

Abstract
Here we disclose a cascade strategy for naphthyne formation that capitalizes on the traditional benzyne generation (i.e., from an ortho-silyl aryl triflate) and the thermal hexadehydro-Diels–Alder (HDDA) reaction. In this transformation, three distinct aryne species work in tandem, two of which can be formally considered as a 1,2-benzidyne, and each undergoes a different type of trapping event. Many examples were explored by varying the naphthyne capture chemistry as well as the 1,2-benzdiyne equivalent. This strategy enables rapid construction of various naphthalene products and has potential for the synthesis of extended polycyclic arenes.
Graphical Abstract
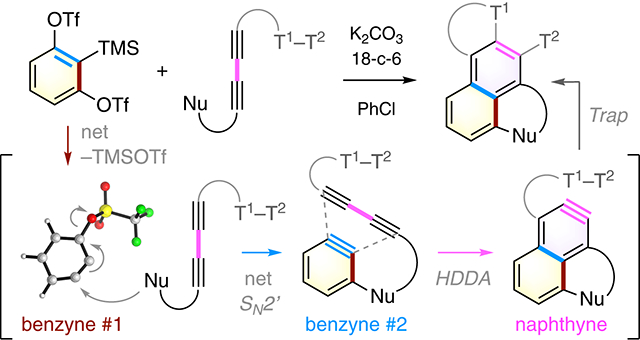
The development of mild and versatile methods for generation of arynes has enabled myriad synthetic applications of these long-standing reactive intermediates.1 The fluoride ion induced aryne formation from ortho-silyl aryl triflates (the Kobayashi protocol; cf. 1 to 2, Figure 1a) is perhaps the most enabling of the classical methods that has fueled the advances in modern aryne chemistry.2 The thermal generation of benzynes from, minimally, a triyne substrate like 3 represents a complementary strategy (Figure 1a).3 This so-called hexadehydro-Diels–Alder (HDDA) reaction also has considerable generality.4 In the work reported here, we have developed a strategy that capitalizes on both the Kobayashi protocol and the HDDA reaction to conveniently generate naphthynes, arynes of greater structural complexity, from relatively simple building blocks.
Figure 1.

Marrying of classical and HDDA benzyne generation to produce naphthyne 13. Ts = p-toluenesulfonyl. Tf = trifluoromethylsulfonyl. TMS = trimethylsilyl
Two key design principles drove our study. First, Li and coworkers have pioneered the use of 1,2-benzdiyne equivalent 4 (Figure 1b).5 Upon treatment with a silaphilic nucleophile/base (e.g., fluoride or carbonate ion), benzyne 5 can be generated in which substituent X can both direct nucleophilic addition to the meta-position and serve as a leaving group to regenerate a second benzyne. This net SN2’ reaction to form 6 (an example in which a thioamide has served as the initial benzyne trapping nucleophile) is followed by intramolecular capture by the amide nitrogen atom to produce the benzothiazole 7. Second, arynes have been shown to function as diynophiles in HDDA reactions. For example, benzyne 8 can engage with a 1,3-diyne unit to form naphthyne 9 (Figure 1c).6 This process can be extended to form higher homologs, including a tetracyne, 10.7 Therefore, we hypothesized that nucleophile 11, bearing a pendant 1,3-diyne moiety, attacks the 1,2-benzdiyne equivalent 5 to give the benzyne 12 (Figure 1d). Subsequent HDDA reaction produces naphthyne 13, which is then trapped to form product 14. Notably, this cascade involves three distinct reactive benzyne species, each of which needs to undergo a different type of transformation [cf. maroon (1,2-benzdiyne #1) vs. blue (1,2-benzdiyne #2) vs. magenta (HDDA naphthyne), throughout].
We opted to test the feasibility of this idea using 4-Tf and diyne 15 bearing a sulfonamide functional group, a nucleophile demonstrated in Li’s work to be an effective participant in the capture of the initial benzyne generated from this 1,2-benzdiyne equivalent (Figure 2).5 Formation of benzyne 5-Tf was expected to be the rate-limiting step due to the relatively short lifetime of arynes. The DFT optimized geometry for 5-Tf [SMD(chlorobenzene)/B3LYP/6–31G**] indicated that the triflate group renders appreciable distortion (116° vs. 137°) of the two sp-hybridized benzyne carbons. As a result, the sulfonamide anion 17 should selectively add to carbon b to form the transient aryl anion 18.8 Elimination of the triflate anion would then produce a second benzyne 19 as long as protonation of the anionic carbon in 18 was sufficiently slow. In 19, the 1,3-diyne moiety is separated by a three-atom tether from the proximal benzyne sp-carbon, so the intramolecular HDDA reaction was expected to proceed readily to give the naphthyne 20.3,9 The pendent silyl ether group should then capture the naphthyne faster than another external reagent (e.g., 17) to arrive at the product naphthalene 16 via nucleophilic addition and retro-Brook rearrangement. To tolerate the silyl ether group, we screened several weak, non-fluoride bases. Unfortunately, diyne 15 was susceptible to decomposition under mildly basic conditions, perhaps initiated by deprotonation of a propargylic proton.10 Therefore, we examined diyne 21, having gem-dimethyl substitution to replace the labile propargylic C–H bonds. To our delight, desired naphthalene 23 was formed in 27% yield using readily available potassium carbonate/18-crown-6 ether as the base (entry 1, tabular insert in Figure 2). When 4-Ts instead of 4-Tf was used as the 1,2-benzdiyne equivalent, the formation of benzyne 5-Ts was noticeably slower, and 23 was isolated in 45% yield (entry 2). Attempts to further optimize the yield of 23 (altering base, solvent, and/or temperature; Table S1) were not successful. However, we noticed that in the entry 1 result, we recovered 68% of diyne 21 (84% brsm), suggesting that the cascade following the initial trapping by 21 (cf. 18 to 16 via 19 and 20) was efficient. We suspected that the para-toluenesulfonamide (TsNH) used in entries 1 and 2 was not a sufficiently efficient trap for benzyne 5. Therefore, we explored modification of the pKa of the sulfonamide by changing to the trifluoromethylsulfonyl (Tf) group11 to increase the concentration of the triflate stabilized analog of anion 17. This should accelerate the rate of the initial benzyne trapping event. Indeed, under the same reaction conditions, triflamide 22 efficiently reacted with either 4-Tf or 4-Ts to afford naphthalene 24 in 72% or 76% yield, respectively (entries 3 and 4).
Figure 2.

Proposed mechanism and optimization of the sulfonamide substrate.
Encouraged by this result, we proceeded by examining this transformation with a series of diynes 25a-i, each of which bears a different trapping functionality potentially capable of capturing naphthyne 26 intramolecularly (Figure 3). First, we tested several heteroatom-based nucleophiles, 25a-d. Internal silyl ether traps with tethers different that in 21 and 22 gave good yields (27a and 27b). Moreover, a free alcohol was accommodated, leading to 27c. This reaction afforded a significant major by-product, namely 27c’ (the trimethylsilyl analog of 24). We presume that the hydroxy group in diyne 25c was being silylated either by the carbonate-TMS ester or, perhaps more likely, directly by the 1,2-benzdiyne equivalent 4. Thus, the free alcohol 25c or its corresponding TMS silyl ether gave 27c and 27c’, respectively. Quenching the reaction mixture directly with TBAF provided 27c in 55% yield. The intramolecular sulfonamide trapping leading to 27d also provided some mechanistic inferences. Consistent with our initial findings (Figure 2), the TfNHR nitrogen in 25d is a faster trap than the TsNHR, which further supports the idea that the active nucleophile is indeed the deprotonated sulfonamide anion (cf. 17-Tf vs. 17-Ts).
Figure 3.

Examples of naphthyne formation followed by intramolecular trapping by appended T1–T2 groups; together, these demonstrate that a variety of electron rich species can capture the naphthyne intermediates.
Several additional classes of intramolecular trapping reactions were explored. An appropriately placed, pendant phenyl substituent efficiently gave the intramolecular Diels–Alder (IMDA) trapped adduct 27e in 75% yield.12 Formation of the aromatic ene reaction product 27f13 was accompanied, unexpectedly, by the formation of the 2:1 adduct 27f’, which most likely arose from Diels–Alder reaction of benzyne 5-Tf with the electron rich arene ring in 27f. Once again, the regioselectivity of that second event can be explained by the distortion model (cf. 5-Tf, Figure 2)—the more electron rich carbon (para to the oxygen) preferentially engaged the more electrophilic benzyne carbon (meta to the triflate group) in an asynchronous, concerted cycloaddition. The gem-dimethylated analog 25g led to a different outcome; the aromatic ene pathway was completely suppressed and the IMDA product 27g was produced. These differences could be nicely accounted for by DFT calculations of the competing transition state geometries (see Figure S1). A dibenzofuran motif can be established by use of an ortho-methoxyphenyl substituent as a trapping group (cf. 27h). When 4-Ts was used as the bisbenzyne equivalent, the formation of 27h was accompanied by the byproduct 27h’, as a single constitutional isomer (see later discussion, accompanying Figure 4). This reveals a non-innocent role of the tosylate anion leaving group from the SN2’ process with 5-Ts. When 4-Tf was used as the precursor instead, 27h was isolated in 80% yield due to the weaker nucleophilicity of triflate anion. Finally, if the rate of intramolecular ortho-methoxy trapping was further decreased, as exemplified by the case of 25i, the steric repulsion caused by the additional ring fusion thwarted formation of the dibenzofuran product.; only the tosylate-trapped product 27i was observed. This suggests that the transition state for methoxy attack at the proximal benzyne carbon requires a close to planar geometry to achieve effective orbital overlap. As a result, the naphthyne was primarily trapped by the external tosylate anion to provide binaphthol derivative 27i as a single constitutional isomer in 40% yield plus a small amount of the isomeric products of DA reaction with a chlorobenzene solvent molecule (see Supporting Information).
Figure 4.

Calculated distortion angles of naphthynes 26h, 26i, and A-C.
The unanticipated products 27h’ and 27i, were formed with high regioselectivity during trapping by the external nucleophile. This was unexpected, because there are no obvious structural features (substituents, steric effect) that might account for this selectivity. Nonetheless, we computed the [SMD(chlorobenzene)/B3LYP/6–31G**] geometries of naphthynes 26h and 26i, which lead to 27h’ and 27i, respectively (Figure 4). The distortion of these two naphthynes is quite large and in a direction consistent with the observed nucleophilic attack by the tosylate anion at carbon a, the atom with the larger internal bond angle. To try to understand the contributing factor(s) for this distortion, we also computed the structures of several analogs. Removal of the aryl substituent (cf. A) has only a minor impact on the distortion (∠a–∠b). When the sulfonamide nitrogen was replaced by a methylene group (cf. B), again only a minor effect was seen. However, when the strain of the five membered ring that bridges the peri-positions on the naphthyne was removed (cf. C), the distortion essentially disappeared. Thus, it is the five-membered ring that is responsible for strongly perturbing the geometry of the naphthyne, thereby inducing attack by TsO– at the more electrophilic carbon in 26h and 26i.
To demonstrate a different element of modularity in the overall strategy, we prepared four different 1,2-benzdiyne equivalents (28, 30, 32, and 34; Figure 5). Symmetrical 1,2-benzdiyne equivalents 28, 30, and 32, when reacted with one of the diynylsulfonamides 25, gave the corresponding naphthalene products 29, 31, and 33, respectively, in good yields. The reaction of unsymmetrical precursor 34 and 22 afforded 35 as the only constitutional isomer, indicating faster initial benzyne generation through preferential loss of the triflate vs. tosylate.5e When 34 was paired with the diyne 25h, no dibenzofuran product was seen, again likely reflecting the inability of the o-methoxyphenyl group to adopt a sufficiently planar geometry for good orbital overlap, now because of the presence of the methyl substituent at the naphthyne peri-position. On the other hand, a significant amount of chlorobenzene (solvent) trapped products (multiple isomers detected by NMR and MS analyses) of the naphthyne was observed. To reduce the number of possible regio- and diastereo- isomers, this reaction was carried out in p-xylene, which underwent a clean DA reaction with the naphthyne 37 to produce 38 in 75% yield. This result, along with the observed formation of 27i, opens the possibility of using an external reagent for capturing naphthynes 26. It is surprising that we did not detect products arising from addition of the diynylsulfonamide 25h to the intermediate naphthyne 37. Indeed, this type of 2:1 adduct was not observed for any of the reactions in our studies.
Figure 5.

Reactions of other 1,2-benzdiyne equivalents. Chlorobenzene was the solvent for the first five examples; p-xylene was used for the formation of 38.
We show a practical aspect of this transformation in the gram-scale reaction depicted in Figure 6a. There, the loading of 4-Ts was lowered to 1.5 equivalent, and slow addition of 4-Ts was not crucial. The yield of 24 (67%) was comparable to that of the smaller scale outcome (76%, Figure 2). If an additional 1,3-diyne unit is present, as in, for example, sulfonamide 39, an anthracyne intermediate can be formed by a second HDDA reaction of the naphthyne, which was then trapped by the tethered silyl ether to give the anthracene derivative 40 (Figure 6b).7 This result demonstrates the potential of this tandem strategy for rapid construction of polycyclic arenes.
Figure 6.

(a) Large scale reaction, and (b) synthesis of larger polycyclic arenes.
In summary, we have developed a one-pot cascade strategy for naphthyne formation using a 1,3-diyne containing a nucleophilic nitrogen atom and a 1,2-benzdiyne equivalent. This transformation requires only mildly basic conditions and allows rapid construction of various naphthalene products via three reactive aryne species (Figures 3 and 5). DFT calculations revealed that a five-membered ring that bridges the peri-positions on the naphthyne can induce significant distortion of the two sp-hybridized carbons in 2,3-naphthynes (Figure 4). Moreover, we have demonstrated the potential application of this process in the synthesis of polycyclic arenes (Figure 6b). Future studies will focus on the use of different nucleophiles and of different linkers14 that connect the 1,3-diyne to the benzyne for the final, HDDA step, as well as applications to the synthesis of extended π-systems.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Foundation (CHE-1665389). Computations were performed using resources available through the University of Minnesota Supercomputing Institute (MSI). NMR data were obtained with an instrument acquired through the NIH Shared Instrumentation Grant program (S10OD011952).
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures for all new reactions; spectroscopic characterization data for all new compounds; results of DFT calculations (3D structures, geometries, and energies); copies of NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Hoffman RW Dehydrobenzene and Cycloalkynes; Academic: New York, 1967. [Google Scholar]; (b) Tadross PM; Stoltz BM A Comprehensive History of Arynes in Natural Product Total Synthesis. Chem. Rev 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]; (c) Gampe CM; Carreira EM Arynes and Cyclohexyne in Natural Product Synthesis. Angew. Chem., Int. Ed 2012, 51, 3766–3778. [DOI] [PubMed] [Google Scholar]; (d) Perez D; Pena D; Guitian E Aryne Cycloaddition Reactions in the Synthesis of Large Polycyclic Aromatic Compounds. Eur. J. Org. Chem 2013, 2013, 5981–6013. [Google Scholar]; (e) Takikawa H; Nishii A; Sakai T; Suzuki K Aryne-Based Strategy in the Total Synthesis of Naturally Occurring Polycyclic Compounds. Chem. Soc. Rev 2018, 47, 8030–8056. [DOI] [PubMed] [Google Scholar]
- (2).Himeshima Y; Sonoda T; Kobayashi H Fluoride-Induced 1,2-Elimination of o-(Trimethylsilyl)phenyl Triflate to Benzyne under Mild Conditions. Chem. Lett 1983, 8, 1211–1214. [Google Scholar]
- (3).(a) Miyawaki; Suzuki; Kawano; Ueda Cycloaromatization of a Non-Conjugated Polyenyne System. Tetrahedron Lett 1997, 38, 3943–3946. [Google Scholar]; (b) Bradley; Johnson Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2 + 4] Cycloaromatization to a Benzyne Intermediate. J. Am. Chem. Soc 1997, 119, 9917–9918. [Google Scholar]; (c) Tsui JA; Sterenberg BT A Metal Templated 4 + 2 Cycloaddition Reaction of an Alkyne and a Diyne To Form a 1,2-Aryne. Organometallics 2009, 28, 4906–4908. [Google Scholar]
- (4).(a) Hoye TR; Baire B; Niu D; Willoughby PH; Woods BP The Hexadehydro-Diels-Alder Reaction. Nature 2012, 490, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yun SY; Wang K-P; Lee N-K; Mamidipalli P; Lee D Alkane C-H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc 2013, 135, 4668–4671. [DOI] [PubMed] [Google Scholar]; (c) For a review: Diamond OJ; Marder Methodology and Applications of the Hexadehydro-Diels-Alder (HDDA) Reaction. Org. Chem. Front 2017, 4, 891–910. [Google Scholar]
- (5).(a) Shi; Qiu; Wang; Xu; Li Domino Aryne Precursor: Efficient Construction of 2,4-Disubstituted Benzothiazoles. J. Am. Chem. Soc 2015, 137, 5670–5673. [DOI] [PubMed] [Google Scholar]; (b) Qiu D; He J; Yue X; Shi J; Li Y Diamination of Domino Aryne Precursor with Sulfonamides. Org. Lett 2016, 18, 3130–3133. [DOI] [PubMed] [Google Scholar]; (c) Li L; Qiu D; Shi J; Li Y Vicinal Diamination of Arenes with Domino Aryne Precursors. Org. Lett 2016, 18, 3726–3729. [DOI] [PubMed] [Google Scholar]; (d) Yoshida S; Nakamura Y; Uchida K; Hazama Y; Hosoya T Aryne Relay Chemistry en Route to Aminoarenes: Synthesis of 3-Aminoaryne Precursors via Regioselective Silylamination of 3-(Triflyloxy)arynes. Org. Lett 2016, 18, 6212–6215. [DOI] [PubMed] [Google Scholar]; (e) Xu H; He J; Shi J; Tan L; Qiu D; Luo X; Li Y Domino Aryne Annulation via a Nucleophilic–Ene Process. J. Am. Chem. Soc 2018, 140, 3555–3559. [DOI] [PubMed] [Google Scholar]; (f) Lv C; Wan C; Liu S; Lan Y; Li Y Aryne Trifunctionalization Enabled by 3-Silylaryne as a 1,2-Benzdiyne Equivalent. Org. Lett 2018, 20, 1919–1923. [DOI] [PubMed] [Google Scholar]; (g) For a review: Shi J; Li Y; Li Y Aryne Multifunctionalization with Benzdiyne and Benztriyne Equivalents. Chem. Soc. Rev 2017, 46, 1707–1719. [DOI] [PubMed] [Google Scholar]
- (6).(a) Miyawaki K; Kawano T; Ueda I Multiple Cycloaromatization of Novel Aromatic Enediynes Bearing a Triggering Device on the Terminal Acetylene Carbon. Tetrahedron Lett. 1998, 39, 6923–6926. [Google Scholar]; (b) Miyawaki K; Kawano T; Ueda I Domino Thermal Radical Cycloaromatization of Non-Conjugated Aromatic Hexa- and Heptaynes: Synthesis of Fluoranthene and Benzo[a]Rubicene Skeletons. Tetrahedron Lett. 2000, 41, 1447–1451. [Google Scholar]; (c) Yoshida S; Shimizu K; Uchida K; Hazama Y; Igawa K; Tomooka K; Hosoya T Construction of Condensed Polycyclic Aromatic Frameworks Through Intramolecular Cycloaddition Reactions Involving Arynes Bearing an Internal Alkyne Moiety. Chem. Eur. J 2017, 23, 15332–15335. [DOI] [PubMed] [Google Scholar]
- (7).Xiao X; Hoye TR The Domino Hexadehydro-Diels–Alder Reaction Transforms Polyynes to Benzynes to Naphthynes to Anthracynes to Tetracynes (and Beyond?). Nat. Chem 2018, 10, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Cheong PH-Y; Garg NK; Houk KN Indolyne and Aryne Distortions and Nucleophilic Regioselectivities. J. Am. Chem. Soc 2010, 132, 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Medina JM; Mackey JL; Garg NK; Houk KN The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions. J. Am. Chem. Soc 2014, 136, 15798–15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Ajaz A; Bradley AZ; Burrell RC; Li WHH; Daoust KJ; Bovee LB; DiRico KJ; Johnson RP Concerted vs Stepwise Mechanisms in Dehydro-Diels-Alder Reactions. J. Org. Chem 2011, 76, 9320–9328. [DOI] [PubMed] [Google Scholar]; (b) Skraba-Joiner SL; Johnson RP; Agarwal J Dehydropericyclic Reactions: Symmetry-Controlled Routes to Strained Reactive Intermediates. J. Org. Chem 2015, 80, 11779–11787. [DOI] [PubMed] [Google Scholar]; (c) Xiao X; Woods BP; Xiu W; Hoye TR Benzocyclobutadienes: An Unusual Mode of Access Reveals Unusual Modes of Reactivity. Angew. Chem. Int. Ed 2018, 57, 9901–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen M; He CQ; Houk KN Mechanism and Regioselectivity of an Unsymmetrical Hexadehydro-Diels-Alder (HDDA) Reaction. J. Org. Chem 2019. 84, 1959–1963. [DOI] [PubMed] [Google Scholar]
- (10).(a) Wang T; Naredla RR; Thompson SK; Hoye TR The Pentadehydro-Diels-Alder Reaction. Nature 2016, 532, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) dos Passos Gomes G; Morrison AE; Dudley GB; Alabugin IV Optimizing Amine-Mediated Alkyne–Allene Isomerization to Improve Benzannulation Cascades: Synergy between Theory and Experiments. Eur. J. Org. Chem 2019, 2019, 512–518.s [Google Scholar]
- (11).Bordwell FG Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res 1988, 21, 456–463. [Google Scholar]
- (12).(a) Miller RG; Stiles M Reaction of Benzyne with Benzene and Naphthalene. J. Am. Chem. Soc 1963, 85, 1798–1800. [Google Scholar]; (b) Tabushi I; Yamada H; Yoshida Z; Oda R Reactions of Benzyne with Substituted Benzenes. Bull. Chem. Soc. Jpn 1977, 50, 285–290. [Google Scholar]; (c) Pogula VD; Wang T; Hoye TR Intramolecular [4 + 2] Trapping of a Hexadehydro-Diels-Alder (HDDA) Benzyne by Tethered Arenes. Org. Lett 2015, 17, 856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Niu D; Hoye TR The Aromatic Ene Reaction. Nat. Chem 2014, 6, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
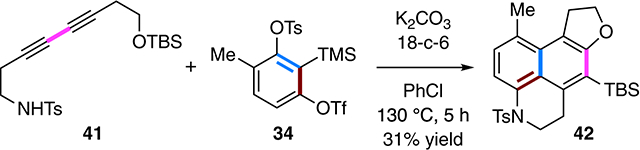
(14).One such linker is the sulfonamidoethyl moiety that connects the nucleophile and 1,3-diyne in 41. This substrate, which lacks acidified propargylic protons, was able to capture the benzyne derived from 34 to produce the adduct 42. This reaction is also notable because it represents a rare example of the HDDA reaction in which the diynophile and the 1,3-diyne are separated by a four-atom linker (cf. ref 9c).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.