

Fifty years after the first publication on Rett syndrome, Banerjee et al. review the molecular, cellular and circuit neurobiology of the disorder. They summarize recent advances in therapeutic interventions explored in preclinical models, as well as lessons learnt from past clinical trials and how these might inform future therapeutic approaches.
Keywords: development, synapse, plasticity, autism, disorders
Abstract
With the recent 50th anniversary of the first publication on Rett syndrome, and the almost 20 years since the first report on the link between Rett syndrome and MECP2 mutations, it is important to reflect on the tremendous advances in our understanding and their implications for the diagnosis and treatment of this neurodevelopmental disorder. Rett syndrome features an interesting challenge for biologists and clinicians, as the disorder lies at the intersection of molecular mechanisms of epigenetic regulation and neurophysiological alterations in synapses and circuits that together contribute to severe pathophysiological endophenotypes. Genetic, clinical, and neurobiological evidences support the notion that Rett syndrome is primarily a synaptic disorder, and a disease model for both intellectual disability and autism spectrum disorder. This review examines major developments in both recent neurobiological and preclinical findings of Rett syndrome, and to what extent they are beginning to impact our understanding and management of the disorder. It also discusses potential applications of knowledge on synaptic plasticity abnormalities in Rett syndrome to its diagnosis and treatment.
Introduction
Rett syndrome (OMIM 312750) is a severe and progressive neurodevelopmental disorder characterized by a wide range of neurologic and behavioural features. With an incidence of 1:10 000–15 000, Rett syndrome is the second most common cause of severe intellectual disability in females and, during its period of developmental regression, a substantial proportion of affected individuals meet diagnostic criteria for autism spectrum disorder (ASD) (Chahrour and Zoghbi, 2007; Guy et al., 2011; Percy, 2011; Banerjee et al., 2012; Katz et al., 2016; Leonard et al., 2016). Rett syndrome results in >90% cases from sporadic pathogenic mutations in the X-linked methyl-CpG-binding protein 2 gene (MECP2, human; Mecp2, mouse; located at Xq28). Other genes, most notably CDKL5 and FOXG1, have been associated with atypical and rarely classic forms of Rett syndrome (Neul et al., 2010; Sajan et al., 2017). While Rett syndrome has not been included as a separate entity within the framework of ASD in the Diagnostic and Statistical Manual of Mental Disorders – Fifth Edition (DSM-5) (American Psychiatric Association, 2013), discoveries following the identification of the link between Rett syndrome and MECP2 mutations have contributed towards deepening our understanding of the genetic and pathophysiological mechanisms of Rett syndrome and of the neurobiology of ASD and neurodevelopmental disorders in general. In this context, Rett syndrome has become one of the best disease models of abnormal synaptic plasticity. Thus, half a century after the first publication on Rett syndrome, and almost 20 years since the first report on MECP2 mutations in Rett syndrome, it gives us an opportunity to both look back at the long and winding road the field has travelled, lessons that have been learned, and how they have impacted our understanding and management of the disorder. We also discuss how our knowledge on synaptic plasticity abnormalities in Rett syndrome could improve diagnosis and treatment of the disorder.
Clinical features and genetics of Rett syndrome: a dynamic disorder associated with MECP2 mutations
Rett syndrome was originally described in German in 1966 by the Viennese paediatrician Andreas Rett (Rett, 1966, 2016). This initial description of 22 female children, which included the core features of the disorder mentioned below, was expanded in 1983 in an article in Annals of Neurology by the Swedish child neurologist Bengt Hagberg. This study, which reported 35 cases, established the first link between Rett syndrome and autistic features (Hagberg et al., 1983). Four clinical manifestations constitute the core diagnostic features of Rett syndrome: loss of expressive language; loss of fine motor (i.e. hand) skills; impairment in ambulation; and presence of hand stereotypic movements. These characteristic features, and others more variable in frequency and severity, emerge at different times during the dynamic course of Rett syndrome. After a relatively normal early postnatal period, head growth deceleration and global cognitive and motor delay become apparent and, typically between 1.5 and 3 years, variable loss of spoken language and hands skills develop. Sometimes, ambulation also regresses. Developmental regression is the distinctive diagnostic feature of Rett syndrome; although recovery of function is common, it is usually partial (Neul et al., 2010). Following loss of skills, other clinical manifestations emerge. They include, among others, breathing and other autonomic abnormalities, seizures, scoliosis, and abnormal muscle tone (that constitute supportive criteria for individuals with two or three core features, which are labelled as ‘atypical’) (Neul et al., 2010). Gastrointestinal and bone density anomalies, both related directly or indirectly to MECP2 deficiency, in conjunction with the abovementioned orthopaedic problems make Rett syndrome a multi-system disorder that requires multi-disciplinary management. The complex evolution of Rett syndrome continues after the relatively stable (‘pseudo-stationary’) post-regression period, when most of the associated features become evident, and extends into adolescence and adulthood, which are characterized by progressive decline, particularly in the motor domain (i.e. parkinsonian features could also be present at later stages). Behavioural problems can be obvious at different periods in the evolution of Rett syndrome (Buchanan et al., 2018). Autistic features are severe in a subset of individuals with Rett syndrome (∼20–50%) almost exclusively during the regression period, with longstanding deficits in social communication and interaction skills in only 10–15% of affected females (Neul et al., 2014; Percy and Glaze, 2017). The complex clinical course of Rett syndrome has been described in different ways, including the four stages proposed by Hagberg (2002), which suggest a continuous disruption in brain development and function. This profile, in conjunction with data discussed in the following sections, suggests a disruption in synaptic function.
From the aetiological viewpoint, a major breakthrough was the report in 1999 by Huda Zoghbi’s laboratory of the association between mutations in MECP2, a gene located at the Xq28 site, and most cases of Rett syndrome (Amir et al., 1999). Development of mouse models of Rett syndrome followed quickly, first from the laboratories of Adrian Bird and Rudolf Jaenisch (Chen et al., 2001; Guy et al., 2001), opening a floodgate of studies using these preclinical animal models. More recently, patient-derived induced pluripotent stem cell (iPSC) models of human neurons with and without mutations in MECP2 have provided powerful systems for discovering Rett syndrome mechanisms and potential therapeutics (Marchetto et al., 2010; Li et al., 2013; Mellios et al., 2017).
Rett syndrome is associated with more than 500 pathogenic or likely pathogenic MECP2 mutations involving mainly exons 3 and 4 and the C-terminal region (Gold et al., 2018). However, eight point MECP2 mutations, four missense and four nonsense (R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C); C-terminal deletions; and exonic deletions account for the vast majority of cases (Neul et al., 2008; Glaze et al., 2010). Mapping mutations in the crystal structure of the protein reveal that most of these mutations are located in three key domains: a methyl-binding domain (MBD), a transcriptional-repression domain (TRD), and nuclear localization signal (NLS) domain (Gold et al., 2018). While genotype–phenotype correlations are limited, some general profiles are recognized: a group with milder presentation and more protracted course, and a more severe group, including nonsense mutations affecting the transcriptional repression domain of MECP2 (Cuddapah et al., 2014). Intermediate severity MECP2 mutations have also been reported (Archer et al., 2007), while ∼5% of classical and 25% of variant Rett syndrome patients do not have mutations in the MECP2 (Neul et al., 2014; Gold et al., 2018).
Molecular function of MECP2: regulation of transcription
The fundamental mechanisms of MECP2 function have long been known, yet understanding phenotypic symptoms of MECP2 abnormality is complicated by its complex targets and by pleiotropic effects in target genes and signalling pathways (Ip et al., 2018). However, several recent studies have substantially clarified the mode of action of MECP2 and the effects of MECP2 mutations. MECP2 was originally identified based on its ability to bind methylated DNA, and was hence considered an epigenetic transcriptional repressor (Lewis et al., 1992; Lyst and Bird, 2015). It was subsequently described as a transcriptional activator or repressor, depending on the molecular context (Chahrour et al., 2008). Functionally, it is now clear that activity-dependent phosphorylation in specific residues of MECP2 mediates its interaction with several important transcriptional co-repressors including the NCoR/SMRT complex (Ebert et al., 2013). While several missense mutations in the MBD of MECP2 are associated with Rett syndrome and variable clinical severity, missense mutations (e.g. R306C) distal to the TRD, in the NCoR interaction domain (NID), abolish the interaction of MECP2 with the NCoR/SMRT co-repressor (Lyst et al., 2013), also causing a disruption in MECP2 function and Rett syndrome with a relatively milder phenotype (Cuddapah et al., 2014). The role of MECP2 in regulating BDNF levels, a major synaptic modulator and one of the first identified targets of MECP2, is an example of the complex mechanisms underlying synaptic activity and transcription. Adequate BDNF levels seem to be defined by brain regional, temporal, and functional state requirements. Whether the latter are reflected by BDNF plasma measurements is still unclear but worth exploring in the context of the development of new treatments (Li and Pozzo-Miller, 2014). During early life experiences, DNA methyltransferase (DNMT3A) transiently binds across transcribed regions of lowly expressed genes, specifying methylated CA (mCA) patterns on which MECP2 can further fine-tune long-lasting cell type-specific transcription via its repression effects (Stroud et al., 2017). Consistent with this pattern, MECP2 represses expression, especially of long mCA-enriched genes (Gabel et al., 2015). The effects of MECP2 seem to be genome-wide and gene body-dependent, rather than promoter/enhancer-specific, and subtle, but correlate well with DNA methylation density, affecting several brain-specific long genes (Kinde et al., 2016; Lagger et al., 2017; Stroud et al., 2017). A recent study reinforces the importance of the MBD and NID by showing that introduction of a truncated protein containing only the MBD and NID domains into MECP2-deficient mice reverses neurological symptoms (Tillotson et al., 2017). Although research on nonsense (truncating) MECP2 mutations has been more limited, it is critical in the context of the study by Tillotson et al. (2017). It is also important because mutations involving the TRD, particularly, the nuclear localization signal within this domain, are associated with the most severe Rett syndrome phenotypes (Cuddapah et al., 2014).
At the same time, several lines of evidence suggest additional mechanisms of MECP2 function. MECP2 is known to interact with 5-hydroxymethylcytosine (5hmC)-bearing DNA (Mellén et al., 2012) and can bind to hmCG, albeit with a lower affinity than hmCA (Kinde et al., 2016). 5hMC is associated with functional demethylation of expressed genes (Mellén et al., 2017), potentially enabling MECP2 to achieve de-repression of transcription. Cell type-specific biotin tagging of MECP2, combined with knock-in mice carrying Rett syndrome-associated mutations (R106W and T158M), reveals gene expression changes specific to each mutation and to excitatory or inhibitory cell types, including reductions in the transcription of long genes and post-transcriptional compensation at the cellular level (Johnson et al., 2017). Furthermore, MECP2 can bind to RNA and regulate alternative splicing and miRNA processing (Young et al., 2005). Importantly, early deficits in neuronal differentiation and migration in MECP2 mutant mice and in cerebral organoids derived from Rett syndrome patients arise from upregulation of miR-199 and miR-214, and their effects on extracellular signal-regulated kinase (ERK/MAPK) and protein kinase B (PKB/AKT) signalling (Mellios et al., 2017). MECP2 deficiency also misregulates miR-15a, affecting brain derived neurotrophic factor (BDNF) expression and dendritic morphogenesis (Gao et al., 2015).
Neurobiology of Rett syndrome: a disorder of plasticity at synapses and circuits
Since the early post-mortem characterizations of brain abnormalities in Rett syndrome, later expanded by in vivo neuroimaging, there has been strong evidence for a generalized dendritic and synaptic disorder (Kaufmann and Moser, 2000; Kaufmann et al., 2016). This work has been greatly enhanced by the availability of mouse models of Rett syndrome. Indeed, studies of synapses and neuronal subtypes were the initial focus for assessments of synaptic function in Mecp2 knockout mice (Banerjee et al., 2012; Zoghbi and Bear, 2012; Sahin and Sur, 2015). Global brain-wide activity mapping using immediate early gene c-Fos expression has revealed distinct functional deficits including decreased activity levels in key nodes of the default-mode network in forebrain and midbrain areas and hyperexcitability in hindbrain areas (Kron et al., 2012). Thus, the effects of Mecp2 deletion need to be examined keeping in mind spatio-temporal variations in development and function of different brain regions (Katz et al., 2016). Furthermore, the balance and spatiotemporal relationship between excitation and inhibition appear to be an important facet in Rett syndrome, instead of deficits of either excitation or inhibition alone.
The effects of MECP2 deficiency on both excitation and inhibition can be found on at least three different levels. The first level is anatomical, where increased perisomatic GABAergic terminals (Durand et al., 2012; Krishnan et al., 2015; but see He et al., 2014) and decreased excitatory projections (Sceniak et al., 2016, but see Li et al., 2016) have been found in the cortex and hippocampus. Second, at the level of specific synapses: while the potency of inhibitory synapses in the cerebral cortex of Mecp2 knockout mice appear not to be changed (Dani et al., 2005; Nelson et al., 2006; Wood et al., 2009), or decreased (Calfa et al., 2015) as measured with the amplitude of miniature inhibitory postsynaptic currents (mIPSC), there is no clear result on excitatory synapses. Increases (Calfa et al., 2015), decreases (Dani et al., 2005; Chao et al., 2007; Wood et al., 2009) or no change (Nelson et al., 2006) have all been reported for excitatory miniature excitatory postsynaptic currents (mEPSC). A reduction of sensory-evoked feedforward excitatory drive has generally been reported in Mecp2 knockout mice, and this reduction has also been shown to affect several neuronal subclasses including parvalbumin (parvalbumin-positive) expressing inhibitory interneurons (Banerjee et al., 2016). Brainstem circuits, on the other hand, shows reduced GABAergic inhibition and hyperexcitable expiratory neurons resulting in autonomic disturbances such as apnoeas and respiratory irregularities (Abdala et al., 2016). Altered cellular excitability and deficits in synaptic plasticity can, in turn, significantly affect higher-order processes such as impaired spatial and contextual memory formation (Kee et al., 2018). Third, the efficacy of synapses is affected by the internal status of the postsynaptic target neuron. At least one such mechanism in Mecp2 knockout mice and patients with Rett syndrome has been identified: decreased cross-membrane chloride gradient resulting from reduced potassium chloride co-transporter 2 (KCC2) expression alters the GABAAR reversal potential and reduces the potency and efficacy of GABAergic inhibition (Duarte et al., 2013; Banerjee et al., 2016).
Alterations in excitation-inhibition (E/I) balance have been shown to have consequences for cortical plasticity in neural circuits. Consistent with a reduction in inhibition received by pyramidal neurons, hence controlling the strength and spatial range of immature inhibition (Lo et al., 2017), monocular deprivation induced ocular dominance plasticity has been found to extend into adulthood in Mecp2 heterozygous female mice (Tropea et al., 2009; Castro et al., 2014). Parvalbumin-specific deletion in mice leads to immature adult visual cortical plasticity (Banerjee et al., 2016), which is rescued by enhancing inhibition via intracerebral infusion of the GABAA receptor agonist diazepam (He et al., 2014). Similarly, pup gathering behaviour and associated auditory cortical plasticity are also impaired in Mecp2 heterozygous female mice (Krishnan et al., 2017). The direction of changes in E/I balance favours excitation: intracellular measurements in cortical neurons reveal that whereas inhibition and excitation are both reduced in Mecp2 knockout mice, inhibition is reduced to a greater degree, thus enhancing the E/I ratio (Banerjee et al., 2016). Indeed, patients with Rett syndrome (Jian et al., 2007; Glaze et al., 2010; Cardoza et al., 2011) and Mecp2 knockout mice have reduced threshold and increased propensity for seizures, along with altered network oscillations (Calfa et al., 2011; McLeod et al., 2013; Goffin et al., 2014; Zhang et al., 2014). While some of the changes in Mecp2 knockout mice can be explained as results of homeostatic maladaptive compensation (Nelson and Valakh, 2015), deleting Mecp2 from parvalbumin-positive neurons alone causes reduction of inhibitory as well as excitatory drive to visual cortex neurons (Banerjee et al., 2016), similar to brain-wide deletion of Mecp2, indicating that reduced inhibition is almost certainly a primary effect of MECP2 deficiency in sensory cortices (cf.Robertson and Baron-Cohen, 2017).
One important way the field has advanced is in going beyond simple synaptic measurements in ex vivo brain slices and probing alterations in circuits in intact animals in vivo. Some of these mechanisms can be used as a new biological variable to study pathophysiological mechanisms contributing to the maladaptive processes in Rett syndrome. In these studies, sensory modalities have served as a window to provide novel and fundamental insights into biological processes of pathophysiology of Rett syndrome (Robertson and Baron-Cohen, 2017). Deficits in maturation of cell type-specific inhibition have emerged as an important facet of the circuit defects in Rett syndrome. Loss of Mecp2 from forebrain GABAergic neurons recapitulates diverse and prominent features of Rett syndrome (Chao et al., 2010). Interestingly, GABAergic neuron-specific conditional knockout leads to a wide range of symptoms similar to global-knockout of Mecp2 (Ure et al., 2016), whereas excitatory neuron-specific conditional deletion leads to milder, and specific symptoms, namely, stereotypies and social anxiety (Meng et al., 2016). Furthermore, interneuron-specific re-expression of Mecp2 has been shown to ameliorate some of the deficits seen in Rett syndrome (Goffin et al., 2014; Ure et al., 2016). Deconstructing MECP2 deficit in specific GABAergic neuronal populations using cell-specific manipulations resulted in parsing out these non-overlapping neurological features in parvalbumin-positive or somatostatin expressing interneurons (Ito-Ishida et al., 2015). Behaviourally, parvalbumin-specific conditional Mecp2 knockout mice show deficits in sensory, social, and motor dysfunction, whereas somatostatin conditional knockout mice show seizure and stereotyped behaviour (Ito-Ishida et al., 2015).
Using cell-specific conditional mutant Rett syndrome mouse models, sensitive assays have been developed to study neural signatures of sensory perception. Deletion of Mecp2 from a small population of parvalbumin-positive and not somatostatin-positive neurons alone recapitulate effects of global Mecp2 deletion on pyramidal neurons, indicating parvalbumin-positive neurons play a crucial role in propagating the effect of MECP2 deficit in a non-cell autonomous manner to a distributed circuit affecting cortical coding in mouse primary visual cortex (V1). Deleting Mecp2 globally or from parvalbumin-positive neurons causes a significant increase in response variability, reduced signal-to-noise ratio, and a reduction in response reliability to natural visual scene, significantly altering network correlation structure in V1 (Banerjee et al., 2016). Interestingly, loss- and gain-of-function mutations in Mecp2 are thought to underlie two distinct neurological syndromes yet both have strikingly similar phenotypic features in animal models (Lombardi et al., 2015), and Rett syndrome and MECP2 duplication syndrome share many neurobehavioural features (Neul et al., 2010; Van Esch, 2012). However, limited attempts have been made so far to understand the exact nature of underlying mechanisms leading to these changes (Lim et al., 2017). Altered visual processing has also been found in a mouse model of MECP2 duplication syndrome (MECP2 Tg1) showing higher visual acuity and contrast sensitivity at 8 and 14 weeks (Zhang et al., 2017). Mecp2 overexpressing mice show abnormally elevated synchrony in the firing rate of hippocampal CA1 pyramidal neurons, an impaired homeostatic response to perturbations of E/I balance, and decreased excitatory input onto inhibitory neurons (Lu et al., 2016). Mice overexpressing Mecp2 also demonstrate altered structural plasticity, including altered stability of dendritic spine clusters and enhanced motor learning (Jiang et al., 2013; Ash et al., 2018).
Although the data reviewed here emphasize the individual neuron and circuit bases of MECP2 deficiency and Rett syndrome, contributions from astrocytes and, perhaps also, microglia should not be overlooked. Their role in regulating glutamate levels and other aspects of synaptic function has already been postulated as a mechanism underlying Rett syndrome (Kaufmann et al., 2016). The ability to gain insight into glial abnormalities through in vivo neuroimaging (Horská et al., 2009) also make glia an important target for the development of synaptic biomarkers in Rett syndrome.
Despite our detailed understanding of synaptic alterations in Rett syndrome, many questions still remain about abnormalities in synaptic plasticity in Rett syndrome and MECP2 deficiency. Do the initial synaptic deficits become less modifiable or enhanced by inefficient compensatory processes? What is the timeline of these processes in humans with MECP2 deficiency? Additionally, is it possible to improve E/I imbalances of different nature in different brain regions (i.e. cortical hypoexcitability, brainstem hyperexcitability) using the same therapeutic strategy?
Management of Rett syndrome: current practices and development of new treatments
Considering the multi-systemic involvement in Rett syndrome, a coordinated multidisciplinary approach to medical care and management is the preferred option. At an early age, shortly after the diagnosis, intensive early intervention as applied to other neurodevelopmental disorders is recommended (Warren et al., 2011). Recently, this approach has received empirical support from a recent environmental enrichment trial consisting of motor learning and exercise, combined with social, cognitive and sensory enrichment. The 6-month study on 12 females with Rett syndrome, under age 6, resulted in improvements in gross motor function and increased plasma BDNF levels but no changes in growth, sleep, or behaviour (Downs et al., 2018). Currently, there are no specific treatments for Rett syndrome; therefore, medical management is symptomatic. In addition to traditional drug treatments [e.g. antiepileptic drugs for seizures, selective serotonin reuptake inhibitors (SSRIs) for anxiety], preventive approaches that include nutritional management, with emphasis on caloric intake and vitamin D levels, prevention of gastrointestinal and orthopaedic complications, and personalized rehabilitation therapies, are becoming standards of care. For details on management, see corresponding chapters in Kaufmann et al. (2017).
The last few years have been a very active period for clinical trials in Rett syndrome. In contrast with previous studies, this new era attempts to take advantage of the neurobiology of the disorder. Specifically, most trials have been based on proof-of-principle research in Mecp2-deficient mice. A few studies have focused on a specific neurotransmitter or circuit [preliminary positive results in preliminary study of dextromethorphan, an NMDA receptor antagonist, mainly for cognition and seizures (Smith-Hicks et al., 2017); negative outcome of desipramine, a noradrenaline reuptake inhibitor, for breathing abnormalities (Mancini et al., 2018); ongoing study of sarizotan, a serotonin 1A receptor agonist, for breath holding (NCT02790034)]. Completed trials have targeted more general mechanisms, specifically IGF-1 function, and they have included both full-length IGF-1 (i.e. mecasermin) and a modified IGF-1 active N-terminus peptide (i.e. trofinetide) (Khwaja et al., 2014; Glaze et al., 2017; Neuren Pharmaceuticals, 2017; O’Leary et al., 2018). While the outcome of the mecasermin trials has been mixed, suggesting selective (e.g. communication) or not long-lasting effects, the two phase 2 trofinetide trials have been labelled as positive. The paediatric study, which showed more robust outcomes, demonstrated global but variable improvements that were more prominent in the behavioural domain. Other planned trials, to begin during the next year, will test new classes of drugs with presumed generalized neural function. These drugs with the potential of, at least, optimizing function and facilitating intrinsic compensatory mechanisms include a sigma 1 receptor agonist, a modulator of mitochondrial function and neuronal homeostasis (Anavex Life Sciences Corp., 2018); and cannabidiol (CBD), a compound isolated from cannabis with broad neurobehavioural effects but without psychoactive action (GW Pharmaceuticals, 2018). The next trofinetide trial, the first ever phase 3 (pivotal) study looking for a disorder-specific drug approval in Rett syndrome, will also begin in the next year (Acadia Pharmaceuticals and Neuren Pharmaceuticals, 2018).
Completing this general overview, it is important to note the first gene (MECP2) therapy trial in Rett syndrome (Rett Syndrome Research Trust, 2018) was modelled after the successful (i.e. improved survival and motor function) single dose intravenous adeno-associated virus serotype 9 delivery of complementary DNA (encoding the SMN protein) in spinal muscular atrophy type 1 (Mendell et al., 2017). Despite these exciting developments, the Rett syndrome field is experiencing the same challenges as other neurodevelopmental disorders pursuing neurobiologically-based treatments: inadequate outcome measures and markers of response (Katz et al., 2016; Kaufmann et al., 2016; Budimirovic et al., 2017). To this critical shortcoming must be added the need for innovative study designs that can monitor safety and efficacy in dynamic ways at early stages of drug development. This is particularly important because preclinical work with mouse and other experimental models has been, in general, unable to point out specific symptoms and signs of Rett syndrome and related disorders to be improved by a particular drug (Kaufmann et al., 2016; Berry-Kravis et al., 2018). The heterogeneity of Rett syndrome, manifested as variable severity of common symptoms, leads to variable positive responses (e.g. in some individuals greater decrease in disruptive behaviours; in others, greater improvement in non-verbal communication), as demonstrated in the recent mecasermin and trofinetide trials discussed above. How to capture these beneficial effects at every step of the drug development pathway is a major concern in Rett syndrome trials. (For details about clinical trials in Rett syndrome, including outcome measure and study design issues, see Katz et al., 2016; Kaufmann et al., 2016.)
Disrupted synaptic plasticity: implications for diagnosis and treatment of Rett syndrome
The preceding sections have summarized evidence supporting the notion that Rett syndrome is a disorder of synaptic function and, more specifically, of synaptic plasticity. Many specific issues, including to what extent maladaptive synaptic responses could exacerbate or perpetuate initial deficits, remain unclear. More important is the extent to which data from animal studies with MECP2 deficiency could be extended to individuals with Rett syndrome and to which particular stage of the disease or clinical manifestation. Despite all these uncertainties, approaching Rett syndrome as a synaptic plasticity disorder may have practical implications.
To date the diagnosis of Rett syndrome is still clinical and based predominantly on the history of developmental regression, with the associated challenge of caregivers’ recollections. Presence of MECP2 mutations is supportive, but not confirmatory because of the limited genotype–phenotype correlations in Rett syndrome (Neul et al., 2014). Females with MECP2 mutations may present with a wide range of phenotypes other than Rett syndrome, without necessarily loss of skills (Neul et al., 2010). Early neurophysiological studies indicate that visual evoked potential (N1-P1) amplitude in Rett syndrome is decreased in individuals during the post-regression period and suggest that this change evolves during regression (LeBlanc et al., 2015). Although electrophysiological markers of brain abnormalities prior to or during regression could be more informative and lead to early interventions, these data show the promise of synaptic biomarkers as complementary diagnostic tools. Other examples of potential biomarkers of disease stage or progression are the levels of N-acetylaspartate (NAA) and myoinositol, magnetic resonance spectroscopy neuronal and glial markers, respectively. While the former decreases with respect to control levels in Rett syndrome beginning at age 5–6 years, the latter shows a continuous increase from early childhood (Horská et al., 2009). Thus, NAA levels could become a reliable marker of the pseudo-stationary post-regression period, characterized by appearance of seizures and autonomic disturbances. On the other hand, myoinositol levels would reflect the degree of glial abnormalities associated with synaptic disruption. Biomarkers of disease progression may have prognostic relevance and assist in deciding on more aggressive interventions.
The natural history of Rett syndrome, in conjunction with our knowledge on synaptic abnormalities, indicate that age, and more importantly disease stage, are key factors when designing drug trials. The recently reported early intervention trial (Downs et al., 2018) suggests that intense and prolonged sensory-motor stimulation can improve motor function, when instituted shortly after diagnosis and up to the early post-regression period. These data are in correspondence with the timing of spontaneous but partial recovery of spoken language and hand skills observed in most patients with Rett syndrome (Hagberg, 2002). Whether brain responsiveness to stimulation extends beyond this stage in Rett syndrome is unknown; however, these data raise the possibility of enhancing or complementing drug effects by combining drug administration with sensory-motor stimulation. Although most clinical trials aim at testing drugs as early as possible in the evolution of Rett syndrome, safety and logistical considerations determine that some of them will only enrol older children, teens, or adults. Since severity of symptoms, including breath-holding, seizures, and motor dysfunction, is particularly high in late childhood and adolescence (Tarquinio et al., 2017, 2018), combination of drugs with non-pharmacological therapies or other interventions for overcoming decreased or aberrant plasticity seems to be compelling. The experience with the GABAB agonist arbaclofen in fragile-X syndrome suggests age-dependent efficacy with greater efficacy in childhood than at later ages (Berry-Kravis et al., 2017). While using different designs, the paediatric and adolescence/adult trofinetide trials in Rett syndrome suggest similar differences.
Figure 1.
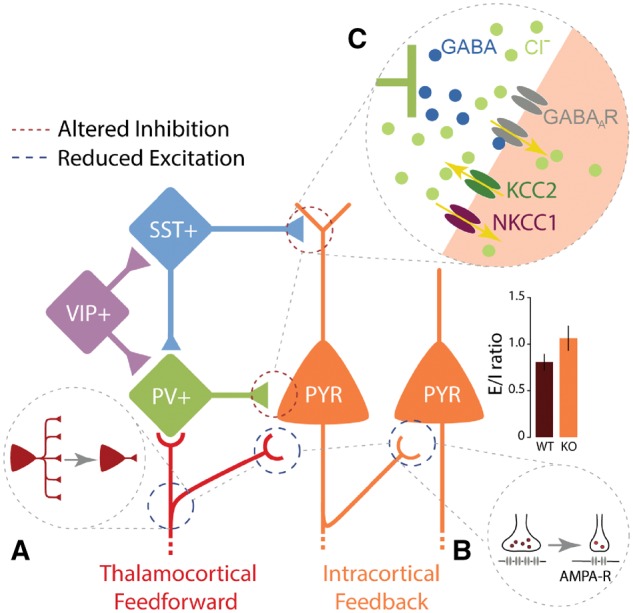
Altered synapse-specific computation in Rett syndrome. While different synapses in distinct parts of the brain are differentially modulated upon loss of MECP2, three essential ideas capture synaptic-specific changes that alter excitatory and inhibitory (E/I) balance and increase E/I ratio in animal models of Rett syndrome. (A) A significant reduction of thalamocortical feedforward excitatory drive to supragranular excitatory pyramidal neurons (PYR) and inhibitory parvalbumin (PV+) interneurons in the cortex has been found (Banerjee et al., 2016). (B) Weakening of excitatory drive could be resulted from a long-term depression (LTD) like mechanism at glutamatergic synapses (thalamocortical as well as intracortical), upon a downregulation of AMPA-type glutamate receptors (AMPA-R). A third mechanism (C) is a reduction of inhibitory currents can be due to altered GABAA receptor (GABAAR) mediated reversal potential in excitatory neurons. This is caused by a developmental delay in the maturation of GABAergic inhibition and a reduction in chloride exporter KCC2 expression (Banerjee et al., 2016). SST = somatostatin; VIP = vasoactive intestinal polypeptide.
Important seminal work in Rett syndrome animal models demonstrating the possibility to achieve prolonged survival and reversibility of disease phenotypes with gene reinstatement, even into adulthood, seemingly makes Rett syndrome one of the more tractable neurodevelopmental disorders as far as potential for disease modification and improvement (Guy et al., 2007; Gadalla et al., 2017). Viral gene therapy approaches can potentially rescue a radically truncated version of MECP2 that has recently been shown to be able to reverse neurological symptoms (Tillotson et al., 2017). The planned gene delivery trial, the first attempt at correcting the primary defect in Rett syndrome, raises the key question of lower age limit for major reversal or prevention of the phenotype. Taking into consideration that in the USA, the median age of diagnosis is 2.7 years for classic Rett syndrome and 3.8 years for atypical Rett syndrome, would newborn MECP2 mutation screening be necessary for successful corrective interventions? Alternatively, will it be possible to reverse most abnormal synaptic plasticity already established by the time of diagnosis? A final consideration about the ‘double jeopardy’ effect of MECP2 deficit (Kaufmann et al., 2005) is that, even if abnormal developmental plasticity is not ameliorated, the need of MECP2 for adult learning and memory indicates that it is never too late for attempting to improve neurological function in Rett syndrome.
Conclusion
It has been more than 50 years since the discovery of Rett syndrome, yet the current standard of care for patients remains limited to supportive and symptomatic therapies. Drug treatment consists mainly of off-label prescriptions due to the lack of approved medications for the disorder. However, with the current collaborative efforts of academic investigators, patient organizations and advocacy groups, and industry partners, the future of clinical trials in Rett syndrome looks promising, trending towards probing disease-modifying therapies and rational, innovative clinical trial designs. The availability of multiple treatment options is encouraging since, most likely, not all individuals with Rett syndrome will respond to the same degree to different drugs. Our review also argues for an essential role of MECP2 in the development and maintenance of neuronal circuits, and its specific role in distinct cellular subtypes. Understanding this neurobiology is crucial in enhancing the effectiveness of animal models in designing robust and reproducible preclinical translational studies. Taking into consideration synaptic abnormalities in Rett syndrome is also key for the design and interpretation of drug and non-pharmacological trials, as well as for the development of strong clinical biomarkers.
Funding
This work was supported by a Marie Skłodowska-Curie Fellowship (CIRCDYN) and a NARSAD Young Investigator award from the Brain & Behavior Research Foundation (A.B.), NIH grant MH085802 and the Simons Foundation Autism Research Initiative through the Simons Center for the Social Brain (M.S.) and NIH grant U54 HD061222 (W.E.K.) and the Rettsyndrome.org foundation (W.E.K., M.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
In 2018, W.E.K. was a consultant to Anavex, AveXis, EryDel, GW Pharmaceuticals, Neuren/Acadia, Newron Pharmaceuticals, Ovid, Stalicla, and Zynerba. As of January 1, 2019, W.E.K. is the Chief Medical Officer of Anavex Life Sciences Corp.
References
- Abdala AP, Toward MA, Dutschmann M, Bissonnette JM, Paton JFR. Deficiency of GABAergic synaptic inhibition in the Kölliker-Fuse area underlies respiratory dysrhythmia in a mouse model of Rett syndrome. J Physiol 2016; 594: 223–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acadia Pharmaceuticals and Neuran Pharmaceuticals. ACADIA Pharmaceuticals and Neuren Pharmaceuticals announce exclusive License Agreement for the North American development and commercialization of Trofinetide in Rett syndrome. 2018. www.neurenpharma.com/irm/PDF/1759_0/NeurenandACADIAannounceagreementforNorthAmerica (29 August 2018, date last accessed).
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185–8. [DOI] [PubMed] [Google Scholar]
- Anavex Life Sciences Corp. Anavex Life Sciences to initiate Phase 2 study of ANAVEX®2–73 in Parkinson’s disease Dementia and provides clinical study update for ANAVEX®2–73 in Rett syndrome. 2018. https://www.anavex.com/anavex-life-sciences-initiate-phase-2-study-anavex2–73-parkinsons-disease-dementia-provides-clinical-study-update-anavex2–73-rett-syndrome/ (29 August 2018, date last accessed).
- Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J et al. Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J Med Genet 2007; 44: 148–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash RT, Fahey PG, Park J, Zoghbi HY, Smirnakis SM. Increased axonal bouton stability during learning in the mouse model of MECP2 duplication syndrome. eNeuro 2018. doi: 10.1523/ENEURO.0056-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Castro J, Sur M. Rett syndrome: genes, synapses, circuits, and therapeutics. Front Psychiatry 2012; 3: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K et al. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci USA 2016; 113: E7287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodev Disord 2017; 9: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov 2018;17:280–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan CB, Stallworth JL, Scott AE, Glaze DG, Lane JB, Skinner SA et al. Behavioral profiles in Rett syndrome: data from the natural history study. Brain Dev 2018. doi: 10.1016/j.braindev.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budimirovic DB, Berry-Kravis E, Erickson CA, Hall SS, Hessl D, Reiss AL et al. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J Neurodev Disord 2017; 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfa G, Li W, Rutherford JM, Pozzo-Miller L. Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus 2015; 25: 159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfa G, Percy AK, Pozzo-Miller L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp Biol Med 2011; 236: 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoza B, Clarke A, Wilcox J, Gibbon F, Smith PEM, Archer H et al. Epilepsy in Rett syndrome: association between phenotype and genotype, and implications for practice. Seizure 2011; 20: 646–9. [DOI] [PubMed] [Google Scholar]
- Castro J, Garcia RI, Kwok S, Banerjee A, Petravicz J, Woodson J et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett syndrome. Proc Natl Acad Sci USA 2014; 111: 9941–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008; 320: 1224–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron 2007; 56: 422–37. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010; 468: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007; 56: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001; 27: 327–31. [DOI] [PubMed] [Google Scholar]
- Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet 2014; 51: 152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA 2005; 102: 12560–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J, Rodger J, Li C, Tan X, Hu N, Wong K et al. Environmental enrichment intervention for Rett syndrome: an individually randomised stepped wedge trial. Orphanet J Rare Dis 2018; 13: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte ST, Armstrong J, Roche A, Ortez C, Perez A, O’Callaghan Mdel M et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One 2013; 8: e68851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A et al. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 2012; 76: 1078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S et al. Activity-dependent phosphorylation of MECP2 threonine 308 regulates interaction with NcoR. Nature 2013; 499: 341–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015; 522: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KKE, Vudhironarit T, Hector RD, Sinnett S, Bahey NG, Bailey MES et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther Methods Clin Dev 2017; 5: 180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Su J, Guo W, Polich ED, Magyar DP, Xing Y et al. Inhibition of miR-15a promotes BDNF expression and rescues dendritic maturation deficits in MeCP2-deficient neurons. Stem Cells 2015; 33: 1618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaze DG, Neul JL, Percy A, Feyma T, Beisang A, Yaroshinsky A et al. A double-blind, randomized, placebo-controlled clinical study of Trofinetide in the treatment of Rett syndrome. Pediatr Neurol 2017; 76: 37–46. [DOI] [PubMed] [Google Scholar]
- Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO et al. Epilepsy and the natural history of Rett syndrome. Neurology 2010; 74: 909–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Brodkin ES, Blendy JA, Siegel SJ, Zhou Z. Cellular origins of auditory event-related potential deficits in Rett syndrome. Nat Neurosci 2014; 17: 804–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold WA, Krishnarajy R, Ellaway C, Christodoulou J. Rett syndrome: a genetic update and clinical review focusing on comorbidities. ACS Chem Neurosci 2018; 9: 167–76. [DOI] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 2011; 27: 631–52. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007; 315: 1143–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 2001; 27: 322–6. [DOI] [PubMed] [Google Scholar]
- GW Pharmaceuticals. GW Pharmaceuticals to report third quarter financial results and host conference call on 7 August, 2018. 2018. http://ir.gwpharm.com/news-releases/news-release-details/gw-pharmaceuticals-plc-reports-fiscal-third-quarter-2018 (29 August 2018, date last accessed).
- Hagberg B. Clinical manifestations and stages of rett syndrome. Ment Retard Dev Disabil Res Rev 2002; 8: 61–5. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol 1983; 14: 471–9. [DOI] [PubMed] [Google Scholar]
- He LJ, Liu N, Cheng TL, Chen XJ, Li YD, Shu YS et al. Conditional deletion of Mecp2 in Parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat Commun 2014; 5: 5036. [DOI] [PubMed] [Google Scholar]
- Horská A, Farage L, Bibat G, Nagae LM, Kaufmann WE, Barker PB et al. Brain metabolism in Rett syndrome: age, clinical, and genotype correlations. Ann Neurol 2009; 65: 90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 2018; 19: 368–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in Parvalbumin-and Somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 2015; 88: 651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Paediatr Neurol 2007; 11: 310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Ash RT, Baker SA, Suter B, Ferguson A, Park J et al. Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci 2013; 33: 19518–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Zhao Y-T, Fasolino M, Lamonica JM, Kim YJ, Georgakilas G et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med 2017; 23: 1203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM, Bird A, Coenraads M, Gray SJ, Menon DU, Philpot BD et al. Rett syndrome: crossing the threshold to clinical translation. Trends Neurosci 2016; 39: 100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Johnston MV, Blue ME. MeCP2 expression and function during brain development: implications for Rett syndrome’s pathogenesis and clinical evolution. Brain Dev 2005; 27 (Suppl 1): S77–S87. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 2000; 10: 981–91. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Percy AK, Clarke A, Leonard H, Naidu S. Rett syndrome. London: Mac Keith Press; 2017. [Google Scholar]
- Kaufmann WE, Stallworth JL, Everman DB, Skinner SA. Neurobiologically-based treatments in Rett syndrome: opportunities and challenges. Expert Opin Orphan Drugs 2016; 4: 1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee SE, Mou X, Zoghbi HY, Ji D. Impaired spatial memory codes in a mouse model of Rett syndrome. Elife 2018; 7: pii: e31451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja OS, Ho E, Barnes KV, O’Leary HM, Pereira LM, Finkelstein Y et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci USA 2014; 111: 4596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Wu DY, Greenberg ME, Gabel HW. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc Natl Acad Sci USA 2016; 113: 15114–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K, Lau BYB, Ewall G, Huang ZJ, Shea SD. MECP2 regulates cortical plasticity underlying a learned behaviour in adult female mice. Nat Commun 2017; 8: 14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K, Wang BS, Lu J, Wang L, Maffei A, Cang J et al. MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc Natl Acad Sci USA 2015; 112: E4782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with Ketamine treatment. J Neurosci 2012; 32: 13860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017; 13: e1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JJ, DeGregorio G, Centofante E, Vogel-Farley VK, Barnes K, Kaufmann WE et al. Visual evoked potentials detect cortical processing deficits in Rett syndrome. Ann Neurol 2015; 78: 775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol 2016; 13: 37–51. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992; 69: 905–14. [DOI] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014; 76: 737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu X, Pozzo-Miller L. Excitatory synapses are stronger in the hippocampus of Rett syndrome mice due to altered synaptic trafficking of AMPA-type glutamate receptors. Proc Natl Acad Sci USA 2016; 113: E1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013; 13: 446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Z, Downs J, Wong K, Ellaway C, Leonard H. Expanding the clinical picture of the MECP2 duplication syndrome. Clin Genet 2017; 91: 557–63. [DOI] [PubMed] [Google Scholar]
- Lo SQ, Sng JCG, Augustine GJ. Defining a critical period for inhibitory circuits within the somatosensory cortex. Sci Rep 2017; 7: 7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi LM, Baker SA, Zoghbi HY. MECP2 disorders: from the clinic to mice and back. J Clin Invest 2015; 125: 2914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Ash RT, He L, Kee SE, Wang W, Yu D et al. Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a Rett syndrome mouse model. Neuron 2016; 91: 739–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 2015; 16: 261–75. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci 2013; 16: 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini J, Dubus J-C, Jouve E, Roux J-C, Franco P, Lagrue E et al. Effect of desipramine on patients with breathing disorders in Rett syndrome. Ann Clin Transl Neurol 2018; 5: 118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010; 143: 527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod F, Ganley R, Williams L, Selfridge J, Bird A, Cobb SR. Reduced seizure threshold and altered network oscillatory properties in a mouse model of Rett syndrome. Neuroscience 2013; 231: 195–205. [DOI] [PubMed] [Google Scholar]
- Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N, Kim A et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012; 151: 1417–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellén M, Ayata P, Heintz N. 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc Natl Acad Sci USA 2017; 114: E7812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 2017; 23: 1051–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017; 377: 1713–22. [DOI] [PubMed] [Google Scholar]
- Meng X, Wang W, Lu H, He L, Chen W, Chao ES et al. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 2016; 5: pii: e14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol 2006; 16: 710–6. [DOI] [PubMed] [Google Scholar]
- Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015; 87: 684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO et al. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology 2008; 70: 1313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68: 944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Lane JB, Lee H-S, Geerts S, Barrish JO, Annese F et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodev Disord 2014; 6: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuren Pharmaceuticals. Neuren’s Phase 2 trial of trofinetide demonstrates significant clinical benefit in pediatric Rett syndrome. Neuren (NEU) - ASX Announcement 22 March 2017. Available from: www.rettsyndrome.org/document.doc?id=574 (29 August 2018, date last accessed).
- O’Leary HM, Kaufmann WE, Barnes KV, Rakesh K, Kapur K, Tarquinio DC et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol 2018; 5: 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy AK. Rett syndrome. Arch Neurol 2011; 68: 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy AK, Glaze D. The natural history of Rett syndrome: Building on recent experience In: Kaufmann WE, editor. Rett Syndrome. London: Mac Keith Press; 2017. p. 14–23. [Google Scholar]
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr 1966; 116: 723–6. [PubMed] [Google Scholar]
- Rett A. On a remarkable syndrome of cerebral atrophy associated with hyperammonaemia in childhood. Wien Med Wochenschr 2016; 166: 322–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rett Syndrome Research Trust. AveXis reports on Rett gene therapy program: AVXS-201. 2018. reverserett.org/avexis-reports-rett-gene-therapy-program-avxs-201.
- Robertson CE, Baron-Cohen S. Sensory perception in autism. Nat Rev Neurosci 2017; 18: 671–84. [DOI] [PubMed] [Google Scholar]
- Sahin M, Sur M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science; 2015. doi: 10.1126/science.aab3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan SA, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR, Glaze DG et al. Genet Med 2017; 19: 13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Lang M, Enomoto AC, James Howell C, Hermes DJ, Katz DM. Mechanisms of functional hypoconnectivity in the medial prefrontal cortex of Mecp2 null mice. Cereb Cortex 2016; 26: 1938–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Hicks CL, Gupta S, Ewen JB, Hong M, Kratz L, Kelley R et al. Randomized open-label trial of dextromethorphan in Rett syndrome. Neurology 2017; 89: 1684–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD et al. Early-life gene expression in neurons modulates lasting epigenetic states. Cell 2017; 171: 1151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarquinio DC, Hou W, Berg A, Kaufmann WE, Lane JB, Skinner SA et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain 2017; 140: 306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarquinio DC, Hou W, Neul JL, Berkmen GK, Drummond J, Aronoff E et al. The course of awake breathing disturbances across the lifespan in Rett syndrome. Brain Dev 2018; 40: 515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D et al. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 2017; 550: 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA 2009; 106: 2029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure K, Lu H, Wang W, Ito-Ishida A, Wu Z, He L et al. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. Elife 2016; 5: pii: e14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H. MECP2 duplication syndrome. Mol Syndromol 2012; 2: 128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren Z, McPheeters ML, Sathe N, Foss-Feig JH, Glasser A, Veenstra-VanderWeele J. A systematic review of early intensive intervention for autism spectrum disorders. Pediatrics 2011; 127: e1303–11. [DOI] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci 2009; 29: 12440–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci USA 2005; 102: 17551–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Yu B, Liu J, Jiang W, Xie T, Zhang R et al. Altered visual cortical processing in a mouse model of MECP2 duplication syndrome. Sci Rep 2017; 7: 6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Peterson M, Beyer B, Frankel WN, Zhang ZW. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J Neurosci. 2014; 34: 2754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012; 4. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]