

Abstract
The treatment of tumors driven by overexpression or amplification of MYC oncogenes remains a significant challenge in drug discovery. Here, we present a new strategy towards the inhibition of MYC via the disruption of the protein-protein-interaction between MYC and its chromatin cofactor WDR5. Blocking the association of these proteins is hypothesized to disrupt the localization of MYC to chromatin, thus disrupting the ability of MYC to sustain tumorigenesis. Utilizing a high-throughput screening campaign and subsequent structure-guided design, we identify small molecule inhibitors of this interaction with potent in vitro binding affinity, and report structurally related negative controls that can be used to study the effect of this disruption. Our work suggests that disruption of this protein-protein interaction may provide a path toward an effective approach for the treatment of multiple tumors, and anticipate that the molecules disclosed can be used as starting points for future efforts toward compounds with improved drug-like properties.
Keywords: WDR5, MYC, RBBP5, structure-based design, high-throughput screening
Graphical Abstract
INTRODUCTION
MYC oncogenes encode a family of related transcription factors (C-, L- and N-MYC) that are overexpressed in a significant number of malignancies.1,2 The oncogenic effects of these MYC proteins are caused by their action as sequence-specific DNA binding transcription factors that drive the expression of genes required for cell growth, proliferation, metabolism, genome instability, and apoptosis.3 MYC proteins are intrinsically disordered, with minimal secondary structure; as such, they lack traditional binding sites for direct inhibition and have been considered to be ‘undruggable.’4 Despite significant effort and investment toward the identification of compounds that can directly affect MYC function, few successes have been reported to date.5,6 New approaches to MYC-targeted therapies may focus on the inhibition of the protein-protein interactions (PPI’s) of MYC with critical partner proteins.6,7 For example, much work on the MYC-MAX heterodimer has been reported,8–12 and the direct interactions of MYC with other proteins, such as Aurora A, have recently garnered interest.13–15 We have previously reported on the identification of WD repeat-containing protein 5 (WDR5) as a crucial partner in the facilitated recruitment of MYC to chromatin,16 thus making it a critical co-factor for MYC-driven tumorigenesis. Further we have defined the WDR5-MYC interaction site as a potentially tractable target for small molecule inhibition of MYC-driven tumors.16,17
WD-repeat proteins are a ubiquitous family of scaffolding proteins, containing β-propeller domains that form donut-shaped structures which participate in many multi-protein complexes.18,19 In yeast, WD-repeats are reported to participate in more interactions than any other protein domain.20 The direct targeting of these multi-protein complexes make the WD-repeat family an interesting, although clinically unvalidated, target class in drug discovery.21 WDR5 is one such regulatory scaffolding protein that has reported involvement in protein complexes related to chromatin structure and epigenetic machinery.22 The protein has two major binding sites, previously referred to as the WDR5 Binding Motif (WBM) and WDR5 Interaction Motif (WIN) sites (Figure 1A and B, respectively), that have each been demonstrated to be key interfaces for its role in complex formation.23–25
Figure 1.

An overlay of WDR5 bound to MYC and MLL peptides (PDB: 4Y7R & 3EG6 respectively). A) Visualization of the MYC MbIIIb peptide bound to a cleft of WDR5, referred to as the ‘WBM’ binding site; B) The MLL1 peptide binds on the reverse face of WDR5 with an arginine residue deep into the ‘WIN’ site binding pocket.
We previously described that the association of WDR5 with MYC occurs at the WBM site, and confirmed by co-immunoprecipitation that WDR5 binds to the central-portion of MYC via the evolutionarily conserved ‘MYC box’ MbIIIb motif. The binding surface is a shallow, hydrophobic cleft containing multiple basic residues, that binds to a 10-amino acid chain containing a “EEIDVV” motif that is conserved in all extant MYC proteins. This same binding cleft has been shown to also bind other proteins; those reported to-date include RBBP5 in the MLL1/SET complex,26,27 and KANSL2, a protein critical for the histone acetyltransferase (HAT) activity of the NSL complex.28 These proteins (MYC, RBBP5, KANSL2) contain a highly similar binding motif (IDVV, LDVV, VDVT respectively) that engage WDR5 in a structurally homologous way. On the opposite face of the protein is the WIN-binding site, which is well studied and contains a deep binding pocket that binds mixed lineage leukemia 1 (MLL1) via a conserved arginine containing motif known as the WIN-motif.23–25 The ability of MLL1 to act as a histone methyltransferase (HMT) and catalytically methylate histone H3 lysine 4 (H3K4) has been demonstrated to be reliant on the formation of a four-protein complex, including WDR5, RBBP5, ASH2L and DPY30 (WRAD complex).26,29,30 Multiple small molecules that disrupt this complex by binding to WDR5 at the WIN binding site have now been published, including both peptidomemetics31–33 and small molecule inhibitors.34–38
Due to the tertiary structure of the protein, targeting the WIN site is expected to be much more straightforward than discovering potent small molecules against the WBM site. It has been demonstrated that targeting the central cavity of WD-repeat proteins leads to inhibitors that have significantly higher potency than compounds that occupy cavities on the outer surfaces,39 highlighting the challenge presented in targeting this MYC-binding site. For example, CDC20 and Cdc4 are WD-repeat proteins for which inhibitors are reported that bind at such ‘external’ sites with affinities of >10 μM and 6.2 μM, respectively.40,41 Interestingly, the Cdc4 inhibitor, SCF-12, is reported to occlude substrate binding into the central cavity through a long-range allosteric effect.40 To date, however, there are no reports of a small molecule probe that directly binds to the WBM cleft used by MYC and RBBP5.
Here, we describe the discovery of the small molecules that bind to the WBM-binding cleft. Utilizing structure-based design approaches based upon our previously published structure of WDR5, we have improved the affinity of the screening hits. Further, we characterize the utility of the lead compounds for their ability to disrupt binding of WDR5 to MYC and RBBP5, and demonstrate that they are capable of functional inhibition of the WRAD complex.
RESULTS AND DISCUSSION
Identification and validation of hits
A high-throughput screen (HTS) was performed using a competition fluorescence polarization-based (FP) assay to assess interruption of the binding of a FITC-labeled peptide probe derived from the MYC MbIII protein sequence to WDR5 (aa. 23–334).16 Compounds from the Vanderbilt Discovery and VICB collections (~250,000 total) were screened at 50 μM, and 410 preliminary ‘hit’ compounds that demonstrated greater than 15% inhibition of the interaction were identified (a hit rate of 0.16%). After duplicate screening, 110 compounds were confirmed as hits; each of these was subsequently evaluated using a ten-point dose-response protocol to calculate binding affinity from fresh DMSO compound dilutions. 76 compounds afforded a measurable Kd value and were taken forward as MYC-site hits (0.03% confirmed hit-rate).
As the HTS was based upon a competition assay, we also validated the hits in an orthogonal biophysical assay, collecting SOFAST 1H-15N heteronuclear multiple quantum coherence (HMQC) spectra of WDR5 to determine direct binding of the compounds. Peak shifts that correspond to binding at the WBM-binding site were identified by obtaining an HMQC spectrum of uniformly 15N labeled WDR5 with the unlabeled MYC-derived peptide. Identification of this chemical shift perturbation (CSP) pattern allowed us to monitor compound binding to this site by NMR. Screening hits which did not cause any CSP to the NMR spectra of WDR5 were removed from consideration for follow-up chemistry. Validated hits induced either peak shift (Figure 2A) or peak broadening (Figure 2B). These findings suggest that different HTS hits presumably exhibited an intermediate and fast exchange respectively, based upon the NMR chemical shift timescale. These exchange regimes suggest Kd values in the single-digit micromolar (intermediate exchange) or high micromolar (fast exchange), range consistent with the primary FP assay, ranging from 8–175 μM. Multiple confirmed hits contain a bi-aryl sulfonamide motif, with many of the examples also containing an acidic motif that may interact with the charged residues that surround the MYC binding site of WDR5 (Exemplar hits shown in Figure 3).
Figure 2.

Example 1H–15N SOFAST-HMQC spectra showing the observable effect of compounds that A) induce peak shift; and B) induce peak broadening. Apo-WDR5 (Blue), Hit-WDR5 (Red)
Figure 3.

Representative hits that bind to WDR5 at the WBM-site. Kd values obtained by a fluorescence polarization assay utilizing a FITC-labeled peptide based upon the MbIIIb sequence.
X-ray co-crystal structure of WDR5 bound with a WBM-site hit
We obtained an X-ray co-crystal structure of the leading hit, 1, bound to WDR5 by co-crystallization. The overlay of this structure with those of published peptides confirmed that the small molecule bound into the WBM site that mediates the binding of WDR5 to MYC and RBBP5 (Figure 4A). The chlorine atom on the aniline ring is well positioned to mimic the hydrophobic side-chain of the isoleucine of the IDVV motif, while the methoxy group provides a potential vector for compound growth. Key interactions responsible for compound binding include: 1) a hydrogen bond from an oxygen of the sulfonamide to the backbone NH of ASN225; and 2) a halogen bonding interaction from the aromatic bromine to the carbonyl of TRP273 (Figure 4B). These interactions appear to cause a shift of the ASN225 containing loop towards the ligand.
Figure 4.

A) An overlay of hit compound 1 with the structure of bound MYC-derived MbIIIb peptide (PDB: 4Y7R), demonstrating the frame shift of the loop enabling retainment of the H-bond interaction with the backbone; WDR5:1 complex (green), WDR5:peptide complex (grey). B) Zoom of the binding site of the co-crystal structure of WDR5 and 1 (PDB: 6U6W). Shown as dashes are the halogen bonding interaction between the bromine atom and Trp273 and the hydrogen bond between the sulfonamide oxygen and Asn225; key amino acid residues are shown as sticks and labeled.
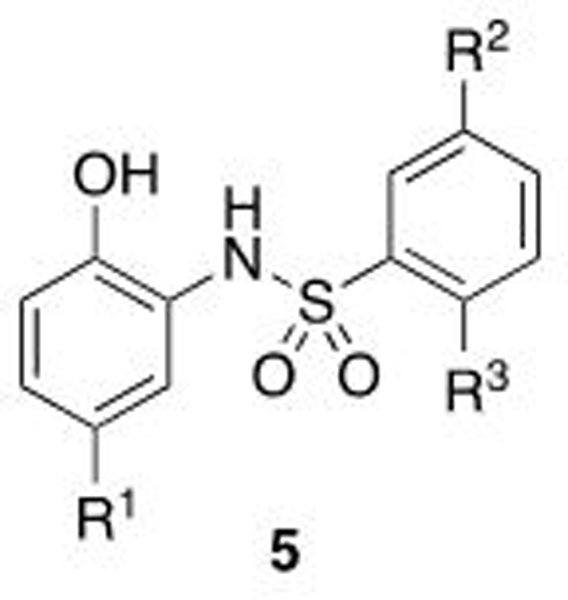
Early SAR studies
After confirmation of the hit compounds, the focus of our initial synthetic efforts was to investigate the two halogen substituents, the aniline ring chlorine and the sulfonyl ring bromide. The bromine atom forms a key halogen bonding interaction in the X-ray structure. Indeed, when this bromine atom is removed (5a, Table 1) all binding affinity for WDR5 is lost, while replacement with either a chlorine or an iodide both affords a compound with reduced Kd (5b, 5c). Interestingly, removing any one of the four substituents that decorate the biaryl-sulfonamide substructure proved highly deleterious (5d, 5e), demonstrating that this substitution pattern shows good shape complementarity for this binding surface. Similarly, we observed a consistent, significant improvement in affinity when switching from a methoxy R3 group to a phenol (5f).
Table 1.
Early SAR of HTS hit, 1.
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | FPA Kd (μM)a |
| 1 | Cl | Br | OMe | 4.71 ± 1.59 |
| 5a | Cl | H | OMe | > 40 |
| 5b | Cl | Cl | OMe | 8.41 ± 0.57 |
| 5c | Cl | I | OMe | 8.39 ± 2.47 |
| 5d | Cl | Br | H | 36.1 ± 7.06 |
| 5e | H | Br | OMe | > 40 |
| 5f | Cl | Br | OH | 1.25 ± 0.46 |
| 5g | Ph | Br | OMe | 37.0 ± 6.79 |
| 5h | t-Bu | Br | OMe | 3.78 ± 1.92 |
| 5i | c-Pr | Br | OH | 0.818 ± 0.44 |
| 5j | OCF3 | Br | OH | 0.549 ± 0.04 |
| 5k | SF5 | Br | OH | 0.110 ± 0.08 |
| 5l | 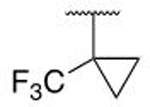 |
Br | OH | 0.547 ± 0.11 |
| 5m | 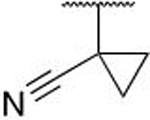 |
Br | OH | 0.852 ± 0.36 |
| 5n | 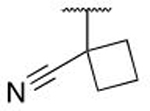 |
Br | OH | 0.289 ± 0.19 |
| 5o | 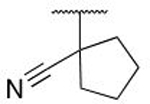 |
Br | OH | 0.614 ± 0.33 |
| 5p | 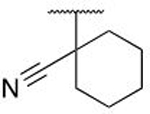 |
Br | OH | 0.229 ± 0.03 |
Data displayed is the average of at least 3 independent replicates ± standard deviation.
Early in the project we observed compounds that were able to bind below the lower limit of detection of the HTS FP assay that used the FITC-labeled MYC peptide (to Kd < 2.5 μM). The data for hits shown in Figure 3 corresponds to this peptide-based probe used throughout the screening campaign. We later designed and synthesized fluorescein-labeled small-molecule probes with higher affinity for WDR5 compared to the peptide, and thus offered lower limits of detection in the FP assay and correspondingly our interpretation of SAR. For clarity, data shown in Tables 1–3 utilize only a single generation of small-molecule fluorescein probe (see structure in Supplemental Figure S1), hence the difference in Kd value of 1 reported in Figure 3, from the high throughput screening campaign, and Table 1.
Table 3.
Structures and biochemical data for salicylamide derived WDR5 WBM-site inhibitors
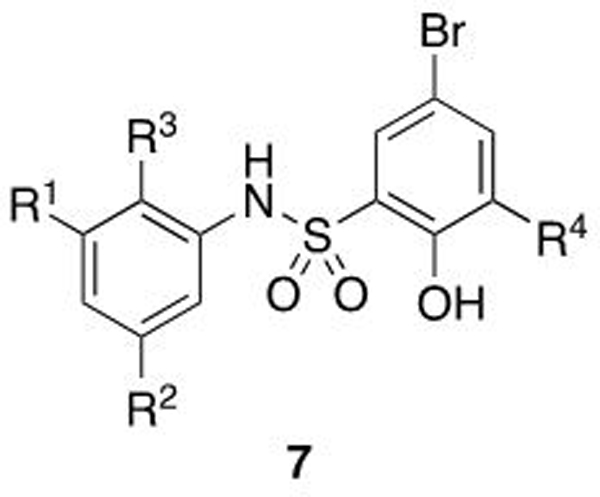 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | FPA Kd (μM) a |
| 7a | 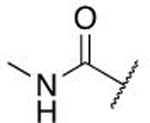 |
 |
OH | H | 0.746 ± 0.033 |
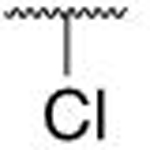
| 7b | Cl | 0.298 ± 0.087 | |||
| 7c | 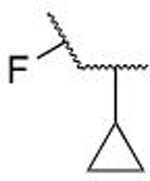 |
OH | H | 1.42 ± 1.59 | |
| 7d | Cl | 0.238 ± 0.089 | |||
| 7e | 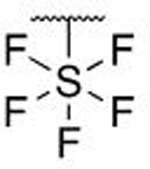 |
H | H | 0.848 ± 0.187 | |
| 7f | OH | H | 0.204 ± 0.030 | ||
| 7g | Cl | 0.190 ± 0.032 | |||
| 7h |  |
H | H | 1.08 ± 0.43 | |
| 7i | Cl | 0.128 ± 0.020 | |||
| 7j | OH | H | 0.566 ± 0.024 | ||
| 7k | Cl | 0.069 ± 0.034 | |||
| 7l | 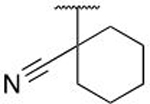 |
OH | H | 0.779 ± 0.484 | |
| 7m | Cl | 0.362 ± 0.040 | |||
| 7n b |  |
 |
OH | H | 0.046 ± 0.014 |
| 7o | 0.757 ± 0.017 | ||||
| 7p | 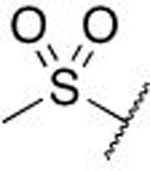 |
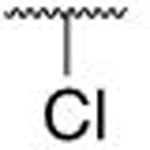 |
H | H | 1.88 ± 0.48 |
| 7q | Cl | 0.220 ± 0.031 | |||
| 7r | OH | H | 1.07 ± 0.30 | ||
| 7s | Cl | 0.123 ± 0.05 | |||
| 7t | 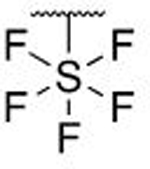 |
H | H | 0.511 ± 0.050 | |
| 7u | Cl | 0.093 ± 0.001 | |||
| 7v | OH | H | 0.167 ± 0.049 | ||
| 7w | Cl | 0.045 ± 0.013 | |||
| 7x | 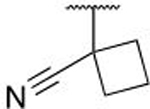 |
H | Cl | 0.215 ± 0.058 | |
| 7y | OH | H | 0.349 ± 0.174 | ||
| 7z | Cl | 0.110 ± 0.057 | |||
Data displayed is the average of at least 3 independent replicates ± standard deviation;
Data is the average of two independent replicates.
On the aniline ring, a chlorine atom occupies the same region as the isoleucine of the native IDVV motif, and as such the chlorine was substituted for a range of hydrophobic groups that were hypothesized to more optimally occupy this region of the protein. Replacement of the halogen with phenyl was unfavorable and resulted in a loss of binding affinity (5g), while the tert-butyl analog (5h) has similar affinity to the parent chlorine. X-ray crystallography demonstrated that larger groups tended to sit in the same space in the binding pocket, but doing so forces the rest of the molecule to shift upward (Figure 5A); smaller aliphatic groups complement this pocket much better (5i, 5j). Pentafluorosulfanyl groups have been reported as metabolically inert tert-butyl analogs,42 and their use herein affords the most potent example of this phenolic class, 5k, with ~5-fold improved binding affinity in comparison to the trifluoromethyl cyclopropane, 5l.43
Figure 5.

X-Ray co-crystallography of WDR5 bound to WBM-site inhibitors. A) An overlay of the X-Ray co-crystal structure of 5g (green, PDB: 6U80) and 1 (grey). There are few major changes in the protein orientation, but 5g diminished binding affinity compared to 1, suggesting the upward frame-shift affords an unfavorable position of the small molecule ligand; B) Structure of 6b bound to WDR5 (PDB: 6U8B), key residues are shown as sticks, highlighting hydrogen and halogen bonding interactions between the ligand and protein; C) Structure of 5n bound to WDR5, demonstrating the orientation of the nitrile toward the protein (PDB: 6U8O); D) Structure of 6g bound to WDR5 (PDB: 6U8L). E) Structure of 7k bound to WDR5 (PDB: 6U5Y); F) Structure of 7u bound to WDR5 (PDB: 6U5M).
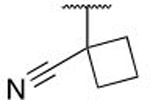
The cyano-spirocyclic examples (5m–5p) were initially designed to act as a handle for further growth of the compounds along the edge of this pocket, with the hydrocarbon chain hypothesized, using molecular docking studies, to complement the hydrophobic region. However, we observed by X-ray crystallography that the cyano moiety of 5n is instead oriented toward the protein, albeit with no obvious directional interaction (Figure 5C). The 3- and 5-membered ring examples, 5m and 5o, have slightly diminished affinity in comparison to the 4- and 6-membered rings. We hypothesize that the shape of C3- and C5-carbocycles produce a less favorable orientation of the nitrile, although we do not have crystallographic evidence of such an altered binding pose.
Optimization to salicylic acid motif
The X-ray co-crystal structure of 1 allowed us to employ structure-based design for the development of further optimized compounds, although a significant challenge to this effort relates to the shallow nature of this binding interface, which contains few areas that may traditionally be referred to as a ‘pocket.’44 As well as the early optimization of the hydrophobic moiety in the ‘ILE’ region of the peptide binding site, a further breakthrough came from the addition of a carboxylic acid directed toward Q289 affording salicylic acid derivative 6b (Kd = 0.43 μM, Table 2). This engagement of Q289 afforded an increase in binding affinity compared to 1, and the positive interaction was confirmed by X-ray crystallography (Figure 5B). Similarly, a significant potency increase was obtained by the addition of a chlorine ortho to the phenol of the sulfonyl ring (6d, Table 2); gaining additional hydrophobic contacts with WDR5 in the interface occupied by a valine side chain of Myc and RBBP5 (IDVV).
Table 2.
Optimization of salicylic acid derivatives, 6.
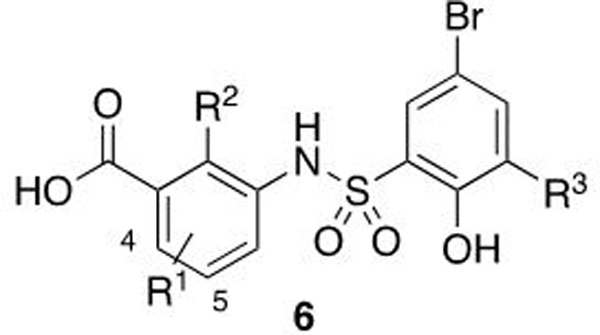 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | FPA Kd (μM)a |
| 6a | 5-Cl | H | H | 2.88 ± 0.68 |
| 6bb | OH | H | 0.431 ± 0.08 | |
| 6c | OH | H | 0.223 ± 0.03 | |
| 6d | OH | Cl | 0.163 ± 0.02 | |
| 6e | 5-cPr | OH | H | 0.184 ± 0.09 |
| 6f | OH | Cl | 0.207 ± 0.06 | |
| 6g | 4-F-5-cPr | OH | H | 0.122 ± 0.04 |
| 6h | OH | Cl | 0.050 ± 0.01 | |
| 6i | 5-OCF3 | H | H | 0.194 ± 0.02 |
| 6j | OH | H | 0.137 ± 0.01 | |
| 6k | OH | Cl | 0.051 ± 0.032 | |
| 6l | 5-SF5 | H | H | 0.064 ± 0.020 |
| 6m | H | Cl | 0.029 ± 0.024 | |
| 6n | OH | H | 0.047 ± 0.025 | |
| 6o | OH | Cl | 0.031 ± 0.018 | |
| 6p | 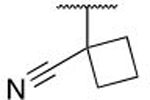 |
H | H | 0.229 ± 0.140 |
| 6q | OH | H | 0.067 ± 0.040 | |
| 6r | OH | Cl | 0.030 ± 0.019 | |
| 6s | 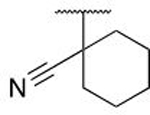 |
OH | H | 0.195 ± 0.028 |
| 6t | OH | Cl | 0.038 ± 0.003 | |
Data displayed is the average of at least 3 independent replicates ± standard deviation.
Compound contains a para-methoxy bromide on the sulfonyl ring.
The cyclopropyl derivatives (6e & 6f) demonstrated an improvement versus the parent chloro-containing compound, and the addition of an ortho-fluoro group afforded a further increase (6g). This enhanced affinity could be explained by the good shape complementarity of this compound to the binding site as demonstrated crystallographically (Figure 5E). In line with the SAR trends of Table 1, the hydrophobic chloro-replacement trifluoromethoxy, pentafluorosulfanyl, and cyano-spirocycle derivatives afforded low nanomolar potency with improvements 5 to 15-fold upon the addition of the acid. Generally, we observed a significant decrease in binding affinity when removing the phenol of the salicylic acid moiety, 6a, 6i, 6p; however, this decrease was not observed for the -SF5 analogs (6l, 6m to 6n, 6o).
Salicylic Acid Replacement
With this series of salicylic acid-based compounds, we were able to reach low-nanomolar levels of potency. However, we suspected inherent issues related to permeability and plasma protein binding due to the presence of the carboxylic acid, limiting their use in whole cell mechanism of action (MOA) studies. To avoid these problems, we explored the replacement of the carboxylic acid with a methyl amide (Table 3). Within this salicylamide series, a reduction in potency compared to the direct acid analogs was observed; however, by combining optimal SAR observations in the ILE262 region we were able to identify analogs which retained binding affinities below 100 nM. The hydrophobic ring of the cyclobutyl-cyano moiety (7k), adopts a similar binding pose as the cyclopropane of 6g (Figure 5D), bolstering our hypothesis of improved affinity when designing analogs that grow from the ipso carbon. Interestingly, while in the salicylic acid series, the cyclohexyl derivatives 6s, 6t demonstrated high binding affinity, this trend did not hold for the amide derivatives 7l, 7m.
During this exercise we explored alternate amide groups, including 7n, 7o, but, with the exception of 7n, we observed very flat SAR when exploring different amine partners (data not shown). We hypothesize that the amide group is directed toward solvent and does not make positive interactions with the protein, which led to the choice of this vector for attaching the small molecule FITC-probes used for the assay (structure of probe shown in Supplemental Figure S1). In a separate salicylate replacement effort, we were able to demonstrate that the acid/amide moiety could be exchanged with a sulfone; for example, methyl sulfone containing compounds 7p-7z were synthesized. Their design was driven in part to enhance physicochemical properties which might overcome some of the limitations observed in the salicylic acid subseries. Incorporation of the best-in-class pieces led to compounds such as 7w and 7z that bind with high potency to WDR5 and are comparable to analogs in the acid and amide series. Indeed, examples lacking the aniline-ring phenol were superior to the corresponding amide (compare 7b vs 7q, 7e vs 7t). Co-crystallization of WDR5 with 7x demonstrates that the sulfone also engages Q289 via a hydrogen bond (Figure 5F). Similar to the amides, the methyl group is directed toward bulk solvent offering a potential vector for future derivatization to tune the physicochemical properties.
Phenol substitutions
While we were able to discover small molecules that bind to WDR5 with excellent affinity, considering the shallow nature of the binding site, we recognize that these molecules retain functionality, such as phenols, that would likely occlude their development. Tables 2 and 3 already detail the synthesis of selected compounds with the aniline-ring phenol removed. Separately, we explored several strategies to remove or replace the para-bromophenol of the sulfonyl ring. A number of phenol and bromine replacement strategies failed (not shown), however, we were able to show, using optimized aniline-ring groups “capping” the phenol as a methoxy (e.g. 8, 9) can lead to inhibitors with sub-micromolar affinity in selected cases (Figure 6). Further, combining the best-in-class aniline groups to generate 6-bromoquinoline-8-sulfonamides afforded nanomolar analogs that do not contain the para-bromophenol motif. In this class, we observe that the sulfone and acid derivatives retain promising affinity in comparison to their direct ortho-chlorophenol analogs. Interestingly, the reduction in affinity observed when replacing the bromophenol appears more pronounced for the methyl amides (compare 7j, 7k, vs 9, 13), while the sulfone examples 8, 10 and 11 remain similar to 7x-7z. Notably, 15 is an exception.
Figure 6.

Selected compounds featuring removal or replacement of the sulfonyl ring phenol.
Pharmaceutical properties
We explored the tier 1 DMPK profile of a number of compounds within this class (Table 4). These data demonstrate that all tested analogs have good solubility, and the amide and sulfone series have significantly improved MDCK permeability in comparison to their salicylic acid counterparts. There was no evidence of P-gp mediated efflux with the tested analogs. A limitation of this compound class is the severe plasma protein binding exhibited by all tested compounds; indeed, salicylic acid itself is known to be a substrate for human serum albumin.45,46 Efforts to overcome this hurdle to develop an improved chemical probe are ongoing.
Table 4.
Pharmaceutical property profile of select compounds.a
| Compound | Solubility (μM) |
MDCK A-B Papp (106/s) |
MDCK B-A Papp (106/s) |
Fu (%) |
|---|---|---|---|---|
| 5f | >100 | 36.4 | - | <0.0026 |
| 5n | >100 | 27.8 | - | <0.0005 |
| 5k | >100 | 14.7 | - | - |
| 6g | >100 | 0.61 | - | <0.0008 |
| 6r | >100 | 0.33 | - | <0.0003 |
| 7a | >100 | 16.7 | 14.1 | <0.0004 |
| 7c | >100 | 12.9 | 3.4 | <0.0002 |
| 7j | >100 | 45.2 | - | <0.0006 |
| 7k | >100 | 14.0 | - | <0.0006 |
| 7n | >100 | 58.4 | - | 0.003 |
| 7p | >100 | 4.79 | - | 0.004 |
| 7q | >100 | 3.89 | - | 0.0007 |
| 7t | >100 | 16.1 | - | 0.002 |
| 7w | >100 | 18.6 | - | <0.0003 |
All experiments performed at Q2 Solutions Ltd.
WDR5 stabilization by thermal shift
In order to further demonstrate effective, on-target binding of our inhibitors to WDR5, we used differential scanning fluorimetry (DSF) to examine a subset of compounds (Figure 7). For the salicylic acid subseries (Figure 7A), we observe that the most potent examples by FPA, (6o, 6r, 6t) also impart the largest ΔTm, and thereby stabilization of WDR5. A negative control compound, 6-NC, contains a regioisomeric bromine designed such that the key halogen bonding bromine atom is moved around the phenyl ring, causing a significant decrease in potency (see Supplemental Figure S1.) This is a direct regioisomer of 6q, and in this assay 6-NC has minimal effect (ΔTm = 0.42 °C). When comparing a compound sub-set that all contain the spirocyclobutane moiety (Figure 7B), the addition of the carboxylic acid (6q vs 5n) offers a significant increase in binding affinity by FPA, but only a marginal improvement in thermal shift is observed. However, the additional hydrophobic interaction brought by the addition of the chlorine on the sulfonyl-ring affords compounds with an increased ΔTm (6r, 7k). Figure 7C highlights that the binding of 7k demonstrates a concentration-dependent response. A summary of the ΔTm values is displayed in Figure 7D, and the effect of further tested examples is shown in Supplemental Table S1. We have also confirmed that the binding of the 7k in the absence of WDR5 protein does not have any effect on the background fluorescence at any of these concentrations (Supplemental Figure S2), and that these compounds do not display significant binding at the ‘WIN’ binding site (Supplemental Figure S4).38
Figure 7.

Assessment of compounds by thermal shift, all compounds measured at 10 μM in triplicate, average values are displayed; RFU = relative fluorescence unit. A) A comparison of compounds in the salicylic acid subseries. B) A comparison of compounds that all contain the spirocyclobutane substructure. C) A ten-point dilution of compound 7k demonstrates the dose-dependent effect on WDR5 stabilization. D) The measured ΔTm values for compounds displayed in panels A–C.
Confirmation of target engagement in cell lysates
To assess whether compounds disrupt the association between WDR5 and interacting partners at the WBM-site, (MYC and RBBP5), we immunoprecipitated WDR5 from cell lysates treated with compounds. We first chose a representative selection of compounds from the acid and amide subseries with a range of binding affinities to WDR5 as measured by FPA. Initially, we treated cell lysates with 50 μM compound, and observed varying degrees of disruption between both WDR5-c-MYC and WDR5-RBBP5 as compared to both the vehicle control and the representative negative control compounds that bind with markedly lower affinity to WDR5 from both acid and amide subseries (Figure 8A, B). As anticipated, the degree of disruption between WDR5-c-MYC and WDR5-RBBP5 was similar for each compound. Furthermore, the degree of disruption also corresponded to the compound’s respective binding affinity to WDR5, where compounds with a higher affinity to WDR5 disrupted the WBM-site protein interactions to a greater degree, further highlighting the efficacy of the acid compound subseries. Similarly, exemplar compounds from the sulfone series, as well as those with phenol replacements can also disrupt binding of c-MYC to WDR5 in cellular lysates (Supplemental Figure S3), in a manner that is associated with the affinity of their binding to WDR5.
Figure 8.

Compound target engagement in cell lysates. Levels of c-MYC and RBBP5 proteins in association with WDR5 were assessed after treatment with compound in lysates from WDR5-FLAG and c-MYC-HA co-overexpressing HEK293 cells. A fraction of the lysate before addition of compound is shown as a control lane. An α-FLAG immunoprecipitation was used to isolate WDR5, which is not detectable in the input due to the excess of WDR5 in the IP fraction. c-MYC and RBBP5 were blotted in independent experiments. Data are representative of minimum two independent biological repeats. α-FLAG immunoprecipitation from lysates treated with 50 μM compound from the A) acid subseries and B) amide subseries. C) A dose response treatment of compounds 6r and 7k from 6.25 to 50 μM, as compared to 50 μM treatment with respective inactive controls 6-NC and 7-NC and the vehicle control (VC). Control cells express c-MYC-HA and a GFP-FLAG construct. Panels D), E) and F) present quantification of two independent experiments, normalized to vehicle control.
Further, to assess the effects of lower compound concentrations on the disruption of the WDR5-c-MYC and WDR5-RBBP5 associations, we also conducted a dose response treatment with compounds 6r and 7k, alongside their corresponding negative controls 6-NC and 7-NC (Figure 8C). We observed a dose-dependent disruption of the association between both WDR5-c-MYC and WDR5-RBBP5, as compared to the vehicle control and to the respective negative control compounds from each subseries. The dose-dependency was most clearly demonstrated in response to 7k treatment when assessing the association between WDR5 and RBBP5. The improved efficacy of the acid compound 6r was also evident, as a marked disruption between both WDR5-c-MYC and WDR5-RBBP5 was clearly observed even at the lowest concentration tested.
Overall, these compounds demonstrate biochemical binding affinity caused by the displacement of a FITC-labeled probe from recombinant, purified WDR5, and binding to WDR5 in cell lysates by thermal shift and co-immunoprecipitation. We are also able to demonstrate a functional effect of this binding, as these compounds inhibit the biochemical histone methyltransferase (HMT) activity of MLL-1 in the full WRAD complex (Supplemental Table S2). The results of this assay suggest that compounds disrupting the WDR5:RBBP5 interface of the multi-protein WRAD complex may offer an alternative strategy for the targeting of tumors driven by abrogation of MLL-proteins, avoiding the WIN binding site. Interestingly, we observe that the potency of functional inhibition in the HMT assay is much closer to the biochemical binding affinity of inhibitors at the WBM site, versus previously disclosed inhibitors of WIN site from both our group and others.32,33,38 The inherent weaker affinity of the WDR5:RBBP5 interaction may present a significantly lower barrier for a small molecule disruptor to overcome in comparison to that of the WDR5:MLL interface.47
Chemistry
All compounds were synthesized in modular fashion using a sulfonamide coupling to join elaborated anilines and sulfonyl chlorides. This coupling used pyridine as a base, which favors N- versus O-linked reaction. For examples 1, 5a–e, 5g, 5h, both coupling partners were commercially available, allowing for rapid initial SAR exploration (Scheme 1A), while the more elaborate aniline pieces often required multi-step synthesis. Chlorosulfonation of the 4-bromophenol derivatives afforded corresponding sulfonyl chlorides, 18a,b (Scheme 1B). Examples 5j and 5k both originated from corresponding phenols, which were nitrated and reduced to the required aniline intermediates 20. The Suzuki-Miyaura coupling using cyclopropylboronic acid was reliant on protection to form intermediate 23. The phenol was benzyl protected, and the aniline masked as the corresponding nitro group. After cross-coupling the subsequent hydrogenation cleanly affords the anilino-phenol, 23 (Scheme 1D). The trifluoromethylcyclopropane example was synthesized using methodology from Merck,48,49 cross-coupling first to the CF3-styrene, 21, and cyclopropanation using a sulfonium ylide as a diazomethane equivalent (Scheme 1E). As previously described, deprotection by hydrogenation affords the desired intermediate 26.
Scheme 1.

Synthesis of sulfonamides, and the requisite aniline building blocks to compounds 5. Reagents and conditions: a) Pyridine, DCM, r.t.; b) HSO3Cl, 0 °C– r.t.; c) HNO3. H2SO4, DCM, 0 °C– r.t.; d) H2, Pd/C, MeOH, r.t.; e) ArSO2Cl, pyridine, DCM, r.t.; f) Benzyl bromide, K2CO3, MeCN, Δ; g) Cyclopropylboronic acid, Pd(OAc)2, PCy3.HBF4, K3PO4, Toluene, 80 °C; h) 3,3,3-trifluoroprop-1-ene, Pd(dppf)Cl2.DCM, K2CO3, dioxane, water, 70 °C; i) Ph2MeSBF4, LiHMDS, THF, −78 °C.
The cyano-spirocycles were all synthesized by cyclization of O-protected intedmediates 27 with the corresponding dibromoalkane using sodium hydride in DMSO and the common bromide intermediates 28 were used to furnish all of the required aniline intermediates (Scheme 2). Toward products 5m-p, the bromide was displaced with tert-butyl carbamate to furnish the desired aniline, 29a-d, after deprotection. Intermediates 28 could instead be converted to the respective phenyl ester using palladium-mediated carbonylation to generate 30 or to the methyl sulfone by palladium-mediated coupling with sodium methanethiolate and then oxidation with potassium peroxymonosulfate to 32. The synthesis of products 6q, 6r and 12 was completed by heating the phenyl ester, 31, with lithium hydroxide. The salicylamide formation to 7j-o, 9 and 13 proceeded by heating the ester directly with the corresponding amine; such amide formations did not require prior hydrolysis.
Scheme 2.

Synthesis of cyano-spirocycle products utilizing common bromide intermediate, 27. Reagents and conditions: a) dialkylbromide, NaH, DMSO, r.t.; b) tert-butylcarbamate, Pd2(dba)3, Xantphos, Cs2CO3, Toluene, 90 °C; c) BBr3, DCM, −78 °C; d) TFA, DCM, r.t.; e) ArSO2Cl, Pyridine, DCM, r.t.; f) Phenyl formate, phenol, Pd(OAc)2, P(tBu)3.HBF4, NEt3, Toluene; g) HNO3, TBAB, DCE/H2O, 0–23 °C; h) H2, Pd/C, MeOH, r.t.; i) 2M LiOH, THF, r.t.; j) Amine, DIPEA, THF, Δ; k) NaSMe, Pd2(dba)3, Xantphos, NEt3, THF, Δ; l) Oxone, EtOH, H2O, r.t.; m) HNO3, H2SO4, DCM, 0 °C.
Pentafluorosulfanyl analogs were synthesized using a similar chemical approach as previously used for the cyano-spirocycles (Scheme 3). The -SF5 group in all instances was incorporated in the commercially available intermediatedes. 34 underwent same palladium-mediated carbonylation and demethylation to 35, but the nitration of the phenyl ester was unsuccessful despite a condition screen. However, nitration of the benzoic acid and subsequent esterification to the methyl ester afforded access to intermediate 36 in good yield (Scheme 3A). The corresponding sulfones were introduced via palladium-cross coupling of the aryl bromide with dimethyl disulfide, then oxidation to 38; nitration with a nitric acid followed by hydrogenation furnished 39.
Scheme 3.

Synthesis of pentafluorosulfonyl products from commercial bromide intermediate, 30. Reagents and conditions: a) Phenyl formate, phenol, Pd(OAc)2, PtBu3.HBF4, Et3N, MeCN, 90 °C; b) BBr3, DCM, −78 °C; c) LiOH, THF, 65 °C; d) HNO3, H2SO4, DCM, 0 °C; e) H2SO4, MeOH, 65 °C; f) H2, Pd/C, MeOH, r.t.; g) ArSO2Cl, pyridine, DCM, r.t.; h) Amine, THF, 65 °C; i) Br2, FeCl3, AcOH, 0 °C; j) S2(CH3)2, Cu, pyridine, 115 °C, k) Oxone, EtOH, H2O, r.t.; k)
The synthesis of the densely substituted analogs 6g, 6h, 7c, and 7d is outlined in Scheme 4. The readily available acid 40 was esterified, and the subsequent bromination was found to be largely selective para- to the phenol. The inseparable, minor dibrominated byproduct was separable after subsequent nitration to 43. As described above, protection of the phenol was required for cross-coupling of cyclopropylboronic acid, and a similar sequence of hydrogenation, sulfonamide coupling, and hydrolysis or amidation affords the screened compounds.
Scheme 4.

Synthesis of 6g, 6h, 7c, and 7d. Reagents and Conditions: a) TMS diazomethane, THF, MeOH, 0 °C; b) HBF4.Et2O, NBS, MeCN, −10 °C c) HNO3, H2SO4, DCM, 0 °C; d) BnBr, K2CO3, MeCN, Δ; e) Cyclopropylboronic acid, Pd(OAc)2, PCy3.HBF4, K3PO4, Toluene, 80 °C; f) H2, Pd/C, MeOH, r.t.; g) ArSO2Cl, pyridine, DCM, r.t.; h) LiOH, THF, 65 °C; i). H2NMe, THF, 65 °C.
The synthesis of analogs that lack a phenol on the aniline ring is shown in Scheme 5. The sulfone intermediates are all synthesized by palladium-mediated cross coupling with sodium methanesulfinate (Scheme 5A – C). Toward 7t and 7u, a Curtis rearrangement of the acid intermediate was utilized to generate aniline 49. Spiro-cyclobutanes were synthesized from toluene-derived starting materials, which were brominated, and the nitrile introduced using TMSCN. Cyclisation with 1,3-dihalopropanes affords intermediates 51 and 53, that were used for sulfonamide formation.
Scheme 5.

Synthesis of sulfone analogs 7p, 7q, 7t, 7u and 7x (A – C), and spirocyclobutane anlogs 6p, 7h, 7i (D). Reagents and Conditions: a) MeSO2Na, CuI, L-Proline, NaOH, DMSO, 80 °C; b) ArSO2Cl, pyridine, DCM, r.t.; c) 2M LiOH, THF, 65 °C; d) DPPA, t-BuOH, DME, 100 °C; e) TFA, DCM, r.t.; f) Boc2O, MeOH, r.t.; g) NBS, AIBN, CCl4, 80 °C; h) TMSCN, K2CO3, MeCN, 0–50 °C; i) 1,3-dibromopropane, NaH, DMSO, r.t.; j) HCl, dioxane, 50 °C; k) 1,3-diiodopropane, KOtBu, THF, r.t.; l) tert-butylcarbamate, Pd2(dba)3, Xantphos, Cs2CO3, Toluene, 90 °C; m) H2NMe, THF, 65 °C.
CONCLUSIONS
WD-repeat proteins are an emerging class of targets for a range of therapeutic areas, although there has been, to date, no clinical validation of these proteins with approved drugs. Inhibitors of WDR5 are being sought as highly attractive options for the treatment of multiple cancer types. To date, there have been several inhibitors published that bind to WDR5 at the ‘WIN’ binding site, but no inhibitors of the ‘WBM’ site have been disclosed. An inhibitor of this site directly prevents WDR5 from being able to bind to MYC, RBBP5, and KANSL2 among others, and thus could be of therapeutic interest in cancers driven by oncogenic effects of these proteins. Further, this approach may offer an alternative pathway to affect the HMT activity of MLL1 while not directly preventing the binding of other protein partners into the MLL1 binding region.
We performed a high throughput screen of the Vanderbilt library and obtained a series of biaryl sulfonamide hits that were unambiguously confirmed as inhibitors of this binding site by NMR and X-ray crystallography. Using rational design, we optimized hit compound 1, yielding multiple analogs binding at less than 30 nM. To our knowledge, the data reported herein represents the first published examples of a small molecule inhibitor binding to an external cleft of a WD-repeat protein with such levels of affinity. Binding to the protein was confirmed by secondary assays, including NMR and DSF. Biochemical activity at this cleft was demonstrated in a secondary HMT assay, whereby the small molecule was able to successfully disrupt the WRAD multi-protein complex, and thus the methyltransferase function of MLL1. These compounds bind on the opposite face of WDR5 to the MLL1 interaction. We observe no major changes in the protein X-ray structure that would suggest an allosteric modification in the MLL1 binding pocket, yet these compounds are still able to prevent the arginine methylation of this complex. Intriguingly, the difference in activity observed between the binding and HMT assays is significantly less than that previously reported for direct WIN-site inhibitors.37 Further, some SAR correlations are evident with subseries of compounds, even in this limited dataset, prompting significant interest and further studies within our laboratories.
These inhibitors were demonstrated to be highly soluble, and permeability was significantly improved by switching from a salicylic acid to salicylamide or methyl sulfone motif, all of which are able to form a H-bond interaction with GLN289. Compounds from this series are able to disrupt the interaction of WDR5 with both c-MYC and RBBP5 by co-immunoprecipitation in cellular lysates in a manner that is correlated with binding affinity. However, the low free fraction shown by most analogs limits their utility in whole cells. Ongoing efforts seek to improve the drug-like character of these compounds to discover molecules that can be used as probes of the interruption of the WBM-site.
EXPERIMENTAL
Protein Expression and Purification.
Truncated WDR5 (a.a. 22–334) was cloned into a pET vector with a 6xHis-SUMO tag fused at the N-terminus. The plasmids WDR5 was transformed into E. coli BL21 (DE3) cells. The overnight culture was used to start a 10 L fermentation (BioFlo 415, New Brunswick Scientific) grown at 37 °C. For NMR samples, uniformly 15N-labeled protein was produced in minimal M9 medium, where 15NH4Cl (Cambridge Isotope Laboratories) and D-glucose were used as sole nitrogen and carbon sources. When the cell density reached OD600 = 2.5, the temperature was lowered to 30 °C. The protein was expressed overnight with 1mM isopropyl-β-D-thiogalactoside (IPTG). Myc peptide (DEEEIDVVSVE) was ordered (Genscript) as HPLC purified synthetic polypeptide. It was dissolved in DMSO for further use.
Cell pellets were dissolved in lysis buffer (1XPBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and10% glycerol), and broken by homogenization (APV-2000, APV). The lysate was cleared by centrifugation and filtering, and then applied to an affinity column (140 mL, ProBond, Invitrogen). Bound protein was eluted by an imidazole gradient. The His-SUMO-tag was removed by SUMO protease cleavage during dialysis and the subsequent subtractive second nickel-column. WDR5 protein was then purified by size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using NMR or crystallization buffer.
HTS Screening.
The previously described Myc peptide,16 was labelled with FITC and used as the probe for FPA assays. The probe was ordered form Genscript. 5 μM WDR5 protein and 5 μM probe were used in the HTS, and the buffer condition was 1XPBS plus 300 mM NaCl, pH 6.0, 0.5mM TCEP, 0.1% CHAP, and 5% DMSO. Vanderbilt Discovery Collection (VDC) and VICB collection compounds (~250,000) were tested at 50 μM concentration, and the compounds was put into 781 386-well plates with necessary positive and negative controls in each plate. After adding all reagents, the plates were shaken for ~2 minutes, and incubated for 60 minutes before first plate to be read on plate reader (Biotek). The reading settings of the plate reader were 50 flashes, low lamp energy, and 7.75 read height. All the plates were screened, and those plates (totaling 17) with Z’ less than 0.3 were repeated. Compounds, which showed >15% inhibition, were selected for the confirmation screen. Confirmed compounds then were used for a 10-point dose response study.
Hit Validation by NMR.
NMR samples contained 2 mg/mL (~67 μM) 15N-labeled WDR5 in 25 mM phosphate buffer, pH=6.0, 100 mM NaCl, and 1 mM DTT. HTS hits were used at 100 μM. Nuclear magnetic resonance screening was conducted using a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5mm single-axis z-gradient cryoprobe and a Bruker Sample Jet sample changer. Two-dimensional, gradient-enhanced 1H-15N heteronuclear multiple-quantum coherence (SOFAST-HMQC)51 spectra were collected at 25 °C and used to track chemical shift perturbation upon compound binding. Spectra were processed and visualized using Topspin (Bruker BioSpin).
Protein Crystallization, Data Collection, and Structure Refinement.
WDR5 was concentrated to 10 mg/mL (~300 μM) in the buffer of 20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT. Co-crystals were obtained at 18 °C using the hanging drop method. The crystallization condition was 0.1 M Bis-Tris pH 6.0, 0.2 M ammonium acetate, 28% to 32% PEG3350. Soaking was also applied to some compounds using crystals of WDR5 that contain an MLL-site binder. Crystals were flash frozen in liquid nitrogen directly.
Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D and G beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. Data were indexed, integrated, and scaled with HKL2000.52 Molecular replacement was achieved with Phaser44 as implemented in CCP4.4553 using a previously determined WDR5 structure (PDB code 3EG6). Refinement of the structural models was conducted with PHENIX54 and included rounds of manual model building in COOT.55 All structure images were prepared with PyMOL.56
Cloning and plasmids.
Full length human WDR5 with an N-terminal FLAG tag was cloned into the NgoMIV and SalI sites as a NgoMIV/XhoI fragment. pBabe-GFP was constructed by PCR amplifying the GFP fragment from pEGFP-C2 (Clontech) and cloning the product into the EcoRI and BamHI sites of pBabe Puro. Full length human c-MYC with a C-terminal double HA tag was cloned into the BamHI and EcoRI sites of pBabe-IGH.16 HEK293 cells stably expressing MYC2HA were made by retroviral transduction followed by selection in Hygromycin (50 μg/mL). The mixed population was then infected with pBabe-Puro expressing GFP or WDR5 with selection in puromycin (1 μg/mL). For retroviral transductions, HEK293T cells were transfected with the appropriate pBabe vector, the pCL10A packaging vector, and pMax-GFP to estimate transfection efficiency. Viral supernatant was collected and used to infect HEK293 class over three days.
Cell culture.
HEK293 cells were maintained in DMEM supplemented with 10% FBS. Hygromycin B (50 μg/mL) and puromycin (100 ng/mL) were added to media to maintain plasmid expression. Both cell lines were tested and confirmed negative of mycoplasma using the VenorGem PCR test kit (Sigma Aldrich). After thawing from liquid nitrogen, cells were passaged at least twice before use in experiments, and passaged for a maximum of 25 times.
WDR5 Immunoprecipitation.
HEK293 cells were harvested and lysates were prepared on ice in lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X100 and supplemented with protease and phosphatase inhibitors). Equal amounts of protein lysate were subject to immunoprecipitation with M2 agarose overnight (for WDR5-c-MYC) or for 2 hours with magnetic M2 beads (for WDR5-RBBP5) at 4 °C. Immune complexes were recovered, washed in lysis buffer, and resolved by SDS-PAGE. Immunoblotting was performed using the indicated primary antibodies, incubated with labeled secondary antibodies and membranes were scanned using the Odyssey imager (LiCor).
Antibodies.
The following primary antibodies were used for this study: α-c-MYC (#5605), α-RBBP5 (#13171), α-WDR5 (#13105), α-FLAG (#8146) all purchased from Cell Signaling.
Pharmaceutical property determinations
All experiments performed at Q2 Solutions Ltd. (https://www.q2labsolutions.com) using commercially available, standard assay formats. Kinetic solubility determinations: Method QUI-SOL-001. MDCK permeability determinations: Method QUI-PERM-003. Protein binding assessment: Method QUI-PERM-002.
General Chemistry
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Hydrogenation reactions are performed using an atmospheric balloon. Normal phase flash silica gel-based column chromatography is performed using ready-to-connect cartridges from ISCO, on irregular silica gel, particle size 15–40 μM using a Teledyne ISCO Combiflash Rf system. Preparative reversed-phase HPLC was performed on a Gilson instrument equipped with a Phenomenex Kinetex C18 column, using gradients of MeCN in H2O and 0.1% TFA. Compounds that are obtained as a TFA salt after purification were afforded as free base, by dissolving the salt in EtOAc and washing with sat. aq. K2CO3. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at either 400 MHz or 600 MHz on a Bruker spectrometer, as stated. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual non-deuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; br, broad. All compounds were of 95% purity or higher, unless otherwise noted, as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LCMS experiments were performed with the following parameters: Phenomenex Kinetex 2.6 μm XB-C18 100 Å, LC column 50 × 2.1 mm; a gradient of 5%–95% MeCN in H2O, and 0.1% TFA, over a 1.2 min or 2.0 min gradient. HRMS experiments were conducted at the University of Notre Dame Mass Spectrometry and Proteomics Facility.
General Procedure A: Sulfonamide coupling
The corresponding aniline (1 eq) and sulfonyl chloride (1.2–1.5 eq) were combined in CH2Cl2 (at 0.2 M]) at r.t., and pyridine (3 eq) was added; the mixture was stirred for 1–16 h. The mixture was concentrated in vacuo and purified by ISCO flash chromatography, unless stated.
General Procedure B: Hydrogenation
Corresponding intermediate (1 eq) was dissolved in MeOH, EtOAc was added to aid dissolution where required. Pd/C (5% C by wt., 10 mol%) was added and the mixture stirred under a H2 atmosphere for 16 h. The mixture was filtered through celite, concentrated, and purified by ISCO flash chromatography if required.
General Procedure C: Nitration
Corresponding intermediate (1 eq) was dissolved in DCM(at [0.4 M]) and cooled to 0 °C in an ice/water bath. To this was added a pre-mixed solution of HNO3 (conc., 1.5 eq) and H2SO4 (conc., 1.5 eq) drop wise. The mixture was stirred for 1–16 h, allowing to warm to r.t., then poured over ice and extracted with DCM. The crude material was purified by ISCO flash chromatography.
General Procedure D: BBr3 mediated demethylation
The intermediate (1 eq) was dissolved in anhydrous DCM (at [0.4 M]) and cooled to −78 °C in an acetone/dry ice bath under an inert atmosphere. To this was added BBr3 ([1.0 M in DCM], 2 eq) drop-wise, and the mixture stirred for 1 h at −78 °C, then allowed to warm to r.t.. The mixture was poured over an ice-water slurry and extracted with EtOAc, washing with water, brine. Crude product was purified by ISCO flash chromatography.
General Procedure E: Ester hydrolysis
The ester intermediate (1 eq) was stirred with LiOH (2 M, 5 eq) in THF (at 0.2 M]) for 1–16 h at 65 °C, unless stated at r.t.. The mixture was acidified with hydrochloric acid, extracted with EtOAc and washed with brine. The crude material was purified by preparative HPLC.
General Procedure F: Methyl amide synthesis
To the salicylate ester intermediate (1 eq) was added methylamine ([2.0M in THF], 10 eq) and the mixture heated at 65 °C for 1–16 h. The mixture was concentrated in vacuo and purified by preparative HPLC.
General Procedure G: Benzyl protection
The intermediate phenol (1 eq), benzyl bromide (1.05 eq) and potassium carbonate (1.1 eq) were combined in acetonitrile and heated to reflux for 18 h. Upon cooling the mixture was concentrated in vacuo, re-dissolved in EtOAc and washed with water, brine. Crude material was purified by ISCO flash chromatography.
5-Bromo-N-(5-chloro-2-hydroxyphenyl)-2-methoxybenzenesulfonamide (1)
2-Amino-4-chlorophenol (144 mg, 1.0 mmol) was reacted with 5-bromo-2-methoxybenzenesulfonyl chloride, 16a (428 mg, 1.5 mmol) following General Procedure A, affording the title compound as a colorless solid (283 mg, 0.72 mmol, 72%). 1H NMR (400 MHz, MeOH-d4) δH 7.83 (d, J = 2.5 Hz, 1H), 7.66 (dd, J = 8.9, 2.5 Hz, 1H), 7.31 (d, J = 2.5 Hz, 1H), 7.08 (d, J = 8.9 Hz, 1H), 6.88 (dd, J = 8.6, 2.6 Hz, 1H), 6.67 (d, J = 8.6 Hz, 1H), 3.91 (s, 3H); LCMS tR = 1.44 min, m/z = 393.7 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+H]+ calculated for C13H12BrClNO4S 393.9353, found 393.9344.
N-(5-Chloro-2-hydroxyphenyl)-2-methoxybenzenesulfonamide (5a)
2-Amino-4-chlorophenol (14 mg, 0.10 mmol) was reacted with 2-methoxybenzenesulfonyl chloride (31 mg, 0.15 mmol) following General Procedure A, affording the title compound as a colorless solid (25 mg, 0.08 mmol, 80%). 1H NMR (400 MHz, CDCl3) δH 7.79 (dd, J = 7.9, 1.7 Hz, 1H), 7.57 (ddd, J = 8.7, 7.3, 1.7 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.03 (t, J = 7.6 Hz, 1H), 6.99 (dd, J = 8.7, 2.5 Hz, 1H), 6.86 – 6.79 (m, 3H), 6.58 (s, 1H), 4.07 (s, 3H); LCMS tR = 1.54 min, m/z = 313.9 [M+H]+; Purity (AUC) ≥95%.
5-Chloro-N-(5-chloro-2-hydroxyphenyl)-2-methoxybenzenesulfonamide (5b)
2-Amino-4-chlorophenol (14 mg, 0.1 mmol) was reacted with 5-chloro-2-methoxybenzenesulfonyl chloride (36 mg, 0.15 mmol) following General Procedure A, affording the title compound as a colorless solid (21 mg, 0.06 mmol, 60%). 1H NMR (400 MHz, CDCl3) δH 8.50 (s, 1H), 7.47 (dd, J = 6.6, 2.6 Hz, 2H), 7.34 (dd, J = 8.9, 2.6 Hz, 1H), 7.11 (dd, J = 8.8, 2.5 Hz, 1H), 7.04 (s, 1H), 6.89 (d, J = 8.8 Hz,1H), 6.70 (d, J = 8.8 Hz, 1H), 3.65 (s, 3H); LCMS tR = 1.03 min, m/z = 377.3 [M+H]+; Purity (AUC) ≥95%.
N-(5-Chloro-2-hydroxyphenyl)-5-iodo-2-methoxybenzenesulfonamide (5c)
2-Amino-4-chlorophenol (14 mg, 0.1 mmol) was reacted with 5-iodo-2-methoxybenzenesulfonyl chloride (50 mg, 0.15 mmol) following General Procedure A, affording the title compound as a pale brown solid (5 mg, 0.01 mmol, 11%). 1H NMR (400 MHz, MeOH-d4) δ 7.98 (d, J = 2.3 Hz, 1H), 7.82 (dd, J = 8.8, 2.3 Hz, 1H), 7.27 (d, J = 2.6 Hz, 1H), 6.95 (d, J = 8.8 Hz, 1H), 6.86 (dd, J = 8.6, 2.6 Hz, 1H), 6.67 (d, J = 8.6 Hz, 1H), 3.90 (s, 3H); LCMS tR = 1.03 min, m/z = 440.1 [M+H]+; Purity (AUC) ≥95%.
3-Bromo-N-(5-chloro-2-hydroxyphenyl)benzenesulfonamide (5d)
2-Amino-4-chlorophenol (17 mg, 0.12 mmol) was reacted with 3-bromobenzenesulfonyl chloride (46 mg, 0.18 mmol) following General Procedure A, affording the title compound as a colorless solid (22 mg, 0.72 mmol, 51%). 1H NMR (400 MHz, MeOH-d4) δH 7.92 (t, J = 1.8 Hz, 1H), 7.75 – 7.66 (m, 2H), 7.37 (t, J = 8.0 Hz, 1H), 7.33 (d, J = 2.6 Hz, 1H), 6.95 (dd, J = 8.7, 2.6 Hz, 1H), 6.66 (d, J = 8.7 Hz, 1H); LCMS tR = 1.50 min, m/z = 363.6 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(2-hydroxyphenyl)-2-methoxybenzenesulfonamide (5e)
2-Aminophenol (17 mg, 0.16 mmol) was reacted with 16a (68 mg, 0.24 mmol) following General Procedure A, affording the title compound as a colorless solid (22 mg, 0.06 mmol, 38%). 1H NMR (400 MHz, CDCl3) δH 7.88 (d, J = 2.5 Hz, 1H), 7.63 (dd, J = 8.8, 2.5 Hz, 1H), 7.07 (ddd, J = 8.9, 7.4, 1.6 Hz, 1H), 6.95 (d, J = 8.9 Hz, 1H), 6.91 (dd, J = 8.0, 1.4 Hz, 1H), 6.86 (dd, J = 8.0, 1.6 Hz, 1H), 6.83 (s, 1H), 6.73 (td, J = 7.7, 1.4 Hz, 1H), 6.39 (br s, 1H); LCMS tR = 1.41 min, m/z = 359.3 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-chloro-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5f)
Step A: 5-Bromo-2-hydroxybenzenesulfonyl chloride (18a)
4-Bromophenol, 17a (1.73 g, 10 mmol) was added portion wise to ice-cold chlorosulfonic acid (4.77 mL, 70 mmol) and the mixture stirred for 16 h, warming to r.t. The mixture was carefully poured over a slurry of ice, DCM and brine, and extracted with DCM. Purification by flash chromatography affords title compound as a pale brown oily solid (1.50 g, 5.53 mmol, 55%). 1H NMR (400 MHz, CDCl3) ∂H 7.95 (d, J = 2.4 Hz, 1H), 7.71 (dd, J = 8.9, 2.4 Hz, 1H), 7.04 (d, J = 8.9 Hz, 1H).
Step B: 5-Bromo-N-(5-chloro-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide.
2-Amino-4-chlorophenol (14 mg, 0.1 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording title compound as a colorless solid (21 mg, 0.06 mmol, 55%). 1H NMR (400 MHz, CDCl3) δH 7.66 (d, J = 2.5 Hz, 1H), 7.53 (dd, JJ = 8.8, 2.5 Hz, 1H), 7.08 (d, J = 8.2 Hz, 2H), 6.88 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H); LCMS tR = 1.53 min, m/z = 377.8, 379.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(4-hydroxy-[1,1’-biphenyl]-3-yl)-2-methoxybenzenesulfonamide (5g)
3-Amino-[1,1’-biphenyl]-4-ol (24.3 mg, 0.13 mmol) was reacted with 16a following General Procedure A to afford title compound as a colorless solid (28 mg, 0.06 mmol, 49%). 1H NMR (400 MHz, CDCl3) δH 7.98 (t, J = 1.9 Hz, 1H), 7.57 (dd, J = 8.8, 2.5 Hz, 1H), 7.40 – 7.35 (m, 5H), 7.31 – 7.27 (m, 1H), 7.18 (dd, J = 8.4, 2.2 Hz, 1H), 6.88 (t, J = 7.7 Hz, 2H), 3.96 (s, 3H); LCMS tR = 1.644 min, m/z = 435.3 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(tert-butyl)-2-hydroxyphenyl)-2-methoxybenzenesulfonamide (5h)
2-Amino-4-(tert-butyl)phenol (22 mg, 0.13 mmol) was reacted with 16a following General Procedure A to afford title compound as a colorless solid (26 mg, 0.06 mmol, 48%). 1H NMR (400 MHz, CDCl3) δH 7.86 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 8.8, 2.5 Hz, 1H), 7.07 (dd, J = 8.6, 2.4 Hz, 1H), 6.95 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 8.5 Hz, 1H), 6.76 (d, J = 2.5 Hz, 2H), 6.27 (s, 1H), 4.05 (s, 3H), 1.10 (s, 9H); LCMS tR = 1.70 min, m/z = 415.3 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-cyclopropyl-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5i)
Step A: 1-(Benzyloxy)-4-bromo-2-nitrobenzene (22)
2-Nitro-4-bromophenol, 21 (10.9 g, 50 mmol) was reacted according to General Procedure G, to afford 22 as a yellow solid (14.2 g, 46.1 mmol, 92%). LCMS tR = 1.25 min, m/z = 307.2 [M+H]+.
Step B: 1-(Benzyloxy)-4-cyclopropyl-2-nitrobenzene
1-(benzyloxy)-4-bromo-2-nitrobenzene, 22 (1.54 g, 5.0 mmol), cyclopropylboronic acid (515 mg, 6.0 mmol), Pd(OAc)2 (56 mg, 0.25 mmol), PCy3.HBF4 (184 mg, 0.50 mmol), K3PO4 (2.65 g, 12.5 mmol) were taken in toluene (25 mL) and water (5 mL) and stirred at 110 °C in a sealed tube for 16 h. The cooled mixture was filtered through celite, diluted with EtOAc and washed with water. Purification by flash chromatography affords a yellow solid (1.22 g, 4.53 mmol, 91%). LCMS tR = 1.27 min, does not ionize by ESI.
Step C: 2-Amino-4-cyclopropylphenol (23)
1-(Benzyloxy)-4-cyclopropyl-2-nitrobenzene (539 mg, 2.0 mmol) was reacted following General Procedure B to afford 23 as a colorless solid (262 mg, 1.75 mmol, 88%). 1H NMR (400 MHz, MeOH-d4) δH 6.59 (d, J = 8.2 Hz, 1H), 6.50 (d, J = 2.2 Hz, 1H), 6.36 (dd, J = 8.2, 2.2 Hz, 1H), 1.80 – 1.70 (m, 1H), 0.87 – 0.78 (m, 2H), 0.58 – 0.49 (m, 2H); LCMS tR = 1.00 min, m/z = 150.3 [M+H]+.
Step D: 5-Bromo-N-(5-cyclopropyl-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5i)
2-Amino-4-cyclopropylphenol, 23 (15 mg, 0.1 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording title compound as a colorless solid (17 mg, 0.04 mmol, 44%). 1H NMR (400 MHz, CDCl3) δH 8.40 (s, 1H), 7.61 (d, J = 2.5 Hz, 1H), 7.52 (dd, J = 8.8, 2.5 Hz, 1H), 6.90 (dd, J = 8.4, 2.2 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.60 (d, J = 2.2 Hz, 1H), 6.56 (s, 1H), 5.53 (s, 1H), 1.80 – 1.68 (m, 1H), 0.91 – 0.81 (m, 2H), 0.46 (dt, J = 6.6, 4.7 Hz, 2H); LCMS tR = 1.56 min, m/z = 383.8, 385.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-2-hydroxy-N-(2-hydroxy-5-(trifluoromethoxy)phenyl)benzenesulfonamide (5j)
Step A. 2-Amino-4-(trifluoromethoxy)phenol (20a)
2-Nitro-4-(trifluoromethoxy)phenol (900 mg, 4.03 mmol) was reacted following General Procedure B, affording a crude brown solid (693 mg, 3.59 mmol, 89%) that was taken forward without purification. LCMS tR = 0.83 min, m/z = 194.1 [M+H]+.
Step B. 5-Bromo-2-hydroxy-N-(2-hydroxy-5-(trifluoromethoxy)phenyl)benzenesulfonamide (5j)
2-amino-4-(trifluoromethoxy)phenol (19 mg, 0.1 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording a colorless solid (4 mg, 0.009 mmol, 9%). 1H NMR (400 MHz, MeOH-d4) δH 7.80 (d, J = 2.4 Hz, 1H), 7.58 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 8.8, 2.5 Hz, 1H), 7.43 (d, J = 2.3 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF −60.3; LCMS tR = 1.57 min, m/z = 429.2 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-2-hydroxy-N-(2-hydroxy-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (5k)
Step A: 2-Nitro-4-(pentafluorosulfanyl)phenol
4-Hydroxyphenyl sulfur pentafluoride (1.1 g, 5 mmol) was dissolved in DCM (25 mL) and cooled to 0 °C before the addition of nitronium tetrafluoroborate (863 mg, 6.5 mmol) and stirred for 18 h. The mixture was neutralized with sat. aq. NaHCO3 and extracted with DCM to afford a crude yellow oil (479 mg, 1.81 mmol, 36%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δH 10.78 (s, 1H), 8.57 (d, J = 2.7 Hz, 1H), 7.96 (dd, J = 9.2, 2.7 Hz, 1H), 7.27 (d, J = 9.2 Hz, 1H); 19F NMR (376 MHz, CDCl3) δF 82.43 (d, J = 151 Hz, 1F), 63.76 (d, J = 151 Hz, 4F); LCMS tR = 1.03 min, does not ionize by ESI.
Step B: 2-Amino-4-(pentafluorosulfanyl)phenol (20b)
2-Nitro-4-(pentafluorosulfanyl)phenol (479 mg, 1.81 mmol) was reacted following General Procedure B, affording a brown gum (99 mg, 0.42 mmol, 23%). LCMS tR = 0.55 min, m/z = 236.2 [M+H]+.
Step C: 5-Bromo-2-hydroxy-N-(2-hydroxy-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (5k)
2-Amino-4-(pentafluorosulfanyl)phenol, 20b (24 mg, 0.1 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording 5k as a colorless solid (12 mg, 0.026 mmol, 26%). 1H NMR (400 MHz, CDCl3) δH 7.63 (d, J = 2.6 Hz, 1H), 7.59 – 7.54 (m, 1H), 7.57 – 7.52 (m, 1H), 7.31 (d, J = 2.6 Hz, 1H), 6.96 (d, J = 9.0 Hz, 1H), 6.89 (d, J = 9.0 Hz, 1H), 6.70 (br s, 2H); 19F NMR (376 MHz, CDCl3) δF 84.3 (p, J = 151 Hz, 1F), 64.0 (d, J = 151 Hz, 4F); LCMS tR = 1.66 min, m/z = 1.66 min, m/z = 471.2 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-2-hydroxy-N-(2-hydroxy-5-(1-(trifluoromethyl)cyclopropyl)phenyl)benzenesulfonamide (5l)
Step A: 2-(4-(Benzyloxy)-3-nitrophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane
4-Hydroxy-3-nitrophenylboronic acid, pincaol ester, 24 (980 mg, 3.7 mmol), was reacted following General Procedure G. Purification by flash chromatography affords yellow solid (1215 mg, 3.4 mmol, 92%). MS (ESI) m/z = 373.1 [M+NH4]+.
Step B: 1-(Benzyloxy)-2-nitro-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (25)
2-(4-(Benzyloxy)-3-nitrophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (710 mg, 2.0 mmol), 2-bromo-3,3,3-trifluoroprop-1-ene (830 μL, 8.0 mmol), Pd(dppf)Cl2.DCM (163 mg, 0.2 mmol) and K2CO3 (553 mg, 4.0 mmol) were taken in dioxane:water (10:1, 10 mL) and heated to 70 °C for 3 h under an inert atmosphere. The cooled mixture was diluted with EtOAc and washed with water, brine. Purification by flash chromatography affords a straw-colored oil (433 mg, 1.34 mmol, 67%). 1H NMR (400 MHz, CDCl3) δH 7.96 (d, J = 2.4 Hz, 1H), 7.61 – 7.55 (m, 1H), 7.48 – 7.32 (m, 5H), 7.13 (d, J = 8.8 Hz, 1H), 6.01 (d, J = 1.6 Hz, 1H), 5.80 (d, J = 1.6 Hz, 1H), 5.28 (s, 2H); 19F NMR (376 MHz, CDCl3) δF −65.1; LCMS tR = 1.26 min, does not ionize by ESI.
Step C: 1-(Benzyloxy)-2-nitro-4-(1-(trifluoromethyl)cyclopropyl)benzene
To a mixture of 1-(benzyloxy)-2-nitro-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (65 mg, 0.20 mmol) and Ph2MeSBF4 (115 mg, 0.40 mmol) in anhydrous THF at −78 °C, was added LiHMDS [1.0 M in THF] (0.8 mL, 0.8 mmol). The mixture was stirred for 12 h, warming to r.t., then diluted with EtOAc and washed with water. Purification by flash chromatography affords a pale-yellow gum (32 mg, 0.09 mmol, 47%). 1H NMR (400 MHz, CDCl3) δH 7.96 (d, J = 2.3 Hz, 1H), 7.61 (dd, J = 8.7, 2.3 Hz, 1H), 7.51 – 7.35 (m, 5H), 7.11 (d, J = 8.7 Hz, 1H), 5.27 (s, 2H), 1.45 – 1.39 (m, 2H), 1.09 – 1.01 (m, 1H); 19F NMR (376 MHz, CDCl3) δF −70.3; LCMS tR = 1.26 min, does not ionize by ESI.
Step D: 5-Bromo-2-hydroxy-N-(2-hydroxy-5-(1-(trifluoromethyl)cyclopropyl)phenyl)benzenesulfonamide (5l)
1-(benzyloxy)-2-nitro-4-(1-(trifluoromethyl)cyclopropyl)benzene (32 mg, 0.09 mmol) was reacted following General Procedure B, affording a crude colorless solid (20 mg, 0.09 mmol) that was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording a colorless solid (13 mg, 0.029 mmol, 31%). 1H NMR (400 MHz, MeOH-d4) δH 7.72 (d, J = 2.5 Hz, 1H), 7.49 (dd, J = 8.8, 2.5 Hz, 1H), 7.36 (d, J = 2.2 Hz, 1H), 6.99 (dd, J = 8.3, 2.2 Hz, 1H), 6.83 (d, J = 8.8 Hz, 1H), 6.70 (d, J = 8.3 Hz, 1H), 1.30 – 1.20 (m, 2H), 0.93 – 0.87 (m, 2H); 19F NMR (376 MHz, CDCl3) δF −71.9; LCMS tR = 1.67 min, m/z = 451.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(1-cyanocyclopropyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5m)
Step A: 1-(4-Methoxy-3-nitrophenyl)cyclopropane-1-carbonitrile (28a)
1-(4-Methoxyphenyl) cyclopropanecarbonitrile, 27 (1 mL, 6.3 mmol) was reacted following General Procedure C, affording a yellow solid (1178 mg, 5.40 mmol, 86%). 1H NMR (400 MHz, CDCl3) δH 7.69 (d, J = 2.5 Hz, 1H), 7.60 (dd, J = 8.8, 2.5 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 3.97 (s, 3H), 1.83 – 1.71 (m, 2H), 1.45 – 1.36 (m, 2H); LCMS tR = 0.87 min, does not ionize by ESI.
Step B: 1-(4-Hydroxy-3-nitrophenyl)cyclopropane-1-carbonitrile
1-(4-methoxy-3-nitrophenyl)cyclopropane-1-carbonitrile (218 mg, 1.0 mmol) was reacted following General Procedure D to afford a cream solid (161 mg, 0.79 mmol, 79%). 1H NMR (400 MHz, CDCl3) δH 10.54 (s, 1H), 8.01 (d, J = 2.5 Hz, 1H), 7.60 (dd, J = 8.8, 2.5 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), 1.87 – 1.71 (m, 2H), 1.48 – 1.32 (m, 2H). LCMS tR = 0.86 min, does not ionize by ESI.
Step C: 1-(3-amino-4-hydroxyphenyl)cyclopropane-1-carbonitrile (29a)
1-(4-hydroxy-3-nitrophenyl)cyclopropane-1-carbonitrile (102 mg, 0.5 mmol) was reacted following General Procedure B, affording a cream solid (74 mg, 0.43 mmol, 85%). 1H NMR (400 MHz, CDCl3) δH 6.73 (d, J = 2.2 Hz, 1H), 6.67 (d, J = 8.2 Hz, 1H), 6.57 (dd, J = 8.2, 2.2 Hz, 1H), 4.72 (s, 1H), 3.71 (s, 2H), 1.65 – 1.58 (m, 2H), 1.32 – 1.27 (m, 2H); m/z (ESI) = 175.3 [M+H]+.
Step D: 5-Bromo-N-(5-(1-cyanocyclopropyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5m)
1-(3-amino-4-hydroxyphenyl)cyclopropane-1-carbonitrile (27 mg, 0.16 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording a colorless solid (47 mg, 0.12 mmol, 72%). 1H NMR (400 MHz, CDCl3) δH 8.51 (s, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 8.9, 2.4 Hz, 1H), 7.07 (dd, J = 8.5, 2.4 Hz, 1H), 6.92 – 6.85 (m, 3H), 6.80 (s, 1H), 1.67 – 1.61 (m, 2H), 1.26 – 1.20 (m, 2H); LCMS tR = 1.42 min, m/z = 408.9, 410.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(1-cyanocyclobutyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5n)
Step A: 2-(4-hydroxy-3-nitrophenyl)acetonitrile
4-Hydroxybenzylacetontrile (2.66 g, 20 mmol) was reacted following General Procedure C to afford a yellow-orange solid (3.42 g, 19.2 mmol, 96%). 1H NMR (400 MHz, CDCl3) δH 10.57 (s, 1H), 8.10 (d, J = 2.4 Hz, 1H), 7.57 (dd, J = 8.7, 2.4 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 3.76 (s, 2H).
Step B: 2-(4-(benzyloxy)-3-nitrophenyl)acetonitrile
2-(4-hydroxy-3-nitrophenyl)acetonitrile (1.78 g, 10 mmol) was reacted following General Procedure G to afford a yellow-orange solid (2.31 g, 8.6 mmol, 86%). 1H NMR (400 MHz, CDCl3) δH 7.82 (d, J = 2.4 Hz, 1H), 7.49 (dd, J = 8.7, 2.4 Hz, 1H), 7.47 – 7.30 (m, 6H), 7.15 (d, J = 8.7 Hz, 1H), 5.26 (s, 2H), 3.74 (s, 2H).
Step C: 1-(4-(benzyloxy)-3-nitrophenyl)cyclobutane-1-carbonitrile
To a solution of 2-(4-(benzyloxy)-3-nitrophenyl)acetonitrile (134 mg, 0.5 mmol) and 1,3-dibromopropane (61 μL, 0.6 mmol) in DMSO (5 mL) was added carefully added NaH (60% in mineral oil, 60 mg, 1.5 mmol) and the mixture stirred for 16 h. The mixture was diluted with EtOAc:Et2O (1:1, 50 mL) and washed with water (3 × 100 mL), brine (100 mL). Purification by flash chromatography affords a yellow-brown oil (80 mg, 0.26 mmol, 52%); m/z (ESI) = 309.0 [M+H]+.
Step D: 1-(3-amino-4-hydroxyphenyl)cyclobutane-1-carbonitrile (29b)
1-(4-(benzyloxy)-3-nitrophenyl)cyclobutane-1-carbonitrile (80 mg, 0.26 mmol) was reacted following General Procedure B to afford a colorless oil (26 mg, 0.14 mmol, 53%). 1H NMR (400 MHz, MeOH-d4) δH 6.84 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 8.2 Hz, 1H), 6.66 (dd, J = 8.2, 2.3 Hz, 1H), 2.77 – 2.66 (m, 2H), 2.66 – 2.56 (m, 2H), 2.42 – 2.28 (m, 1H), 2.13 – 2.00 (m, 1H); m/z (ESI) = 189.1 [M+H]+.
Step E: 5-Bromo-N-(5-(1-cyanocyclobutyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5n)
1-(3-amino-4-hydroxyphenyl)cyclobutane-1-carbonitrile, 29b (26 mg, 0.14 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording a colorless solid (22.5 mg, 0.05 mmol, 38%). 1H NMR (400 MHz, CDCl3) δH 7.65 (d, J = 2.4 Hz, 1H), 7.56 (dd, J = 8.8, 2.4 Hz, 1H), 7.22 (dd, J = 8.5, 2.4 Hz, 1H), 6.98 (d, J = 2.4 Hz, 1H), 6.95 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 6.71 (s, 1H), 2.82 – 2.71 (m, 2H), 2.52 – 2.33 (m, 2H), 2.08 – 2.01 (m, 2H); LCMS tR = 1.60 min, m/z = 439.8, 441.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(1-cyanocyclopentyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5o)
Step A: 1-(4-(benzyloxy)-3-nitrophenyl)cyclopentane-1-carbonitrile
To a solution of 2-(4-(benzyloxy)-3-nitrophenyl)acetonitrile (134 mg, 0.5 mmol) and 1,4-dibromobutane (72 μL, 0.6 mmol) in DMSO (5 mL) was added carefully added NaH (60% in mineral oil, 60 mg, 1.5 mmol) and the mixture stirred for 16 h. The mixture was diluted with EtOAc:Et2O (1:1, 50 mL) and washed with water (3 × 100 mL), brine (100 mL). Purification by flash chromatography affords a yellow-brown oil (103 mg, 0.32 mmol, 64%). 1H NMR (400 MHz, CDCl3) δH 7.89 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 8.8, 2.5 Hz, 1H), 7.48 – 7.31 (m, 5H), 7.13 (d, J = 8.8 Hz, 1H), 5.26 (s, 2H), 2.55 – 2.44 (m, 2H), 2.14 – 1.88 (m, 6H); LCMS tR = 1.96 min, m/z = 323.1 [M+H]+.
Step B: 1-(3-amino-4-hydroxyphenyl)cyclopentane-1-carbonitrile (29c)
1-(4-(benzyloxy)-3-nitrophenyl)cyclopentane-1-carbonitrile (103 mg, 0.32 mmol) was reacted following General Procedure B to afford a colorless oil (50 mg, 0.25 mmol, 78%). 1H NMR (400 MHz, MeOH-d4) δH 6.86 (t, J = 1.4 Hz, 1H), 6.68 (d, J = 1.4 Hz, 2H), 2.44 – 2.29 (m, 2H), 2.13 – 2.00 (m, 2H), 2.00 – 1.89 (m, 4H); m/z (ESI) = 203.2 [M+H]+.
Step C: 5-Bromo-N-(5-(1-cyanocyclopentyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5o)
1-(3-amino-4-hydroxyphenyl)cyclopentane-1-carbonitrile, 29c (50 mg, 0.25 mmol) was reacted with 18a (41 mg, 0.15 mmol) following General Procedure A, affording a colorless solid (12 mg, 0.04 mmol, 11%). 1H NMR (400 MHz, CDCl3) δH 7.61 (d, J = 2.5 Hz, 1H), 7.50 (dd, J = 8.8, 2.5 Hz, 1H), 7.17 (dd, J = 8.5, 2.3 Hz, 1H), 6.98 (d, J = 2.3 Hz, 2H), 6.90 (d, J = 5.2 Hz, 1H), 6.87 (d, J = 5.2 Hz, 1H), 2.40 – 2.26 (m, 2H), 2.01 – 1.81 (m, 6H); LCMS tR = 1.65 min, m/z = 453.8, 455.8 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(1-cyanocyclohexyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5p)
Step A: 1-(4-(benzyloxy)-3-nitrophenyl)cyclohexane-1-carbonitrile
To a solution of 2-(4-(benzyloxy)-3-nitrophenyl)acetonitrile (134 mg, 0.5 mmol) and 1,5-dibromopentane (82 μL, 0.6 mmol) in DMSO (5 mL) was added carefully added NaH (60% in mineral oil, 60 mg, 1.5 mmol) and the mixture stirred for 16 h. The mixture was diluted with EtOAc:Et2O (1:1, 50 mL) and washed with water (3 × 100 mL), brine (100 mL). Purification by flash chromatography affords a yellow-brown oil (91 mg, 0.27 mmol, 54%). LCMS tR = 1.98 min, m/z = 337.0 [M+H]+.
Step B: 1-(3-amino-4-hydroxyphenyl)cyclohexane-1-carbonitrile (29d)
2221-(4-(Benzyloxy)-3-nitrophenyl)cyclohexane-1-carbonitrile (91 mg, 0.27 mmol) was reacted following General Procedure B to afford a colorless oil (39 mg, 0.18 mmol, 67%). 1H NMR (400 MHz, MeOH-d4) δH 6.91 (d, J = 1.9 Hz, 1H), 6.72 (d, J = 1.9 Hz, 1H), 6.71 (s, 1H), 2.12 – 2.02 (m, 3H), 1.92 – 1.69 (m, 6H), 1.43 – 1.26 (m, 1H); m/z (ESI) = 217.1 [M+H]+.
Step C: 5-Bromo-N-(5-(1-cyanocyclohexyl)-2-hydroxyphenyl)-2-hydroxybenzenesulfonamide (5p)
1-(3-Amino-4-hydroxyphenyl)cyclohexane-1-carbonitrile, 29d (39 mg, 0.18 mmol) was reacted with 18a (54 mg, 0.20 mmol) following General Procedure A, affording a colorless solid (24 mg, 0.05 mmol, 30%). 1H NMR (400 MHz, MeOH-d4) δH 7.79 (d, J = 2.5 Hz, 1H), 7.51 (dd, J = 8.8, 2.5 Hz, 1H), 7.37 (d, J = 2.4 Hz, 1H), 7.07 (dd, J = 8.5, 2.4 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 1.98 (dt, J = 12.8, 1.9 Hz, 2H), 1.91 – 1.63 (m, 9H), 1.43 – 1.27 (m, 1H); LCMS tR = 1.72 min, m/z = 467.8, 469.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-chlorobenzoic acid (6a)
Methyl 3-amino-5-chlorobenzoate (143 mg, 0.5 mmol) was reacted with 18a following General Procedure A, which without purification was hydrolyzed following General Procedure E, to afford title compound as a colorless solid (86 mg, 0.21 mmol, 42% over two steps). 1H NMR (400 MHz, MeOH-d4) δH 7.83 (d, J = 2.5 Hz, 1H), 7.65 (d, J = 2.6 Hz, 1H), 7.55 (d, J = 2.6 Hz, 1H), 7.53 (dd, J = 8.7, 2.5 Hz, 1H), 7.42 (d, J = 2.6 Hz, 1H), 6.87 (d, J = 8.7 Hz, 1H); LCMS tR = 1.31 min, m/z = 427.8, 429.7 [M+Na]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-methoxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (6b)
Step A: Methyl 3-((5-bromo-2-methoxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate
Methyl 3-amino-5-chloro-2-hydroxybenzoate (202 mg, 1.0 mmol) was reacted with 16a following General Procedure A, to afford a colorless solid (370 mg, 0.82 mmol, 82%). 1H NMR (DMSO-d6) δH 7.81 – 7.76 (m, 2H), 7.55 (d, J = 2.7 Hz, 1H), 7.49 (d, J = 2.6 Hz, 1H), 7.19 (dt, J = 8.6, 1.0 Hz, 1H), 3.88 (s, 3H), 3.76 (s, 3H); LCMS tR = 1.77 min, m/z = 450, 452 [M+H]+.
Step B: 3-((5-Bromo-2-methoxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (6b)
Methyl 3-((5-bromo-2-methoxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate (370 mg, 0.82 mmol) was reacted following General Procedure E, to afford a colorless solid (260 mg, 0.59 mmol, 72%). 1H NMR (DMSO-d6) δH 9.52 (s, 1H), 7.80 – 7.75 (m, 2H), 7.52 (d, J = 2.7 Hz, 1H), 7.43 (d, J = 2.7 Hz, 1H), 7.18 (d, J = 9.6 Hz, 1H), 3.77 (s, 3H); LCMS tR = 1.55 min, m/z = 436.0, 438.0 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+Na]+ calculated for C14H11BrClNNaO6S 457.9071, found 457.9075.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (6c)
Step A: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate
Methyl 3-amino-5-chloro-2-hydroxybenzoate (605 mg, 3.0 mmol) was reacted with 18a following General Procedure A, to afford a colorless solid (918 mg, 2.1 mmol, 70%). LCMS tR = 1.15 min, m/z = 437.8 [M+H]+.
Step B: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid, 6c
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate (918 mg, 2.10 mmol) was reacted following General Procedure E, to afford a colorless solid (425 mg, 1.0 mmol, 48%). 1H NMR (400 MHz, MeOH-d4) δH 7.81 (d, J = 2.5 Hz, 1H), 7.62 (d, J = 2.6 Hz, 1H), 7.55 (d, J = 2.6 Hz, 1H), 7.53 (dd, J = 8.7, 2.5 Hz, 1H), 6.87 (d, J = 8.7 Hz, 1H); LCMS tR = 1.53 min, m/z = 421.7, 423.7 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+Na]+ calculated for C13H9BrClNNaO6S 443.8915, found 443.8909.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (6d)
Step A: 5-Bromo-3-chloro-2-hydroxybenzenesulfonyl chloride (18b)
Chlorosulfonic acid (4.66 mL, 70 mmol) was cooled to 0 °C and 2-chloro-4-bromophenol (2.08 g, 10 mmol) was added portion-wise and stirred for 24 h. The mixture was carefully poured over a slurry of ice, DCM and brine, and extracted with DCM. Purification by flash chromatography affords title compound as a colorless solid (2.19 g, 7.15 mmol, 72%). 1H NMR (400 MHz, CDCl3) δH 7.92 (1H, d, J = 2.4 Hz), 7.86 (1H, d, J = 2.4 Hz).
Step B: Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate
Methyl 3-amino-5-chloro-2-hydroxybenzoate (192 mg, 0.95 mmol) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride, 18b following General Procedure A, to afford a colorless solid (374 mg, 0.91 mmol, 91%). 1H NMR (400 MHz, CDCl3) δH 11.03 (s, 1H), 7.71 – 7.68 (m, 2H), 7.64 (d, J = 2.5 Hz, 1H), 7.62 (d, J = 2.5 Hz, 1H), 3.95 (s, 3H); LCMS tR = 1.22 min, m/z = 471.6 [M+H]+.
Step C: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (6d)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate (298 mg, 0.63 mmol) was reacted following General Procedure E, to afford a colorless solid (231 mg, 0.51 mmol, 80%). 1H NMR (400 MHz, CDCl3) δH 7.76 (s, 2H), 7.63 (d, J = 2.5 Hz, 1H), 7.61 (d, J = 2.6 Hz, 1H); LCMS tR = 1.68 min, m/z = 457.6 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoic acid (6e)
Step A: Methyl 5-bromo-2-hydroxy-3-nitrobenzoate
Methyl 2-hydroxy-4-bromobenzoate (9.24 g, 40 mmol) was reacted following General Procedure C, to afford nitrated product as a crude yellow solid (10.3 g, 37.3 mmol, 93%), which was used without purification. 1H NMR (400 MHz, CDCl3) δH 8.27 (d, J = 2.5 Hz, 1H), 8.24 (d, J = 2.5 Hz, 1H), 4.03 (s, 3H); LCMS tR = 1.02 min, m/z = 276, 278 [M+H]+.
Step B: Methyl 2-(benzyloxy)-5-bromo-3-nitrobenzoate
Methyl 5-bromo-2-hydroxy-3-nitrobenzoate (8.9 g, 32.2 mmol) was reacted following General Procedure G. Purification by flash column chromatography affords a yellow solid (10.6g, 28.8 mmol, 89%).
Step C: Methyl 2-(benzyloxy)-5-cyclopropyl-3-nitrobenzoate
Methyl 2-(benzyloxy)-5-bromo-3-nitrobenzoate (1.1 g, 3.0 mmol), cyclopropylboronic acid (515 mg, 6 mmol), Pd(OAc)2 (33.7 mg, 0.15 mmol), PCy3.HBF4 (110 mg, 0.30 mmol) and K3PO4 (1.59 g, 7.5 mmol) were taken in Toluene:H2O (10:1, 15 mL) and heated to 80 °C for 16 h. The cooled mixture was diluted with EtOAc and washed with water, brine. Purification by flash chromatography affords a brown oil (895 mg, 2.73 mmol, 91%). 1H NMR (400 MHz, CDCl3) δH 7.77 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 2.5 Hz, 1H), 7.52 – 7.47 (m, 2H), 7.46 – 7.35 (m, 3H), 3.91 (s, 3H), 2.03 – 1.94 (m, 1H), 1.13 – 1.07 (m, 2H), 0.83 – 0.74 (m, 2H).
Step D: Methyl 3-amino-5-cyclopropyl-2-hydroxybenzoate
Methyl 2-(benzyloxy)-5-cyclopropyl-3-nitrobenzoate (895 mg, 2.73 mmol) was reacted following General Procedure B, to afford an off-white solid (495 mg, 2.39 mmol, 88%). 1H NMR (400 MHz, CDCl3) δH 10.78 (s, 1H), 7.15 (d, J = 2.1 Hz, 1H), 6.94 (d, J = 2.1 Hz, 1H), 3.96 (s, 3H), 1.83 (tt, J = 8.4, 5.1 Hz, 1H), 0.96 – 0.86 (m, 2H), 0.68 – 0.59 (m, 2H); LCMS tR = 1.28 min, m/z = 208.2 [M+H]+.
Step E: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoate
Methyl 3-amino-5-cyclopropyl-2-hydroxybenzoate (207 mg, 1.0 mmol) and 18a were reacted following General Procedure A to afford a cream solid (395 mg, 0.89 mmol, 89%). 1H NMR (400 MHz, MeOH-d4) δH 7.76 (d, J = 2.5 Hz, 1H), 7.51 (dd, J = 8.8, 2.5 Hz, 1H), 7.36 (d, J = 2.3 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 3.93 (s, 3H), 1.84 (tt, J = 8.4, 5.1 Hz, 1H), 0.98 – 0.90 (m, 2H), 0.54 (dt, J = 6.5, 4.7 Hz, 2H); LCMS tR = 1.67 min, m/z = 442.1, 444.1 [M+H]+.
Step F: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoic acid (6e)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoate (44 mg, 0.10 mmol) was reacted following General Procedure E to afford a colorless solid (33 mg, 0.078 mmol, 78%). 1H NMR (400 MHz, DMSO-d6) δH 11.25 (s, 1H), 8.85 (s, 1H), 7.68 (d, J = 2.6 Hz, 1H), 7.59 (dd, J = 8.7, 2.6 Hz, 1H), 7.30 (d, J = 2.3 Hz, 1H), 7.06 (d, J = 2.3 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H), 1.84 (tt, J = 8.4, 5.1 Hz, 1H), 0.97 – 0.77 (m, 2H), 0.51 – 0.37 (m, 2H); LCMS tR = 1.51 min, m/z = 427.8, 429.8 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+H]+ calculated for C16H15BrNO6S 427.9798, found 427.9796.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoic acid (6f)
Step A: Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoate
Methyl 3-amino-5-cyclopropyl-2-hydroxybenzoate (41 mg, 0.20 mmol) and 18b were combined following General Procedure A, to afford a colorless solid (76 mg, 0.16 mmol, 80%). 1H NMR (400 MHz, DMSO-d6) δH 10.62 (s, 1H), 9.42 (s, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.34 (d, J = 2.3 Hz, 1H), 7.09 (d, J = 2.3 Hz, 1H), 3.88 (s, 3H), 1.89 (ddt, J = 13.5, 8.5, 4.3 Hz, 1H), 1.03 – 0.76 (m, 2H), 0.61 – 0.32 (m, 2H); LCMS tR = 1.76 min, m/z = 475.8, 477.7 [M+H]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoic acid (6f)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-2-hydroxybenzoate (24 mg, 0.05 mmol) was reacted following General Procedure E to afford a colorless solid (19 mg, 82%). 1H NMR (400 MHz, MeOH-d4) δH 7.61 (dd, J = 2.4, 1.7 Hz, 1H), 7.53 (dd, J = 10.1, 2.4 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.30 (d, J = 2.3 Hz, 1H), 1.85 (tt, J = 8.4, 5.1 Hz, 1H), 0.98 – 0.90 (m, 2H), 0.59 – 0.52 (m, 2H); LCMS tR = 1.53 min, m/z = 462.8, 464.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoic acid (6g)
Step A: Methyl 2-fluoro-6-hydroxybenzoate (41)
2-Fluoro-6-hydroxybenzoic acid, 40 (5.0 g, 32 mmol) was dissolved in THF (160 mL) and MeOH (32 mL) and cooled to 0 °C. TMS diazomethane [2.0 M in hexanes] (17.6 mL, 35.2 mmol) was added over 20 mins and the mixture stirred for 3 h at 0 °C. The mixture was concentrated, redissolved in EtOAc, and washed with water and brine. Purification by flash chromatography affords a colorless liquid (3.8 g, 22.3 mmol, 70%). 1H NMR (400 MHz, CDCl3) δH 11.26 (s, 1H), 7.40 – 7.36 (m, 1H), 6.79 (br d, 1H), 6.63 – 6.58 (m, 1H), 3.99 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −105.1; LCMS tR = 1.33 min, m/z = 171.1 [M+H]+.
Step B: Methyl 3-bromo-2-fluoro-6-hydroxybenzoate (42)
41 (3.8 g, 22.3 mmol) was dissolved in MeCN (144 mL) and cooled to −10 °C in a salt/ice bath. HBF4.Et2O (3.37 mL, 24.6 mmol) was added, followed by N-bromosuccinimide (4.8 g, 26.8 mmol) portion wise, then stirred for 1 h at −10 °C. The mixture was concentrated in vacuo, then redissolved in EtOAc and washed with sat. aq. Na2S2O8, sat. aq. NaCl and dried (MgSO4). Purification by flash chromatography affords a cream solid (2.49 g, 10 mmol, 45%). 1H NMR (400 MHz, CDCl3) δH 11.26 (s, 1H), 7.58 (dd, 1H, 9.1, 7.5 Hz), 6.76 (dt, 7.5, 0.9 Hz), 4.01 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −86.7; LCMS tR = 1.59 min, m/z = 249, 251 [M+H]+; Note: Material contains di-brominated by-product which is inseparable by chromatography, but is readily removed after Step C.
Step C: Methyl 3-bromo-2-fluoro-6-hydroxy-5-nitrobenzoate (43)
42 (2.49 g, 10 mmol) was reacted following General Procedure C, to afford a pale-yellow solid (1.92 mg, 65%). 1H NMR (400 MHz, CDCl3) δH 8.44 (d, 1H, J = 7.0 Hz), 4.03 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −88.1; LCMS tR = 1.55 min, m/z = 294, 296 [M+H]+.
Step D: Methyl 2-(benzyloxy)-5-bromo-6-fluoro-3-nitrobenzoate (44)
43 (588 mg, 2.0 mmol) was reacted following General Procedure G to afford a pale-yellow solid (615 mg, 80%). 1H NMR (400 MHz, CDCl3) δH 8.25 (d, 1H, J = 7.0 Hz), 7.42 – 7.38 (m, 5H), 5.16 (s, 2H), 3.88 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −86.8; LCMS tR = 1.75 min, m/z = 406, 408 [M+Na]+.
Step E: Methyl 2-(benzyloxy)-5-cyclopropyl-6-fluoro-3-nitrobenzoate
Methyl 2-(benzyloxy)-5-bromo-6-fluoro-3-nitrobenzoate (103 mg, 1.2 mmol), cyclopropylboronic acid (155 mg, 1.8 mmol), Pd(OAc)2 (13.5 mg, 0.06 mmol), PCy3.HBF4 (44.2 mg, 0.12 mmol) and K3PO4 (637 mg, 3.0 mmol) were taken in Toluene:H2O (10:1, 6 mL) and heated to 80 °C for 16 h. The cooled mixture was diluted with EtOAc and washed with water, brine. Purification by flash chromatography affords a cream solid (150 mg, 0.43 mmol, 43%). 1H NMR (400 MHz, CDCl3) δH 7.59 (d, 1H, J = 7.6 Hz), 7.45 – 7.37 (m, 5H), 5.12 (s, 2H), 3.88 (s, 3H), 2.09 – 2.06 (m, 1H), 1.11 – 1.07 (m, 2H), 0.81 – 0.77 (m, 2H); 19F NMR (376 MHz, CDCl3) δF −108.7; LCMS tR = 1.85 min, m/z = 363.1 [M+H]+.
Step F: Methyl 3-amino-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (45)
Methyl 2-(benzyloxy)-5-cyclopropyl-6-fluoro-3-nitrobenzoate (145 mg, 0.42 mmol) was reacted following General Procedure B, to afford an off-white solid (66 mg, 70%). 1H NMR (400 MHz, CDCl3) δH 11.17 (s, 1H), 6.46 (d, J = 7.1 Hz, 1H), 4.01 (s, 3H), 2.12 – 1.92 (m, 1H), 1.07 – 0.82 (m, 2H), 0.70 – 0.50 (m, 2H); 19F NMR (376 MHz, CDCl3) δF −125.0; LCMS tR = 1.04 min, m/z = 226.1 [M+H]+.
Step G: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate
45 (45 mg, 0.2 mmol) and 18a were reacted following General Procedure A to afford a pale-brown solid (69 mg, 0.15 mmol, 75%). 1H NMR (400 MHz, MeOH-d4) δH 11.34 (s, 1H), 10.13 (s, 1H), 9.00 (s, 1H), 7.61 (dd, J = 8.7, 2.6 Hz, 1H), 7.57 (d, J = 2.5 Hz, 1H), 6.97 (d, J = 8.7 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 3.82 (s, 3H), 1.90 – 1.78 (m, 1H), 0.95 – 0.77 (m, 2H), 0.39 – 0.24 (m, 2H); LCMS tR = 1.82 min, m/z = 460, 462 [M+H]+.
Step H: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoic acid (6g)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (23 mg, 0.05 mmol) was reacted following General Procedure E, to afford colorless solid (9 mg, 40%). 1H NMR (400 MHz, MeOH-d4) δH 7.71 (d, J = 2.5 Hz, 1H), 7.53 (dd, J = 8.8, 2.5 Hz, 1H), 7.13 (d, J = 7.4 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 3.96 (s, 3H), 2.01 – 1.93 (m, 1H), 0.99 – 0.91 (m, 2H), 0.59 – 0.51 (m, 2H); 19F NMR (376 MHz, MeOH-d4) δF −116.6; LCMS tR = 1.53 min, m/z = 467.7, 469.7 [M+NH4]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoic acid (6h)
Step A: Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate
Methyl 3-amino-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (45 mg, 0.2 mmol) and 18b were reacted following General Procedure A to afford a pale-brown solid (69 mg, 0.15 mmol, 75%). 1H NMR (400 MHz, MeOH-d4) δH 7.73 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.14 (d, J = 7.4 Hz, 1H), 3.97 (s, 3H), 2.03 – 1.94 (m, 1H), 1.01 – 0.93 (m, 2H), 0.63 – 0.55 (m, 2H); 19F NMR (376 MHz, MeOH-d4 δF −115.5; LCMS tR = 1.94 min, m/z = 494.2, 496.2 [M+H]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoic acid (6h)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (23 mg, 0.05 mmol) was reacted following General Procedure E, to afford a colorless solid (10 mg, 42%). 1H NMR (400 MHz, DMSO-d6) δH 7.94 (d, J = 2.5 Hz, 1H), 7.61 (d, J = 2.5 Hz, 1H), 6.78 (d, J = 7.7 Hz, 1H), 1.89 (td, J = 8.4, 4.2 Hz, 1H), 1.00 – 0.83 (m, 2H), 0.48 – 0.37 (m, 2H); 19F NMR (376 MHz, DMSO-d6) δF −116.3; LCMS tR = 1.74 min, m/z = 477.7, 479.7 [M+H]+; Purity (AUC) ≥95%. HRMS (ESI/TOF) [M+Na]+ calculated for C16H12BrClFNNaO6S 501.9133, found 501.9121.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(trifluoromethoxy)benzoic acid (6i)
Step A: Methyl 3-bromo-5-(trifluoromethoxy)benzoate
3-Bromo-5-(trifluoromethoxy)benzoic acid (2.0 g, 7.0 mmol), was dissolved in THF (35 mL) and MeOH (7 mL) and cooled to 0 °C. TMS diazomethane [2.0 M in Et2O] (3.85 mL, 7.7 mmol) was added drop-wise, then the mixture stirred for 16 h. The mixture was concentrated and purified by flash chromatography to afford a colorless solid (1.82 g, 6.09 mmol, 82%). 1H NMR (400 MHz, CDCl3) δH 8.12 (t, J = 1.6 Hz, 1H), 7.83 (t, J = 2.4 Hz, 1H), 7.57 (t, J = 1.6 Hz, 1H), 3.95 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −57.98; m/z (ESI) = 300.1 [M+H]+.
Step B: Methyl 3-amino-5-(trifluoromethoxy)benzoate
Methyl 3-bromo-5-(trifluoromethoxy)benzoate (1.79 g, 5.98 mmol) tert-butyl carbamate (841 mg, 7.18 mmol), Pd2dba3 (110 mg, 0.12 mmol), Xantphos (208 mg, 0.36 mmol) and Cs2CO3 (5.85 g, 17.9 mmol) were combined in Toluene (24 mL) in a sealed tube and stirred at 90 °C for 16 h. The mixture was filtered through celite, concentrated and the residue re-dissolved in DCM (50 mL). TFA (10 mL) was added and the mixture stirred at r.t. for 2 h. The mixture was washed with sat. aq. Na2CO3, concentrated and purified by flash chromatography to afford a brown solid (1.18 g, 5.02 mmol, 84%). m/z (ESI) = 236.1 [M+H]+.
Step C: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(trifluoromethoxy)benzoate
Methyl 3-amino-5-(trifluoromethoxy)benzoate (118 mg, 0.5 mmol) and 18a were reacted following General Procedure A to afford a pale-brown solid (179 mg, 0.38 mmol, 76%). 1H NMR (400 MHz, CDCl3) δH 7.72 – 7.69 (m, 1H), 7.68 (d, J = 2.5 Hz, 1H), 7.64 (t, J = 1.7 Hz, 1H), 7.52 (dd, J = 8.8, 2.5 Hz, 1H), 7.27 (s, 1H), 6.89 (d, J = 8.9 Hz, 1H), 3.93 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −58.0; LCMS tR = 1.11 min, m/z = 486.9, 488.9 [M+NH4]+.
Step D: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(trifluoromethoxy)benzoic acid (6i)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(trifluoromethoxy)benzoate (91 mg, 0.19 mmol) was reacted following General Procedure E to afford a colorless solid (64 mg, 0.14 mmol, 72%). 1H NMR (400 MHz, MeOH-d4) δH 7.86 (d, J = 2.5 Hz, 1H), 7.79 (dd, J = 2.1, 1.4 Hz, 1H), 7.53 – 7.49 (m, 2H), 7.35 (dq, J = 2.1, 1.1 Hz, 1H), 6.85 (d, J = 8.7 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF −59.7; LCMS tR = 1.34 min, m/z = 477.7, 479.7 [M+Na]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoic acid (6j)
Step A: Methyl 2-hydroxy-5-(trifluoromethoxy)benzoate
2-Hydroxy-5- (trifluoromethoxy)benzoic acid (2.0 g, 9.00 mmol) was dissolved in DCM (30 mL) and MeOH (30 mL) and cooled to 0 °C. To the flask was added EDCI (2.59 g, 13.5 mmol), followed by DMAP (220.0 mg, 1.80 mmol, 0.2 eq). The reaction was stirred for 16 h, warming to r.t.. The mixture was washed with water and dried (Na2SO4). Purification by flash chromatography afforded title compound (1.35 g, 5.74 mmol, 63%). LCMS tR = 1.62 min, m/z = 237.2 [M+H]+.
Step B. Methyl 2-hydroxy-3-nitro-5-(trifluoromethoxy)benzoate
Methyl 2-hydroxy-5- (trifluoromethoxy)benzoate (1.36 g, 5.74 mmol) was reacted following General Procedure C. Purification by flash chromatography afforded title compound (1.61 g, quant.). LCMS tR = 1.51 min, m/z = 252.1 [M+H]+.
Step C: Methyl 3-amino-2-hydroxy-5-(trifluoromethoxy)benzoate
Methyl 2- hydroxy-3-nitro-5-(trifluoromethoxy)benzoate (1.61 g, 5.74 mmol) was reacted following General Procedure B. The crude product was used for the next step without further purification (1.40 g, 5.59 mmol, 97% over 2 steps). LCMS tR = 1.42 min, m/z = 251.2 [M+H]+.
Step D: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoate
Methyl 3-amino-2-hydroxy-5-(trifluoromethoxy)benzoate (1.26 g, 5.0 mmol) and 18a were reacted following General Procedure A to afford a pale-brown solid (1.02 g, 2.2 mmol, 43%). 1H NMR (400 MHz, CDCl3) δH 11.10 (s, 1H), 8.68 (s, 1H), 7.68 (d, J = 2.4 Hz, 1H), 7.64 – 7.60 (m, 1H), 7.51 – 7.46 (m, 3H), 6.88 (d, J = 8.9 Hz, 1H), 3.96 (s, 3H); 19F NMR (376 MHz, CDCl3) δF −58.6; LCMS tR = 1.20 min, m/z = 485.8, 487.8 [M+H]+.
Step E: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoic acid (6j)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoate (1.02 g, 2.09 mmol) was reacted following General Procedure E to afford a colorless solid (575 mg, 1.22 mmol, 58%). 1H NMR (400 MHz, MeOH-d4) δH 7.82 (d, J = 2.5 Hz, 1H), 7.58 – 7.55 (m, 1H), 7.54 (dd, J = 8.8, 2.5 Hz, 1H), 7.45 – 7.41 (m, 1H), 6.86 (d, J = 8.8 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF −60.3; LCMS tR = 1.57 min, m/z = 471.8, 473.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoic acid (6k)
Step A. Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoate
Methyl 3-amino-2-hydroxy-5-(trifluoromethoxy)benzoate (53.4 mg, 0.21 mmol) was reacted with 18b following General Procedure A to afford title compound as a solid (26.4 mg, 31%). LCMS tR = 1.80 min, m/z = 521.7 [M+H]+.
Step B. 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoic acid, 6k
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(trifluoromethoxy)benzoate (10.5 mg, 0.02 mmol) was reacted following General Procedure E to afford a colorless solid (7.0 mg, 68%). 1H NMR (400 MHz, MeOH-d4) δ 7.74 (q, J = 2.4 Hz, 2H), 7.54 (d, J = 2.3 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δ −60.3; LCMS tR = 1.66 min, m/z = 507.6 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoic acid (6l)
Step A: Methyl 3-bromo-5-(pentafluorosulfanyl)benzoate
3-Bromo-5-(pentafluorosulfanyl) benzoic acid (1.05 g, 3.21 mmol) was dissolved in THF (16 mL) and MeOH (3.2 mL) and cooled to 0 °C. TMS diazomethane [2.0 M in Et2O] (1.77 mL, 3.53 mmol) was added dropwise, then the mixture stirred for 16 h. The mixture was concentrated and purified by flash chromatography to afford a colorless solid (1.02 mg, 2.97 mmol, 93%). 1H NMR (400 MHz, CDCl3) δH 8.36 (dd, J = 2.1, 1.3 Hz, 1H), 8.33 (t, J = 1.6 Hz, 1H), 8.07 (t, J = 2.0 Hz, 1H), 3.98 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 81.67 (d, J = 151 Hz, 1F), 62.94 (d, J = 151 Hz, 4F).
Step B: Methyl 3-amino-5-(pentafluorosulfanyl)benzoate
Methyl 3-bromo-5-(pentafluorosulfanyl)benzoate (890 mg, 2.61 mmol), tert-butyl carbamate (367 mg, 3.13 mmol), Pd2dba3 (48 mg, 0.05 mmol), Xantphos (91 mg, 0.16 mmol) and Cs2CO3 (2.55 g, 7.83 mmol) were combined in Toluene (10 mL) in a sealed tube and stirred at 90 °C for 16 h. The mixture was filtered through celite, concentrated and the residue was redissolved in DCM (25 mL). TFA (5 mL) was added and the mixture stirred at r.t. for 2 h. The mixture was washed with sat. aq. Na2CO3, concentrated and purified by flash chromatography to afford a brown solid (585 mg, 2.11 mmol, 81%). 1H NMR (400 MHz, CDCl3) δH 7.82 – 7.77 (m, 1H), 7.49 – 7.47 (m, 1H), 7.22 (t, J = 2.1 Hz, 1H), 3.95 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 83.9 (p, J = 150 Hz, 1F), 62.47 (d, J = 150 Hz, 4F).
Step C: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoate
Methyl 3-amino-5-(pentafluorosulfanyl)benzoate (139 mg, 0.50 mmol) and 18a were reacted following General Procedure A to afford a colorless solid (184 mg, 0.36 mmol, 72%). 1H NMR (400 MHz, CDCl3) δH 8.69 (d, J = 5.0 Hz, 2H), 8.26 (t, J = 1.7 Hz, 1H), 7.92 (br s, 1H), 7.74 – 7.69 (m, 2H), 7.56 (dd, J = 8.8, 2.4 Hz, 1H), 6.93 (d, J = 8.8 Hz, 1H), 3.98 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 81.6 (p, J = 151 Hz, 1F), 62.7 (d, J = 151 Hz); LCMS tR = 1.14 min, m/z = 528.8, 530.9 [M+NH4]+.
Step D: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoic acid (6l)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoate (68 mg, 0.13 mmol) was reacted following General Procedure E to afford a colorless solid (8 mg, 0.016 mmol, 12%). 1H NMR (600 MHz, MeOH-d4) δH 8.02 – 7.98 (m, 2H), 7.87 – 7.83 (m, 2H), 7.51 (dd, J = 8.8, 2.5 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF 81.4 (p, J = 149 Hz, 1F), 60.9 (d, J = 149 Hz, 4F); LCMS tR = 1.40 min, m/z = 519.7, 521.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoic acid (6m)
Step A: Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoate
Methyl 3-amino-5-(pentafluorosulfanyl)benzoate (139 mg, 0.50 mmol) and 18b were reacted following General Procedure A to afford title compound as a colorless solid (131 mg, 0.24 mmol, 48%). LCMS tR = 1.67 min, m/z = 569.5 [M+NH4]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoic acid (6m)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoate (72 mg, 0.13 mmol) was reacted following General Procedure E to afford a colorless solid (18 mg, 0.034 mmol, 26%). 1H NMR (400 MHz, MeOH-d4) δH 8.08 – 8.04 (t, J = 1.9 Hz, 1H), 8.02 (t, J = 1.9 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.84 (t, J = 1.9 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF 81.4 (p, J = 149 Hz, 1F), 60.98 (d, J = 149 Hz, 4F); LCMS tR = 1.10 min, m/z = 533.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (6n)
Step A: Phenyl 2-methoxy-5-(pentafluorosulfanyl)benzoate
(3-Bromo-4-methoxyphenyl)pentafluorosulfane, 34 (939 mg, 3.0 mmol), Pd(OAc)2 (34 mg, 0.15 mmol), P(tBu)3.HBF4 (174 mg, 0.60 mmol) and phenol (282 mg, 3.0 mmol) were taken in MeCN (13.7 mL) in a 40 mL reaction vial and phenyl formate (500 μL) and Et3N (1.25 mL, 9.0 mmol) were added. The reaction was run in 5-parallel reactions. The sealed vials were heated to 90 °C for 18 h, then combined after filtration through celite, concentrated, redissolved in EtOAc (100 mL) and washed with H2O (100 mL). Purification by flash chromatography affords a pale brown liquid (4.36 g, 12.3 mmol, 82%).
1H NMR (400 MHz, CDCl3) δH 8.41 (d, J = 2.9 Hz, 1H), 7.93 (dd, J = 9.2, 2.9 Hz, 1H), 7.48 – 7.40 (m, 2H), 7.31 – 7.26 (m, 2H), 7.07 (d, J = 9.2 Hz, 1H), 6.93 (tt, J = 7.3, 1.1 Hz, 1H), 4.00 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 84.52 (d, J = 150 Hz, 1F), 64.07 (d, J = 150 Hz, 4F); MS (ESI) m/z = 355.2.
Step B: Phenyl 2-hydroxy-5-(pentafluorosulfanyl)benzoate (35)
Phenyl 2-methoxy-5-(pentafluorosulfanyl)benzoate (2.80 g) was reacted following General Procedure D, to afford a colorless solid (1.81 g, 5.31 mmol, 67%). 1H NMR (400 MHz, CDCl3) δH 10.88 (s, 1H), 8.50 (d, J = 2.8 Hz, 1H), 7.92 (dd, J = 9.2, 2.8 Hz, 1H), 7.53 – 7.45 (m, 2H), 7.40 – 7.31 (m, 1H), 7.29 – 7.20 (m, 3H), 7.11 (d, J = 9.2 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ 84.58 (d, J = 151 Hz, 1F), 63.90 (d, J = 151 Hz, 4F); MS (ESI) m/z = 341.3.
Step C: 2-Hydroxy-5-(pentafluorosulfanyl)benzoic acid
Phenyl 2-hydroxy-5-(pentafluorosulfanyl)benzoate, 35 (1.81 g, 5.32 mmol) was reacted following General Procedure E, to afford title compound (1.15 g, 4.35 mmol, 82%). 1H NMR (400 MHz, MeOHd4) δH 8.27 (d, J = 2.9 Hz, 1H), 7.91 (dd, J = 9.2, 2.9 Hz, 1H), 7.07 (d, J = 9.2 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF 83.68 (d, J = 149 Hz, 1F), 62.35 (d, J = 149 Hz, 4F).
Step D: 2-Hydroxy-3-nitro-5-(pentafluorosulfanyl)benzoic acid
2-Hydroxy-5-(pentafluorosulfanyl)benzoic acid (300 mg, 1.14 mmol) was reacted following General Procedure C, to afford title compound (159 mg, 0.51 mmol, 45%). 1H NMR (400 MHz, MeOH-d4) δH 8.60 (d, J = 2.8 Hz, 4H), 8.53 (d, J = 2.8 Hz, 4H). 19F NMR (376 MHz, MeOH-d4) δF 80.9 (p, J = 149 Hz), 62.4 (d, J = 149 Hz).
Step E: Methyl 2-hydroxy-3-nitro-5-(pentafluorosulfanyl)benzoate
To a mixture containing 2-hydroxy-3-nitro-5-(pentafluorosulfanyl)benzoic acid (60 mg, 0.19 mmol) in MeOH (1 mL), two drops of H2SO4 were added. The reaction mixture was heated to reflux and stirred overnight. The cooled mixture was concentrated and the residue dissolved in DCM and washed with water. The organic phase was concentrated to afford crude title compound which was used without purification (60 mg, 0.19 mmol, 96%). 1H NMR (400 MHz, CDCl3) δH 8.55 (d, J = 2.9 Hz, 1H), 8.51 (d, J = 2.9 Hz, 1H), 4.09 (s, 3H).
Step F: Methyl 3-amino-2-hydroxy-5-(pentafluorosulfanyl)benzoate (36)
Methyl 2-hydroxy-3-nitro-5-(pentafluorosulfanyl)benzoate (223 mg, 0.69 mmol) was reacted following General Procedure B, to afford title compound (170 mg, 0.58 mmol, 84%). 1H NMR (400 MHz, Chloroform-d) δ 11.26 (s, 1H), 7.64 (d, J = 2.6 Hz, 1H), 7.20 (d, J = 2.6 Hz, 1H), 3.98 (br s, H); 19F NMR (376 MHz, Chloroform-d) δ 85.67 (p, J = 151 Hz, 1F), 63.71 (d, J = 150 Hz, 4F); LCMS tR = 1.06 min, m/z = 294.2 [M+H]+.
Step G: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfaneyl)benzoate
36 (50 mg, 0.17 mmol) and 18a were reacted following General Procedure A to afford title compound as a colorless solid (54 mg, 0.10 mmol, 60%). 1H NMR (400 MHz, CDCl3) δH 8.06 (d, J = 2.7 Hz, 1H), 8.03 (d, J = 2.7 Hz, 1H), 7.70 (d, J = 2.5 Hz, 1H), 7.48 (dd, J = 8.8, 2.5 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 3.99 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 83.5 (p, J = 151 Hz, 1F), 63.7 (d, J = 151 Hz, 4F). LCMS: tR = 1.24 min, m/z = 527.9, 529.9 [M+H]+.
Step H: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (6n)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfaneyl)benzoate (27 mg, 0.046 mmol) was reacted following General Procedure E, to afford a colorless solid (6 mg, 0.12 mmol, 26%). 1H NMR (400 MHz, MeOH-d4) δH 8.02 (d, J = 2.7 Hz, 1H), 7.97 (d, J = 2.7 Hz, 1H), 7.81 (d, J = 2.5 Hz, 1H), 7.52 (dd, J = 8.8, 2.5 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF 83.0 (p, J = 149 Hz, 1F), 62.2 (d, J = 149 Hz, 4F); LCMS tR = 1.07 min, m/z = 537.3 [M+Na]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (6o)
Step A: Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate
Methyl 3-amino-2-hydroxy-5-(pentafluorosulfanyl)benzoate (50 mg, 0.17 mmol) and 18b were reacted following General Procedure A, to afford a colorless solid (21 mg, 0.04 mmol, 22%). 1H NMR (400 MHz, CDCl3) δH 8.72 – 8.67 (m, 1H), 8.11 (d, J = 2.6 Hz, 1H), 8.02 (d, J = 2.6 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 3.99 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 83.3 (p, J= 151 Hz, 1F), 63.7 (d, J = 151 Hz, 4F). LCMS: tR = 1.19 min, m/z = 562.1 [M+H]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (6o)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate (20 mg, 0.04 mmol) was reacted following General Procedure E, to afford title compound as a colorless solid (8 mg, 0.015 mmol, 41%). 1H NMR (400 MHz, Acetone-d6) δH 8.12 (d, J = 2.7 Hz, 1H), 8.09 (d, J = 2.7 Hz, 1H), 7.86 (d, J = 2.4 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δ 82.94 (p, J = 149 Hz), 62.21 (d, J = 149 Hz); LCMS tR = 1.111 min, m/z = 548.1, 550.1 [M+H]+, Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)benzoic acid (6p)
Step A: Methyl 3-bromo-5-(cyanomethyl)benzoate
To a solution of methyl 3-bromo-5-methylbenzoate, 52 (10.0 g, 44 mmol) in CHCl3 (200 mL) was added NBS (8.6 g, 48 mmol) followed by AIBN (730 mg, 4.4 mmol) and the mixture heated to reflux overnight. Upon cooling the mixture was filtered through celite and concentrated under reduced pressure. To the crude intermediate (6 g, 19.5 mmol) in THF (100 mL) was added TMSCN (1.93 g 19.5 mmol) and stirred for 30 min, then TBAF (1.0 M in THF, 19.5 mL) added. The mixture was stirred at r.t. for 16 h, then concentrated and purified by flash chromatography to afford title compound (2.0 g, 7.9 mmol, 18%). 1H NMR (400 MHz, CDCl3) δΗ 8.17 (s, 1H), 7.96 (s, 1H), 7.73 (s, 1H), 3.97 (s, 3H), 3.82 (s, 2H).
Step B: Methyl 3-bromo-5-(1-cyanocyclobutyl)benzoate
To a solution of methyl 3-bromo-5-(cyanomethyl)benzoate (500 mg, 2 mmol) and 1,3-diiodopropane (1.18 g, 4 mmol) in THF (30 mL) was added t-BuOK (448 mg, 4 mmol) and the mixture stirred at r.t. for 16 h. The mixture was diluted with EtOAc and washed with sat. aq. NH4Cl, concentrated, and purified by flash chromatography to afford title compound (353 mg, 1.2 mmol, 60%). 1H NMR (400 MHz, CDCl3) δΗ 8.13 (t, J = 1.7 Hz, 1H), 8.01 (t, J = 1.7 Hz, 1H), 7.74 (t, J = 1.7 Hz, 1H), 3.95 (s, 3H), 2.82 – 2.91 (m, 2H), 2.64 (td, J = 9.4, 12.0 Hz, 2H), 2.40 – 2.55 (m, 1H), 2.12 (td, J = 4.6, 7.0 Hz, 1H).
Step C: Methyl 3-amino-5-(1-cyanocyclobutyl)benzoate (53)
Methyl 3-bromo-5-(1-cyanocyclobutyl)benzoate (1.1 g, 3.7 mmol), tert-butylcarbamate (500 mg, 4.3 mmol), Cs2CO3 (2.33 g, 7.2 mmol) in DMF (20 mL) was added Pd2dba3 (171 mg, 0.19 mmol), Xantphos (220 mg, 0.38 mmol) and the mixture heated to 150 °C for 16 h. The cooled mixture was filtered through celite and concentrated. The residue was dissolved in DCM (20 mL) and TFA (5 mL) added, the mixture stirred for 1 h, then washed with sat. aq. NaHCO3 and concentrated. Purification by flash chromatography affords title compound (300 mg, 1.3 mmol, 35%). 1H NMR (400 MHz, CDCl3) δΗ 7.45 (s, 1H), 7.26 – 7.35 (m, 1H), 6.89 (t, J = 1.9 Hz, 1H), 3.91 (s, 3H), 2.74 – 2.93 (m, 2H), 2.55 – 2.69 (m, 2H), 2.42 (td, J = 8.8, 11.5 Hz, 1H), 1.94 – 2.13 (m, 1H); MS (ESI) m/z = 231.1 [M+H]+.
Step D: Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)benzoate
Methyl 3-amino-5-(1-cyanocyclobutyl)benzoate, 53 (230 mg, 1.0 mmol) and 18a were reacted according to General Procedure A. Purification by flash chromatography afforded a colorless solid (435 mg, 0.94 mmol, 94%). MS (ESI) m/z = 484.0 [M+NH4]+.
Step E: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)benzoic acid (6p)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)benzoate (435 mg, 0.94 mmol) was reacted following General Procedure E (at r.t.) to afford a colorless solid (297 mg, 0.66 mmol, 70%). 1H NMR (400 MHz, MeOH-d4) δH 7.85 (d, J = 2.5 Hz, 1H), 7.79 (t, J = 1.8 Hz, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.50 (t, J = 1.8 Hz, 1H), 7.47 (dd, J = 8.8, 2.5 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H), 2.77 (ddt, J = 12.4, 8.8, 3.3 Hz, 2H), 2.61 – 2.49 (m, 2H), 2.38 (dp, J = 11.7, 8.8 Hz, 1H), 2.07 (dtt, J = 11.7, 9.2, 4.5 Hz, 1H); LCMS tR = 1.44 min, m/z = 467.8, 469.8 [M+NH4]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (6q)
Step A: 1-(3-Bromo-4-methoxyphenyl)cyclobutane-1-carbonitrile
2-(3-Bromo-4-methoxyphenyl)acetonitrile (4.52 g, 20 mmol) and 1,3-dibromopropane (2.23 mL, 22 mmol) were dissolved in DMSO (100 mL), to this mixture was added NaH (60% dispersion in mineral oil, 2.0 g, 50 mmol) portion-wise. The solution was stirred for 16 h at r.t., then diluted with EtOAc:Et2O (200 mL, 1:1) and washed with water (3 × 400 mL). The combined aqueous layers were back-extracted with further EtOAc (200 mL). The combined organics were washed with sat. aq. NaCl, concentrated and purified by ISCO flash column chromatography (120 g, 0–25% EtOAc in hexanes) to afford a colorless oil (3.65 g, 13.7 mmol, 68%). 1H NMR (400 MHz, CDCl3) δH 7.61 (d, J = 2.4 Hz, 1H), 7.34 (dd, J = 8.6, 2.4 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 3.93 (s, 3H), 2.88 – 2.78 (m, 2H), 2.66 – 2.54 (m, 2H), 2.44 (dt, J = 11.6, 8.7 Hz, 1H), 2.16 – 2.04 (m, 1H); LCMS tR = 1.10 min, no mass observed.
Step B: Phenyl 5-(1-cyanocyclobutyl)-2-methoxybenzoate
1-(3-Bromo-4-methoxyphenyl)cyclobutane-1-carbonitrile (531 μL, 3.43 mmol), Pd(OAc)2 (39 mg, 0.17 mmol), P(tBu4).HBF4 (199 mg, 0.69 mmol) and phenol (323 mg, 3.43 mmol) were taken up in anhydrous MeCN (13.7 mL) in a sealed tube. Phenyl formate (687 μL, 6.86 mmol) and NEt3 (1.43 mL, 10.3 mmol) were added and the mixture heated to 90 °C for 18 h. This reaction was performed in quadruplicate. Upon cooling the four reactions were combined, filtered through celite, and concentrated. The residue was dissolved in EtOAc and washed with water. Purification by flash chromatography afforded a colorless solid (3.72 g, 12.1 mmol, 88%).1H NMR (CDCl3) δH 8.04 (d, J = 2.6 Hz, 1H), 7.61 (dd, J = 8.7, 2.6 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.33 – 7.23 (m, 3H), 7.09 (d, J = 8.7 Hz, 1H), 3.98 (s, 3H), 2.93 – 2.83 (m, 2H), 2.66 (dt, J = 11.8, 9.0 Hz, 2H), 2.55 – 2.41 (m, 1H), 2.19 – 2.06 (m, 1H); LCMS tR = 1.71 min, m/z = 308.1 [M+H]+.
Step C: Phenyl 5-(1-cyanocyclobutyl)-2-hydroxybenzoate
Phenyl 5-(1-cyanocyclobutyl)-2-methoxybenzoate (3.72 g, 12.1 mmol) was reacted following General Procedure D to afford a colorless oil (3.47 g, 11.84 mmol, 98%). 1H NMR (400 MHz, CDCl3) δH 10.55 (s, 1H), 8.09 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.7, 2.5 Hz, 1H), 7.52 – 7.43 (m, 2H), 7.34 (d, J = 7.4 Hz, 1H), 7.25 – 7.20 (m, 2H), 7.09 (d, J = 8.7 Hz, 1H), 2.91 – 2.81 (m, 2H), 2.72 – 2.59 (m, 2H), 2.53 – 2.38 (m, 1H), 2.16 – 2.05 (m, 1H) (Method A) LCMS: tR = 1.20 min, m/z = 294.1 [M+H]+.
Step D: Phenyl 5-(1-cyanocyclobutyl)-2-hydroxy-3-nitrobenzoate
Phenyl 5-(1-cyanocyclobutyl)-2-hydroxybenzoate (2.93 g, 10 mmol) was dissolved in DCE/H2O (1:1, 20 mL) and cooled to 0 °C in an ice/water bath. Tetrabutylammonium bromide (161 mg, 0.50 mmol) was added, followed by HNO3 (conc., 1.3 mL, 20 mmol). The mixture was stirred vigorously at 60 °C for 16 h, then cooled, diluted with CH2Cl2, and washed with water. Purification by flash chromatography afforded a pale-yellow solid (2.71 g, 8.0 mmol, 80%). 1H NMR (400 MHz, CDCl3) δH 8.42 (d, J = 2.5 Hz, 1H), 8.31 (d, J = 2.6 Hz, 1H), 7.55 – 7.48 (m, 2H), 7.43 – 7.35 (m, 1H), 7.32 – 7.24 (m, 3H), 3.00 – 2.89 (m, 2H), 2.76 – 2.63 (m, 2H), 2.61 – 2.49 (m, 1H), 2.25 – 2.11 (m, 1H); LCMS tR = 1.18 min, m/z = 339.0 [M+H]+.
Step E: Phenyl 3-amino-5-(1-cyanocyclobutyl)-2-hydroxybenzoate
Phenyl 5-(1-cyanocyclobutyl)-2-hydroxy-3-nitrobenzoate (2.71 g, 8.0 mmol) was reacted according to General Procedure B. Purification by flash chromatography afforded a cream solid (2.15 g, 7.0 mmol, 87%). 1H NMR (400 MHz, CDCl3) δH 10.70 (d, J = 0.7 Hz, 1H), 7.52 – 7.46 (m, 2H), 7.48 (d, J = 2.3 Hz, 1H), 7.38 – 7.32 (m, 1H), 7.27 – 7.22 (m, 2H), 6.98 (dd, J = 2.3, 0.7 Hz, 1H), 2.90 – 2.77 (m, 2H), 2.71 – 2.58 (m, 2H), 2.54 – 2.39 (m, 1H), 2.16 – 2.07 (m, 1H); LCMS (Method B) tR = 1.08 min, m/z = 309.2 [M+H]+.
Step F: Phenyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate
Phenyl 3-amino-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (308 mg, 1.0 mmol) and 18a were reacted according to General Procedure A. Purification by flash chromatography afforded a colorless solid (393 mg, 0.72 mmol, 72%). 1H NMR (400 MHz, CDCl3) δH 8.63 (dt, J = 4.4, 1.7 Hz, 2H), 7.90 (d, J = 2.3 Hz, 1H), 7.82 (d, J = 2.3 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.49 – 7.45 (m, 2H), 7.38 – 7.35 (m, 2H), 7.23 – 7.17 (m, 2H), 6.88 (d, J = 8.8 Hz, 1H), 2.94 – 2.84 (m, 3H), 2.68 – 2.56 (m, 2H), 2.55 – 2.43 (m, 1H), 2.21 – 2.09 (m, 1H); LCMS tR = 1.86 min, m/z = 559.8, 561.7 [M+NH4]+.
Step G: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (6q)
Phenyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (108 mg, 0.20 mmol) was reacted following General Procedure E (at r.t. instead of 65 °C) to afford a colorless solid (61 mg, 0.16 mmol, 80%). 1H NMR (400 MHz, CDCl3) δH 7.85 (d, J = 2.5 Hz, 1H), 7.68 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 8.8, 2.5 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 2.82 – 2.72 (m, 2H), 2.60 – 2.49 (m, 2H), 2.46 – 2.32 (m, 1H), 2.16 – 2.03 (m, 1H); LCMS tR = 1.44 min, m/z = 466.8, 468.7 [M+H]+; Purity (AUC) ≥95%.; HRMS (ESI/TOF) [M+Na]+ calculated for C18H15BrN2NaO6S 488.9726, found 488.9746.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (6r)
Step A: Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate
Phenyl 3-amino-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (154 mg, 0.50 mmol) and 18b were reacted following General Procedure A to afford a brown solid (208 mg, 72%). 1H NMR (400 MHz, MeOH-d4) δH 7.79 (d, J = 2.5 Hz, 1H), 7.78 (d, J = 2.5 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.72 (d, J = 2.5 Hz, 1H), 2.05 (dd, J = 8.3, 3.5 Hz, 2H), 1.99 – 1.87 (m, 2H), 1.89 – 1.78 (m, 1H), 1.80 – 1.69 (m, 3H), 1.46 – 1.31 (m, 1H); LCMS tR = 1.97 min, m/z = 595.7, 597.7 [M+H]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (6r)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (58 mg, 0.10 mmol) was reacted following General Procedure E at room temperature to afford a colorless solid (12 mg, 48%). 1H NMR (400 MHz, MeOH-d4) δH 7.76 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 2.82 – 2.72 (m, 2H), 2.62 – 2.51 (m, 2H), 2.46 – 2.32 (m, 1H), 2.14 – 2.03 (m, 1H); LCMS tR = 1.56 min, m/z = 500.7, 502.8 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+Na]+ calculated for C18H14BrClN2NaO6S 522.9337, found 522.9358.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoic acid (6s)
Step A: 1-(3-Bromo-4-methoxyphenyl)cyclohexane-1-carbonitrile
2-(3-Bromo-4-methoxyphenyl)acetonitrile (4.52 g, 20 mmol) and 1,5-dibromopentane (3.0 mL, 22 mmol) were dissolved in DMSO (100 mL), to this mixture was added NaH (60% dispersion in mineral oil, 2.0 g, 50 mmol) portion-wise. The solution was stirred for 16 h at r.t., then diluted with EtOAc:Et2O (200 mL, 1:1) and washed with water (3 × 400 mL). The combined aqueous layers were back-extracted with further EtOAc (200 mL). The combined organics were washed with sat. aq. NaCl, concentrated, and purified by ISCO flash column chromatography (120 g, 0–25% EtOAc in hexanes) to afford a colorless solid (4.35 g, 14.8 mmol, 74%). 1H NMR (400 MHz, CDCl3) δH 7.62 (d, J = 2.5 Hz, 1H), 7.42 (dd, J = 8.6, 2.5 Hz, 1H), 6.90 (d, J = 8.6 Hz, 1H), 3.90 (s, 3H), 2.18 – 2.10 (m, 2H), 1.91 – 1.79 (m, 6H), 1.78 – 1.65 (m, 2H); LCMS (Method B) tR = 1.10 min, no mass observed.
Step B: Phenyl 5-(1-cyanocyclohexyl)-2-methoxybenzoate
1-(3-Bromo-4-methoxyphenyl)cyclohexane-1-carbonitrile (883 mg, 3.0 mmol), Pd(OAc)2 (39 mg, 0.17 mmol), P(tBu4).HBF4 (199 mg, 0.69 mmol) and phenol (323 mg, 3.43 mmol) were taken up in anhydrous MeCN (13.7 mL) in a sealed tube. Phenyl formate (687 μL, 6.86 mmol) and NEt3 (1.43 mL, 10.3 mmol) were added and the mixture heated to 90 °C for 18 h. This reaction was performed in quadruplicate. Upon cooling the four reactions were combined, filtered through celite, and concentrated. The residue was dissolved in EtOAc and washed with water. Purification by flash chromatography afforded a colorless solid (3.53 g, 10.5 mmol, 88%). 1H NMR (CDCl3) δH 8.07 (d, J = 2.7 Hz, 1H), 7.73 (dd, J = 8.8, 2.7 Hz, 1H), 7.49 – 7.40 (m, 2H), 7.33 – 7.22 (m, 3H), 7.08 (d, J = 8.8 Hz, 1H), 3.97 (s, 3H), 2.22 (d, J = 11.6 Hz, 2H), 1.98 – 1.74 (m, 8H); LCMS (Method A): tR = 1.86 min, m/z = 336.1 [M+H]+.
Step C: Phenyl 5-(1-cyanocyclohexyl)-2-hydroxybenzoate
Phenyl 5-(1-cyanocyclohexyl)-2-methoxybenzoate (3.52 g, 10.5 mmol) was reacted following General Procedure D to afford a colorless oil (3.03 g, 9.49 mmol, 90%). 1H NMR (CDCl3) δH 10.55 (s, 1H), 8.19 (d, J = 2.6 Hz, 1H), 7.70 (dd, J = 8.8, 2.6 Hz, 1H), 7.54 – 7.45 (m, 2H), 7.40 – 7.33 (m, 1H), 7.28 – 7.22 (m, 2H), 7.10 (d, J = 8.9 Hz, 1H), 2.23 (d, J = 12.2 Hz, 2H), 2.03 – 1.72 (m, 8H); LCMS (Method A): tR = 1.75 min, m/z = 322.0 [M+H]+.
Step D: Phenyl 3-amino-5-(1-cyanocyclohexyl)-2-hydroxybenzoate
Phenyl 5-(1-cyanocyclohexyl)-2-hydroxybenzoate (3.03 g, 9.5 mmol) was dissolved in DCE/H2O (1:1, 20 mL) and cooled to 0 °C in an ice/water bath. Tetrabutylammonium bromide (153 mg, 0.47 mmol) was added, followed by HNO3 (conc., 1.2 mL, 19.0 mmol). The reaction was stirred vigorously at 60 °C for 18 h, then cooled, diluted with CH2Cl2, and washed with water. The crude material was carried forward without purification for hydrogenation, following General Procedure B. Upon purification a colorless oil was obtained (1.65 g, 4.9 mmol, 52%). 1H NMR (CDCl3) δH 10.55 (s, 1H), 8.19 (d, J = 2.6 Hz, 1H), 7.70 (dd, J = 8.8, 2.6 Hz, 1H), 7.52 – 7.46 (m, 2H), 7.39 – 7.32 (m, 1H), 7.28 – 7.22 (m, 2H), 7.10 (d, J = 8.8 Hz, 1H), 2.23 (d, J = 12.2 Hz, 3H), 1.99 – 1.72 (m, 8H); LCMS (Method B) tR = 1.28 min, m/z = 337.1 [M+H]+.
Step E: Phenyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate
Phenyl 3-amino-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (308 mg, 0.91 mmol) and 18a (313 mg, 1.1 mmol) were reacted according to General Procedure A. Purification by flash chromatography afforded a colorless solid (443 mg, 0.78 mmol, 85%).
1H NMR (400 MHz, MeOH-d4) δH 7.85 (d, J = 2.5 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.52 (dd, J = 8.8, 2.5 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 2.08 – 1.87 (m, 4H), 1.88 – 1.77 (m, 1H), 1.74 (td, J = 12.9, 10.5, 6.3 Hz, 4H), 1.46 – 1.31 (m, 1H); LCMS tR = 1.29 min, m/z = 588.3, 590.3 [M+NH4]+.
Step F: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoic acid, 6s
Phenyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (29 mg, 0.05 mmol) was reacted following General Procedure E, to afford a colorless solid (17 mg, 69%). 1H NMR (400 MHz, MeOH-d4) δH 7.85 (d, J = 2.5 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.52 (dd, J = 8.8, 2.5 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 2.08 – 1.87 (m, 4H), 1.88 – 1.77 (m, 1H), 1.74 (td, J = 12.9, 10.5, 6.3 Hz, 4H), 1.46 – 1.31 (m, 1H); LCMS tR = 1.65 min, m/z = 494.8, 496.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoic acid (6t)
Step A: Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate
Phenyl 3-amino-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (308 mg, 0.91 mmol) and 18b (336 mg, 1.1 mmol) were reacted according to General Procedure A. Purification by flash chromatography afforded a colorless solid (455 mg, 0.75 mmol, 82%). LCMS (Method B) tR = 1.39 min, m/z = 622.3 [M+NH4]+.
Step B: 3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoic acid (6t)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (30 mg, 0.05 mmol) was reacted following General Procedure E, to afford a colorless solid (17 mg, 64%). 1H NMR (400 MHz, MeOH-d4) δH 7.79 (d, J = 2.5 Hz, 1H), 7.78 (d, J = 2.5 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.72 (d, J = 2.5 Hz, 1H), 2.05 (dd, J = 8.3, 3.5 Hz, 2H), 1.99 – 1.87 (m, 2H), 1.89 – 1.78 (m, 1H), 1.80 – 1.69 (m, 3H), 1.46 – 1.31 (m, 1H); LCMS tR = 1.74 min, m/z = 530.7 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+Na]+ calculated for C20H18BrClN2NaO6S 550.9650, found 550.9681.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxy-N-methylbenzamide (7a)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoate (109 mg, 0.25 mmol) was reacted with methylamine following General Procedure F, to afford a colorless solid (89 mg, 82%). 1H NMR (400 MHz, DMSO-d6) δH 7.80 (d, J = 2.5 Hz, 1H), 7.54 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 8.8, 2.5 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 2.88 (s, 3H); LCMS tR = 1.57 min, m/z = 434.8, 436.8 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+Na]+ calculated for C14H12BrClN2NaO5S 456.9231, found 456.9247.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxy-N-methylbenzamide (7b)
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-chloro-2-hydroxybenzoic acid (23 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (5 mg, 21 %). 1H NMR (400 MHz, CDCl3) δH 10.94 (br s, 1H), 7.72 (dd, J = 9.2, 2.4 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.4 Hz, 1H), 7.63 (dd, J = 2.4, 1.2 Hz, 2H), 7.47 (s, 1H), 7.12 (d, J = 2.4 Hz, 1H), 6.27 (br s, 1H), 3.00 (d, J = 4.9 Hz, 3H); LCMS tR = 1.69 min, m/z = 470.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxy-N-methylbenzamide (7c)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (23 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (21 mg, 91%). 1H NMR (400 MHz, DMSO-d6) δH 11.27 (s, 1H), 8.88 (s, 1H), 8.44 – 8.35 (m, 1H), 7.60 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.3 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 2.79 (d, J = 4.2 Hz, 3H), 1.94 – 1.79 (m, 1H), 0.95 – 0.81 (m, 2H), 0.45 – 0.34 (m, 2H); 19F NMR (376 MHz, DMSO-d6) δF −120.4; LCMS tR = 1.69 min, m/z = 458.8, 460.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxy-N-methylbenzamide (7d)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-cyclopropyl-6-fluoro-2-hydroxybenzoate (49 mg, 0.1 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (36 mg, 73%). 1H NMR (400 MHz, MeOH-d4) δH 7.73 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 2.93 (s, 3H), 2.21 – 1.86 (m, 1H), 1.05 – 0.87 (m, 2H), 0.70 – 0.51 (m, 2H); 19F NMR (376 MHz, MeOH-d4) δF −121.7; LCMS tR = 1.83 min, m/z = 492.7, 494.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-N-methyl-5-(pentafluorosulfanyl)benzamide (7e)
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(pentafluorosulfanyl)benzoic acid, 6l (25 mg, 0.05 mmol), was dissolved in DCM (0.5 mL), DIPEA (26 μL, 0.15 mmol) added and the mixture cooled to 0 °C before the addition of HATU (29 mg, 0.075 mmol). The mixture was stirred for 30 mins, then methylamine (2.0 M in THF, 125 μL, 0.25 mmol) was added and stirred for 16 h. The mixture was diluted with DCM, washed with water and purified by prep-HPLC to afford a colorless solid (6 mg, 0.01, 23%). 1H NMR (400 MHz, MeOH-d4) δH 7.99 (dt, J = 5.8, 1.5 Hz, 2H), 7.85 (dd, J = 2.4, 1.3 Hz, 2H), 7.50 (dd, J = 8.8, 2.5 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 3.92 (s, 3H); 19F NMR (376 MHz, MeOH-d4) δF 81.2 (p, J = 149 Hz, 1F), 61.0 (d, J = 149 Hz, 4F); LCMS tR = 1.33 min, m/z = 510.7, 512.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-N-methyl-5-(pentafluorosulfanyl)benzamide (7f)
Methyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfaneyl)benzoate (26 mg, 0.05 mmol) was reacted following General Procedure F, to afford a colorless solid (7 mg, 0.013 mmol, 26%). 1H NMR (400 MHz, CDCl3) δH 8.34 (s, 1H), 8.05 (s, 2H), 7.68 (d, J = 2.4 Hz, 1H), 7.50 (dd, J = 8.8, 2.4 Hz, 1H), 7.28 (s, 1H), 6.87 (d, J = 8.8 Hz, 1H), 4.00 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 83.3 (d, J = 150 Hz), 63.7 (d, J = 150 Hz); LCMS tR = 1.89 min, m/z = 527.9, 529.9 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-N-methyl-5-(pentafluorosulfanyl)benzamide (7g)
Methyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate (18 mg, 0.03 mmol) was reacted following General Procedure F, to afford title compound as a colorless solid (3 mg, 0.005 mmol, 17%). 1H NMR (400 MHz, CDCl3) δH 8.04 (d, J = 2.4 Hz, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 2.3 Hz, 1H), 7.53 (d, J = 2.3 Hz, 1H), 6.42 (br s, 1H), 3.04 (d, J = 4.9 Hz, 3H); 19F NMR (376 MHz, CDCl3) δF 84.0 (p, J = 150 Hz), 64.1 (d, J = 150 Hz); LCMS tR = 1.16 min, m/z = 561.1, 563.1 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-N-methylbenzamide (7h)
Step A: 3-Amino-5-(1-cyanocyclobutyl)-N-methylbenzamide
53 (230 mg, 1.0 mmol) was taken in a solution of methylamine ([2.0 M in THF), 2 mL) and heated to 65 °C for 3 h. The mixture was concentrated to afford title compound (202 mg, 0.88 mmol, 88%) which was used without purification. MS (ESI) m/z = 230.1.
Step B: 3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-N-methylbenzamide, 7h
3-Amino-5-(1-cyanocyclobutyl)-N-methylbenzamide (23 mg, 0.1 mmol) and 18a (33 mg, 0.12 mmol) were reacted according to General Procedure A. Purification by reverse phase HPLC afforded a colorless solid (12 mg, 0.026 mmol, 26%). 1H NMR (400 MHz, DMSO-d6) δH 8.47 (s, 1H), 7.76 (d, J = 2.5 Hz, 1H), 7.60 – 7.42 (m, 3H), 7.28 (s, 1H), 6.88 (d, J = 8.8 Hz, 1H), 3.18 (s, 3H), 2.76 (d, J = 4.5 Hz, 3H), 2.74 – 2.65 (m, 2H), 2.38 – 2.21 (m, 2H), 2.04 – 1.92 (m, 2H); LCMS tR = 1.41 min, m/z = 463.8, 465.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-N-methylbenzamide (7i)
3-Amino-5-(1-cyanocyclobutyl)-N-methylbenzamide (23 mg, 0.1 mmol) and 18b (37 mg, 0.12 mmol) were reacted according to General Procedure A. Purification by reverse phase HPLC afforded a colorless solid (17 mg, 0.034 mmol, 34%). 1H NMR (400 MHz, DMSO-d6) δH 8.50 (d, J = 4.9 Hz, 1H), 7.91 (d, J = 2.5 Hz, 1H), 7.79 (d, J = 2.5 Hz, 1H), 7.55 (t, J = 1.8 Hz, 1H), 7.53 (d, J = 1.6 Hz, 1H), 7.28 (t, J = 1.9 Hz, 1H), 2.76 (d, J = 4.5 Hz, 3H), 2.77 – 2.65 (m, 2H), 2.37 – 2.22 (m, 1H), 1.99 (dtt, J = 9.1, 7.0, 4.5 Hz, 1H); LCMS tR = 1.50 min, m/z = 497.8, 499.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-methylbenzamide (7j)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (31 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (18 mg, 75%). 1H NMR (400 MHz, MeOH-d4) δH 7.82 (d, J = 2.5 Hz, 1H), 7.56 – 7.47 (m, 3H), 6.84 (d, J = 8.8 Hz, 1H), 2.91 (s, 3H), 2.81 – 2.71 (m, 2H), 2.65 – 2.51 (m, 2H), 2.38 (dt, J = 11.5, 8.6 Hz, 1H), 2.12 – 2.02 (m, 1H); LCMS tR = 1.54 min, m/z = 479.8, 481.8 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+H]+ calculated for C19H19BrN3O5S 480.0223, found 480.0239.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-methylbenzamide (7k)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (29 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (13 mg, 54%). 1H NMR (400 MHz, DMSO-d6) δH 11.07 (br s, 1H), 9.39 (br s, 2H), 9.12 (d, J = 4.6 Hz, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.71 (d, J = 2.1 Hz, 1H), 7.71 (d, J = 2.5 Hz, 1H), 7.35 (d, J = 2.1 Hz, 1H), 2.83 (d, J = 4.6 Hz, 3H), 2.75 – 2.65 (m, 2H), 2.58 – 2.46 (m, 2H), 2.25 (dt, J = 11.3, 8.6 Hz, 1H), 2.05 – 1.88 (m, 1H); LCMS tR = 1.66 min, m/z = 513.7, 515.8 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+H]+ calculated for C19H19BrClN3O5S 513.9834, found 513.9848.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxy-N-methylbenzamide (7l)
Phenyl 3-((5-bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (29 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (16 mg, 63%). 1H NMR (400 MHz, DMSO-d6) δH 11.30 (s, 1H), 9.09 (s, 1H), 8.88 (s, 1H), 7.71 (d, J = 2.6 Hz, 1H), 7.65 (d, J = 2.1 Hz, 1H), 7.58 (dd, J = 8.7, 2.1 Hz, 1H), 7.50 – 7.45 (m, 3H), 6.91 (d, J = 8.7 Hz, 1H), 2.81 (d, J = 4.3 Hz, 3H), 1.99 (d, J = 11.7 Hz, 2H), 1.90 – 1.51 (m, 8H); LCMS tR = 1.72 min, m/z = 507.8, 509.8 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxy-N-methylbenzamide (7m)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclohexyl)-2-hydroxybenzoate (30 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (21 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δH 9.09 (d, J = 4.4 Hz, 1H), 7.92 (d, J = 2.3 Hz, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.46 (d, J = 2.2 Hz, 1H), 2.82 (d, J = 4.4 Hz, 3H), 2.02 (d, J = 12.7 Hz, 2H), 1.88 – 1.51 (m, 7H), 1.26 (d, J = 12.7 Hz, 1H); LCMS tR = 1.84 min, m/z = 543.7 [M+H]+; Purity (AUC) ≥95%; HRMS (ESI/TOF) [M+H]+ calculated for C21H22BrClN3O5S 542.0147, found 542.0161.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-(1H-1,2,4-triazol-3-yl)benzamide (7n)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (27 mg, 0.05 mmol), 3-amino-1,2,4-triazole (21 mg) and NEt3 (35 μL, 0.25 mmol) were heated to 90 °C in dioxane (1 mL) for 16 h. The mixture was concentrated and purified by prep-HPLC to afford a colorless solid (15 mg, 56%) after neutralization with sat. aq. K2CO3. LCMS tR = 1.41 min, m/z = 532.8, 534.7 [M+H]+; Purity (AUC) ≥95%.
3-((5-Bromo-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-(2-methoxyethyl)benzamide (7o)
Phenyl 3-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (27 mg, 0.05 mmol), (2-methoxyethyl)ethylamine (22 μL, 0.25 mmol) and NEt3 (35 μL, 0.25 mmol) were heated to 90 °C in dioxane (1 mL) for 16 h. The mixture was concentrated and purified by prep-HPLC to afford a colorless solid (16 mg, 61%). 1H NMR (400 MHz, MeOH-d4) δH 7.80 (d, J = 2.5 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.53 (d, J = 2.2 Hz, 1H), 7.50 (dd, J = 8.7, 2.5 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 3.55 (d, J = 1.4 Hz, 4H), 3.36 (s, 3H), 2.81 – 2.70 (m, 2H), 2.62 – 2.51 (m, 2H), 2.44 – 2.30 (m, 1H), 2.11 – 1.97 (m, 1H); LCMS tR = 1.57 min, m/z = 523.8, 525.7 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-N-(3-chloro-5-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7p)
Step A: 3-Chloro-5-(methylsulfonyl)aniline
1-chloro-3-iodo-5-nitrobenzene (400 mg, 1.41 mmol), sodium methanesulfinate (174 mg, 1.7 mmol), CuI (27 mg, 0.14 mmol), L-proline (32 mg, 0.28 mmol) and NaOH (11 mg, 0.28 mmol) were taken in DMSO (3 mL) under an inert atmosphere and heated to 85 °C for 16 h. Upon cooling, the mixture was diluted with H2O and extracted with DCM. The organic phase was dried and concentrated to afford a colorless solid, which without purification was reacted following General Procedure B, to afford a yellow solid (108 mg, 0.53 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δH 7.23 (s, 1H), 7.08 (s, 1H), 6.86 (s, 1H), 4.11 (s, 2H), 3.04 (s, 3H).
Step B: 5-Bromo-N-(3-chloro-5-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7p)
3-Chloro-5-(methylsulfonyl)aniline (20 mg, 0.10 mmol) was reacted with 18a following General Procedure A, to afford a colorless solid (20 mg 0.046 mmol, 48%). 1H NMR (400 MHz, MeOH-d4) δH 7.88 (d, J = 2.5, 1H), 7.65 (t, J = 1.7 Hz, 1H), 7.58 (t, J = 1.7 Hz, 1H), 7.52 (dd, 8.8, 2.5 Hz, 1H), 7.47 (t, 1.7 Hz, 1H), 6.85 (d, J= 8.8 Hz, 1H), 3.06 (s, 3H); LCMS tR = 1.63 min, m/z = 458.7 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-N-(3-chloro-5-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7q)
3-Chloro-5-(methylsulfonyl)aniline (50 mg, 0.24 mmol) was reacted with 18b following General Procedure A, to afford a colorless solid (38 mg 0.085 mmol, 35%). 1H NMR (400 MHz, CDCl3) δH 7.79 (t, J = 1.9 Hz, 1H), 7.70 (d, J = 2.2 Hz, 2H), 7.52 (d, J = 1.9 Hz, 2H), 3.05 (s, 3H); LCMS: tR = 1.07 min, m/z = 492.8 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-chloro-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7r)
Step A: 4-Chloro-2-(methylthio)-6-nitrophenol
To a solution of 2-bromo-4-chloro-6-nitrophenol (1000 mg, 3.96 mmol) in pyridine (10 mL) was added Cu (500 mg, 7.9 mmol) and dimethyl disulfide (713 μL, 7.9 mmol). The mixture was heated to 110 °C for 16 h. The cooled mixture was filtered through celite, suspended in 3 M HCl and extracted with DCM and concentrated. Purification by flash chromatography affords title compound (630 mg, 2.87 mmol, 72%). MS (ESI) m/z = 221.2 [M+H]+.
Step B: 4-Chloro-2-(methylsulfonyl)-6-nitrophenol
4-Chloro-2-(methylthio)-6-nitrophenol (1024 mg, 4.66 mmol) was dissolved in EtOH:H2O (1:1, 15 mL) and oxone (2.86 g, 9.32 mmol) added. The mixture was stirred at r.t. for 24 h. The mixture was diluted with water and extracted with DCM, and dried (MgSO4) to afford a yellow solid (1155 mg, 4.58 eq, 98%), which was used without further purification. 1H NMR (400 MHz, CDCl3) δH 11.40 (s, 1H), 8.39 (d, J = 2.7 Hz, 1H), 8.31 (d, J = 2.7 Hz, 1H), 7.26 (s, 1H), 3.34 (s, 3H).
Step C: 2-Amino-4-chloro-6-(methylsulfonyl)phenol
4-chloro-2-(methylsulfonyl)-6-nitrophenol (68 mg, 0.20 mmol) was reacted following General Procedure B to afford title compound (60 mg, 0.2 mmol, 98%) which was taken forward without purification. 1H NMR (400 MHz, CDCl3) δH 7.00 (d, J = 2.3 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H), 3.12 (s, 3H). MS (ESI) m/z =
Step D: 5-Bromo-N-(5-chloro-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7r)
Amino-4-chloro-6-(methylsulfonyl)phenol (21 mg, 0.09 mmol) and 5-bromo-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, to afford a colorless solid (11 mg, 0.024 mmol, 26%). 1H NMR (400 MHz, CDCl3) δH 9.13 (s, 1H), 8.33 (s, 1H), 7.72 (d, J = 2.5 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.54 (dd, J = 8.9, 2.4 Hz, 1H), 7.48 (d, J = 2.5 Hz, 1H), 7.18 (s, 1H), 6.91 (d, J = 8.9 Hz, 1H), 3.08 (s, 3H); LCMS: tR = 1.71 min, m/z = 475.1 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-N-(5-chloro-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7s)
Amino-4-chloro-6-(methylsulfonyl)phenol (21 mg, 0.09 mmol) and 5-bromo-3-chloro-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, affording a colorless solid (12.2 mg, 0.024 mmol, 28%). 1H NMR (400 MHz, CDCl3) δH 7.76 (d, J = 2.5 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.45 (d, J = 2.5 Hz, 1H), 3.11 (s, 3H); LCMS: tR = 1.06 min, m/z = 509.1, 511.1 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-2-hydroxy-N-(3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (7t)
Step A: Methyl 3-(methylsulfonyl)-5-(pentafluorosulfanyl)benzoate
Methyl 3-bromo-5-(pentafluorosulfanyl)benzoate (600 mg, 1.91 mmol), sodium methanesulfinate (146 mg, 1.4 mmol), CuI (23 mg, 0.12 mmol), L-proline (27 mg, 0.24 mmol) and NaOH (10 mg, 0.24 mmol) were taken in DMSO (2.5 mL) under an inert atmosphere and heated to 85 °C for 16 h. Upon cooling, the mixture was diluted with H2O and extracted with DCM. The organic phase was dried and concentrated to afford a colorless solid (164.1 mg, 0.48 mmol, 25% yield), which was taken forward without purification. 1H NMR (400 MHz, CDCl3-d) δH 8.74 (d, J = 1.7 Hz, 1H), 8.68 (t, J = 1.7 Hz, 1H), 8.50 (t, J = 1.7 Hz, 1H), 4.02 (s, 3H), 3.15 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 80.4 (t, J = 151 Hz), 63.0 (d, J = 151 Hz). LCMS tR = 0.98 min, m/z = 341.1 [M+H]+.
Step B: 3-(Methylsulfonyl)-5-(pentafluorosulfanyl)benzoic acid
Methyl 3-(methylsulfonyl)-5-(pentafluorosulfanyl)benzoate (162 mg, 0.54 mmol) was reacted following General procedure E, to afford a colorless solid (117.7 mg, 0.36 mmol, 67%). 1H NMR (400 MHz, CDCl3) δH 8.79 (s, 1H), 8.73 (s, 1H), 8.56 (s, 1H), 3.17 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 80.2 (d, J = 151 Hz, 1F), 63.0 (d, J = 151 Hz, 4F).
Step C: 3-(Methylsulfonyl)-5-(pentafluorosulfanyl)aniline
3-(Methylsulfonyl)-5-(pentafluorosulfanyl)benzoic acid (117 mg, 0.36 mmol), diphenylphosphoryl azide (119 mg, 0.43 mmol) and tert-butanol (172 μL, 1.8 mmol) were taken in DME (1.8 mL) and heated to 100 °C for 16 h. The cooled mixture was diluted with EtOAc and washed with sat. aq. NH4Cl, sat. aq. NaHCO3, water, then concentrated. The residue was dissolved in DCM (5 mL), TFA (1 mL) added and the mixture stirred for 1 h at r.t. The mixture was concentrated and purified by flash chromatography to afford a colorless solid (51.2 mg, 0.17 mmol, 48%). 1H NMR (400 MHz, CDCl3) δH 7.61 (t, J = 1.8 Hz, 1H), 7.32 (t, J = 1.8 Hz, 1H), 7.25 (t, J = 1.8 Hz, 1H), 4.29 (s, 2H), 3.07 (s, 3H);19F NMR (376 MHz, CDCl3) δ 79.4 (p, J = 151 Hz, 1F), 59.53 (d, J = 150.5 Hz, 4F).
Step D: 5-Bromo-2-hydroxy-N-(3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (7t)
3-(Methylsulfonyl)-5-(pentafluorosulfanyl)aniline (20 mg, 0.07 mmol) and 18a were reacted following General Procedure A, to afford a colorless solid (8.4 mg, 0.016 mmol, 23%). 1H NMR (400 MHz, CDCl3) δH 8.08 (s, 1H), 7.82 (s, 1H), 7.77 (s, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.65 (s, 1H), 7.54 (dd, J = 8.8, 2.5 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 3.08 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 80.5 (d, J = 151 Hz), 62.9 (d, J = 151 Hz); LCMS: tR = 1.128 min, m/z = 548.8 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-2-hydroxy-N-(3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (7u)
3-(Methylsulfonyl)-5-(pentafluorosulfanyl)aniline (20 mg, 0.07 mmol) and 18b were reacted following General Procedure A, to afford a colorless solid (7 mg, 0.013 mmol, 19%). 1H NMR (400 MHz, CDCl3) δH 8.08 (s, 1H), 7.82 (s, 1H), 7.77 (s, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.65 (s, 1H), 6.90 (d, J = 8.8 Hz, 1H), 3.08 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 80.5 (d, J = 151 Hz, 1F), 62.9 (d, J = 151 Hz, 4F); LCMS: tR = 1.201 min, m/z = 584.7 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-2-hydroxy-N-(2-hydroxy-3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (7v)
Step A: 2-Bromo-4-(pentafluorosulfanyl)phenol
To 4-(pentafluorosulfaneyl)phenol (800 mg, 3.63 mmol) in acetic acid (4 mL) was added FeCl3 (59 mg, 0.36 mmol). The mixture was cooled to 0 °C and then Br2 (812 mg, 5.45 mmol) was added. The mixture was allowed to reach room temperature and stirred for 2 h. Then, the solvent was removed under reduced Stpressure and the crude material was purified by flash chromatography to afford title compound (439 mg, 1.5 mmol, 41%). 1H NMR (400 MHz, CDCl3) δH 7.90 (d, J = 2.6 Hz, 1H), 7.63 (dd, J = 9.0, 2.6 Hz, 1H), 7.06 (d, J = 9.0 Hz, 1H).
Step B: 2-(Methylthio)-4-(pentafluorosulfanyl)phenol
To 2-bromo-4-(pentafluorosulfanyl)phenol (439 mg, 1.47 mmol) in pyridine (4 mL) was added Cu (500 mg, 7.9 mmol) and dimethyl disulfide (713 μL, 7.9 mmol). The mixture was heated to 110 °C for 16 h. The cooled mixture was filtered through celite, suspended in 3 M HCl and extracted with DCM and concentrated. Purification by flash chromatography affords title compound (84 mg, 0.32 mmol, 32%). 1H NMR (400 MHz, CDCl3) δH 7.89 (d, J = 2.7 Hz, 1H), 7.63 (dd, J = 9.0, 2.7 Hz, 1H), 7.05 – 6.97 (m, 2H), 2.37 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 85.5 (d, J = 151 Hz), 64.3 (d, J = 151 Hz); MS (ESI) m/z = 266.0 [M+H]+.
Step C: 2-(Methylsulfonyl)-4-(pentafluorosulfanyl)phenol
2-(methylthio)-4-(pentafluorosulfanyl)phenol (84 mg, 0.32 mmol) was dissolved in EtOH:H2O (1:1, 1 mL) and oxone (197 g, 9.32 mmol) added. The mixture was stirred at r.t. for 24 h. The mixture was diluted with water and extracted with DCM, and dried (MgSO4) to afford title compound (44 mg, 0.25 mmol, 78%). 1H NMR (400 MHz, CDCl3) δH 9.25 (s, 1H), 8.11 (d, J = 2.7 Hz, 1H), 7.91 (dd, J = 9.2, 2.7 Hz, 1H), 7.13 (d, J = 9.2 Hz, 1H), 3.18 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 83.2 (d, J = 151 Hz), 64.0 (d, J = 151 Hz).
Step D: 2-(Methylsulfonyl)-6-nitro-4-(pentafluorosulfanyl)phenol
2-(Methylsulfonyl)-4-(pentafluorosulfanyl)phenol (74 mg, 0.25 mmol) was reacted following General Procedure C to afford a yellow solid (68 mg, 0.20 mmol, 80%).1H NMR (400 MHz, CDCl3) δH 8.81 (s, 1H), 8.70 (s, 1H), 3.38 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 80.0 (p, J = 152 Hz), 63.9 (d, J = 152 Hz).
Step E: 2-Amino-6-(Methylsulfonyl)-4-(pentafluorosulfaneyl)phenol
2-(Methylsulfonyl)-6-nitro-4-(pentafluorosulfanyl)phenol (235 mg, 0.68 mmol) was reacted following General Procedure B to afford title compound as a pale brown solid (214 mg, 0.68 mmol, quant.), which was taken forward without purification. MS (ESI) m/z = 314.1 [M+H]+.
Step F: 5-Bromo-2-hydroxy-N-(2-hydroxy-3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide, 7v
2-Amino-6-(methylsulfonyl)-4-(pentafluorosulfanyl)phenol (40 mg, 0.13 mmol) and 5-bromo-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, to afford the title compound (24 mg, 0.044 mmol, 34%). 1H NMR (400 MHz, CDCl3) δH 8.03 (d, J = 2.8 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.67 (d, J = 2.8 Hz, 1H), 7.62 (dd, J = 8.8, 2.5 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 3.31 (s, 3H); 19F NMR (376 MHz, MeOH-d4) δF 79.1 (p, J = 149 Hz), 59.4 (d, J = 149 Hz); LCMS tR = 1.01 min, m/z = 547.9, 549.9 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-2-hydroxy-N-(2-hydroxy-3-(methylsulfonyl)-5-(pentafluorosulfanyl)phenyl)benzenesulfonamide (7w)
2-Amino-6-(methylsulfonyl)-4-(pentafluorosulfanyl)phenol (21 mg, 0.09 mmol) and 5-bromo-3-chloro-2-hydroxy-benzenesulfonyl chloride was reacted following General Procedure A, to afford title compound (32 mg, 0.055 mmol, 42%). 1H NMR (400 MHz, CDCl3) δH 8.16 (d, J = 2.5 Hz, 1H), 7.85 (d, J = 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 2.5 Hz, 1H), 3.17 (s, 3H); 19F NMR (376 MHz, CDCl3) δF 79.0 (p, J = 151 Hz, 1F), 60.7 (d, J = 151 Hz, 4F); LCMS tR = 1.01 min, m/z = 566.3, 569.9 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-N-(3-(1-cyanocyclobutyl)-5-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7x)
Step A: 3-Methyl-5-(methylsulfonyl)aniline
3-Bromo-5-methylaniline (2.0 g, 10.75 mmol), sodium methanesulfinate (1.32 g, 12.9 mmol), CuI (205 mg, 1.08 mmol), L-proline (248 mg, 2.15 mmol) and NaOH (86 mg, 0.24 mmol) were taken in DMSO (22 mL) under an inert atmosphere and heated to 85 °C for 16 h. Upon cooling, the mixture was diluted with H2O and extracted with DCM. The organic phase was dried and concentrated to afford title compound (1.2 g, 6.46 mmol, 60% yield). 1H NMR (400 MHz, CDCl3) δH 7.10 (d, J = 1.6 Hz, 1H), 7.01 (d, J = 2.1 Hz, 1H), 6.71 (dd, J = 2.1, 1.6 Hz, 1H), 3.90 (s, 2H), 3.01 (s, 3H), 2.36 – 2.31 (m, 3H); MS (ESI) m/z = 371.4 [2M+H]+.
Step B: tert-Butyl (3-methyl-5-(methylsulfonyl)phenyl) carbamate
A mixture containing 3-methyl-5-(methylsulfonyl)aniline (1196 mg, 6.46 mmol) and di-tert-butyl carbonate (1671 mg, 7.66 mmol) in MeOH (22 mL) was allowed to stir at r.t. for 16 h. The solvent was removed under reduced pressure and the crude material was purified by flash chromatography to afford title compound (826 mg, 2.89 mmol, 45%) 1H NMR (400 MHz, CDCl3) δH 7.71 (t, J = 1.9 Hz, 1H), 7.56 (s, 1H), 7.39 (d, J = 1.9 Hz, 1H), 6.86 (s, 1H), 3.03 (s, 3H), 2.39 (s, 3H), 1.51 (s, 9H). MS (ESI) m/z = 303.2 [M+NH4]+.
Step C: tert-Butyl (3-(bromomethyl-5-(methylsulfonyl)phenyl) carbamate
tert-Butyl (3-methyl-5-(methylsulfonyl)phenyl) carbamate (826 mg, 2.89 mmol) and NBS (618 mg, 3.47 mmol) were taken in CCl4 (25 mL) and the solution was degassed with Ar for 10 min. To this mixture was added AIBN (119 mg, 724 mmol) and the reaction mixture was heated to 80 °C for 16 h. The cooled reaction mixture was filtered. The filtrate was washed first with HCl [1 M] and then with a saturated solution of NaHCO3. The organic phase was dried using a phase separator and the solvent was removed under reduced pressure. The crude material was purified by flash chromatography to give (961 mg, 2.64 mmol, 91%). 1H NMR (400 MHz, Chloroform-d) δ 7.84 (d, J = 1.8 Hz, 1H), 7.60 (s, 1H), 6.88 (s, 1H), 4.47 (s, 2H), 3.06 (s, 3H), 1.52 (s, 9H). MS (ESI) m/z = 381.3, 383.2 [M+NH4]+.
Step D: tert-Butyl (3-(cyanomethyl-5-(methylsulfonyl)phenyl) carbamate
tert-Butyl (3-(bromomethyl-5-(methylsulfonyl)phenyl) carbamate (961 mg, 2.64 mmol) was dissolved in MeCN (5.0 mL) and cooled to 0 °C. K2CO3 (438 mg, 3.17 mmol) and TMSCN (0.420 mL, 3.17 mmol) were added and heated to 50 °C for 16 h. The cooled mixture was diluted with NaOH [3 M] and extracted with EtOAc. The crude material was purified by flash chromatography to title compound (441 mg, 1.42 mmol, 49%). 1H NMR (400 MHz, CDCl3) δH 7.89 (t, J = 1.8 Hz, 1H), 7.77 (s, 1H), 7.55 (t, J = 1.8 Hz, 1H), 6.82 (s, 1H), 3.81 (s, 2H), 3.07 (s, 3H), 1.53 (s, 9H). MS (ESI) m/z = 328.3 [M+NH4]+.
Step E: tert-Butyl (3-(1-cyanocyclobutyl)-5-(methylsulfonyl)phenyl) carbamate
tert-Butyl (3-(cyanomethyl-5-(methylsulfonyl)phenyl) carbamate (421 mg, 1.36 mmol) and 1,3-dibromopropane (151 μL, 1.49 mmol) were dissolved DMSO (7 mL) was NaH (60% dispersion in mineral oil, 140 mg, 3.39mmol) was added with caution. The solution was stirred for 16 h at r.t., then diluted with EtOAc:Et2O (100 mL, 1:1) and washed with water (3 × 100 mL). The combined aqueous layers were back-extracted with further EtOAc (100 mL). The combined organics were washed with sat. aq. NaCl, concentrated and purified by flash column chromatography to afford a colorless oil (311 mg, 0.887 mmol, 65%). 1H NMR (400 MHz, CDCl3) δH 7.94 (s, 1H), 7.80 (s, 1H), 7.56 (s, 1H), 7.30 (s, 1H), 3.06 (s, 3H), 2.87 – 2.76 (m, 2H), 2.69 – 2.56 (m, 2H), 2.43 (dt, J = 11.9, 8.8 Hz, 1H), 2.13 – 2.04 (m, 1H), 1.50 (s, 9H); LCMS tR = 0.99 min, m/z = 368.3 [M+NH4]+.
Step F: 1-(3-Amino-5-(methylsulfonyl)phenyl)cyclobutene-1-carbonitrile hydrochloride
tert-Butyl (3-(1-cyanocyclobutyl)-5-(methylsulfonyl)phenyl) carbamate (311 mg, 0.887 mmol) was dissolved in a solution of HCl (4 M in dioxane, 5 mL) and heated to 50 °C for 1 h. The solvent was removed under reduced pressure to afford title compound (253 mg, 0.88 mmol, quant) which was taken forward without purification. 1H NMR (400 MHz, CDCl3) δH 8.14 (s, 1H), 7.96 (s, 1H), 7.82 (s, 1H), 3.09 (s, 3H), 2.79 (s, 2H), 2.61 (d, J = 11.4 Hz, 2H), 2.44 – 2.34 (m, 1H), 2.07 (s, 1H); LCMS tR = 0.59 min, m/z = 251.0 [M+H]+.
Step G: 5-Bromo-3-chloro-N-(3-(1-cyanocyclobutyl)-5-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide
1-(3-Amino-5-(methylsulfonyl)phenyl)cyclobutene-1-carbonitrile hydrochloride (83 mg, 0.29 mmol) and 5-bromo-3-chloro-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, to afford the title compound (55 mg, 0.11 mmol, 38%). 1H NMR (400 MHz, CDCl3) δH 7.90 (br s, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 1.8 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.62 (d, J = 1.8 Hz, 1H), 7.53 (t, J = 1.8 Hz, 1H), 3.08 (s, 3H), 2.92 – 2.81 (m, 2H), 2.64 – 2.52 (m, 2H), 2.52 – 2.42 (m, 1H), 2.16 – 2.07 (m, 1H); LCMS tR = 1.81 min, m/z = 518.9, 521.9 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-(1-cyanocyclobutyl)-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7y)
Step A: 1-(4-Methoxy-3-(methylthio)phenyl)cyclobutane-1-carbonitrile
1-(3-Bromo-4-methoxyphenyl)cyclobutane-1-carbonitrile (200 mg, 0.75 mmol), sodium methanethiolate (55 mg, 0.79 mmol), Pd2dba3 (17 mg, 0.02 mmol), Xantphos (22 mg 0.04 mmol) were combined in a vial purged with argon. THF (3.75 mL) and Et3N (0.13 mL, 0.94 mmol) were added and the mixture heated to reflux for 18 h. The cooled mixture was filtered, concentrated and purified by flash chromatography to afford a yellow oil (174 mg, 0.746 mmol, 99% yield). 1H NMR (400 MHz, CDCl3) δH 7.21 – 7.13 (m, 2H), 6.87 – 6.80 (m, 1H), 3.90 (s, 3H), 2.88 – 2.75 (m, 2H), 2.59 (dt, J = 9.8, 8.0 Hz, 2H), 2.45 (s, 3H), 2.43 – 2.36 (m, 1H), 2.12 – 2.00 (m, 1H). MS (ESI) m/z = 234.2 [M+H]+.
Step B: 1-(4-Methoxy-3-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile
1-(4-methoxy-3-(methylthio)phenyl)cyclobutane-1-carbonitrile (174 mg, 0.671 mmol) was dissolved in EtOH:H2O (1:1, 1.5 mL) and oxone (413 mg, 1.34 mmol) added. The mixture was stirred at r.t. for 24 h. The mixture was diluted with water and extracted with DCM, and dried (MgSO4) to afford a pale-yellow solid (188 mg, 0.58 mmol, 87% yield) that was taken forward without purification. 1H NMR (400 MHz, CDCl3) δH 8.03 (d, J = 2.5 Hz, 1H), 7.67 (dd, J = 8.6, 2.5 Hz, 1H), 7.10 (d, J = 8.6 Hz, 1H), 2.91 – 2.81 (m, 1H), 2.68 – 2.58 (m, 2H), 2.53 – 2.40 (m, 1H), 2.17 – 2.07 (m, 1H).
Step C: 1-(4-Hydroxy-3-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile
1-(4-Methoxy-3-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile (71 mg, 0.27 mmol) was reacted following General Procedure D, to afford an off-white solid (68 mg, 0.27 mmol, quant.). 1H NMR (400 MHz, CDCl3) δH 8.86 (s, 1H), 7.68 (d, J = 2.5 Hz, 1H), 7.58 (dd, J = 8.7, 2.5 Hz, 1H), 7.10 (d, J = 8.7 Hz, 1H), 3.15 (s, 3H), 2.91 – 2.77 (m, 2H), 2.69 – 2.54 (m, 2H), 2.54 – 2.39 (m, 1H), 2.17 – 2.02 (m, 1H).
Step D: 1-(4-Hydroxy-3-(methylsulfonyl)-5-nitrophenyl)cyclobutane-1-carbonitrile
1-(4-Hydroxy-3-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile (170 mg, 0.68 mmol) was reacted following General Procedure C to afford title compound (145 mg, 0.49 mmol, 84%). %). 1H NMR (400 MHz, Chloroform-d) δ 8.41 (d, J = 2.5 Hz, 1H), 8.33 (dt, J = 2.2, 1.0 Hz, 1H), 3.34 (s, 3H), 2.95 – 2.84 (m, 2H), 2.69 – 2.57 (m, 2H), 2.57 – 2.43 (m, 1H), 2.19 – 2.06 (m, 1H); MS (ESI) m/z = 314.3 [M+NH4]+.
Step E: 1-(3-Amino-4-hydroxy-5-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile
1-(4-Hydroxy-3-(methylsulfonyl)-5-nitrophenyl)cyclobutane-1-carbonitrile (145 mg, 0.489 mmol) was reacted following General Procedure B to afford title compound (122 mg, 0.458 mmol, 94% yield1H NMR (400 MHz, Chloroform-d) δ 7.01 (d, J = 2.2 Hz, 1H), 6.93 (d, J = 2.2 Hz, 1H), 3.13 (s, 3H), 2.83 – 2.72 (m, 2H), 2.62 – 2.50 (m, 2H), 2.41 (dt, J = 11.5, 8.8 Hz, 1H), 2.11 – 1.98 (m, 1H); MS (ESI) m/z = 267.2 [M+H]+.
Step F: 5-Bromo-N-(5-(1-cyanocyclobutyl)-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7y)
1-(3-Amino-4-hydroxy-5-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile (11 mg, 0.04 mmol) and 5-bromo-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, to afford the title compound (3 mg, 0.006 mmol, 15%). 1H NMR (400 MHz, CDCl3) δH 7.76 (d, J = 2.5 Hz, 1H), 7.68 (d, J = 2.4 Hz, 1H), 7.60 (dd, J = 8.8, 2.5 Hz, 1H), 7.38 (d, J = 2.4 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H), 3.27 (s, 3H), 2.80 – 2.65 (m, 2H), 2.56 – 2.24 (m, 3H), 2.15 – 1.94 (m, 1H); LCMS tR = 1.44 min, m/z = 501.0, 503.0 [M+H]+; Purity (AUC) ≥95%.
5-Bromo-3-chloro-N-(5-(1-cyanocyclobutyl)-2-hydroxy-3-(methylsulfonyl)phenyl)-2-hydroxybenzenesulfonamide (7z)
1-(3-Amino-4-hydroxy-5-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile (15 mg, 0.056 mmol) and 5-bromo-3-chloro-2-hydroxy-benzenesulfonyl chloride were reacted following General Procedure A, affording the title compound (8 mg, 0.015 mmol, 27% yield). 1H NMR (400 MHz, Acetonitrile-d3) δH 7.81 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 3.16 (s, 3H), 2.77 – 2.70 (m, 2H), 2.53 – 2.45 (m, 2H), 2.34 (dq, J = 11.5, 8.6 Hz, 1H), 2.06 – 1.97 (m, 1H); LCMS tR = 1.02 min, m/z = 552.2, 553.2 [M+NH4]+; Purity (AUC) ≥95%.
5-Bromo-N-(5-chloro-2-hydroxy-3-(methylsulfonyl)phenyl)-2-methoxybenzenesulfonamide (8)
Amino-4-chloro-6-(methylsulfonyl)phenol (50 mg, 0.23 mmol) and 16a were reacted following General Procedure A to afford the title compound (3 mg, 0.006 mmol, 3%). 1H NMR (400 MHz, CDCl3) δH 9.24 (s, 1H), 8.00 (d, J = 2.5 Hz, 1H), 7.82 (d, J = 2.5 Hz, 1H), 7.68 (s, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 6.89 (d, J = 8.8 Hz, 1H), 3.96 (s, 3H), 3.11 (s, 3H). LCMS tR = 1.055 min, m/z = 489.2 [M+NH4]+; Purity (AUC) ≥ 95%.
3-((5-Bromo-2-methoxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-methylbenzamide (9)
Phenyl 3-((5-bromo-2-methoxyphenyl)sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (28 mg, 0.05 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (14 mg, 57%). 1H NMR (400 MHz, MeOH-d4) δH 7.88 (d, J = 2.5 Hz, 1H), 7.65 (dd, J = 8.9, 2.5 Hz, 1H), 7.56 (d, J = 2.2 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.06 (d, J = 8.9 Hz, 1H), 3.90 (s, 3H), 2.89 (s, 3H), 2.81 – 2.70 (m, 2H), 2.62 – 2.50 (m, 2H), 2.38 (dq, J = 11.5, 8.5 Hz, 1H), 2.12 – 1.98 (m, 1H); LCMS tR = 1.74 min, m/z = 493.8, 495.8 [M+H]+; Purity (AUC) ≥95%.
6-Bromo-N-(5-chloro-2-hydroxy-3-(methylsulfonyl)phenyl)quinoline-8-sulfonamide (10)
Amino-4-chloro-6-(methylsulfonyl)phenol (10 mg, 0.45 mmol) and 6-bromoquinoline-8-sulfonyl chloride were reacted following General Procedure A, affording the title compound (4 mg, 0.008 mmol, 18%). 1H NMR (400 MHz, CDCl3) δH 9.10 (dd, J = 4.3, 1.7 Hz, 1H), 8.48 (d, J = 2.2 Hz, 1H), 8.23 (d, J = 2.2 Hz, 1H), 8.20 (dd, J = 8.4, 1.7 Hz, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 8.4, 4.3 Hz, 1H), 7.32 (d, J = 2.5 Hz, 1H), 3.05 (s, 3H); LCMS tR = 1.038 min, m/z = 491.2, 493.2 [M+H]+; Purity (AUC) ≥95%.
6-Bromo-N-(5-(1-cyanocyclobutyl)-2-hydroxy-3-(methylsulfonyl)phenyl)quinoline-8-sulfonamide (11)
1-(3-Amino-4-hydroxy-5-(methylsulfonyl)phenyl)cyclobutane-1-carbonitrile (21 mg, 0.079 mmol) and 6-bromoquinoline-8-sulfonyl chloride were reacted following General Procedure A, to afford the title compound (16 mg, 0.030 mmol, 38%). 1H NMR (400 MHz, CDCl3) δH 9.11 (dd, J = 4.4, 1.6 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.25 – 8.17 (m, 2H), 7.93 (d, J = 2.3 Hz, 1H), 7.62 (dd, J = 8.4, 4.3 Hz, 1H), 7.37 (d, J = 2.3 Hz, 1H), 3.06 (s, 3H), 2.82 (ddd, J = 11.9, 8.5, 4.1 Hz, 2H), 2.59 – 2.51 (m, 2H), 2.44 (dq, J = 11.5, 8.7, 8.1 Hz, 1H), 2.14 – 2.04 (m, 1H); LCMS tR = 1.02 min, m/z = 552.2, 553.2 [M+H]+; Purity (AUC) ≥95%.
3-((6-Bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (12)
Step A: Phenyl 3-((6-bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate
Phenyl 3-amino-5-(1-cyanocyclobutyl)-2-hydroxybenzoate 31 mg, 0.10 mmol) and 6-bromoquinoline-8- sulfonyl chloride were reacted following General Procedure D to afford a colorless solid (22 mg, 0.04 mmol, 38%). LCMS tR = 1.18 min, m/z = 578.3, 580.3 [M+H]+.
Step B: 3-((6-Bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoic acid (12)
Phenyl 3-((6-bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (71 mg, 0.12 mmol) was reacted following General Procedure E, to afford title compound (40 mg, 0.08 mmol, 65%). 1H NMR (400 MHz, MeOH-d4) δH 9.07 (dd, J = 4.3, 1.7 Hz, 1H), 8.41 – 8.38 (m, 2H), 8.34 (dd, J = 8.4, 1.7 Hz, 1H), 7.79 (d, J = 2.4 Hz, 1H), 7.65 (dd, J = 8.4, 4.3 Hz, 1H), 7.56 (d, J = 2.4 Hz, 1H), 2.79 – 2.69 (m, 2H), 2.59 – 2.48 (m, 2H), 2.44 – 2.30 (m, 1H), 2.06 (ddt, J = 9.0, 6.9, 4.5 Hz, 1H); LCMS tR = 1.04 min, m/z = 502.3, 504.2 [M+H]+; Purity (AUC) ≥95%.
3-((6-Bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxy-N-methylbenzamide (13)
Phenyl 3-((6-bromoquinoline)-8-sulfonamido)-5-(1-cyanocyclobutyl)-2-hydroxybenzoate (22 mg, 0.04 mmol) was reacted with methylamine following General Procedure F to afford a colorless solid (7 mg, 36%). 1H NMR (400 MHz, MeOH-d4) δH 9.09 (dd, J = 4.3, 1.7 Hz, 1H), 8.42 (d, J = 2.2 Hz, 1H), 8.40 (d, J = 2.2 Hz, 1H), 8.37 (dd, J = 8.4, 1.8 Hz, 1H), 7.70 – 7.66 (m, 1H), 7.68 (d, J = 2.3 Hz, 1H), 7.42 (d, J = 2.2 Hz, 1H), 2.82 (s, 3H), 2.79 – 2.71 (m, 2H), 2.63 – 2.52 (m, 1H), 2.43 – 2.32 (m, 1H), 2.11 – 2.00 (m, 1H); LCMS tR = 1.19 min, m/z = 514.8, 516.7 [M+H]+; Purity (AUC) ≥95%.
3-((6-Bromoquinoline)-8-sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (14)
Step A: Methyl 3-((6-bromoquinoline)-8-sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate
Methyl 3-amino-2-hydroxy-5-(pentafluorosulfanyl)benzoate (35 mg, 0.12 mmol) and 6-bromoquinoline-8-sulfonyl chloride were reacted following General Procedure A, to afford title compound as a colorless solid (28 mg, 0.05 mmol, 42%). MS (ESI) m/z = 563.2 [M+H]+.
Step B: 3-((6-Bromoquinoline)-8-sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoic acid (14)
Methyl 3-((6-bromoquinoline)-8-sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate (14 mg, 0.022 mmol) was reacted following General Procedure E to afford title compound (8 mg, 0.015 mmol, 66%). 1H NMR (400 MHz, MeOH-d4) δH 9.04 (dd, J = 4.3, 1.7 Hz, 1H), 8.45 – 8.39 (m, 2H), 8.37 (dd, J = 8.4, 1.7 Hz, 1H), 8.15 (d, J = 2.8 Hz, 1H), 7.92 (d, J = 2.7 Hz, 1H), 7.67 (dd, J = 8.4, 4.3 Hz, 1H); 19F NMR (376 MHz, MeOH-d4) δF 83.0 (p, J = 150 Hz, 1F), 62.1 (d, J = 150 Hz, 4F); LCMS tR = 1.13 min, m/z = 549.1, 551.2 [M+H]+, Purity (AUC) ≥95%.
3-((6-Bromoquinoline)-8-sulfonamido)-2-hydroxy-N-methyl-5-(pentafluorosulfanyl)benzamide (15)
Phenyl 3-((6-bromoquinoline)-8-sulfonamido)-2-hydroxy-5-(pentafluorosulfanyl)benzoate (14 mg, 0.022 mmol) was reacted following General Procedure F to afford title compound (9 mg, 0.015 mmol, 69%). 1H NMR (400 MHz, CDCl3) δH 9.11 (dd, J = 4.4, 1.7 Hz, 1H), 8.43 (d, J = 2.2 Hz, 1H), 8.22 (d, J = 2.4 Hz, 1H), 8.20 – 8.12 (m, 2H), 7.58 (dd, J = 8.4, 4.3 Hz, 1H), 7.41 (d, J = 2.5 Hz, 1H), 6.41 – 6.36 (m, 1H), 2.94 (d, J = 4.8 Hz, 3H); 19F NMR (376 MHz, CDCl3) δF 84.6 (p, J = 150 Hz, 1F), 64.1 (d, J = 150 Hz, 4F); LCMS tR = 1.19 min, m/z = 563.2 [M+H]+; Purity (AUC) ≥95%.
Supplementary Material
Acknowledgements
The authors thank Alexey Kuznetsov and Caden Schlund for their contributions to this series of compounds during this project. The authors thank our co-workers at the High-Throughput Screening Core facility at Vanderbilt University, TN, for compound management services and use of their instrumentation, and the Vanderbilt University Biomolecular NMR Facility for instrumentation. This facility receives support from an NIH SIG Grant (1S-10RR025677–01) and Vanderbilt University matching funds. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by the Argonne National Laboratory, was supported by the U.S. Department of Energy (contract no. DE-AC02–06CH11357). Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant (grant no. 085P1000817). This work was supported by a grant from the NIH/NCI (CA200709 to W.P.T). This work was also supported by grants from the Robert J. Kleberg, Jr., and Helen C. Kleberg Foundation [to W.P.T. and S.W.F.), The TJ Martell Foundation [to W.P.T. and S.W.F.], the Edward P. Evans Foundation [to W.P.T.], Alex’s Lemonade Stand Foundation [to W.P.T.], and St. Baldrick’s Foundation [to W.P.T.].
Abbreviations Used
- WDR5
WD repeat-containing protein 5
- WBM
WDR5-binding motif
- WIN
WDR5-interaction site
- RBBP5
retinoblastoma binding protein 5
- KANSL2
KAT8 Regulatory NSL Complex Subunit 2
- MLL
mixed lineage leukemia
- HMT
histone methyl transferase
- HTS
high-throughput screen
- FP
fluorescence polarization
- FPA
fluorescence polarization assay
- FITC
fluorescein isothiocyanate
- CSP
chemical shift perturbation
- DSF
differential scanning fluorimetry
- RFU
relative fluorescence unit
Footnotes
PDB ID Codes
Atom coordinates and structure factors for WDR5-ligand complexes can be accessed in the PDB via the following accession codes: 1, 6U6W; 5g, 6U80; 5n, 6U8O; 6b, 6U8B; 6g, 6U8L; 7k, 6U5Y; 7t, 6U5M.
REFERENCES
- (1).Spencer CA; Groudine M. Control of C-Myc Regulation in Normal and Neoplastic Cells. Adv. Cancer Res 1991, 56, 1–48. [DOI] [PubMed] [Google Scholar]
- (2).Beroukhim R; Mermel CH; Porter D; Wei G; Raychaudhuri S; Donovan J; Barretina J; Boehm JS; Dobson J; Urashima M; McHenry KT; Pinchback RM; Ligon AH; Cho Y-J; Haery L; Greulich H; Reich M; Winckler W; Lawrence MS; Weir BA; Tanaka KE; Chiang DY; Bass AJ; Loo A; Hoffman C; Prensner J; Liefeld T; Gao Q; Yecies D; Signoretti S; Maher E; Kaye FJ; Sasaki H; Tepper JE; Fletcher JA; Tabernero J; Baselga J; Tsao M-S.; Demichelis F; Rubin MA; Janne PA; Daly MJ; Nucera C; Levine RL; Ebert BL; Gabriel S; Rustgi AK; Antonescu CR; Ladanyi M; Letai A; Garraway LA; Loda M; Beer DG; True LD; Okamoto A; Pomeroy SL; Singer S; Golub TR; Lander ES; Getz G; Sellers WR; Meyerson M. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463 (7283), 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tansey WP Mammalian MYC Proteins and Cancer. New J. Sci 2014, 2014, 1–27. [Google Scholar]
- (4).Dang CV; Reddy EP; Shokat KM; Soucek L. Drugging the “undruggable” Cancer Targets. Nat. Rev. Cancer 2017, 17 (8), 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).McKeown MR; Bradner JE Therapeutic Strategies to Inhibit MYC. Cold Spring Harb. Perspect. Med 2014, 4 (10), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Whitfield JR; Beaulieu M-E; Soucek L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol 2017, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kalkat M; Resetca D; Lourenco C; Chan P-K; Wei Y; Shiah Y-J; Vitkin N; Tong Y; Sunnerhagen M; Done SJ; Boutros PC; Raught B; Penn LZ MYC Protein Interactome Profiling Reveals Functionally Distinct Regions That Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72 (5), 836–848.e7. [DOI] [PubMed] [Google Scholar]
- (8).Berg T; Cohen SB; Desharnais J; Sonderegger C; Maslyar DJ; Goldberg J; Boger DL; Vogt PK Small-Molecule Antagonists of Myc/Max Dimerization Inhibit Myc-Induced Transformation of Chicken Embryo Fibroblasts. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (6), 3830–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yin X; Giap C; Lazo JS; Prochownik EV Low Molecular Weight Inhibitors of Myc–Max Interaction and Function. Oncogene 2003, 22 (40), 6151–6159. [DOI] [PubMed] [Google Scholar]
- (10).Wang H; Chauhan J; Hu A; Pendleton K; Yap JL; Sabato PE; Jones JW; Perri M; Yu J; Cione E; Kane MA; Fletcher S; Prochownik EV; Disruption of Myc-Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecule 10074-G5. Oncotarget 2013, 4 (6), 936–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kiessling A; Sperl B; Hollis A; Eick D; Berg T. Selective Inhibition of C-Myc/Max Dimerization and DNA Binding by Small Molecules. Chem. Biol 2006, 13 (7), 745–751. [DOI] [PubMed] [Google Scholar]
- (12).Hart JR; Garner AL; Yu J; Ito Y; Sun M; Ueno L; Rhee J-K; Baksh MM; Stefan E; Hartl M; Bister K; Vogt PK; Janda KD Inhibitor of MYC Identified in a Kröhnke Pyridine Library. Proc. Natl. Acad. Sci 2014, 111 (34), 12556–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gustafson WC; Meyerowitz JG; Nekritz EA; Chen J; Benes C; Charron E; Simonds EF; Seeger R; Matthay KK; Hertz NT; Eilers M; Shokat KM; Weiss WA Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer Cell 2014, 26 (3), 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Richards MW; Burgess SG; Poon E; Carstensen A; Eilers M; Chesler L; Bayliss R. Structural Basis of N-Myc Binding by Aurora-A and Its Destabilization by Kinase Inhibitors. Proc. Natl. Acad. Sci 2016, 113 (48), 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bayliss R; Burgess SG; Leen E; Richards MW A Moving Target: Structure and Disorder in Pursuit of Myc Inhibitors. Biochem. Soc. Trans 2017, 45 (3), 709–717. [DOI] [PubMed] [Google Scholar]
- (16).Thomas LR; Wang Q; Grieb BC; Phan J; Foshage AM; Sun Q; Olejniczak ET; Clark T; Dey S; Lorey S; Alicie B; Howard GC; Cawthon B; Ess KC; Eischen CM; Zhao Z; Fesik SW; Tansey WP Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Thomas LR; Foshage AM; Weissmiller AM; Tansey WP The MYC-WDR5 Nexus and Cancer. Cancer Res. 2015, 75 (19), 4012–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Smith TF; Gaitatzes C; Saxena K; Neer EJ The WD Repeat: A Common Architecture for Diverse Functions. Trends Biochem. Sci 1999, 24 (5), 181–185. [DOI] [PubMed] [Google Scholar]
- (19).Xu C; Min J. Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2 (3), 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stirnimann CU; Petsalaki E; Russell RB; Mü Ller CW WD40 Proteins Propel Cellular Networks. Trends Biochem. Sci 2010, 35, 565–574. [DOI] [PubMed] [Google Scholar]
- (21).Song R; Wang Z-D; Schapira M. Disease Association and Druggability of WD40 Repeat Proteins. J. Proteome Res 2017, 16 (10), 3766–3773. [DOI] [PubMed] [Google Scholar]
- (22).Guarnaccia A; Tansey W. Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med 2018, 7 (2), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Patel A; Vought VE; Dharmarajan V; Cosgrove MS A Conserved Arginine-Containing Motif Crucial for the Assembly and Enzymatic Activity of the Mixed Lineage Leukemia Protein-1 Core Complex. J. Biol. Chem 2008, 283 (47), 32162–32175. [DOI] [PubMed] [Google Scholar]
- (24).Patel A; Dharmarajan V; Cosgrove MS Structure of WDR5 Bound to Mixed Lineage Leukemia Protein-1 Peptide. J. Biol. Chem 2008, 283 (47), 32158–32161. [DOI] [PubMed] [Google Scholar]
- (25).Song J-J; Kingston RE WDR5 Interacts with Mixed Lineage Leukemia (MLL) Protein via the Histone H3-Binding Pocket. J. Biol. Chem 2008, 283 (50), 35258–35264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Southall SM; Wong PS; Odho Z; Roe SM; Wilson JR Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33 (2), 181–191. [DOI] [PubMed] [Google Scholar]
- (27).Odho Z; Southall SM; Wilson JR Characterization of a Novel WDR5-Binding Site That Recruits RbBP5 through a Conserved Motif to Enhance Methylation of Histone H3 Lysine 4 by Mixed Lineage Leukemia Protein-1. J. Biol. Chem 2010, 285 (43), 32967–32976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dias J; Van Nguyen N; Georgiev P; Gaub A; Brettschneider J; Cusack S; Kadlec J; Akhtar A. Structural Analysis of the KANSL1/WDR5/KANSL2 Complex Reveals That WDR5 Is Required for Efficient Assembly and Chromatin Targeting of the NSL Complex. Genes Dev. 2014, 28 (9), 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Avdic V; Zhang P; Lanouette S; Groulx A; Tremblay V; Brunzelle J; Couture JF Structural and Biochemical Insights into MLL1 Core Complex Assembly. Structure 2011, 19 (1), 101–108. [DOI] [PubMed] [Google Scholar]
- (30).Li Y; Han J; Zhang Y; Cao F; Liu Z; Li S; Wu J; Hu C; Wang Y; Shuai J; Chen J; Cao L; Li D; Shi P; Tian C; Zhang J; Dou Y; Li G; Chen Y; Lei M. Structural Basis for Activity Regulation of MLL Family Methyltransferases. Nature 2016, 530 (7591), 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Karatas H; Townsend EC; Cao F; Chen Y; Bernard D; Liu L; Lei M; Dou Y; Wang S. High-Affinity, Small-Molecule Peptidomimetic Inhibitors of MLL1/WDR5 Protein–Protein Interaction. J. Am. Chem. Soc 2013, 135 (2), 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Karatas H; Li Y; Liu L; Ji J; Lee S; Chen Y; Yang J; Huang L; Bernard D; Xu J; Townsend EC; Cao F; Ran X; Li X; Wen B; Sun D; Stuckey JA; Lei M; Dou Y; Wang S. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)–Mixed Lineage Leukemia (MLL) Protein–Protein Interaction. J. Med. Chem 2017, 60 (12), 4818–4839. [DOI] [PubMed] [Google Scholar]
- (33).Cao F; Townsend EC; Karatas H; Xu J; Li L; Lee S; Liu L; Chen Y; Ouillette P; Zhu J; Hess JL; Atadja P; Lei M; Qin ZS; Malek S; Wang S; Dou Y. Targeting MLL1 H3K4 Methyltransferase Activity in Mixed-Lineage Leukemia. Mol. Cell 2014, 53 (2), 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; Barsyte-Lovejoy D; Li F; Poda G; Schapira M; Wu M; Dong A; Senisterra G; Stukalov A; Huber KVM; Schönegger A; Marcellus R; Bilban M; Bock C; Brown PJ; Zuber J; Bennett KL; Al-awar R; Delwel R; Nerlov C; Arrowsmith CH; Superti-Furgaet G. Pharmacological Targeting of the WDR5-MLL Interaction in C/EBPα N-Terminal Leukemia. Nat. Chem. Biol 2015, 11 (8), 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Getlik M; Smil D; Zepeda-Velázquez C; Bolshan Y; Poda G; Wu H; Dong A; Kuznetsova E; Marcellus R; Senisterra G; Dombrovski L; Hajian T; Kiyota T; Schapira M; Arrowsmith CH; Brown PJ; Vedadi M; Al-awar R. Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem 2016, 59 (6), 2478–2496. [DOI] [PubMed] [Google Scholar]
- (36).Li D-D; Chen W-L; Wang Z-H; Xie Y-Y; Xu X-L; Jiang Z-Y; Zhang X-J; You Q-D; Guo X-K High-Affinity Small Molecular Blockers of Mixed Lineage Leukemia 1 (MLL1)-WDR5 Interaction Inhibit MLL1 Complex H3K4 Methyltransferase Activity. Eur. J. Med. Chem 2016, 124, 480–489. [DOI] [PubMed] [Google Scholar]
- (37).Wang F; Jeon KO; Salovich JM; Macdonald JD; Alvarado J; Gogliotti RD; Phan J; Olejniczak ET; Sun Q; Wang S; Camper D; Yuh YP; Shaw JG; Sai J; Rossanese OW; Tansey WP; Stauffer SR; Fesik SW Discovery of Potent 2-Aryl-6,7-Dihydro-5H-Pyrrolo[1,2-a]Imidazoles as WDR5-WIN-Site Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem 2018, 61 (13), 5623–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Aho ER; Wang J; Gogliotti RD; Howard GC; Phan J; Acharya P; Macdonald JD; Cheng K; Lorey SL; Lu B; Wenzel S; Foshage AM; Alvarado J; Wang F; Shaw JG; Zhao B; Weissmiller AM; Thomas LR; Vakoc CR; Hall MD; Heibert SW; Liu Q; Stauffer SR; Fesik SW; Tansey WP Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26 (11), 2916–2928.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schapira M; Tyers M; Torrent M; Arrowsmith CH WD40 Repeat Domain Proteins: A Novel Target Class? Nat. Rev. Drug Discov 2017, 16, 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Orlicky S; Tang X; Neduva V; Elowe N; Brown ED; Sicheri F; Tyers M. An Allosteric Inhibitor of Substrate Recognition by the SCFCdc4 Ubiquitin Ligase. Nat. Biotechnol 2010, 28 (4), 733–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sackton KL; Dimova N; Zeng X; Tian W; Zhang M; Sackton TB; Meaders J; Pfaff KL; Sigoillot F; Yu H; Luo X; King RW Synergistic Blockade of Mitotic Exit by Two Chemical Inhibitors of the APC/C. Nature 2014, 514 (7524), 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Altomonte S; Zanda M. Synthetic Chemistry and Biological Activity of Pentafluorosulphanyl Organic Molecules. J. Fluor. Chem 2012, 143, 57–93. [Google Scholar]
- (43).Barnes-Seeman D; Jain M; Bell L; Ferreira S; Cohen S; Chen X-H; Amin J; Snodgrass B; Hatsis P. Metabolically Stable Tert-Butyl Replacement. ACS Med. Chem. Lett 2013, 4, 514–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nicholls A; McGaughey GB; Sheridan RP; Good AC; Warren G; Mathieu M; Muchmore SW; Brown SP; Grant JA; Haigh JA; Nevins N; Jain AN; Kelley B. Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem 2010, 53 (10), 3862–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kramer SE; Routh JI The Binding of Salicylic Acid and Acetylsalicylic Acid to Human Serum Albumin. Clin. Biochem 1973, 6 (C), 98–105. [DOI] [PubMed] [Google Scholar]
- (46).Yang F; Zhang Y; Liang H. Interactive Association of Drugs Binding to Human Serum Albumin. Int. J. Mol. Sci 2014, 15 (3), 3580–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Patel A; Dharmarajan V; Vought VE; Cosgrove MS On the Mechanism of Multiple Lysine Methylation by the Human Mixed Lineage Leukemia Protein-1 (MLL1) Core Complex. J. Biol. Chem 2009, 284 (36), 24242–24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lapointe BT; Fuller PH; Gunaydin H; Liu K; Sciammetta N; Trotter BW; Zhang H; Barr KJ; MacLean JKF; Molinari DF; Simov V. Substituted Pyrazole Compounds as RORgammaT Inhibitors and Uses Thereof WO2016/130818, 2016. [Google Scholar]
- (49).Zhang H; Barr, Kenneth J; Lapointe, Blair T; Gunaydin H; Liu K; Trotter B,W. Heteroaryl Substituted Benzoic Acids as RORgammaT Inhibitors and Uses Thereof. WO2017/075185, 2017. [Google Scholar]
- (50).Cyr P; Boissarie P; Marinier A. Mild and Diazo-Free Synthesis of Trifluoromethyl-Cyclopropanes Using Sulfonium Ylides. Org. Lett 2019, 21, 2265–2268. [DOI] [PubMed] [Google Scholar]
- (51).Schanda P; Kupče Ē; Brutscher B. SOFAST-HMQC Experiments for Recording Two-Dimensional Deteronuclear Correlation Spectra of Proteins within a Few Seconds. J. Biomol. NMR 2005, 33 (4), 199–211. [DOI] [PubMed] [Google Scholar]
- (52).Otwinowski Z; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (53).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; Mccoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Adams PD; Grosse-Kunstleve RW; Hung L-W; Ioerger TR; Mccoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr. Sect. D 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- (55).Emsley P; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (56).Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.