

Abstract
Bacterial communities of marine sponges are believed to be an important partner for host survival but remain poorly studied. Sponges show difference in richness and abundance of microbial population inhabiting them. Three marine sponges belonging to the species of Pione vastifica, Siphonochalina siphonella and Suberea mollis were collected from Red sea in Jeddah and were investigated using high throughput sequencing. Highly diverse communities containing 105 OTUs were identified in S. mollis host. Only 61 and 43 OTUs were found in P. vastifica and S. siphonella respectively. We identified 10 different bacterial phyla and 31 genera using 27,356 sequences. Most of the OTUs belong to phylum Proteobacteria (29%–99%) comprising of Gammaproteobacteria, Alphaproteobacteria, and Deltaproteobacteria where later two were only detected in HMA sponge, S. mollis. A number of 16S rRNA sequences (25%) were not identified to phylum level and may be novel taxa. Richness of bacterial community and Shannon, Simpson diversity revealed that sponge S. mollis harbors high diversity compared to other two LMA sponges. Dominance of Proteobacteria in sponges may indicate an ecological significance of this phylum in the Red sea sponges. These differences in bacterial composition may be due to difference in location site or host responses to environmental conditions. To the best of our knowledge, the microbial communities of these sponges have never been studied before and this is first attempt to unravel bacterial diversity using PCR-based 454-pyrosequencing method.
Keywords: Red sea, Marine sponges, 454 pyrosequencing, Bacterial diversity, Proteobacteria
1. Introduction
Microbial communities associated with sponges are diverse and make a mutulistic relationship either as pathogen or symbionts (Wilkinson, 1983, Bavestrello et al., 2000). Associated bacteria may help in nitrogen fixation, removal of waste products and also production of secondary bioactive metabolites (Webster et al., 2001). Sponges have attracted research interest to study associated microbial population and their invaluable metabolites that can be used in pharmaceutical and biotechnological applications. Mutualistic relationship is important ecologically for marine sponges and their associated microbes. As sponge provide place for colonization, shelter from predators and nutrients to microbes and in turn by products of sponges are eliminated by these microbes. Bioactive compounds excreted by these microbes also help their host against different microbial disease (Hentschel et al., 2001).
Culture-dependent and independent techniques used to study sponge-microbe interaction. Cultivation techniques are limited as most of bacteria are not easy to culture and remain untapped. Different culture-independent techniques were used widely to study symbiotic microbiome from various sponge samples (Sharp et al., 2007, Webster et al., 2004). In spite of these various methods pyrosequencing may allow deep and quick analysis of microbial population associated with different samples with much high throughput. This technology enables to determine new and novel taxa and also provide enormous sequences of DNA reads and open new avenue for microbial communities of different ecological regions (Huse et al., 2007, Sogin et al., 2006).
Saudi Arabia is largest country bordering and covering around 80% of eastern site of Red sea. Coastal area of Red Sea in Saudi Arabia provide habitats for various communities of mangrove, corals, sea grass and sponges (Price et al., 1998). Sponges from Red Sea have been studied for biologically active compounds as well as for study of microbial communities (Radwan et al., 2010). Previous studies of sponges from Red sea using next generation sequencing have revealed bacterial diversity where new and rare taxa were detected. Using pyrosequencing, they have characterized 2000 different bacterial species belonging to 26 different phyla, 4 novel taxa and 300 archaeal species were recorded from single sponge (Lee et al., 2011).
Three marine sponge species included in this study are well known for their biotechnological importance. P. vastifica is biotechnology and ecologically important species capable of producing antimicrobial compounds (Afifi and Khabour, 2017). Triterpenoidal active metabolites were isolated from sponge S. siphonella. These compounds showed potent cytotoxic activities against cancer cell lines. S. mollis afforded brominated phenolic compounds and purealdin L, aerothionin, and dichloroverongiaquinol compound exhibiting potent antioxidant activity (Abbas et al., 2014). These three sponges didn't show any record of study related to bacterial diversity using culturomics or uncultured methods. Recently we have used only culture based method to study bacterial diversity from P. vastifica and S. siphonella (Bibi et al., 2018). Therefore, this study provides unique information regarding microbial communities and comparison between bacterial communities among these three marine sponges using Next Generation Sequencing (NGS) approach.
2. Materials and methods
2.1. Study site and sample collection
Nine sponge specimens (i.e. three replicates from three sponge samples) located at the depth of 30–40 m and were within few meters distance to each other were collected in single dive in November 2016 at obhur in Red Sea (20°23′8.9664″N and 38°7′21.2124″E) Jeddah, Saudi Arabia. The specimens were inhabiting area that was exposed to sunlight and temperature of the water was 25 °C. For identification of these sponge samples, Dr. Abdulmohsin Al-Sofyani from marine science department King Abdul-Aziz University provided his expertise. These sponges were identified as belong to Pione vastifica, Siphonochalina siphonella and Suberea mollis. After collection, sponges were put inside sterile ziploc plastic bag containing seawater and transferred immediately to the laboratory and were kept at −20 °C until process further.
2.2. Extraction of DNA from sponges
Sponge samples were washed twice with autoclaved distilled water to remove loosely attached bacteria. Sponge specimens were chopped into small pieces and DNA was extracted from three sponge samples using the PowerSoil DNA isolation kit, Mo Bio laboratories (Carlsbad, CA) according to the manufacturer's protocol. Using NanoDrop (ND-1000, Thermo Fisher, USA), quality and quantity of DNA was measured and samples were stored at −20 °C until use.
2.3. Library preparation and emulsion-based PCR (emPCR)
Library was prepared using DNA according to the 454 Seq Sys Amplicon Library Prep Method Manual. Briefly, 20 ng aliquot of each DNA sample was used for a 25ul PCR reaction. The 16S universal primers 27F (5′GAGTTTGATCMTGGCTCAG3′), 518R (5′WTTACCGCGGCTGCTGG3′) were used for amplification of 16S rRNA genes (V1 to V3 regions). FastStart High Fidelity PCR System (Roche, Basel, Switzerland) was used according to the manufacturer’s recommendations. Fidelity PCR System (Roche) was used for PCR under the following conditions: 94 °C for 3 min followed by 35 cycles of 94 °C for 15 s; 55 °C for 45 s and 72 °C for 1 min; and a final elongation step at 72 °C for 8 min. Using AMPure beads (Beckman coulter) PCR products were purified. Using the Picogreen assay quantification assay (Life Technologies) libraries were further quantified. To clonally amplify the purified library, emPCR and amplification was carried out as described previously (Udayangani et al., 2017).
2.4. Next generation sequencing using Roche 454 GS-FLX
After PCR amplification, the emulsion was broken chemically and amplified DNA libraries were recovered from the beads after washing by filtration. Using the biotinylated primer A, all positive beads were purified. The magnetic beads were separated from the DNA library beads and single-stranded template DNA bead bound fragments were recovered from double-stranded after melting. The single-stranded DNA was amplified using sequencing primer. Finally, all beads carrying amplified single-stranded DNA were counted using a Particle Counter (Beckman Coulter). Using Genome Sequencer FLX (454 Life Sciences, Branford, CT, USA), sequencing was performed. A 70–75 mm Pico Titer plate (454 Life Sciences) fitted with a 4 lane gasket was loaded with each sample in 2 regions. For precise OTU analysis, data containing sequence error were removed. After this process, clustering was performed. Using CD-HIT-OTU, data removed contains reads that have length shorter than 40% of the library, reads on sequence similarity. OTUs of the remained reads were generated by cluster cutoff value of 97%. The singleton and doubleton created in this process was not used for further analysis.
2.5. Statistical analysis
For OTU analysis and obtaining taxonomy information QIIME (version 1.8) (Caporaso et al., 2010) was used. The major sequence of each OTU is referred to Greengenes and Silva databases. Taxonomic information is obtained with UCLUST taxonomy assigner method. In order to check the diversity and evenness in microbe community, Shannon and Simpson index were calculated. Also alpha diversity was calculated with Rarefaction curve and Chao1 value. Beta diversity (diversity among samples within the group) was calculated based on Weighted UniFrac distance. Genetic relationship among samples was visualized based on PCoA and UPGMA tree. We used R packages heatmap (Kolde, 2012) to generate heatmap figure and Venn diagram programs (Chen, 2012) was used to produce Venn diagrams.
2.6. Nucleotide sequence accession numbers
The pyrosequencing reads were deposited to the European Nucleotide Archive under accession number ERS2923991, ERS2923992 and ERS2923995 for S. mollis, P. vastifica and S. siphonella respectively.
3. Results
3.1. Bacterial diversity and taxonomic composition in sponges
Three sponge samples were collected from different locations (30–40 m in depth) from North Obhur in the Red Sea. Sponges were identified based on their morphological characteristics by a sponge taxonomist (Table 1). We obtained a total of 55,638 raw sequences from 3 sponge's samples using Roche 454-FLX titanium. A total 27,356 reads were used for diversity and taxonomic analyses. The average reads numbers per sample were ±10,840–28,126 respectively. Bacterial diversity in three sponge species i.e. P. vastifica, S. siphonella and S. mollis were studied at different levels of their taxonomic classification to find bacterial communities. After trimming, denoising and removal of chimera sequences, 27,356 sequences were obtained and further subjected to downstream analysis. Using sequences showing 97% similarities, these sequences were clustered into total of 105, 61 and 43 OTUs respectively for S. mollis P. vastifica and S. siphonella. Numbers of OTUs were higher in sponge S. mollis (105 OTUs) while minimum number (43 OTUs) was observed for sponge S. siphonella.
Table 1.
List of sponge species collected and estimation of diversity.
| Sponge | Sample code | Class | Order | Family | Total reads | Totala OTUs | Chaob | Shannonc | Simpsond |
|---|---|---|---|---|---|---|---|---|---|
| P. vastifica | O1 | Demospongiae | Hadromerida | Clionaidae | 16,672 | 61 | 64.3 | 3.8 | 0.91 |
| S. siphonella | Sk | Demospongiae | Haplosclerida | Callyspongiidae | 10,840 | 43 | 48 | 2.1 | 0.59 |
| S. mollis | G | Demospongiae | Verongiida | Aplysinellidae | 28,126 | 105 | 120 | 4.46 | 0.91 |
OTUs: Operational Taxonomic Unit is an operational definition of a species or group of species often used when only DNA sequence data is available.
Chao1: returns the Chao1 richness estimate for an OTU definition.
Shannon: The Shannon index takes into account the number and evenness of species.
Simpson: The Simpson index represents the probability that two randomly selected individuals in the habitat will belong to the same species.
Alpha diversity indicating richness of various taxa and it varies among three sponge species studied. Rarefaction analysis for sequences obtained for three sponge samples showed high sequence coverage values. This is shown by a clear saturated plateau in three sponges (Fig. 1A). It indicates good coverage and sequencing (100%) from these sponge samples. Alpha diversity indices based on OTUs using Chao1 present high richness of bacterial taxa in the sponge S. mollis and lowest values were recorded for P. vastifica and S. siphonella respectively (Fig. 1B). Microbial diversity indices such as Shannon and Simpson diversity indicated that sponge S. mollis and P. vastifica were more diverse than sponge S. siphonella (Fig. 1C). Bacterial diversity is generally low in two sponges: P. vastifica and S. siphonella except for sponge S. mollis. Shannon’s diversity index values of 4.4 and 3.8 were higher for S. mollis and P. vastifica respectively as compare to low value of 2.0 for S. siphonella indicating minimum bacterial diversity and containing only 43 OTUs. To assess microbial communities across different samples, beta diversity patterns were calculated by using unweighted UniFrac distance matrices in QIIME. This two dimensional PCoA plot have shown differences in three sponge samples as explained by PC1 and PC2 (Fig. 2). This plot clearly distinguishes samples of P. vastifica, S. siphonella and S. mollis. This difference in sponge phylogeny clearly has shown that bacterial communities have correlation with grouping and taxonomic classification of sponges. This data has shown that sponge studied belong to three different genera and harbor different bacterial communities. Especially variability is noteworthy for the sponge samples of S. mollis.
Fig. 1.

Alpha diversity indices of microbial communities from 3 sponge samples. Microbial richness was calculated using observed species (A), chao 1 estimator (B) from the sponges P. vastifica (red), S. mollis (blue) and S. siphonella (orange) and using Shannon and Simpson indices (C).
Fig. 2.
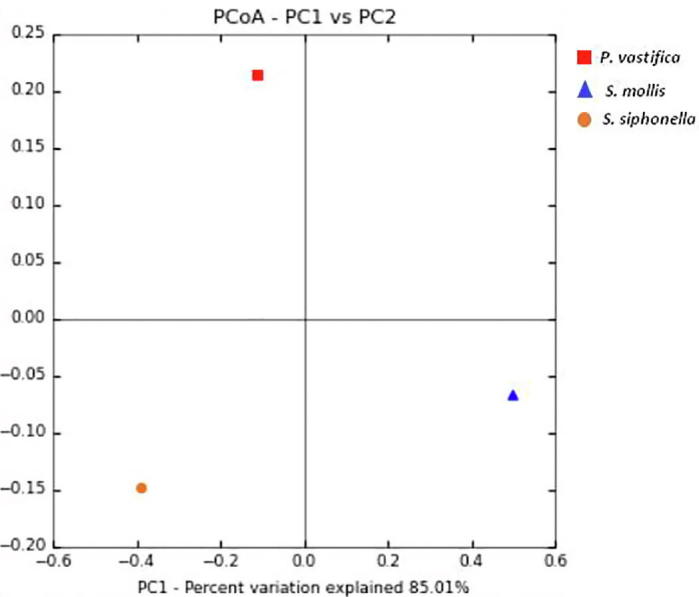
2D Principal coordinate analysis (PCoA) plot based on weighted UniFrac distance matrices.
3.2. Taxonomic richness and composition of bacterial community
Using filtered sequence reads of the 16S rRNA gene, we found 10 different bacterial phyla such as Chloroflexi, Acidobacteria, Bacteroidetes, Actinobacteria, Deinococcus-Thermus, Proteobacteria, Cyanobacteria, Firmicutes, Nitrospirae, and the candidate phylum Poribacteria) from three sponge species. The dominant groups of each sponge sample are shown in Fig. 3. In three sponge species, Proteobacteria (29%–99%) was the dominant phylum consisting of Alphaproteobacteria, Gammaproteobacteria, and Deltaproteobacteria where members of Gammaproteobacteria show dominance. The 16S rRNA sequence of approximately 4% OTU s belong to Alphaproteobacteria and only detected in sponge host S. mollis while absent from two other LMA sponges. In sponge S. mollis, Alphaproteobacteria comprises of two families: Rhodobacteraceae and Rhodospirillaceae and most of these sequences remain unknown at genus level and may present a novel species. Members of Gammaproteobacteria were dominant and detected in three sponge species. In this study, both LMA sponges showed high proportion (99.9%) of class Gammaproteobacteria. Seven different genera: Pseudoalteromonas, Cobetia, Halomonas, Kushneria, Salinicola, Psychrobacter and Pseudomonas were observed to this class. Dominance of Halomonas and Psychrobacter was detected in P. vastifica and S. siphonella respectively.
Fig. 3.

Bacterial community dynamics based on the taxonomic classification of the 16S pyrosequencing reads at the specie level. (x-axis: sample name; y-axis: OTU s proportions).
While bacterial communities in HMA sponge S. mollis were diverse as compare to two other sponge species. Proteobacteria (24%) and Chloroflexi (20%) were predominant groups following Actinobacteria (8%), Acidobacteria (4%), Bacteroidetes (0.58%), Nitrospirae (0.46%), Deinococcus-Thermus (0.31%), candidate phylum Poribacteria (0.06%) and Firmicutes (0.04%). In addition to Gammaproteobacteria and Alphaproteobacteria, Deltaproteobacteria were only restricted to S. mollis host. Members of phylum Actinobacteria in host S. mollis belong to four different genera Iamia, Streptomyces, Psychroflexus and Salinimicrobium. The Acidobacteria was another dominant phylum following Actinobacteria. This phylum comprises of five subgroups i.e Gp3, Gp6 and Gp9-11. Bacteroidetes was another phylum further comprised of Psychroflexus, Salinimicrobium and Salisaeta. The heat map showed distribution of different taxa reported from three sponge hosts (Fig. 4). At all the taxonomic levels, bacterial communities of host S. mollis were more diverse as compare to two other sponge samples in this study. Many unclassified genera belonging to different families were present in all sponge samples, exhibiting complex bacterial community structure. Overall, 203 different genera were detected at genus level taxonomic classification from total sequence reads. Among them 78 OTUs were commonly detected only in sponge sample S. mollis.
Fig. 4.

Heat map showing the richness of 16S rRNA gene sequence reads of bacterial OTUs. All sponge samples are indicated along the x-axis. OTUs are indicated along the y-axis and abundance of each OTU is indicated by colors ranging from white (low abundance or absent) to dark blue (high abundance).
3.3. Similarity and distribution of bacterial community
We have found differences in bacterial communities detected among three different sponge hosts belong to the same class (Demospongiae). Differences between bacterial communities can be clearly evident from clustering of sponge samples based on UPGMA tree in Fig. 5. The UPGMA cluster analysis of three sponges collected from the Red Sea showed similarities between two sponge species i. e P. vastifica and S. siphonella species and third sponge S. mollis grouped separately representing different bacterial communities (Fig. 5). The proportion of OTUs shared among these bacterial communities at genus level for three sponge samples has been demonstrated using a Venn diagram (Fig. 6). The results show that only 5 bacterial OTUs have been shared: for genera Pseudoalteromonas, Cobetia, Halomonas, Psychrobacter and Pseudomonas were commonly detected in three sponge samples at level of greater than 1% concentration (Fig. 6). While 25 unique OTUs belong to different taxon: Gp10, Gp11, Gp3, Gp6, Gp9, Iamia, Acidimicrobiales, Streptomyces, Actinobacteria, Psychroflexus, Salinimicrobium, Salisaeta, Rhodothermaceae, Longilinea, Anaerolineaceae, Litorilinea, Chloroflexi, GpIIa, Truepera, Carnobacterium, Nitrospira, Poribacteria, Rhodobacteraceae, Rhodospirillales, Alphaproteobacteria, Deltaproteobacteria, Pseudoalteromonas, Cobetia, Halomonas, Kushneria, Salinicola and Psychrobacter, were identified only in S. mollis species.
Fig. 5.

UPGMA tree plotted using UniFrac distance matrices. The tree showing similarities of OTUs (%) of 16S rRNA gene sequences obtained from different bacterial taxa associated with sponge samples.
Fig. 6.

Venn diagram at the genus level that exhibit the relationship between OTUs detected in three sponge samples.
4. Discussion
High-throughput 454 pyrosequencing of bacterial 16S rRNA (V1 to V3 regions) was carried out in our study to examine bacterial composition in three sponges belonging to same class (Table 1). Class Demospongiae is more diverse class in phylum Porifera and comprising of more than 5000 species. In marine sponges dominance of different phyla of bacteria vary depending upon taxonomy of sponge and their geographical location. High microbial abundance (HMA) sponges generally are rich in bacterial diversity while low microbial abundance (LMA) sponges usually harbor low bacterial taxa (Giles et al., 2013, Hentschel et al., 2003). Previous studies have demonstrated lower diversity of bacterial communities in LMA sponges in comparison with HMA sponges with high diversity (Weisz et al., 2007, Kamke et al., 2010). In our study bacterial richness varied from 43 to 105 OTUs which is consistent with the range reported in previous studies (Giles et al., 2013, Hentschel et al., 2006). Recent survey of marine sponges has shown that they significantly contribute to the microbial population of the Sea where microbial richness ranges from 50 to 3820 distinct OTUs per host (Thomas et al., 2016). Two sponge species: P. vastifica and S. siphonella harbor low OTUs, low bacterial richness and Proteobacteria as the most prominent phylum thus affiliated with LMA sponges. While one sponge, S. mollis contains high abundance and diverse bacterial communities that is typical of HMA sponges. Similarity of sponge species to HMA and LMA was based exclusively on the diversity and richness of microbial communities as no histological examination was performed for the confirmation of microbial abundance. Vertical transmission of bacteria is common feature of HMA sponges (Schmitt et al., 2008). While in LMA sponges bacteria were taken up by sponge through selective mechanism hence dominated by the same group of Proteobacteria (Weisz et al., 2007).
Low bacterial abundance is observed for S. siphonella and P. vastifica sponge species in this study. In a previous study, sponge of same genus Callyspongia vaginalis showed low diversity mainly consisting of species of Proteobacteria (Hentschel et al., 2003). No record is available for bacterial diversity related studies for species of sponge genus Pione. This is a first study to report bacterial communities from P. vastifica. High diversity in the sponge S. mollis was recorded in present study that is concordant with previous study where 206 OTUs were recorded in sponge from genus Suberea (Turon et al., 2018). In all sponge species, Proteobacteria (29%–99%) was the dominant phylum. High proportion of Proteobacteria was also reported in previous studies of marine sponges. This high proportion of Proteobacteria in LMA sponges is consistent with many previous studies where LMA sponges showed high proportion of Proteobacteria as compare to HMA sponges (Jeong et al., 2013). Proteobacteria perform different functions in host including nitrogen fixation and involve in host defense mechanism (Webster et al., 2013, Li et al., 2006). Class Gammaproteobacteria was dominant in three sponge species where high percentage was recovered from LMA sponges in this study. Two genera, Halomonas and Psychrobacter were dominant in P. vastifica and S. siphonella respectively. Results of this uncultured study are concordant with our recent study where strains exhibiting antimicrobial activity belong to these two genera were isolated from P. vastifica and S. siphonella (Bibi et al., 2018). Different species of the genus Halomonas and Psychrobacter from sponges are already known to exhibit antimicrobial activities (Matobole et al., 2017, Bibi et al., 2018) thus playing their role in host defense mechanism.
High microbial abundance was observed in S. mollis in contrast to P. vastifica and S. siphonella. Approximately 4% OTUs were unassigned in S. mollis host at phylum level. Previous studies also reported presence of unassigned OTUs ranging from 34 to 36% and could not be assigned to any bacterial phylum (White et al., 2012, Cleary et al., 2013). A large number of (25%) 16 S rRNA sequences from S. mollis were not matched to bacteria at genus level so may present some novel taxa. Therefore, these sponge species may be a reservoir of novel bacteria that have not been identified so far. Candidate phylum Poribacteria, Chloroflexi, Actinobacteria and Acidobacteria were only detected in sponge S. mollis. Proteobacteria, Chloroflexi, Actinobacteria, Bacteroidetes, Firmicutes, Acidobacteria from marine sponges are already known to produce biologically active compounds (Brinkmann et al., 2017). S. mollis also harbor candidate phylum Poribacteria that was first discover from sponge tissues and widely spread among members of class Demospongiae (Lafi et al., 2009). Poribacteria, Acidobacteria and Chloroflexi are commonly detected in HMA sponges belonging to different phyla (Kamke et al., 2014). To compare bacterial communities UniFrac analysis was performed. No correlation was observed according to distribution of bacterial communities in sponge samples i.e. they were collected from same geographical location so they could harbor similar bacterial communities. Another study reported composition of complex microbial communities in HMA sponges is influenced by both environmental factors and host phylogeny (Erwin et al., 2012). This reveals that environmental factors might play a role in defining difference in pattern and structure of microbial taxa in marine sponges.
5. Conclusions
Our investigation reveals pattern and distribution of bacterial taxa in LMA and HMA sponge species. These sponges were not studied before and our data showed that three sponge samples belong to two different groups i.e. LMA and HMA on the basis of low and high microbial abundance. The community structure of each sponge showed dominance of phylum Proteobacteria where Gammaproteobacteria was dominant as a class. Richness and abundance of bacterial phyla typical of HMA sponges are absent in LMA sponges. This difference might indicate that different factors related to host and environmental may define composition of bacteria in LMA and HMA sponges. This work increases our knowledge about bacterial communities of marine sponge species that are of medical significance from previous literature.
Acknowledgments
Acknowledgements
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH)–King Abdulaziz City for Science and Technology-the Kingdom of Saudi Arabia-award number (12-BIO3106-03). The authors also, acknowledge with thanks Science and Technology Unit, King Abdulaziz University for technical support.
Conflict of interest
There is no conflict of interest.
Footnotes
Peer review under responsibility of King Saud University.
References
- Abbas A.T., El-Shitany N.A., Shaala L.A., Ali S.S., Azhar E.I., Abdel-Dayem U.A., Youssef D.T. Red Sea Suberea mollis sponge extract protects against CCl4-induced acute liver injury in rats via an antioxidant mechanism. J. Evid. Based Complement. Altern. Med. 2014;2014:745606. doi: 10.1155/2014/745606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afifi R., Khabour O.F. Antibacterial activity of the Saudi Red Sea sponges against Gram-positive pathogens. J. King Saud Univ.-Sci. JKSUS. 2017 [Google Scholar]
- Bavestrello G., Arillo A., Calcinai B., Cattaneo-Vietti R., Cerrano C., Gaino E., Penna A., Sara M. Parasitic diatoms inside Antarctic sponges. Biol. Bul. 2000;198(1):29–33. doi: 10.2307/1542801. [DOI] [PubMed] [Google Scholar]
- Bibi F., Alvi S.A., Al-Sofyani A., Yasir M., Kensarah E.A., Azhar E.I. Two marine sponges-associated cultivable bacteria: Diversity and biological activities. Genet. Mol. Res. 2018;17(2) [Google Scholar]
- Brinkmann C., Marker A., Kurtböke D. An overview on marine sponge-symbiotic bacteria as unexhausted sources for natural product discovery. Diversity. 2017;9(4):40. [Google Scholar]
- Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Pena A.G., Goodrich J.K., Gordon J.I., Huttley G.A. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7(5):335. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H.B., 2012. Venn Diagram: Generate high-resolution Venn and Euler plots. R package version. 113.
- Cleary D.F., Becking L.E., Voogd N.J.D., Pires A.C., Polónia A.R., Egas C., Gomes N.C. Habitat-and host-related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiol. Ecol. 2013;85(3):465–482. doi: 10.1111/1574-6941.12135. [DOI] [PubMed] [Google Scholar]
- Erwin P.M., López-Legentil S., González-Pech R., Turon X. A specific mix of generalists: bacterial symbionts in Mediterranean Ircinia spp. FEMS Microbiol. Ecol. 2012;79(3):619–637. doi: 10.1111/j.1574-6941.2011.01243.x. [DOI] [PubMed] [Google Scholar]
- Giles E.C., Kamke J., Moitinho-Silva L., Taylor M.W., Hentschel U., Ravasi T., Schmitt S. Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 2013;83(1):232–241. doi: 10.1111/j.1574-6941.2012.01467.x. [DOI] [PubMed] [Google Scholar]
- Hentschel U., Fieseler L., Wehrl M., Gernert C., Steinert M., Hacker J., Horn M. Microbial diversity of marine sponges. Prog. Mol. Subcell. Biol. 2003;37:59–88. doi: 10.1007/978-3-642-55519-0_3. [DOI] [PubMed] [Google Scholar]
- Hentschel U., Schmid M., Wagner M., Fieseler L., Gernert C., Hacker J. Isolation and phylogenetic analysis of bacteria with antimicrobial activities from the Mediterranean sponges Aplysina aerophoba and Aplysina cavernicola. FEMS Microbiol. Ecol. 2001;35(3):305–312. doi: 10.1111/j.1574-6941.2001.tb00816.x. [DOI] [PubMed] [Google Scholar]
- Hentschel U., Usher K.M., Taylor M.W. Marine sponges as microbial fermenters. FEMS Microbiol. Ecol. 2006;55(2):167–177. doi: 10.1111/j.1574-6941.2005.00046.x. [DOI] [PubMed] [Google Scholar]
- Huse S.M., Huber J.A., Morrison H.G., Sogin M.L., Welch D.M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8(7):R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong I.H., Kim K.H., Park J.S. Analysis of bacterial diversity in sponges collected off Chujado, an island in Korea, using barcoded 454 pyrosequencing: analysis of a distinctive sponge group containing Chloroflexi. J. Microbiol. 2013;51(5):570–577. doi: 10.1007/s12275-013-3426-9. [DOI] [PubMed] [Google Scholar]
- Kamke J., Rinke C., Schwientek P., Mavromatis K., Ivanova N., Sczyrba A., Woyke T., Hentschel U. The candidate phylum Poribacteria by single-cell genomics: new insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS One. 2014;9(1):e87353. doi: 10.1371/journal.pone.0087353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamke J., Taylor M.W., Schmitt S. Activity profiles for marine sponge-associated bacteria obtained by 16S rRNA vs 16S rRNA gene comparisons. ISME J. 2010;4(4):498–508. doi: 10.1038/ismej.2009.143. [DOI] [PubMed] [Google Scholar]
- Kolde, R., 2012. Pheatmap: Pretty Heatmaps. R package version. 061.
- Lafi F.F., Fuerst J.A., Fieseler L., Engels C., Goh W.W.L., Hentschel U. Widespread distribution of Poribacteria in Demospongiae. Appl. Environ. Microbiol. 2009;75(17):5695–5699. doi: 10.1128/AEM.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee O.O., Wang Y., Yang J., Lafi F.F., Al-Suwailem A., Qian P.Y. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 2011;5(4):650. doi: 10.1038/ismej.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.Y., He L.M., Wu J., Jiang Q. Bacterial community diversity associated with four marine sponges from the South China Sea based on 16S rDNA-DGGE fingerprinting. J. Exp. Mar. Biol. 2006;329(1):75–85. [Google Scholar]
- Matobole R., van Zyl L., Parker-Nance S., Davies-Coleman M., Trindade M. Antibacterial activities of bacteria isolated from the marine sponges Isodictya compressa and Higginsia bidentifera collected from Algoa Bay, South Africa. Mar. Drugs. 2017;15(2):47. [Google Scholar]
- Price A.R., Jobbins G., Shepherd A.R.D., Ormond R.F. An integrated environmental assessment of the Red Sea coast of Saudi Arabia. Environ. Conserv. 1998;25(1):65–76. [Google Scholar]
- Radwan M., Hanora A., Zan J., Mohamed N.M., Abo-Elmatty D.M., Abou-El-Ela S.H., Hill R.T. Bacterial community analyses of two Red Sea sponges. Mar. Biotechnol. 2010;12(3):350–360. doi: 10.1007/s10126-009-9239-5. [DOI] [PubMed] [Google Scholar]
- Schmitt S., Angermeier H., Schiller R., Lindquist N., Hentschel U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 2008;74(24):7694–7708. doi: 10.1128/AEM.00878-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp K.H., Eam B., Faulkner D.J., Haygood M.G. Vertical transmission of diverse microbes in the tropical sponge Corticium sp. Appl. Environ. Microbiol. 2007;73(2):622–629. doi: 10.1128/AEM.01493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin M.L., Morrison H.G., Huber J.A., Welch D.M., Huse S.M., Neal P.R., Arrieta J.M., Herndl G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Natl. Acad. Sci. 2006;103(32):12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T., Moitinho-Silva L., Lurgi M., Björk J.R., Easson C., Astudillo-García C., Olson J.B., Erwin P.M., López-Legentil S., Luter H., Chaves-Fonnegra A. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turon M., Cáliz J., Garate L., Casamayor E.O., Uriz M.J. Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 2018;8(1):15201. doi: 10.1038/s41598-018-33545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayangani R.M.C., Dananjaya S.H.S., Nikapitiya C., Heo G.J., Lee J., De Zoysa M. Metagenomics analysis of gut microbiota and immune modulation in zebrafish (Danio rerio) fed chitosan silver nanocomposites. Fish Shellfish Immunol. 2017;66:173–184. doi: 10.1016/j.fsi.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Webster N.S., Luter H.M., Soo R.M., Botté E.S., Simister R.L., Abdo D., Whalan S. Same, same but different: symbiotic bacterial associations in GBR sponges. Front Microbiol. 2013;3:444. doi: 10.3389/fmicb.2012.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster N.S., Negri A.P., Munro M.M., Battershill C.N. Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 2004;6(3):288–300. doi: 10.1111/j.1462-2920.2004.00570.x. [DOI] [PubMed] [Google Scholar]
- Webster N.S., Wilson K.J., Blackall L.L., Hill R.T. Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl. Environ. Microbiol. 2001;67(1):434–444. doi: 10.1128/AEM.67.1.434-444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz J.B., Hentschel U., Lindquist N., Martens C.S. Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar Biol. 2007;152(2):475–483. [Google Scholar]
- White J.R., Patel J., Ottesen A., Arce G., Blackwelder P., Lopez J.V. Pyrosequencing of bacterial symbionts within Axinella corrugata sponges: diversity and seasonal variability. PloS one. 2012;7(6):e38204. doi: 10.1371/journal.pone.0038204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson C.R. Net primary productivity in coral reef sponges. Science. 1983;219(4583):410–412. doi: 10.1126/science.219.4583.410. [DOI] [PubMed] [Google Scholar]