

Dear Editor,
We would like to present an interesting case here.
INTRODUCTION
Non-alcoholic fatty liver disease is a common cause of chronic liver disease in the Western world and is becoming more prevalent in Asian countries. The prevalence of fatty liver (FL) in the general population in Western countries is estimated to be twenty to thirty percent whereas in Asia, it is estimated to be up to eighteen percent [1]. Genetic factors [2] and the interaction with the environments or behavioral factors including lifestyles, diet, alcohol, smoking and underlying medical problems such as diabetes mellitus are the risk factors or the causes of the majority of patients with FL. Inherited metabolic diseases (IMDs) are rare causes of FL and mostly present in infancy or childhood with usually fatal course, e.g., Reye-like syndrome in patients with medium chain acyl dehydrogenase deficiency and many other fatty acid oxidation disorders (FAODs). Wilson’s disease is the only well-known IMD among adult physicians because of its presentation with acute or chronic liver problems in early childhood to young adults [3]. More recently through the newborn screening in the developed countries, the milder forms of FAODs were identified and these groups of people can present with recurring liver problems during illnesses rather than severe fatal disease [4,5]. FAODs can also present with acute liver problems during pregnancy in heterozygous mothers carrying homozygous affected fetuses [6]. The study showed a high prevalence of long chain hydroxy-acyl dehydrogenase (LCHAD) deficiency carriers in pregnant women with acute fatty liver of pregnancy (AFLD). We report here a patient with severe recurring FL in an adult patient with carnitine palmitoyl transferase 1A (CPT1A) deficiency, an ultra-rare FAOD concomitant with severe illnesses. Physicians should be aware that IMDs can first present itself in adulthood.
SUBJECT
We studied a Thai patient who attended the Genetics Clinic as the part of undiagnosed disease program at King Chulalongkorn Memorial Hospital, Bangkok, Thailand. The medical data, pedigree, physical examinations and laboratory results were recorded. The written informed consent was obtained after counseling session of the implications of the study.
GENOMIC DNA PREPARATION AND WHOLE EXOME SEQUENCING (WES)
To perform genetic analysis, genomic DNA was isolated from peripheral blood leukocytes using a Puregene Blood kit (Qiagen, Hilden, Germany). The genomic DNA was sent to Macrogen, Inc. (Seoul, Republic of Korea) for WES. DNA was captured using a SureSelect Human All Exon version 4 kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a Hiseq 4000 instrument (Illumina, San Diego, CA, USA). Sequence reads were aligned against the University of California Santa Cruz human genome assembly hg19 using Burrows-Wheeler Alignment software (http://bio-bwa.sourceforge.net/; PMID20080505). Single nucleotide variants (SNVs) and insertions/deletions (Indels) were detected by SAMTOOLS (http://samtools.sourceforge.net/; PMID21903627) and annotated against dbSNP & the 1000 Genomes Project. After quality filtering, we looked for variants located in the coding regions of known FAODs genes (ACADM, ACADS, ACADVL, CPT1A, CPT2, ETFA, ETFB, ETFB, ETFDH, GLUD1, HADHA, HADHB, HMGCL, HMGCS2, SLC22A5, and SLC25A20) for all potential pathogenic SNVs and Indels. Variant calling exclusion criteria were (a) coverage <10×; (b) quality score <20; (c) minor allele frequency >1% in the 1000 Genomes Project; NHLB Exome Sequencing Project (ESP) and the Exome Aggregation Consortium database (ExAC) and (d) non-coding variants and synonymous exonic variants. The remaining variants were subsequently filtered out if they were present in our in-house database of 719 unrelated Thai exomes.
CASE PRESENTATION
A 29-year-old woman born to non-consanguineous parents was healthy except having a history of uncomplicated miscarriage at three months pregnancy when she was 20 years old. At 21 years of age, she developed progressive cholestatic jaundice, and weight loss while she was five months pregnant. She was diagnosed with severe Grave’s disease and underwent subtotal thyroidectomy. She had a spontaneous miscarriage a few days prior to the surgery. During that period, she also had episodes of hemolysis requiring blood transfusion and her abnormal serum electrolyte levels were consistent with distal renal tubular acidosis. Surprisingly, her cholestasis was subsided without a definite diagnosis or treatment. Two years later, she had another pregnancy resulting in a healthy daughter.
At the age of 27 years, she was admitted to hospital with severe progressive cholestatic jaundice with massive hepatosplenomegaly and a high fever. The investigations revealed that she also had an acute severe pancreatitis. She underwent a liver biopsy that showed more than 90 percent fat containing hepatocytes (macro-vesicular steatosis) with minimal inflammation (Fig. 1). Periodic Acid Schiff stain was negative for polysaccharides or glycogen. Her jaundice was again improved without a definite measurement. A few months later, the worsening jaundice co-occurred with Escherichia coli (E. coli) primary bacteremia that was successfully treated with a regular course of antibiotics. Ursodeoxycholic acid was the only prescribed drug. At 6-month follow-up, her jaundice and hepatosplenomegaly surprisingly completely disappeared. Timeline of the intermittent cholestatic jaundice and triggering events were summarized in Figure 2.
Figure 1.

Liver core biopsy revealed marked macrovesicular steatosis with cholestasis and mild inflammation (hematoxylin and eosin, ×40). (A) Mild portal inflammation with mixed inflammatory cells. (B) Marked cholestasis and marked macrovesicular steatosis with scattered inflammation. (C) Bile ducts are normal in number. Portal and hepatic veins are patent.
Figure 2.
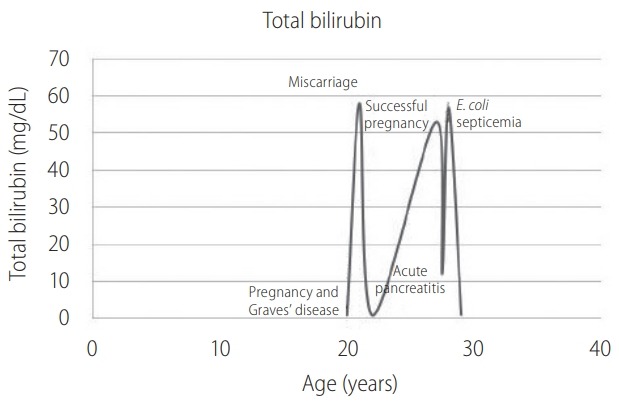
Chronological age, triggering events and levels of bilirubin. Patients’ bilirubin corresponded with triggering events: pregnancy and Graves’ disease at twenty years old; acute pancreatitis at 29 years old and septicemia at thirty years old. E. coli, Escherichia coli.
Medical genetics service was consulted. Severe liver disease worsening intermittently with inter-current illnesses raised the suspicion of FAODs. Plasma carnitine and acylcarnitine profile showed an increased C0 (free carnitine) of 58.74 micromol/L (5.55–20.88) with a very low level of long-chain fatty acids consistent with CPT1A hence a very high C0/C16+C18 ratio (Table 1). Renal tubular acidosis was a known association with this FAOD [7]. Medium chain triglyceride oil was initiated as it was shown to ameliorate the liver and kidney problems in patients with CPT1A deficiency. Other laboratory findings were normo-glycemia and other liver function tests included aspartate transaminase (AST) and AST were normal. We did not test this patient for urine organic acid and ammonia as patient was in the clinical silent when we have a chance to evaluate this patient.
Table 1.
Plasma acylcarnitine profile analyzed with tandem mass spectrometry
| Acylcarnitine profile | Result (μmol/L plasma) | Reference range (μmol/L plasma) |
|---|---|---|
| C0 | 58.74 | 5.55–20.88 |
| C8 | 0.18 | 0.02–0.77 |
| C10 | 0.26 | 0.03–0.87 |
| C14 | 0.02 | 0.00–0.11 |
| C16 | 0.12 | 0.03–0.22 |
| C18 | 0.02 | 0.01–0.13 |
| C0/C16+C18 | 419.57 | 11.50–12.0 |
Using WES, we identified a missense variant, c.1964G>A (p.Arg655His) and an intronic variant, c.988-11G>A. Both have never been previously reported in patients. The former missense variant has a minor allele frequency of 0.00008 in ESP and 0.00001 in ExAC. This variant is predicted to be damaging by the prediction software including SIFT, PolyPhen-2 and MCAP. The second variant is private, not present in databases including the in-house Thai exome one. It is predicted by Human Splicing Finder ver 3.0 (http://www.umd.be/HSF3; PMID: 19339519) to alter a normal splicing acceptor site causing an aberrant transcript in CPT1A. The parental samples were unavailable. The missense variant is classified as variant of uncertain significance (PM2, PP3, PP4) and the intronic variant is classified as variant of uncertain significance (PM2, PP3, PP4) according to ACMG/AMP variant classification. Lack of parent samples and functional study, we cannot confirm the pathogenicity of both variants. But it is highly suspicious by the biochemical analysis and the clinical phenotype.
DISCUSSION
We reported here, to our knowledge, the first adult patient with CPT1A deficiency presented with severe fatty liver during severe inter-current illnesses (acute pancreatitis and acute septicemia) or high physiologic demand (a pregnancy with Graves’ disease). The intermittent nature of the disease affecting the liver in this patient is highly suggestive of a disease of abnormal energy use and production including mitochondrial diseases or FAODs. The biochemical testing revealed a high level of free carnitine and a very low level of long chain fatty acid consistent with the abnormal entry of long chain acyl-carnitine into the mitochondria as the substrates for β-oxidation. This is consistent with CPT1A deficiency. Renal tubular acidosis was a known association with this FAOD [7]. Acute hemolysis requiring transfusion was investigated which revealed an increase osmotic fragility consistent with hereditary ovalocytosis, a prevalent hereditary cause in Southeast Asia. However, the exome sequencing did not identify any variants in SLC4A1, the gene which encodes anion exchange 1 protein or band 3 protein known to cause the Southeast Asian type, ovalocytosis [8]. Hemolysis in this patient could be the manifestation of the CPT1A deficiency during acute metabolic decompensation. WES identified two novel variants in CPTIA. A rare missense variant (c.1964A>G), not previously reported in the patients with CPT1A deficiency as well as the possible splicing intronic variant (c.988-11G>A). They are both predicted to be deleterious. However, we cannot confirm that these two variants are located on the different alleles. The splicing aberration was not tested due to sample unavailability. We did not analyze for the deletion/duplication or structural rearrangement of the gene.
There was no known trigger of Graves’ disease besides pregnancy. Patient was not previously diagnosed with Graves’ disease. Pregnancy can worsen Grave’s disease if that was unknown before. Bacteremia occurred only a month after first hospitalization suspicious of nosocomial related infection. However, E. coli identified in this patient was susceptible to antibiotics. Alcohol, hyper-triglycerides, medications, trauma were all denied as precipitating factors of acute pancreatitis. Gall stone was not seen.
Long chain fatty acids, an important source of energy, are metabolized in the mitochondria via β-oxidation. This requires the mitochondrial outer membrane fatty acid transfer complex including CPT1A to convert activated fatty acids into acylcarnitines [9]. CPT1A is actually the rate limiting step for the entry of long chain fatty acids into the mitochondria for β-oxidation. If there is a reduction of the activity of CPT1A, fatty acids cannot enter the mitochondria for further use. Mitochondrial fatty acid oxidation by the liver provides an alternative source of fuel when glycogen reserves are significantly reduced, most often during fasting or other intercurrent illnesses. Patients with CPT1A deficiency present with fasting-induced hepatic encephalopathy in early childhood that can be potentially fatal. These children who recover are at risk for recurrent episodes of life-threatening illnesses. CPT1A deficiency was reported in a child with jaundice and diffuse macrovesicular steatosis that was spontaneously resolved in months [10]. Initial presentation may occur later in life with similar life-threatening acute hepatic illness as late as a cause of death in a 17-year-old boy [11]. Between episodes of metabolic decompensation, individuals appear normal and long-term liver damage as a result of recurring hepatosteatosis has not been reported. Unlike other long-chain FAODs, cardiac or skeletal muscle involvement in CPT1A deficiency is uncommon as there is a separate isoform of CPT1A, CPTIB, in these organs [12]. Our patient developed severe progressive steato hepatitis at 28 years old concurrent with acute pancreatitis and was partially improved. However, 6 months later, her symptoms were worsening during the episode of severe bacterial infection. Hepatosplenomegaly and cholestatic jaundice disappeared after six months of the last episode.
There has been no previous report of patients with CPT1A deficiency with pregnancy; however, fetal CPT1A deficiency has been associated with AFLD in the mother [13] in the similar fashion of an established maternal risk factor for AFLD with LCHAD deficiency [6]. In those cases with fetal FAODs, the risk of maternal liver diseases is increased in these heterozygous women. The intermediate products of fatty acids that placenta fails to handle may create toxic effects to the mother. These carrier females do not inherent the risk of developing liver diseases inter-gestational periods. Our patient’s first pregnancy was ended with a miscarriage as early as three months into the pregnancy. This probably did not put stress to the metabolism beyond the threshold. However, she developed severe liver disease during her second pregnancy which was also complicated with a highly pathologic increased demand for metabolism from her severe Graves’ disease. She survived after fetal demise and thyroidectomy was performed to control her thyrotoxicosis. Her third pregnancy went unremarkable. We assumed that her CPT1A deficiency was mild and only the pregnancy concurrent with Graves’ disease was severe enough to increase her metabolic demand and resulted in decompensating liver problems during her second pregnancy.
Besides highly expressed in the liver, CPT1A expresses in the kidneys. Patients with CPT1A deficiency are associated with renal tubular acidosis [7]. Renal tubular acidosis is not reported to be associated with other FAODs. The administration of medium chain triglyceride oil can ameliorate the renal manifestations in patients with CPT1A deficiency and prevent the metabolic decompensation during the periods of illnesses.
CPT1A is an ultra-rare disorder with the estimated prevalence of 1:500,000 to 1:1,000,000 from the newborn screening [14]. The natural course of these patients was not fully known as all patients identified through newborn screening program will be treated and prophylactic measures such as avoidance of prolonged fasting, high fat content diet will be implemented to prevent metabolic decompensation. Some founder mutations are highly prevalent in some ethnic groups such as p.Gly710Glu that the carrier rate was reported to be as high as 1:16 in the Hutterite population [15].
The manifestation of recurrent attacks of severe decompensating liver illness might perplex the adult care providers. This case demonstrates the first adult patient predisposing to liver decompensation during severe metabolic crises i.e., pregnancy with Graves’ disease, acute pancreatitis, and severe sepsis. The underlying mechanism is the inherited metabolic condition, CPT1A deficiency, unknown by most of the adult healthcare providers.
Acknowledgments
This study was supported by the Academic Fund of Division of Infectious Disease, Department of Medicine, Chulalongkorn University and the Chulalongkorn Academic Advancement into its 2nd Century Project.
Abbreviations
- AFLD
acute fatty liver of pregnancy
- AST
aspartate transaminase
- CPT1A
carnitine palmitoyl transferase 1A
- ESP
Exome Sequencing Project
- ExAC
Exome Aggregation Consortium database
- FAOD
fatty acid oxidation disorder
- FL
fatty liver
- IMDs
inherited metabolic diseases
- Indels
insertions/deletions
- LCHAD
long chain hydroxy-acyl dehydrogenase
- SNVs
single nucleotide variants
- WES
whole exome sequencing
Ethics approval
This article has been waived for the ethics approval by the standard of the Institutional Research Board of the Faculty of Medicine, Chulalongkorn University and all procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000 (5).
Authors’ contribution
Prasit Phowthongkum was a medical geneticist who saw, formulated the hypothesis and made differential diagnosis to investigate this patient, analyzed the WES and prepared the manuscript.
Kanya Suphapeetiporn and Vorasuk Shotelersuk were responsible for the authorization of the WES result, reviewed the manuscript, commented and finalized the manuscript.
Conflicts of Interest Prasit Phowthongkum, Kanya Suphapeetiporn, and Vorasuk Shotelersuk have no competing interest to be declared financially or non-financially related to the submission of this manuscript.
REFERENCES
- 1.Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM, Seth D, Day CP. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology. 2016;150:1728–1744. doi: 10.1053/j.gastro.2016.01.037. e7. [DOI] [PubMed] [Google Scholar]
- 3.Mahmood S, Inada N, Izumi A, Kawanaka M, Kobashi H, Yamada G. Wilson’s disease masquerading as nonalcoholic steatohepatitis. N Am J Med Sci. 2009;1:74–76. [PMC free article] [PubMed] [Google Scholar]
- 4.Pant M, Oshima K. Cholesteryl ester storage disease: an underdiagnosed cause of cirrhosis in adults. Ann Diagn Pathol. 2017;31:66–70. doi: 10.1016/j.anndiagpath.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33:527–532. doi: 10.1007/s10545-010-9090-x. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Ghaziani TT, Wolf JL. Acute fatty liver disease of pregnancy: updates in pathogenesis, diagnosis, and management. Am J Gastroenterol. 2017;112:838–846. doi: 10.1038/ajg.2017.54. [DOI] [PubMed] [Google Scholar]
- 7.Falik-Borenstein ZC, Jordan SC, Saudubray JM, Brivet M, Demaugre F, Edmond J, et al. Brief report: renal tubular acidosis in carnitine palmitoyltransferase type 1 deficiency. N Engl J Med. 1992;327:24–27. doi: 10.1056/NEJM199207023270105. [DOI] [PubMed] [Google Scholar]
- 8.Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, et al. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Nat Acad Sci U S A. 1991;88:11022–11026. doi: 10.1073/pnas.88.24.11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem. 2011;286:25655–25662. doi: 10.1074/jbc.M111.228692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris AA, Olpin SE, Bennett MJ, Santani A, Stahlschmidt J, McClean P. Cholestatic jaundice assoiciated with carnitine palmitoyltransferast IA deficiency. JIMD Rep. 2013;7:27–29. doi: 10.1007/8904_2012_135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown NF, Mullur RS, Subramanian I, Esser V, Bennett MJ, Saudubray JM, et al. Molecular characterization of L-CPT I deficiency in six patients: insights into function of the native enzyme. J Lipid Res. 2001;42:1134–1142. [PubMed] [Google Scholar]
- 12.Olpin SE, Allen J, Bonham JR, Clark S, Clayton PT, Calvin J, et al. Features of carnitine palmitoyltransferase type I deficiency. J Inherit Metab Dis. 2001;24:35–42. doi: 10.1023/a:1005694320063. [DOI] [PubMed] [Google Scholar]
- 13.Innes AM, Seargeant LE, Balachandra K, Roe CR, Wanders RJ, Ruiter JP, et al. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000;47:43–45. doi: 10.1203/00006450-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Bennett MJ, Santani AB. Carnitine Palmitoyltransferase 1A Deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews(R) [Internet] Seattle (WA): University of Washington; 2005. [Updated 17 Mar 2016]. National Center for Biotechnology Information web site, < https://www.ncbi.nlm.nih.gov/books/NBK1527/>. Accessed 1 Dec 2018. [Google Scholar]
- 15.Prasad C, Johnson JP, Bonnefont JP, Dilling LA, Innes AM, Haworth JC, et al. Hepatic carnitine palmitoyl transferase 1 (CPT1 A) deficiency in North American Hutterites (Canadian and American): evidence for a founder effect and results of a pilot study on a DNA-based newborn screening program. Mol Genet Metab. 2001;73:55–63. doi: 10.1006/mgme.2001.3149. [DOI] [PubMed] [Google Scholar]