

Abstract
Introduction
Due to the high cost and high failure rate of ascertaining amyloid positron emission tomography positivity (PET+) in patients with earlier stage Alzheimer’s disease (AD), an effective pre-screening tool for amyloid PET scans is needed.
Methods
Patients with mild cognitive impairment (n = 33, 24.2% PET+, 42% females, age 74.4 ± 7.5, MMSE 26.8 ± 1.9) and mild dementia (n = 19, 63.6% PET+, 36.3% females, age 73.0 ± 9.3, MMSE 22.6 ± 2.0) were recruited. Amyloid PET imaging, Apolipoprotein E (APOE) genotyping, and plasma amyloid β (Aβ)1–40, Aβ1–42, and total tau protein quantification by immunomagnetic reduction (IMR) method were performed. Receiver operating characteristics (ROC) analysis and Youden’s index were performed to identify possible cut-off points, clinical sensitivities/specificities, and areas under the curve (AUCs).
Results
Amyloid PET+ participants had lower plasma Aβ1–42 levels than amyloid PET-negative (PET−) subjects. APOE ε4 carriers had higher plasma Aβ1–42 than non-carriers. We developed an algorithm involving the combination of plasma Aβ1–42 and APOE genotyping. The success rate for detecting amyloid PET+ patients effectively increased from 42.3 to 70.4% among clinically suspected MCI and mild dementia patients.
Conclusions
Our results demonstrate the possibility of utilizing APOE genotypes in combination with plasma Aβ1–42 levels as a pre-screening tool for predicting the positivity of amyloid PET findings in early stage dementia patients.
Keywords: Amyloid PET, APOE, Plasma Aβ, Biomarkers, MCI, AD
Background
Beta-amyloid (Aβ) plaque deposition in the brain is the pathological hallmark of Alzheimer’s disease (AD) [1–3]. Disease-modifying drugs with anti-amyloid effects are designed to eliminate aggregated Aβ [4–6]. The results of phase I and phase II trials of these anti-amyloid drugs reveal their ability to eliminate Aβ plaques in the brains of patients with prodromal AD [7–9]. To prove the efficacy of these drugs, trials target prodromal AD patients with confirmed Aβ pathology for recruitment. Comprehensive neuropsychological assessment identified participants’ characters and severities [10, 11]. Aβ pathology is confirmed by amyloid positron emission tomography (PET) [12–14]. However, amyloid PET scans are expensive, and the availability of amyloid PET is limited. Widespread use of amyloid PET imaging in the pre-screening phase of clinical trials is thus not feasible. A pre-screening tool with low cost and high efficiency for evaluating the probability of PET positivity/negativity (PET+/PET−) is thus needed.
Cerebrospinal fluid (CSF) biomarkers are an alternative way to diagnose cerebral Aβ pathology. Many studies showed that the Aβ1–42 concentration in the CSF decreases in patients with dementia due to AD [14–17]. The accuracy of discriminating AD from healthy subjects by the CSF Aβ1–42/Aβ1–40 ratio can reach 80% [14]. Furthermore, the negative correlation between the CSF Aβ1–42/Aβ1–40 ratio and Aβ deposition assessed by amyloid PET is strong [18–20]. However, lumbar puncture is an invasive procedure that may cause discomfort and side effects such as headache, back pain, swelling, and bruising. Therefore, CSF biomarkers are not widely assessed in clinical practice. Alternative methods of pre-screening that are comfortable, low risk, and low cost are needed; these pre-screens can be administered before high-cost amyloid PET scans, especially in trials for amyloid modulation therapy.
Blood tests are easy, low cost, low risk, and highly available. A total of 30–50% of blood Aβ protein may come from the brain [21]. However, blood Aβ1–42 and Aβ1–40 levels are extremely low, and it is difficult to determine blood Aβ levels precisely [22–28]. Some reports have indicated that immunomagnetic reduction (IMR) is sensitive enough to assay ultra-low concentration of Aβ1–42 and Aβ1–40 in human plasma [23–27]. Using this technique, the concentrations of plasma Aβ1–42 have been shown to differentiate healthy elderly subjects from subjects with all stages (mild, moderate, and severe) of AD [25]. A previous study demonstrated the accuracy of using the plasma Aβ1–42/Aβ1–40 ratio as a diagnostic parameter in differentiating healthy subjects from patients with either mild or severe AD [25]. In addition, the plasma Aβ1–42/Aβ1–40 ratio increases with increasing amyloid load, as assessed by amyloid PET imaging in normal subjects and patients with dementia due to AD [28]. These results show the feasibility of assaying plasma Aβ1–42 and Aβ1–40 for evaluating whether to perform amyloid PET scans.
Genetic factors influence amyloid aggregation in both normal subjects and patients with AD. It has been reported that the Apolipoprotein E (APOE) ε4 allele is associated with greater Aβ deposition in the brain [29–31]. Once Apolipoprotein E (APOE) protein binds with Aβ, the complex becomes unstable and easily forms fibrillary Aβ [32–34]. The co-existence of APOE and Aβ in amyloid plaques is supported by histopathological findings [35]. Previous studies have found subjects with the APOE ε4 allele have a higher chance of presenting with amyloid PET+ than those without ε4 [14, 36].
In the present study, we sought to develop an algorithm using plasma Aβ1–42, Aβ1–40, tau, and APOE genotypes as a pre-screening tool to enhance the accuracy of predicting amyloid PET+ in clinically suspected mild cognitive impairment (MCI) and mild AD patients.
Methods
Recruitment of subjects
Through the Alzheimer’s Disease Neuroimaging Initiative in Taiwan (T-ADNI), subjects were enrolled at Taipei Veterans General Hospital (Taipei VGH), Linkou Chang Gung Memorial Hospital (CGMH), and Kaohsiung CGMH.
Enrolled subjects were required to be 55 to 90 (inclusive) years of age and to have at least 6 years of education. All subjects were interviewed by neurologists to obtain an extensive clinical history. Demographics, family history, physical examination, neurologic examination, Hachinski ischemic score, vital signs, and blood for screening labs (hematology, chemistry panel, vitamin B12, syphilis rapid plasma reagin, thyroid-stimulating hormone, and free thyroxine) were collected. A standard neuropsychological evaluation was performed. The screening laboratory and magnetic resonance (MR) imaging examinations were used to rule out other major neuropathologies such as tumors, strokes, severe white matter disease, and inflammation, but they were not used to diagnose dementia. All subjects were required to have no history of major brain trauma, brain tumor, stroke, epilepsy, alcoholism, major psychiatric illness, or other systemic diseases that affect cognitive function.
Diagnostic criteria for amnestic mild cognitive impairment (aMCI) and mild dementia were in accordance with the criteria used in Alzheimer’s Disease Neuroimaging Initiative (ADNI). Subjects underwent a series of screening evaluations including the Geriatric Depression Scale, a Mini-Mental State Examination (MMSE), the Chinese version of the Wechsler Memory Scale-III (WMS-III), and the immediate and delayed conditions of the Logical Memory (LM) task. A Clinical Dementia Rating Scale (CDR) score was obtained. All dementia patients and the majority of amnestic MCI patients fulfilled the National Institute on Aging and the Alzheimer’s Association (NIA-AA) recommended criteria for dementia due to AD and for MCI, respectively [10].
Only subjects with CDR scores of 0.5 and MMSE scores of 20–30 were analyzed in this study. Thus, all subjects in this study were clinically suspected MCI or mild AD patients. The demographic information for these early stage AD patients is listed in Table 1. Subjects were divided into two groups according to amyloid PET results.
Table 1.
Demographic information for enrolled clinically suspected early stage AD subjects
| Amyloid PET | Positive | Negative | All |
|---|---|---|---|
| Numbers (% female) | 22 (50.0%) | 30 (36.7%) | 52 (42.3%) |
| Age (years) | 72.1 ± 7.6 | 71.9 ± 9.7 | 72.0 ± 8.8 |
| Education (years) | 12.0 ± 4.3 | 11.0 ± 3.6 | 11.4 ± 3.9 |
| Clinical stage | |||
| aMCI | 8 | 25 | 33 |
| Mild dementia | 14 | 5 | 19 |
| APOE ε4 carrier | 12 | 2 | 14 |
| CDR | 0.5 | 0.5 | 0.5 |
| MMSE | 24.0 ± 2.7 | 27.0 ± 2.2† | 25.8 ± 2.8 |
| Logical memory delayed recall | 5.41 ± 3.92 | 8.40 ± 5.12* | 7.13 ± 4.84 |
Abbreviations: AD Alzheimer’s disease, CDR Clinical Dementia Rating Scale, MMSE Mini-Mental State Examination, PET positron emission tomography
*P value < 0.05
†P value < 0.001
Image data acquisition
The radiosynthesis of 18F-florbetapir [37] and amyloid PET data acquisition [38] were described previously by our group. All PET images were acquired from a single site, and the scanner was calibrated with a Hoffman brain phantom. The 18F-florbetapir PET scan comprised a 10-min acquisition period (acquired in 2 × 5 min frames) beginning 50 min following 10 mCi injection of the 18F-florbetapir tracer. Imaging was performed on a Biograph mCT PET/CT scanner (GE Healthcare, Milwaukee, USA). Each PET image was obtained using the three-dimensional ordered subset expectation maximization (3-D OSEM) reconstruction algorithm (four iterations, 24 subsets; Gaussian filter 2 mm; zoom: three) with CT-based attenuation correction, as well as scatter and random corrections, with a matrix size of 400 × 400 × 148 and a voxel size of 0.68 × 0.68 × 1.5 mm3. Structural MRI scans were acquired using a uniform scanning protocol that minimized and accounted for between-site differences in MRI systems. T1-weighted MRI images were obtained for all subjects to obtain useful anatomical information and enable coregistration with PET images.
Amyloid PET image processing
All PET image data were processed and analyzed using PMOD image analysis software (version 3.7, PMOD Technologies Ltd., Zurich, Switzerland), including MR-based spatial normalization to the Montreal Neurological Institute (MNI) MRI template [39]. Seven volumes of interest (VOIs), the frontal, anterior cingulate, posterior cingulate, precuneus, parietal, occipital, and temporal areas, were selected, and the regional standardized uptake value ratio (SUVR) using the whole cerebellum as the reference region was calculated for each VOI. Moreover, the average SUVR from these seven cerebral cortical VOIs was computed to yield an estimate of global cortical SUVR for further analysis.
The PET images were interpreted by an experienced, blinded nuclear medicine physician (Kun-Ju Lin). A 5-point visual scale was used to classify the amyloid loading, from 0, indicated no tracer retention in cortical gray matter, to 4, indicated high levels of cortical amyloid accumulation. Visual rating scores of 2–4 were considered indicative of amyloid PET+ brains, and ratings of 0–1 were considered negative for amyloid PET [37].
Preparation of human plasma
Subjects were asked to provide a 10-ml non-fasting venous blood sample (K3 EDTA, lavender-top tube). Colleagues were blind to all samples in the laboratory. Blood samples were centrifuged (1500–2500g for 15 min) within 1 h of the draw, and plasma was aliquoted into cryotubes and stored at − 20 °C. Buffy coat leukocytes were collected into another 1.5-ml Eppendorf tube, and genomic DNA was extracted using a DNeasy Blood & Tissue Kit (69506, Qiagen, Valencia CA).
Measurement of plasma Aβ1–42, Aβ1–40, and tau
To warm up frozen human plasma samples, it was first taken to wet ice for approximately 30 min, following by keeping the plasma at room temperature for 5–10 min. For each human plasma sample, duplicated measurements of Aβ1–42 and Aβ1–40 were performed. For Aβ1–42 measurement, 60 μl plasma was mixed with 60 μl IMR Aβ1–42 reagent (MF-AB2-0060, MagQu). For Aβ1–40 measurement, 40 μl plasma was mixed with 80 μl IMR Aβ1–40 reagent (MF-AB0-0060, MagQu). Forty microliters of plasma was mixed with 80 μl IMR tau reagent (MF-TAU-0060, MagQu) for tau measurement. MF-AB0-0060, MF-AB2-0060, and MF-TAU-0060 reagents consisted of magnetic nanoparticles that were conjugated with specific antibody against Aβ1–40 protein at a.a. 1–12 (A3981, SIGMA), Aβ1–42 protein at a.a. 37–42 (ab34376, Abcam), and Tau protein at a.a. 404–441 (T9450, SIGMA), respectively. The reaction signal was recorded and analyzed with an IMR reader (XacPro-S, MagQu). Aβ1–42, Aβ1–40, and tau concentrations were obtained by converting the reaction signal via the standard curve, i.e., the relationship between Aβ1–42, Aβ1–40, or tau concentration and the reaction signal. The standard deviation (SD) of the paired measurements of Aβ1–42, Aβ1–40, and tau concentrations in plasma samples was less than 15%. The reported Aβ1–42, Aβ1–40, and tau concentrations for each plasma sample are the mean value of the duplicated measurements.
APOE genotypes
APOE genotyping was performed by polymerase chain reaction (PCR) amplification of a 500-base-pair fragment of the APOE gene spanning amino acid positions 112 and 158, followed by direct DNA sequencing [40]. Subjects with either one or two ε4 alleles were regarded as ε4 carriers.
Statistical methods
All statistical analysis was performed by using the Statistical Package for the Social Sciences (SPSS) software package (version 17 for Windows®, SPSS Inc., Chicago, IL, USA). Descriptive statistics for demographic, neuropsychological, and plasma biomarker data are presented as the mean ± SD. The threshold for statistical significance was P value < 0.05. Chi-squared tests were used to compare categorical variables between groups. General linear models with age, sex, and education as covariates were used to examine between-group differences in plasma Aβ1–42, Aβ1–40, and tau concentrations. Receiver operating characteristics (ROC) analysis and Youden’s index were performed to identify possible cut-off points, clinical sensitivities/specificities, and areas under the curve (AUCs).
Results
All subjects were clinically suspected MCI or mild AD patients, with CDR scores of 0.5 and MMSE scores of 20–30. The demographic information for the enrolled participants is listed in Table 1.
Amyloid PET results
According to visual rating of amyloid PET images, 22 of 52 (42.3%) subjects were amyloid PET+. Visual rating scores of 1–4 were reported for 30, three one, and 18 subjects, respectively. Rating scores of 2–4 were considered indicative of amyloid positivity. Among the 52 participants, 33 subjects are aMCI and the other 19 subjects are mild dementia. For aMCI patients, 25 (75.8%) subjects are PET− and the other 8 (24.2%) subjects are PET+. For mild dementia patients, 5 (26.3%) subjects are PET− and the other 14 (73.7%) subjects are PET+. The percentage of PET+ in dementia in this study is 73.74%, which is close to 80%.
APOE genotypes and amyloid PET
Fourteen individuals (26.9%) were APOE ε4 carriers. The APOE ε4 carriers had significantly higher incidence of amyloid positivity than non-carriers. A total of 12 of 14 (85.7%) APOE ε4 carriers and 10 of 38 (26.3%) APOE ε4 non-carriers were amyloid PET+, as listed in Table 2.
Table 2.
Numbers, amyloid PET positive percentage, and plasma biomarkers in APOE ε4 carriers and non-carriers
| APOE genotype | ε4 carriers | ε4 non-carriers | |
|---|---|---|---|
| ε4 carriers (ε2ε4, ε3ε4, ε4ε4) | All ε4 non-carriers (ε2ε2, ε2ε3, ε3ε3) | ε4 carriers (ε2ε2, ε2ε3) | |
| Amyloid PET+% | 85.7% | 26.3% | 0 |
| Plasma Aβ1–40 (pg/ml) | 47.1 ± 7.2 | 50.9 ± 7.4 | 50.0 ± 8.5 |
| Plasma Aβ1–42 (pg/ml) | 17.9 ± 2.9 | 16.7 ± 2.9 | 16.5 ± 2.7 |
| Plasma tau (pg/ml) | 24.7 ± 9.9 | 19.3 ± 9.5* | 18.8 ± 8.2 |
| Plasma Aβ1–42/Aβ1–40 | 0.396 ± 0.117 | 0.343 ± 0.111 | 0.349 ± 0.126 |
| Plasma Aβ1–42xtau (pg2/ml2) | 468.2 ± 262.2 | 348.6 ± 242.1 | 331.0 ± 208.6 |
Abbreviations: Aβ Amyloid β, PET positron emission tomography
*P value < 0.05: ε4 carriers vs ε4 non-carriers
Plasma Aβ1–40, Aβ1–42, and tau concentrations and APOE ε4
The mean plasma Aβ1–40 concentration of the 52 participants was 49.86 ± 7.45 pg/ml, the mean Aβ1–42 level was 17.04 ± 2.95 pg/ml, and the mean tau level was 20.76 ± 9.79 pg/ml.
The effect of APOE ε4 on plasma biomarkers was examined. The results are shown in Table 2. Demographic features including age and gender between APOE ε4 carriers and non-carriers were identical. APOE ε4 carriers showed a slightly higher plasma tau level than APOE ε4 non-carriers. Although not significant, APOE ε4 carriers showed a trend of higher levels of plasma Aβ1–42 and higher levels of biomarker combinations such as Aβ1–42/Aβ1–40 and Aβ1–42xtau than APOE ε4 non-carriers. In contrast, the plasma Aβ1–40 level was slightly lower in ε4 carriers.
Plasma Aβ1–40, Aβ1–42, and tau and amyloid PET
Collapsing across APOE genotypes, there were significant differences in both plasma Aβ1–42 between the amyloid PET+ and PET− groups (Fig. 1b), whereas plasma Aβ1–40 and tau showed no between-group difference (Fig. 1a, c). The statistical comparison in individual and combined biomarkers between amyloid PET+ and PET− are listed in Table 3. The ages and gender of PET+ and PET− are identical. ApoE ε4 allele frequency is much higher in PET+ (31.8%) as compared to PET− (3.33%). The amyloid PET+ group showed lower levels of plasma Aβ1–42 (16.3 ± 2.3 vs. 17.6 ± 3.3 pg/ml, P value < 0.05). However, discrimination between amyloid PET+ and PET− patients was not enhanced by using the combinations of Aβ1–42/Aβ1–40 and Aβ1–42xtau (Fig. 1d, e).
Fig. 1.

Plasma a Aβ1–40, b Aβ1–42, c tau, d Aβ1–42/Aβ1–40, and e Aβ1–42xtau for enrolled clinically suspected early stage AD subjects with negative and positive amyloid PET findings. The dashed line in b denotes the cut-off value, 15.58 pg/ml, to discriminate PET− from PET+ according to the ROC curve of all subjects. Abbreviations: Aβ, Amyloid β; AD, Alzheimer’s disease; PET+, amyloid positron emission tomography positivity; PET−, amyloid positron emission tomography negativity; ROC, receiver operating characteristics. *P value < 0.05
Table 3.
APOE ε4 allele frequency and plasma biomarkers between amyloid positive and negative
| Amyloid PET | Positive | Negative |
|---|---|---|
| APOE ε4 allele frequency | 31.8% | 3.33% |
| Plasma Aβ1–40 (pg/ml) | 50.9 ± 7.7 | 49.1 ± 7.3 |
| Plasma Aβ1–42 (pg/ml) | 16.3 ± 2.3 | 17.6 ± 3.3* |
| Plasma Tau (pg/ml) | 18.4 ± 8.5 | 22.5 ± 10.4 |
| Plasma Aβ1–42/Aβ1–40 | 0.334 ± 0.109 | 0.374 ± 0.117 |
| Plasma Aβ1–42xtau | 316.6 ± 207.6 | 427.8 ± 272.1 |
Abbreviations: Aβ Amyloid β, PET positron emission tomography
*P value < 0.05
The plasma Aβ1–42 concentration as a function of amyloid PET SUVR is shown in Fig. 2. For PET− subjects ( ), the plasma Aβ1–42 concentration ranged from 14 to 24 pg/ml with increasing SUVR, with a mean of 17.6 pg/ml. The coefficient of correlation, r, between the plasma Aβ1–42 concentration and SUVR was 0.387, which indicates a moderate positive correlation. However, for PET+ subjects (•), the plasma Aβ1–42 concentration was lower than for amyloid PET− subjects, with a mean of 16.3 pg/ml (r = − 0.068).
), the plasma Aβ1–42 concentration ranged from 14 to 24 pg/ml with increasing SUVR, with a mean of 17.6 pg/ml. The coefficient of correlation, r, between the plasma Aβ1–42 concentration and SUVR was 0.387, which indicates a moderate positive correlation. However, for PET+ subjects (•), the plasma Aβ1–42 concentration was lower than for amyloid PET− subjects, with a mean of 16.3 pg/ml (r = − 0.068).
Fig. 2.
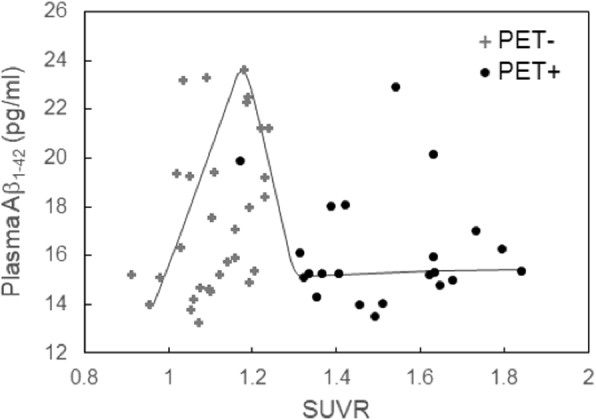
SUVR-dependent plasma Aβ1–42 concentrations for the 52 enrolled clinically suspected early stage AD subjects. Abbreviations: Aβ, Amyloid β; AD, Alzheimer’s disease; SUVR, standardized uptake value ratio; PET+, amyloid positron emission tomography positivity; PET−, amyloid positron emission tomography negativity
Plasma Aβ1–42 as a pre-screening tool for predicting amyloid PET positivity
In a ROC analysis for discriminating PET+ from PET−, the AUC was 0.611. The cut-off value for the inverse of the plasma Aβ1–42 level was 0.064 (pg/ml)−1, which equaled 15.58 pg/ml of plasma Aβ1–42. The corresponding sensitivity and specificity were 59.1% and 60.0%, respectively. The cut-off value is plotted with the dashed line in Fig. 1b.
Given that APOE ε4 status and plasma Aβ1–42 level enhanced the success rate in early stage AD, we further examined the contribution of combining APOE ε4 allele count and plasma Aβ1–42 to the detection of PET+ cases in those clinically suspected MCI and mild AD. The AUC was 0.826 for overall model combined with APOE ε4 status and plasma Aβ1–42 level. To determine the cut-off value of plasma Aβ1–42 in different conditions, we further conducted stratified ROC analysis. First, subjects were divided into two groups according to their APOE ε4 status, i.e., APOE ε4 carriers or non-carriers. Figure 3a plots the observed plasma Aβ1–42 levels for PET− and PET+ patients and for APOE ε4 carriers and non-carriers. We performed the ROC curve for discrimination of PET+ from PET− cases among clinically suspected early stage AD APOE ε4 carriers using (Aβ1–42)−1 as the parameter. The AUC was 0.875. The cut-off value for (Aβ1–42)−1 was found to be 0.054 (pg/ml)−1, which corresponds to 18.68 pg/ml of plasma Aβ1–42. The sensitivity and specificity were 75.0% and 100%, respectively. Remarkably, in APOE ε4 carriers, the success rate for detecting PET+ patients can reach 100% through enrollment of subjects with plasma Aβ1–42 concentrations lower than 18.68 pg/ml.
Fig. 3.

a Plasma Aβ1–42 levels in amyloid PET− and PET+ APOE ε4 carriers and APOE ε4 non-carriers. b Plasma Aβ1–42 levels in amyloid PET− and PET+ APOE ε4 carriers and non-carriers. APOE ε4 non-carriers are divided into APOE ε2 carriers and non-carriers. The dashed and dotted lines are the plasma Aβ1–42 cut-off values, 18.68 pg/ml and 15.58 pg/ml, for differentiating PET− from PET+ in APOE ε4 carriers and non-carriers, respectively. Abbreviations: Aβ, Amyloid β; PET+, amyloid positron emission tomography positivity; PET−, amyloid positron emission tomography negativity
We also performed the ROC curve for differentiating PET+ from PET− in clinically suspected early stage AD APOE ε4 non-carriers using (Aβ1–42)−1 as a parameter. The AUC was 0.718. The cut-off value for (Aβ1–42)−1 was found to be 0.064 (pg/ml)−1, which corresponds to a plasma Aβ1–42 concentration of 15.58 pg/ml, plotted with the dotted line in Fig. 3a. The sensitivity and specificity were 100% and 57.1%, respectively. The success rate for detecting PET+ among APOE ε4 non-carriers was only 26.3% without assaying plasma Aβ1–42. Remarkably, the cut-off value in APOE ε4 carriers (18.68 pg/ml) was higher than that in APOE ε4 non-carriers (15.58 pg/ml). In APOE ε4 non-carriers with plasma Aβ1–42 lower than 15.58 pg/ml, the success rates for detecting PET+ patients were 45.5%.
After considering the effect of APOE ε4 status, there were still 12 PET− APOE ε4 non-carriers with plasma Aβ1–42 lower than 15.58 pg/ml (Fig. 3a). We further considered the effect of APOE ε2. Seven of 38 APOE ε4 non-carriers were APOE ε2 carriers. All seven APOE ε2 carriers among the APOE ε4 non-carriers were found to be PET−, as shown in Fig. 3b. Thus, we may exclude subjects with an APOE ε2 allele and no APOE ε4 allele, i.e., ε2ε2 or ε2ε3, when testing for amyloid PET+.
Algorithm for using plasma Aβ1–42 as a screening tool to increase the rate of positive amyloid PET findings
The attempt to achieve the highest success rate in predicting amyloid PET+ among clinically suspected mild stage AD patients is illustrated in Fig. 4. The contributions of APOE genotype and plasma Aβ1–42 level are considered in the finalized pathway IV. With the inclusion of APOE ε2 status, APOE ε4 allele, and plasma Aβ1–42, the overall success rate for pathway V in predicting amyloid PET+ in clinically suspected MCI and mild dementia patients was 70.4%. The sensitivity, specificity, accuracy, positive predictive value, negative predictive value, and area under the curve for pathways II to V (utilizing various blood biomarkers for PET scan pre-screening) are presented in Table 4. The detailed information of the aMCI subgroup was presented in Additional file 1: Table S1 and Additional file 2: Figure S1.
Fig. 4.

Comparison of the detection accuracy for amyloid PET+ subjects with clinically suspected mild stage AD based on genetic and plasma biomarkers. Abbreviations: Aβ, Amyloid β; AD, Alzheimer’s disease; CDR, Clinical Dementia Rating Scale; MMSE, Mini-Mental State Examination; PET+, amyloid positron emission tomography positivity; PET−, amyloid positron emission tomography negativity
Table 4.
Sensitivity, specificity, positive predictive value, and negative predictive value for pathways II to V, utilizing various blood biomarkers for PET scan pre-screening
| Pathway | II | III | IV | V |
| Biomarker | APOE ε4 | Plasma Aβ1–42 | APOE ε4 + plasma Aβ1–42 | APOE ε4 + ε2 + plasma Aβ1–42 |
| SS | 0.595 | 0.591 | 0.864 | 0.864 |
| SP | 0.933 | 0.600 | 0.600 | 0.733 |
| Accuracy | 0.769 | 0.596 | 0.711 | 0.788 |
| PPV | 0.857 | 0.520 | 0.613 | 0.704 |
| NPV | 0.737 | 0.667 | 0.857 | 0.880 |
| AUC | 0.739 | 0.611 | 0.856 | 0.902 |
Abbreviations: Aβ Amyloid β, NPV negative predictive value, PET positron emission tomography, PPV positive predictive value, SP specificity, SS sensitivity, AUC area under curve
Discussion
Published studies indicate that the occurrence of positive amyloid PET results in clinically suspected early stage AD ranges from 30 to 65%, depending on age, APOE genotype, and other factors [41–43]. Recent clinical trials in clinically suspected AD found a high failure rate for ascertaining amyloid PET+. An effective pre-screening tool for amyloid PET imaging is needed. In the present study, we propose an algorithm using plasma Aβ1–42, Aβ1–40, tau, and APOE allele status to enhance accuracy in detecting amyloid PET+ in clinically suspected early stage AD (Fig. 4). We found that the factors APOE genotype and plasma Aβ1–42 level enhance the success rate for detecting amyloid PET+ patients in clinically suspected aMCI and mild dementia.
In line with previous studies, the current study found a 42.3% occurrence of amyloid positivity in clinically suspected early stage AD patients. This result implies that the failure rate for detecting amyloid PET+ in clinically suspected early stage AD could be more than 50%. Our algorithm, combining APOE genotype and plasma amyloid level, increases the success rate for detecting amyloid PET+ in early-stage AD from 42.3 to 70.4%. Significantly, the success rate is especially enhanced in APOE ε4 non-carriers.
The presence of the APOE ε4 allele may increase the possibility of amyloid PET+ from 42.3 to 70.4% in clinically suspected early stage AD (Fig. 4 pathway II). This result is consistent with evidence that the presence of the APOE ε4 allele is associated with greater amyloid deposition [41, 44, 45]. However, only approximately 30% of APOE ε4 non-carriers were amyloid PET+. This finding implies that APOE ε4 non-carriers are the population responsible for the high failure rate in detecting amyloid PET+ in clinically suspected early stage AD. However, more than 70% of subjects are APOE ε4 non-carriers in the general population. Even in early stage AD, more than 60% of patients are APOE ε4 non-carriers [30, 46, 47]. It is not possible to exclude APOE ε4 non-carriers in studies or trials focused on early stage AD. Other factors should be taken into account in this APOE ε4 non-carrier population to reduce the failure rate for detecting amyloid PET+. It has been reported that the APOE ε2 allele is able to prevent Aβ aggregation or plaque formation [30, 41]. Thus, the APOE ε2 allele should also be accounted for in predicting amyloid PET+ (Fig. 5).
Fig. 5.

Overall receiver operating characteristics (ROC) curves for aMCI subgroups calculated in multivariate logistic models. The area under the curve (AUC) was significantly improved in combined biomarkers compared with single biomarkers (P = 0.014 for APOE ε4 alone vs APOE ε4 + plasma Aβ1–42, P = 0.002 for APOE ε4 alone vs APOE ε4 + APOE ε2 + plasma Aβ1–42, P = 0.002 for plasma Aβ1–42 vs APOE ε4 + plasma Aβ1–42, and P < 0.001 for APOE ε4 + APOE ε2 + plasma Aβ1–42). Abbreviations: Aβ, Amyloid β; AUC, area under the curve; ROC, receiver operating characteristics
Although this is a cross-sectional study, our results showed that the plasma Aβ1–42 concentration may increase as SUVR becomes higher in amyloid PET− patients; however, plasma Aβ1–42 drops steeply at the early stage of amyloid PET+ status, as indicated by the gray solid line in Fig. 2. Our findings echoed previous consensus that PET+ subjects show lower levels of plasma Aβ1–42 than PET− subjects [22, 28, 48, 49]. Although the longitudinal change in plasma biomarker is inconclusive [22], these results may imply that the plasma Aβ1–42 level becomes lower once the formation of Aβ plaques occurs in the brains of early stage AD patients. Remarkably, the correlation between CSF Aβ1–42 and SUVR is not a linear interrelationship and is closer to a hyperbolic model [19, 50]. In the current study, the relationship between the plasma Aβ1–42 concentration and SUVR may be more complicated than the hyperbolic model. Further investigations to clarify this complicated relationship should be conducted in future work. The proposed dynamic changes in plasma Aβ1–42 level have important implications for the selection of participants in drug trials. Amyloid-negative subjects with higher levels of Aβ1–42 may be not recruited for drug trials that target aggregated Aβ, but they are suitable candidates for drug trials targeting soluble Aβ in prodromal AD. A longitudinal study of the progression from PET− to PET+ status in early stage AD is needed to verify our results. Moreover, future research should investigate the biological mechanism by which changes in plasma Aβ lead to the formation of Aβ plaques in the brain.
There is limitation in this work. Given that CSF biomarkers were not used as inclusion criteria, it may happen that some of the aMCI patients do not develop AD lowering prediction rates. Another important question relates to replication/validation in an independent cohort, since the study was carried out in a small sample and individual results showed high variability.
Conclusion
According to the results of the current study, based on a limited sample of clinically suspected aMCI and mild AD subjects, combining APOE genotypes and plasma Aβ1–42 increases the accuracy for detecting amyloid PET+ in early stage AD from 42.3 to 70.4%. Plasma Aβ1–42 cut-off values for discriminating PET+ from PET− patients are proposed for APOE ε4 carriers and non-carriers. The information reported in the current study may help pharmaceutical companies to effectively enroll clinically suspected early stage AD subjects with Aβ plaques in the brain. Future longitudinal studies should be conducted to clarify the biological mechanism for the revolution of the plasma Aβ1–42 level during the progression from amyloid PET− to PET+ stage.
Supplementary information
Additional file 1 : Table S1. Sensitivity, specificity, positive predictive value, and negative predictive value for the pathways II to V, utilizing various blood biomarkers for PET scan pre-screening in aMCI populations.
Additional file 2 : Figure S1. Overall receiver operating characteristic (ROC) curves of each diagnostic pathways calculated in multivariate logistic models. The area under the curve (AUC) was significantly improved in combined biomarkers compared with APOE ε4 alone (P = 0.027 for APOE ε4 + Plasma Aβ1–42, P = 0.005 for APOE ε4 + APOE ε2 + Plasma Aβ1–42). There was still a trend of better AUC compared with Plasma Aβ1–42 alone (P = 0.339 for APOE ε4 + Plasma Aβ1–42, P = 0.130 for APOE ε4 + APOE ε2 + Plasma Aβ1–42). Abbreviations: Aβ, Amyloid β; aMCI, amnestic mild cognitive impairment; AUC, area under the curve, AUC; ROC, receiver operating characteristics
Acknowledgements
We thank Avid Radiopharmaceuticals, Inc. (Philadelphia, PA, USA) for providing the precursor for the preparation of 18F-florbetapir. We also thank Shieh-Yueh at MagQu Co., Ltd. for his help in the assessment of plasma Aβ1–40, Aβ1–42, and tau.
Abbreviations
- Aβ
Amyloid β
- AD
Alzheimer’s disease
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- aMCI
Amnestic mild cognitive impairment
- APOE
Apolipoprotein E
- AUCs
Areas under the curve
- CDR
Clinical Dementia Rating Scale
- CGMH
Chang Gung Memorial Hospital
- CSF
Cerebrospinal fluid
- 3-D OSEM
Three-dimensional ordered subset expectation maximization
- IMR
Immunomagnetic reduction
- LM
Logical memory
- MMSE
Mini-Mental State Examination
- MNI
Montreal Neurological Institute
- MR
Magnetic resonance
- NC
Normal cognition
- NIA-AA
National Institute on Aging and the Alzheimer’s Association
- PET
Positron emission tomography
- PET+
Positron emission tomography positive
- PET-
Positron emission tomography negative
- PCR
Polymerase chain reaction
- ROC
Receiver operating characteristics
- SD
Standard deviation
- SPSS
Statistical Package for the Social Sciences
- SUVR
Standardized uptake value ratio
- T-ADNI
Alzheimer’s Disease Neuroimaging Initiative in Taiwan
- Taipei VGH
Taipei Veterans General Hospital
- VOI
Volumes of interest
- WMS-III
Wechsler Memory Scale-III
Authors’ contributions
This study was designed by KJL and PNW and run by CCH, CCC, YCL, ITH, TCY, WSH, and BHY. SYL, KJL, PCL, CCH, CCC, YCL, ITH, TCY, WSH, BHY, and PNW contributed to the manuscript. All authors read and approved the final manuscript.
Funding
This study was carried out with financial support from the Brain Research Center, National Yang-Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, the Taiwan Alzheimer’s Disease Neuroimaging Initiative (T-ADNI) group, the National Science Council, and the Ministry of Science and Technology, Taiwan (MOST 105-2325-B-182A-005-, MOST 104-2314-B-182A-083-MY2, MOST 104-2314-B-010-016-MY3, MOHW 105-TDU-B-212-133020), Taipei Veterans General Hospital (V104E9-005, V105C-074), and Chang Gung Memorial Hospital under grant (CMRPG3D1802).
Availability of data and materials
The datasets generated during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board, Taipei Veterans General Hospital, and Institutional Review Board, Chang Gung Memorial Hospital, respectively. The study was conducted according to the principles in the Declaration of Helsinki 2013. All participants and guardians of patients with dementia provided written informed consent before participating in this study.
Consent for publication
All authors consent the study for publication. Written and informed consent was obtained from all the included participants.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kun-Ju Lin, Email: lin4857@adm.cgmh.org.tw.
Pei-Ning Wang, Email: pnwang@vghtpe.gov.tw.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13195-019-0566-0.
References
- 1.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Hyman BT, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leinonen V, et al. Amyloid and tau proteins in cortical brain biopsy and Alzheimer’s disease. Ann Neurol. 2010;68:446–453. doi: 10.1002/ana.22100. [DOI] [PubMed] [Google Scholar]
- 4.Aprahamian I, Stella F, Forlenza OV. New treatment strategies for Alzheimer’s disease: is there a hope? Indian J Med Res. 2013;138:449–460. [PMC free article] [PubMed] [Google Scholar]
- 5.Wang CY, et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y) 2017;3:262–272. doi: 10.1016/j.trci.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 2017;3:367–384. doi: 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lannfelt L, et al. Perspectives on future Alzheimer therapies: amyloid-beta protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther. 2014;6:16. doi: 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coric V, et al. Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 2015;72:1324–1333. doi: 10.1001/jamaneurol.2015.0607. [DOI] [PubMed] [Google Scholar]
- 9.Sevigny J, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 10.McKhann GM, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubois B, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/s1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 12.Clark CM, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. Jama. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weston PS, et al. Diagnosing dementia in the clinical setting: can amyloid PET provide additional value over cerebrospinal fluid? J Alzheimers Dis. 2016;54:1297–1302. doi: 10.3233/jad-160302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben Bouallegue F, Mariano-Goulart D, Payoux P. Comparison of CSF markers and semi-quantitative amyloid PET in Alzheimer’s disease diagnosis and in cognitive impairment prognosis using the ADNI-2 database. Alzheimers Res Ther. 2017;9:32. doi: 10.1186/s13195-017-0260-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tapiola T, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 16.Andreasen N, et al. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 17.Holtta M, et al. Evaluating amyloid-beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer’s disease. PLoS One. 2013;8:e66381. doi: 10.1371/journal.pone.0066381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36:297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Landau SM, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol. 2013;74:826–836. doi: 10.1002/ana.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hake A, et al. Florbetapir positron emission tomography and cerebrospinal fluid biomarkers. Alzheimers Dement. 2015;11:986–993. doi: 10.1016/j.jalz.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts KF, et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann Neurol. 2014;76:837–844. doi: 10.1002/ana.24270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hampel H, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639–652. doi: 10.1038/s41582-018-0079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang CC, et al. Biofunctionalized magnetic nanoparticles for specifically detecting biomarkers of Alzheimer’s disease in vitro. ACS Chem Neurosci. 2011;2:500–505. doi: 10.1021/cn200028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S. Y., Chieh J. J., Yang C. C., Liao S. H., Chen H. H., Horng H. E., Yang H. C., Hong C. Y., Chiu M. J., Chen T. F., Huang K. W., Wu C. C. Clinic Applications in Assaying Ultra-Low-Concentration Bio-Markers Using HTS SQUID-Based AC Magnetosusceptometer. IEEE Transactions on Applied Superconductivity. 2013;23(3):1600604–1600604. doi: 10.1109/TASC.2012.2229772. [DOI] [Google Scholar]
- 25.Chiu MJ, et al. Combined plasma biomarkers for diagnosing mild cognition impairment and Alzheimer’s disease. ACS Chem Neurosci. 2013;4:1530–1536. doi: 10.1021/cn400129p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SY, Chiu MJ, Chen TF, Horng HE. Detection of plasma biomarkers using Immunomagnetic reduction: a promising method for the early diagnosis of Alzheimer’s disease. Neurol Ther. 2017;6:37–56. doi: 10.1007/s40120-017-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lue LF, et al. Plasma levels of Abeta42 and tau identified probable Alzheimer’s dementia: findings in two cohorts. Front Aging Neurosci. 2017;9:226. doi: 10.3389/fnagi.2017.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzen KY, et al. Plasma Abeta but not tau is related to brain PiB retention in early Alzheimer’s disease. ACS Chem Neurosci. 2014;5:830–836. doi: 10.1021/cn500101j. [DOI] [PubMed] [Google Scholar]
- 29.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- 32.Bales KR, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 33.Irizarry MC, et al. Modulation of A beta deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci. 2000;920:171–178. doi: 10.1111/j.1749-6632.2000.tb06919.x. [DOI] [PubMed] [Google Scholar]
- 34.Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81:740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-F. [DOI] [PubMed] [Google Scholar]
- 36.Risacher SL, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI) Front Aging Neurosci. 2013;5:11. doi: 10.3389/fnagi.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson KA, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. 2013;9:S72–S83. doi: 10.1016/j.jalz.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin KJ, et al. Whole-body biodistribution and brain PET imaging with [18F]AV-45, a novel amyloid imaging agent--a pilot study. Nucl Med Biol. 2010;37:497–508. doi: 10.1016/j.nucmedbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Hsiao IT, et al. Perfusion-like template and standardized normalization-based brain image analysis using 18F-florbetapir (AV-45/Amyvid) PET. Eur J Nucl Med Mol Imaging. 2013;40:908–920. doi: 10.1007/s00259-013-2350-x. [DOI] [PubMed] [Google Scholar]
- 40.Emi M, et al. Genotyping and sequence analysis of apolipoprotein E isoforms. Genomics. 1988;3:373–379. doi: 10.1016/0888-7543(88)90130-9. [DOI] [PubMed] [Google Scholar]
- 41.Fleisher AS, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68:1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 42.Villemagne VL, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doraiswamy PM, et al. Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology. 2012;79:1636–1644. doi: 10.1212/WNL.0b013e3182661f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drzezga A, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- 45.Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ungar L, Altmann A, Greicius MD. Apolipoprotein E, gender, and Alzheimer’s disease: an overlooked, but potent and promising interaction. Brain Imaging Behav. 2014;8:262–273. doi: 10.1007/s11682-013-9272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ovod V, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13:841–849. doi: 10.1016/j.jalz.2017.06.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura A, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554:249–254. doi: 10.1038/nature25456. [DOI] [PubMed] [Google Scholar]
- 50.Toledo JB, et al. Nonlinear association between cerebrospinal fluid and florbetapir F-18 beta-amyloid measures across the spectrum of Alzheimer disease. JAMA Neurol. 2015;72:571–581. doi: 10.1001/jamaneurol.2014.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 : Table S1. Sensitivity, specificity, positive predictive value, and negative predictive value for the pathways II to V, utilizing various blood biomarkers for PET scan pre-screening in aMCI populations.
Additional file 2 : Figure S1. Overall receiver operating characteristic (ROC) curves of each diagnostic pathways calculated in multivariate logistic models. The area under the curve (AUC) was significantly improved in combined biomarkers compared with APOE ε4 alone (P = 0.027 for APOE ε4 + Plasma Aβ1–42, P = 0.005 for APOE ε4 + APOE ε2 + Plasma Aβ1–42). There was still a trend of better AUC compared with Plasma Aβ1–42 alone (P = 0.339 for APOE ε4 + Plasma Aβ1–42, P = 0.130 for APOE ε4 + APOE ε2 + Plasma Aβ1–42). Abbreviations: Aβ, Amyloid β; aMCI, amnestic mild cognitive impairment; AUC, area under the curve, AUC; ROC, receiver operating characteristics
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.