S radical to the Fe(III)-coordinated hydroxide ligand, a step known generally as “oxygen rebound.” One could envisage also for the possibility of H• abstraction by a ferryl complex in HppE (Scheme S1A) that the demonstrated inversion of the C1 radical – driven in this case by steric contacts with the enzyme – might be essential for avoidance of oxygen rebound. However, for the cases of the HppE variant and (S)-2-HPP analogs that yield ring closure with predominant retention of C1, one might then expect oxygen rebound to compete with cyclization, resulting in formation of some of the C1-hydroxylated product. It is tempting to interpret the fact that we did not, for any combination of variant enzyme and substrate analog, observe such competition from C1 hydroxylation as evidence against H• abstraction by a ferryl complex (Scheme S1A) and in favor of the proposal of Shaik and co-workers that H• abstraction is effected a by a hydrogen-bonded hydroxyl radical (Scheme S1B), a mechanism that could, by virtue of greater separation of the cofactor and C1 radical, potentially preclude oxygen rebound. Definitive resolution of this key mechanistic issue awaits experimental determination of the oxidation state of the cofactor (+IV or +III) immediately before H• abstraction.
CONCLUSIONS
A remarkable robustness of HppE to both sterically unconservative amino-acid substitutions in its active site and stereo-electronically perturbing modifications to its substrate has enabled elucidation of the mechanism of its C–O-coupling step. The results experimentally validate two of the principal conclusions from the recent computational study of Shaik and co-workers, who posited that facile inversion of the C1 radical and radicaloid C–O-coupling produce the cis epoxide of the antibiotic.9 Moreover, the experimental results both identify specific enzyme-substrate/product contacts that drive C1-radical inversion and show that perturbation of these contacts allows the outcome to be tuned from vastly predominant C1-inversion and cis-epoxide formation to vastly predominant C1-retention and trans-epoxide formation. The conclusion of a radicaloid coupling step is likely to apply also to reactions of other non-heme-iron enzymes that form C–O and other carbon-heteroatom bonds. For example, the Fe/2OG enzyme, hyoscyamine 6β-hydroxylase (H6H), installs the epoxide of the plant-derived anesthetic drug, scopolamine, by sequential hydroxylation and cyclization steps.36 Because the radicaloid nature of the epoxidation step would almost certainly require iron coordination of the C6 oxygen that undergoes coupling to C7 in the second step, the analysis predicts that coordination of the heteroatom to the ferryl complex, its Fe(III)–OH successor, or both, might be observable in H6H and, more generally, in other, analogous enzymatic carbon-heteroatom-coupling reactions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (GM113106 to J.M.B. and C.K., GM127079 to C.K., GM119707 to A.K.B., and 1K99GM129460-01 to K.M.D.) We thank Dr. Tapas K. Mal and Dr. Carlos N. Pacheco for helpful discussions of the NMR experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Synthetic methods and spectroscopic data on compounds; procedures for preparation of wild-type and variant HppE enzymes; procedures and data for quantifying enzyme reaction outcomes; Tables assigning NMR resonances of synthetic and enzyme products; methods and data to determine antimicrobial potencies of Fos analogs; NMR spectra of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Gehringer M; Laufer SA, Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J. Med. Chem 2019, 62 (12), 5673–5724. [DOI] [PubMed] [Google Scholar]
- 2.Hendlin D; Stapley EO; Jackson M; Wallick H; Miller AK; Wolf FJ; Miller TW; Chaiet L; Kahan FM; Foltz EL; Woodruff HB, Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science 1969, 166 (3901), 122–123. [DOI] [PubMed] [Google Scholar]
- 3.Marquardt JL; Brown ED; Lane WS; Haley TM; Ichikawa Y; Wong CH; Walsh CT, Kinetics, stoichiometry, and identification of the reactive thiolate in the inactivation of UDP-GlcNAc enolpyruvoyl transferase by the antibiotic fosfomycin. Biochemistry 1994, 33 (35), 10646–10651. [DOI] [PubMed] [Google Scholar]
- 4.Kim DH; Lees WJ; Kempsell KE; Lane WS; Duncan K; Walsh CT, Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 1996, 35 (15), 4923–4928. [DOI] [PubMed] [Google Scholar]
- 5.Thibodeaux CJ; Chang W.-c.; Liu HW, Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev 2012, 112 (3), 1681–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu PH; Murakami K; Seki T; He XM; Yeung SM; Kuzuyama T; Seto H; Liu HW, Protein purification and function assignment of the epoxidase catalyzing the formation of fosfomycin. J. Am. Chem. Soc 2001, 123 (19), 4619–4620. [DOI] [PubMed] [Google Scholar]
- 7.Wang C; Chang W.-c.; Guo Y; Huang H; Peck SC; Pandelia M-E; Lin GM; Liu HW; Krebs C; Bollinger JM Jr., Evidence that the fosfomycin-producing epoxidase, HppE, Is a non-heme-iron peroxidase. Science 2013, 342 (6161), 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krebs C; Galonic Fujimori D; Walsh CT; Bollinger JM Jr., Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res 2007, 40 (7), 484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang BJ; Lu JR; Dubey KD; Dong G; Lai WZ; Shaik S, How do enzymes utilize reactive OH radicals? Lessons from nonheme HppE and Fenton systems. J. Am. Chem. Soc 2016, 138 (27), 8489–8496. [DOI] [PubMed] [Google Scholar]
- 10.Chang W.-c.; Mansoorabadi SO; Liu HW, Reaction of HppE with substrate analogues: evidence for carbon-phosphorus bond cleavage by a carbocation rearrangement. J. Am. Chem. Soc 2013, 135 (22), 8153–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roach PL; Clifton IJ; Hensgens CMH; Shibata N; Schofield CJ; Hajdu J; Baldwin JE, Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature 1997, 387 (6635), 827–830. [DOI] [PubMed] [Google Scholar]
- 12.Burzlaff NI; Rutledge PJ; Clifton IJ; Hensgens CMH; Pickford M; Adlington RM; Roach PL; Baldwin JE, The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature 1999, 401 (6754), 721–724. [DOI] [PubMed] [Google Scholar]
- 13.Tamanaha E; Zhang B; Guo Y; Chang W.-c.; Barr EW; Xing G; St Clair J; Ye SF; Neese F; Bollinger JM Jr.; Krebs C, Spectroscopic evidence for the two C-H-cleaving intermediates of Aspergillus nidulans isopenicillin N synthase. J. Am. Chem. Soc 2016, 138 (28), 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galonic DP; Barr EW; Walsh CT; Bollinger JM Jr.; Krebs C, Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat. Chem. Biol 2007, 3 (2), 113–116. [DOI] [PubMed] [Google Scholar]
- 15.Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr., Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (42), 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB 3rd; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM Jr.; Solomon EI, Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499 (7458), 320–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehmood R; Qi HW; Steeves AH; Kulik HJ, The protein’s role in substrate positioning and reactivity for biosynthetic enzyme complexes: the case of SyrB2/SyrB1. Acs Catalysis 2019, 9 (6), 4930–4943. [Google Scholar]
- 18.McCarthy EL; Booker SJ, Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 2017, 358 (6361), 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao LZ; Stich TA; Fugate CJ; Jarrett JT; Britt RD, EPR-derived structure of a paramagnetic intermediate generated by biotin synthase BioB. J. Am. Chem. Soc 2018, 140 (40), 12947–12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP, Observation of radical rebound in a mononuclear nonheme iron model complex. J. Am. Chem. Soc 2018, 140 (12), 4191–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen BG; Leanza WJ; Beattie TR; Patchett AA; Arison BH; Ormond RE; Kuehl FA; Alberssc. G; Jardetzky O, Phosphonomycin: structure and synthesis. Science 1969, 166 (3901), 123–125. [DOI] [PubMed] [Google Scholar]
- 22.Chang W.-c.; Dey M; Liu PH; Mansoorabadi SO; Moon SJ; Zhao ZBK; Drennan CL; Liu HW, Mechanistic studies of an unprecedented enzyme-catalysed 1,2-phosphono-migration reaction. Nature 2013, 496 (7443), 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z; Liu P; Murakami K; Kuzuyama T; Seto H; Liu HW, Mechanistic studies of HPP epoxidase: configuration of the substrate governs its enzymatic fate. Angew. Chem. Int. Ed 2002, 41 (23), 4529–4532. [DOI] [PubMed] [Google Scholar]
- 24.Woschek A; Wuggenig F; Peti W; Hammerschmidt F, On the transformation of (S)-2-hydroxypropyl-phosphonic acid into fosfomycin in Streptomyces fradiae: a unique method of epoxide ring formation. ChemBioChem 2002, 3 (9), 829–835. [DOI] [PubMed] [Google Scholar]
- 25.Higgins LJ; Yan F; Liu PH; Liu HW; Drennan CL, Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature 2005, 437 (7060), 838–844. [DOI] [PubMed] [Google Scholar]
- 26.Yun D; Dey M; Higgins LJ; Yan F; Liu HW; Drennan CL, Structural basis of regiospecificity of a mononuclear iron enzyme in antibiotic fosfomycin biosynthesis. J. Am. Chem. Soc. 2011, 133 (29), 11262–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peric Simov B; Wuggenig F; Lämmerhofer M; Lindner W; Zarbl E; Hammerschmidt F, Indirect evidence for the biosynthesis of (1S,2S)-1,2-epoxypropylphosphonic acid as a co-metabolite of fosfomycin [(1R,2S)-1,2-epoxypropylphosphonic acid] by Streptomyces fradiae. Eur. J. Org. Chem 2002, 2002 (7), 1139–1142. [Google Scholar]
- 28.McLuskey K; Cameron S; Hammerschmidt F; Hunter WN, Structure and reactivity of hydroxypropylphosphonic acid epoxidase in fosfomycin biosynthesis by a cation- and flavin-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (40), 14221–14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katagiri T; Yamaji S; Handa M; Irie M; Uneyama K, Diastereoselectivity controlled by electrostatic repulsion between the negative charge on a trifluoromethyl group and that on aromatic rings. Chem. Commun 2001, (20), 2054–2055. [DOI] [PubMed] [Google Scholar]
- 30.Blanksby SJ; Ellison GB, Bond dissociation energies of organic molecules. Acc. Chem. Res 2003, 36 (4), 255–263. [DOI] [PubMed] [Google Scholar]
- 31.Kim CK; Lee KA; Bae SY; Han IS; Kim CK, Theoretical studies on the hydride ion affinities of carbocations. Bull. Korean Chem. Soc 2004, 25 (2), 311–313. [Google Scholar]
- 32.Schlosser M, Parametrization of substituents: effects of fluorine and other heteroatoms on OH, NH, and CH acidities. Angew. Chem. Int. Ed 1998, 37 (11), 1497–1513. [DOI] [PubMed] [Google Scholar]
- 33.Washabaugh MW; Stivers JT; Hickey KA, C(α)-proton transfer from 2-(1-hydroxybenzyl)oxythiamin: the unit brønsted slope overestimates the amount of bond formation to the base catalyst in the transition state. J. Am. Chem. Soc 1994, 116 (16), 7094–7097. [Google Scholar]
- 34.Lankin DC; Chandrakumar NS; Rao SN; Spangler DP; Snyder JP, Protonated 3-fluoropiperidines: an unusual fluoro directing effect and a test for quantitative theories of solvation. J. Am. Chem. Soc 1993, 115 (8), 3356–3357. [Google Scholar]
- 35.Chang W.-c.; Guo Y; Wang C; Butch SE; Rosenzweig AC; Boal AK; Krebs C; Bollinger JM Jr., Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science 2014, 343 (6175), 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda J; Okabe S; Hashimoto T; Yamada Y, Molecular-cloning of hyoscyamine 6β-hydroxylase, a 2-oxoglutarate-dependent dioxygenase, from cultured roots of Hyoscyamus-niger. J. Biol. Chem 1991, 266 (15), 9460–9464. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.