

Abstract
Protein kinases are important cellular signaling molecules involved in cancer and a multitude of other diseases. It is well known that inactive kinases display a remarkable conformational plasticity; however, the molecular mechanisms remain poorly understood. Conformational heterogeneity presents an opportunity but also a challenge in kinase drug discovery. The ability to predictively model various conformational states could accelerate selective inhibitor design. Here we performed a proton-coupled molecular dynamics study to explore the conformational landscape of a c-Src kinase. Starting from a completely inactive structure, the simulations captured all major types of conformational states without the use of a target structure, mutation, or bias. The simulations allowed us to test the experimental hypotheses regarding the mechanism of DFG flip, its coupling to the αC-helix movement, and the formation of regulatory spine. Perhaps the most significant finding is how key titratable residues, such as DFG-Asp, αC-Glu, and HRD-Asp, change protonation states dependent on the DFG, αC, and activation loop conformations. Our data offer direct evidence to support a long-standing hypothesis that protonation of Asp favors the DFG-out state and explain as to why DFG flip is also possible in simulations with deprotonated Asp. The simulations also revealed intermediate states, among which a unique DFG-out/α-C state formed as DFG-Asp is moved into a back pocket forming a salt bridge with catalytic Lys, which can be tested in selective inhibitor design. Our finding of how proton coupling enables the remarkable conformational plasticity may shift the paradigm of computational studies of kinases which assume fixed protonation states. Understanding proton-coupled conformational dynamics may hold a key to further innovation in kinase drug discovery.
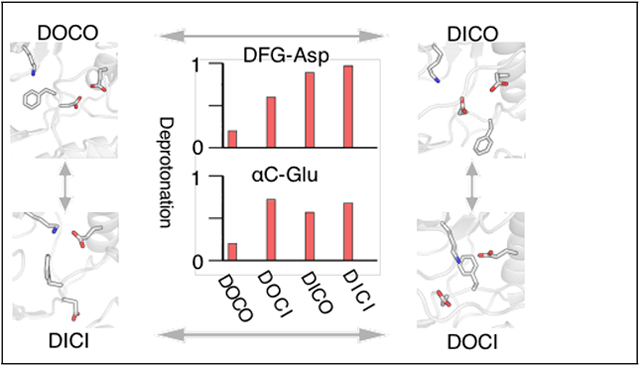
Graphical Abstract
INTRODUCTION
Protein kinases belong to one of the largest enzyme families and play critical roles in cellular signaling processes by phosphorylation of a tyrosine, threonine or serine residue.1 Dysregulation of kinase functions has been linked to cancer, inflammatory and other diseases,2 making kinases important pharmaceutical targets. Since the discovery of Imatinib, a small-molecule inhibitor targeting the tyrosine kinase c-Abl for the treatment of chronic myeloid leukemia, the rate of approval by the US Food and Drug Administration has been steadily increasing.3 Kinases display a remarkable conformational heterogeneity in the inactive form,4 which presents an opportunity but also a challenge in early drug discovery.2,5 In recent years, some highly selective inhibitors have been discovered; however, a general design strategy is lacking, as the molecular mechanism underlying selectivity was understood only after the crystal structure was obtained.5 Thus, the ability to predictively model kinase conformations can potentially fill in the gap and accelerate inhibitor design.
Protein kinases have a well-conserved catalytic or kinase domain,4 composed of a β-sheet rich N-lobe and an α-helical C-lobe, with the ATP and substrate binding sites located in between (Fig. 1). Kinase activation involves the changes of several structural features. The highly conserved Asp-Phe-Gly (DFG) motif is located at the beginning of the activation loop (A-loop). Here we focus on the c-Src tyrosine kinase. In the inactive DFG-out conformation, DFG-Asp (Asp404 in c-Src) sidechain points out of the ATP binding pocket, while the DFG-Phe (Phe405 in c-Src) sidechain points in. In the active DFG-in conformation, the sidechain positions of DFG-Asp and DFG-Phe are swapped, i.e., DFG-Asp points in and DFG-Phe points out. This large conformational change is known as the DFG flip. In the active DFG conformation, DFG-Asp often forms a salt bridge with the catalytic Lys (Lys295 in c-Src) located on the β3 strand of the N-lobe. Another structural feature involved in kinase activation is α-C-helix in the N-lobe. In the inactive αC-out conformation, αC-helix is far from the ATP binding site, while in the active αC-in conformation, αC-helix moves closer, allowing an extremely conserved Glu, αC-Glu (Glu310 in c-Src), to form a salt bridge with the catalytic Lys. Kinase activity also involves the catalytic loop or C-loop, which harbors the His-Arg-Asp (HRD) motif. The HRD-Asp is also known as the catalytic Asp.
Figure 1. Structure and major conformational states of the c-Src kinase.

Cartoon representation of the chicken c-Src kinase domain structure (pdb id 5K9I). The sequences of chicken and human c-Src kinase domains differ only by two amino acids: Met354 and Asp502 in chicken correspond to Thr357 and Glu505 in human, respectively. αC-helix, A-loop, and C-loop are colored orange, red and blue, respectively. Important residues discussed in this work are shown, Asp404, Phe405 (on DFG motif); Glu310 (αC-Glu); Lys295 (catalytic Lys); Met314, Leu325, His384, and Phe405 (R-spine); Arg385 and Arg409 (able to interact with αCGlu). A diagram indicates the four types of conformational states a kinase can adopt.
Active kinases adopt the highly similar DFG-in and αC-in (DICI) structures with an extended A-loop that allows peptide binding.4,6,7 Kornev and Taylor further proposed that kinase activation involves the alignment of two sets of hydrophobic residues connecting the N- and C-lobes, known as the regulatory (R) and catalytic (C) spines.1,8 R-spine is made up of 4 residues, Leu325, Met314, Phe405, and His384 in c-Src, while C-spine is assembled upon ATP binding and will not be further discussed. Contrasting active kinases, inactive kinases display a remarkable conformational plasticity.4 Based on the DFG and αC conformations, the inactive states can be classified into three types, DFG-out and αC-in (DOCI), DFG-in and αC-out (DICO), as well as the completely inactive DFG-out and αC-out (DOCO) type (Fig. 1). The detailed molecular mechanisms underlying the conformational transitions remain poorly understood.
Over the past decade, significant progress has been made in studying kinase conformational dynamics using MD simulations. In a pioneering work, Shan et al. performed unbiased MD to investigate the DFG flip in a tyrosine kinase c-Abl.9 Supported by kinetic experiments and mutagenesis, they proposed that the protonation state of DFG-Asp controls the DFG flip.9 Since this work, numerous MD studies have been published10–18 to investigate tyrosine kinases, particularly c-Src related to c-Abl. Intriguingly, the empirical structure-based pKa calculations by Lovera et. al11 and free energy simulations by Meng et al.12 suggested that the impact of protonation state on the DFG flip is negligible in c-Src.
While previous MD studies focused on the αC movement (DICI v.s. DICO)10,13,16 or the DFG-flip,11,18 knowledge of the entire conformational landscape and mechanisms of transitions remain incomplete. Importantly, previous MD studies have a major limitation in that protonation states of titratable sidechains were fixed. The mechanism of DFG flip was investigated by running independent simulations in which DFG-Asp was fixed in either protonated or deprotonated state.9,12,14,18 Such simulations cannot describe protonation-coupled conformational dynamics. In fact, DFG-Asp may not be the only residue that adopts a conformation-dependent protonation state. Supported by experiments, our most recent work19 demonstrated that Cys and Lys of some kinases may be deprotonated in the DOCO conformation at physiological pH. Additionally, HRD-His may not be neutral as often assumed in the conventional MD.
Inspired by the work of others10–18 and intrigued by the controversy of the protonation-dependent DFG flip, we set out to explore the conformational dynamics of the well-studied c-Src. We hypothesized that the conformational plasticity of kinases is related to the electrostatic networks and by exploiting various pH conditions we may be able to uncover the entire conformational landscape and elucidate the conformational transition mechanisms. To test this hypothesis, we carried out hybrid-solvent continuous constant pH molecular dynamics with pH replica exchange (hereafter abbreviated as CpHMD)20 for c-Src. CpHMD has been validated for a variety of systems, e.g., protein folding,21 enzyme dynamics and inhibitor binding,22,23 as well as conformational transitions of membrane channels and transporters.24–26 In this work, CpHMD simulations initiated from a completely inactive (DOCO) structure were able to directly sample both the DFG flip and αC-helix movement, allowing a detailed characterization of the conformational transitions among all four major states and the coupling to protonation state changes. Simulation data also allowed testing of the experimental hypotheses regarding the pathways of the DFG flip, coupling to αC movement, and R-spine assembly. Our work provided a detailed view of proton-coupled dynamics of kinases, revealing how conformational plasticity is achieved through electrostatic coupling in a tyrosine kinase.
RESULTS AND DISCUSSION
Conformational landscape of c-Src is pH dependent.
CpHMD simulations20 were initiated from the crystal structure of c-Src in the DOCO state (pdb id: 5K9I27). 48 replicas, each assigned with a pH condition from pH 3 to 9.5, underwent constant NPT all-atom molecular dynamics with periodic exchanges of conformational states. All 91 ionizable sidechains were allowed to titration in the simulations. The aggregate sampling time was about 1.8 μs.
Kinase conformational landscapes can be characterized in terms of two order parameters that describe the DFG and αC states. After testing out various order parameters proposed in the literature, we found that a pseudo torsion angle formed by Val402, Ala403, Asp404, and Phe405 (ζ(A403)), which was first proposed by Möbitz using PDB structure analysis,28 can unambiguously discriminate between the DFG-out (ζ ≥120°) and DFG-in (ζ<120°) states. We note, in a previous work,11 a salt bridge between Asp404 and Lys295 was used to define DFG-in conformations; however, this salt bridge does not necessarily indicate a DFG flip (see later discussion). Since Lys295–Glu310 salt-bridge is a characteristic of the active state, we used the distance between Lys295:NZ and Glu310:CD to define the αC-out (R>4.5Å) and αC-in (R<4.5Å) states, as in the work of Sultan et al.16 We note, the distance between Asp404 and Glu310, which was used in some literature28 and databases,29 does not always correlate with the Lys295–Glu310 salt-bridge formation and as such may not be an ideal order parameter.
Fig. 2a shows the free energy surface (FES) as a function of ζ and R for the simulation pH conditions 3–8. The horizontal and vertical lines divide the conformational landscape into four regions: upper right (DOCO), upper left (DOCI), lower right (DICO), and lower left (DICI). Strikingly, the FES is pH dependent and at pH 4 and 5 it displays minima in all four regions, suggesting that all four major conformational states are sampled, including the active DICI state, similar to the scatter plots made with the crystal structures.28 Convergence analysis of the protonation state sampling, conformational populations, FES, and transitions between different states at pH 5 are given in Fig. S1–S4. Comparisons to the crystal structure survey are given in Fig. S5 and S6. Note, the FES for pH 4.5 is similar to pH 5 (Fig. S6).
Figure 2. pH-dependent conformational landscape of c-Src.

(a) FES as a function of the pseudo torsion ζ(Ala403) and distance R between Lys295:NZ and Glu310:CD at different simulation pH. ζ(Ala403) is defined using the CA atoms of Val402, Ala403, Asp404, and Phe405. The free energy is calculated using -kTlnP(ζ, R), where P is probability density, k is Boltzmann constant, and T is simulation temperature (300 K). Horizontal and vertical lines are plotted at 120° and 4.5 Å to define DOCO, DOCI, DICO, and DICI states. The starting structure is indicated by a cross. (b) Representative snapshots representing the FES minima. Lys295, Glu310, Asp404 and Phe405 are shown.
To examine the various conformations, we extracted the snapshots from the minimum regions in the FES for pH 5 (Fig. 2b). The DOCO region displays a shallow valley (ζ of 200–240° and R of 4.5–14 Å), referred to as the DOCO state, and a local minimum (ζ of 150° and R of 6 Å), referred to as the DOCO′ state. In the DOCO state, Asp404 is flipped out, while the Lys295–Glu310 distance samples a wide range. In the non-canonical DOCO′ state, Asp404 is partially flipped in and Glu310 is moved closer to Lys295. A similar conformation can be seen in the crystal structure of an insulin receptor kinase (PDB ID: 1IRK).
The DOCI region displays a local minimum at ζ of 200–240° and R around 3.5 Å. In this DOCI state, Asp404 remains flipped out but the Lys295–Glu310 salt bridge is formed. The DOCI state is also called the Src-like inhibited state, as it was initially discovered for Src kinases, although it was later found for other kinases such as Abl.30 It is interesting to notice that the barrier between DOCI and DOCO states is low, about 1.5 kcal/mol based on the FES, suggesting that αC-helix is highly mobile. The DICO region displays a shallow valley covering ζ of 35–60° and R of 5–10 Å. Snapshots showed that DFG is flipped in, with Asp404 pointing at Lys295 but αC-helix remains further away.
The DICI region shows two local minima. In the canonical DICI state (ζ of 20 and R of about 3.5 Å), DFG is flipped in and both Asp404 and Glu310 form a salt bridge with Lys295. In the non-canonical DICI′ state (ζ of 20° and R of 3.5 Å), DFG is readily flipped in and Glu310 forms a salt bridge with Lys295; however, the Lys295–Asp404 salt bridge is absent. A similar conformation can be seen in the crystal structure of a c-Src kinase (PDB ID: 1YI6). We note, despite the absence of the latter, this state should be considered active, as Asp404 is close enough to the ATP binding pocket such that it can form the magnesium-mediated contacts with ATP, which is necessary for kinase function.8
pH-dependent R-spine formation is consistent with the DFG and αC activation.
We examined whether the simulations captured the dynamical behavior of R-spine and whether the behavior is also pH dependent. The FES in terms of the angle formed by Met314, Phe405, and His384 and the angle formed by Leu325, Phe405, and His384 was plotted for different pH conditions (Fig. 3a and Fig. S10). Depending on the pH, the FES showed 2–4 minimum regions. The minimum in the upper right corner of the FES (the two angles are 160–180°) represent the conformations with an intact R-spine and is most frequently sampled at pH 5, in agreement with the pH-dependent conformational land scape shown in Fig. 2. Snapshots also confirmed that conformations with an intact R-spine are indeed in the DICI state (Fig. 3b, top). We further examined another minimum region, where the two angles are 60–90°. Snapshots showed a unique DOCO conformation, where Asp404 is in a salt bridge with Lys295 but DFG flip has not taken place and αC helix remains out (Fig. 3b, bottom, see later discussion).
Figure 3. R-spine formation is consistent with the active DICI state.

(a) FES at pH 5 as a function of two angles formed between the CB atoms of the R-spine residues. The FES’s at other pH are given in Fig. S8. (b) Representative snapshots from the two minimum regions. The R-spine residues are in green. Asp404, Lys295, and Glu310 are in magenta.
pH-dependent populations of four major conformational states.
To quantify the pH-dependence of conformational dynamics, we calculated the percentage populations of the DOCO, DOCI, DICO, and DICI conformations as defined by the four FES regions (Fig. 2a) at different pH. Consistent with the FES plots, the DICI population displays a maximum at pH4.5–5 and it is reduced to about half, as pH increases to 7, and to nearly zero, as pH decreases to 3 (Fig. 4a). Thus, our data suggests that the active conformation of unphosphorylated c-Src is not the predominate state at physiological pH, consistent with a previous MD study, which showed that phophorylation stabilizes several structural features locking the kinase in the active conformation.31 Note, the shapes of the pH profiles from the self-controlled CpHMD simulations are reliable; however, the population values are likely not accurate due to the limited sampling time.
Figure 4. pH-dependent conformational populations of c-Src and protonation states of important titratable residues.

(a) Percentage population of the DICI conformations as a function of pH. (b) Percentage populations of the DFG-in (black) and αC-in (red) conformations as a function of pH. (c) Probability of deprotonation for Asp404, Glu310, and Asp386 in the four types of conformational states at pH 4 (red), 5 (green), and 6 (blue). Lys295 and His384 sample only the protonated (charged) state below pH 7.
To examine the pH effects on the DFG flip and αC-helix motion individually, we calculated the pH-dependent percentage populations of the DFG-in (DICO and DICI) and αC-in (DOCI and DICI) conformations (Fig. 4b). Similar to the DICI population, the DFG-in population displays a maximum near pH 5 and it decreases to almost zero at pH 3 and reaches a minimum at pH 6.5–7. Interestingly, the pH profile of the αC-in population is identical as that of the DFG-in population in the pH range 3 to 5; however, above pH 5 the population plateaus. These data suggest that the pH dependence of the conformational activation is related to the pH-dependent dynamics of the DFG motif and αC-helix. With increasing pH, the populations of DFG-in and αC-in states increase due to the greater extent of deprotonation for Asp404 and Glu310 (see later analysis and discussion). Intriguingly, the populations of DFG-in and DICI states decrease above pH 5, which can be attributed to the formation of a DOCO intermediate (see later analysis and discussion).
Protonation states of key residues are dependent on the DFG and αC conformations.
To understand the origin of the pH dependence, we examined the protonation states of titratable residues located near regions of large conformational changes, including Asp404, Glu310, Asp386, His384, and Lys295. Interestingly, at pH 4 to 5, where maximum activation was observed in the simulation, Lys295 and His384 are always charged, while Asp404, Glu310, and Asp386 sample both deprotonated and protonated states (Fig. 4c). Note, in all previous MD studies,9–11,13,14,16,18 Glu310 and Asp386 were fixed in deprotonation state while Asp404 was fixed in either protonated or deprotonated state.
Our simulations showed that the protonation states of Asp404, Glu310, and Asp386 are conformation dependent. For Asp404, the deprotonation probabilities are 20%, 60%, 90% and 100% at pH 4; 60%, 80%, 100%, and 100% at pH 5, for DOCO, DOCI, DICO, and DICI conformations, respectively (Fig. 4c). Thus, Asp404 is fully deprotonated in the DFG-in state but it can be either protonated or deprotonated in the DFG-out state, which supports the hypothesis that Asp404 protonation favors the DFG-out state.9 Furthermore, the protonation state of Asp404 is affected by the αC position when DFG is inactive, as evident from the increased fraction of deprotonated Asp404 going from DOCO (20% at pH 4 and 60% at pH 5) to DOCI (60% at pH 4 and 80% at pH5) (Fig. 4c).
For Glu310, the deprotonation probabilities are 20%, 70%, 60% and 70% at pH 4; 80%, 100%, 70%, and 100% at pH 5, for DOCO, DOCI, DICO, and DICI conformations, respectively (Fig. 4c). Thus, Glu310 samples the deprotonated state more frequently in the αC-in vs. the αC-out conformation, irrespective of the DFG conformation. It is worthwhile noticing that both Asp404 and Glu310 remain deprotonated above pH 6, irrespective of the DFG and αC conformations (see later discussion). With regards to the deprotonation probability of Asp386, it follows the increasing order, DOCO < DICI < DOCI/DICO at all three pH conditions. It is important to point out a limitation of the CpHMD simulations. The calculated pKa’s of carboxylic groups forming electrostatic interactions, e.g., Asp404 and Glu310, may be underestimated, by perhaps 1–1.5 units, based on our previous studies.20,22,32 Thus, the calculated peak activation pH may be too low by up to 1–1.5 units, similar to our previous studies of aspartyl proteases.22,32
A-loop can open and close in the inactive states but remains open in the active state.
Besides the DFG motif and αC helix, conformational dynamics of the A-loop also plays an important role in kinase activation and catalysis.6 Crystal structures and solution studies showed that in the inactive state,33 the A-loop is flexible and can adopt many closed as well as open conformations, while in the active state (with or without phosphorylation), the A-loop is extended and open to allow peptide binding.7,34 The closed-to-open conformational switch of the A-loop was also explored in several previous MD studies.31,35–37 Although our simulation started from a DOCO structure with a closed A-loop, the three inactive states sampled both closed and open A-loop, where the loop root-mean-square deviation (RMSD) is greater than 5 Å from the starting structure (Fig. S7). In contrast, the active DICI state sampled only the open A-loop with the RMSD of 6–14 Å (Fig. S7). These data are consistent with the experimental observations and suggest that the A-loop motion is correlated with the dynamics of DFG motif and αC helix. The fact that the A-loop in the simulated DICI state remains open but displays considerable flexibility is consistent with the notion that phosphorylation locks the A-loop in an ordered, extended conformation.7,31,38,39
Protonation state of DFG-Asp in the DFG-out conformation also depends on the A-loop conformation.
While Asp404 is always deprotonated in the DFG-in conformation due to the salt bridge with Lys295, it can be either protonated or deprotonated in the DFG-out conformation depending on the local dielectric environment. For example, Asp404 is in different environments in the two well-known crystal structures of c-Abl, which are both in the Src-like inhibited (DOCI) state but differ significantly in the A-loop conformation.30,33 In the structure with an open A-loop (pdb id: 1OPK33), Asp404 points to a hydrophobic pocket, whereas in the structure with a closed A-loop (pdb id: 1OJ33), Asp404 is exposed to solvent.
Visualization of the simulated DOCI snapshots supports a correlation between the A-loop conformation and the environment of Asp404. In fact, the conformations of the A-loop and DFG motif in the snapshot shown in Fig. 5a bear a high resemblance to those in 1OPK and the snapshot in Fig. 5b resembles 1OPJ. To test the hypothesis that the protonation state of Asp404 may be different in the two substates, DOCI (A-open) and DOCI (A-closed), we calculated the probability of deprotonating Asp404 at pH 3, 4, 5, and 6. Indeed, Asp404 is more protonated in the A-loop open state as compared to the A-loop closed state (Fig. 5c). Thus, the protonation state of Asp404 in the DFG-out state is further modulated by the A-loop conformation which perturbs the local environment of Asp404.
Figure 5. A-loop can open and close in the DOCI state and the e_ect on the protonation state of Asp404.

(a) A simulation snapshot of the DOCI state with an open A-loop (blue) and Asp404 pointing to a hydrophobic pocket, similar to a crystal structure of c-Abl (PDB ID: 1OPK33). (b) A snapshot of the DOCI state with a closed A-loop (red) and Asp404 exposed to solvent, similar to a different c-Abl structure (PDB ID: 1OPJ33). (c) Probability of Asp404 deprotonation in the DOCI state with open (blue) and closed (red) A-loop at different pH. A-loop (residues 404–432) was considered open if the RMS deviation from the starting DOCO crystal structure (PDB ID: 5K9I27) is greater than 5 Å. Asp404, Phe405, Glu310, and Lys295 are shown in the stick model.
DFG flip follows a pathway hypothesized based on crystal structures.
Next, we investigated the mechanism of DFG flip. Based on a crystal structure survey, Möbitz proposed two pathways for the DFG out-to-in transition.28 In pathway 1, the DFG flip is accomplished by a rotation around the backbone Φ angle of DFG-Asp, while in pathway 2, the flip is made possible by a rotation around the angle of DFG-1 (Fig. S6). Considering the presence of crystal structures showing intermediate conformations, Möbitz further proposed28 that pathway 2 is more accessible, particularly for kinases with a small DFG-1 such as Gly, Cys, or Ala. Our calculated FES’ for pH 4–6 as a function of ϕ(Asp404) and ψ(Ala403) are very similar to the scatter plot based on the PDB crystal structures28 (Fig. 6 and Fig. S8). Accordingly, the DFG flip involves either a switch in ϕ(Asp404), i.e., pathway 1, or a gradual change in ψ(Ala403), i.e., pathway 2. The FES for pH 4 and 5 shows two intermediate states in pathway 2. At pH 5, both intermediate states and the final state appear to be more populated. Thus, our data supports the crystal structure based hypothesis28 and further suggests that the increased DFG-in population at pH 5 may be due to the presence the intermediate states which helps lower the barrier of the DFG flip. Note, these intermediates are absent in our survey of the PDB structures of the Src family kinases, likely due to the very small number of them (Fig. S6).
Figure 6. Two pathways of the DFG flip.

Free energy surface of c-Src as a function of ϕ(Asp404) and ψ(Ala403) at different simulation pH conditions. A diagonal line separates the DFG-in and DFG-out conformations. Two pathways for the DFG flip are indicated.
DFG flip involves intermediates and increased deprotonation of DFG-Asp.
Now we switch back to the pseudo dihedral angle ζ(A403) and the Asp404–Glu310 distance to further investigate the DFG flip and its coupling with the αC motion. The FES for pH 5 (Fig. 7a) is very similar to the scatter plot based on the crystal structures of the Aurora A kinase,28 showing a gradual decrease in ζ and initially a slight enlargement of the Asp404–Glu310 distance, before Asp404 completely flips into the ATP binding site and Phe405 flips out. The decrease in ζ(Ala403) is also accompanied by a decrease in ψ(Ala403), consistent with the analysis shown in Fig. 6. Starting from the DFG-out state, the FES shows a pathway with two intermediate states, in which Asp404 is flipped to varying degrees (shown as 2 and 3 in Fig. 7a and 7b). The deprotonated fraction of Asp404 increases from 70% for the starting DFG-out state and the first intermediate to 100% for the second intermediate and the final DFG-in state (Fig. 7b). This data is consistent with the increased degree of Asp404 deprotonation in the DOCO-to-DICO and DOCI-to-DICI transitions at pH 4 and 5 (Fig. 4).
Figure 7. DFG flip, intermediates, and coupling to the deprotonation of DFG-Asp.

(a) FES as a function of ζ(Ala403) and the Cα distance between Asp404 and Glu310 from the simulation at pH 5. Four minimum regions are labeled: 1 (ζ >120°); 2 (130° < ζ <180°); 3 (75° < ζ <130°); and 4 (ζ <75_). Snapshots capturing the DFG flip (blue, Asp404 pointing into the ATP binding site in the front cleft) and an off-pathway DOCO intermediate (magenta, Asp404 pointing into a back pocket) are overlaid on the starting structure (silver). The intermediate is most populated at pH 6 and 7 (Fig. S9). (b) Zoomed-in views of Asp404 and Phe405 in the DFG-out state, partially flipped, and DFG-in state. The probabilities of Asp404 deprotonation for the four minimum regions are given.
A novel DOCO off-pathway intermediate is formed when Asp404 moves into a back pocket.
In contrast to the FES at pH 5, the pathway stops at the partially flipped intermediates and does not progress further below pH 4 or above pH 7 (Fig. S9). Interestingly, at pH 6 and 7, the FES displays a minimum region, where ζ(Ala403) remains large (about the same as the starting crystal structure) but the Asp404–Glu310 distance is significantly widened from about 10 Å in the crystal structure to 14–16 Å (Fig. S9 and dashed circle in Fig. 7a). Snapshots showed that, instead of entering the ATP binding site in the front cleft, Asp404 is moving into the second binding pocket in the back cleft (BP-II29,40) and forms a salt bridge with Lys295. This unique DFG-out conformation is further stabilized by a salt bridge between Glu310 and Arg409 in the A-loop, which locks the αC helix in the out position, similar to the starting crystal structure (Fig. 7a and Fig. 8a). This unique DOCO conformation has not been discussed in the literature, but we identified a similar conformation in the crystal structure of a Src kinase, where the Lys295–Asp404 salt bridge is formed but the coordinates of Arg409 are missing (PDB ID: 2QI8, see Fig. S9).
Figure 8. Salt-bridge interactions of Glu310 impact the DFG flip.

(a) Probability of forming the R409–E310 salt bridge in the DFG-in (blue) and DFG-out (red) states as a function of simulation pH. A representative snapshot of the R409–E310 salt bridge in a DFG-out state is shown on the top. (b) Probability of forming the R385–E310 salt bridge in the DFG-in (blue) and DFG-out (red) states as a function of simulation pH. A representative snapshot of the R385–E310 salt bridge in the DFG-in state is shown on the top. A salt bridge was considered present if the minimum distance between the carboxylate oxygens of Glu and the guanidinium nitrogens of Arg is at most 4 Å.
Salt-bridge interactions of Glu310 impact DFG motion.
Interestingly, the Arg409–Glu310 salt bridge only occurs in the DFG-out conformation and the probability follows a bell-shaped pH profile with a maximum of about 15% at pH 6.5 (Fig. 8a). Recall that the DFG-in population has an inverted bell-shaped pH profile with a minimum at pH 6.5 (Fig. 4a). Thus, our data suggest that above pH 5, the Arg409–Glu310 salt bridge not only stabilizes the inactive αC position but may also stabilize the DFG-out conformation.
In the crystal structures of c-Src kinases,41 Glu310 can also form a salt bridge with Arg385 (HRD-Arg), stabilizing the αC-out state. In our simulations, this salt bridge is additionally correlated with the DFG movement. Below pH 5.5, the salt bridge occurred only in the DFG-out conformations, mostly when Asp404 was rotated clockwise (on-pathway to DFG-in). Above pH 5.5, the salt bridge was found in both DFG-out and DFG-in conformations with the latter dominating above pH 6.5 (Fig. 8b). Thus, the interaction with Arg385 may stabilize the inactive DFG conformations below pH 5.5 but it does not have such effect at higher pH.
CONCLUDING DISCUSSION
Replica-exchange CpHMD simulations were able to capture the conformational activation of the c-Src kinase starting from a completely inactive DOCO crystal structure without prior knowledge of the active state or the use of mutation or biasing potential. Simulations reproduced all four major types of conformations observed in the PDB crystal structures. Strikingly, the DICI state and R-spine formation are most frequently sampled by the simulation with a pH near 5, where DFG-Asp, αC-Glu, and catalytic Asp adopt conformation-dependent protonation states. Our simulations showed that a protonated Asp is only compatible with DFG-out conformations (this is why DFG activation does not occur at low pH), while a deprotonated Asp is compatible with both DFG-in and DFG-out conformations. From the conformational perspective, DFG-in conformations are only compatible with deprotonated Asp, while DFG-out conformations are compatible with both protonated and deprotonated Asp. These data offer direct evidence to support the hypothesis that DFG flip is dependent on the protonation state of Asp9 and explain why the DFG in-to-out flip was observed only with the protonated Asp in some MD simulations.9,14,18
The coupling between protonation equilibria and conformational exchanges can be rationalized by the fact that in the active state DFG-Asp is near the charged catalytic Lys, whereas in an inactive state it can be in a variety of local environment, e.g., buried in a hydrophobic pocket or solvent exposed and proximal to the charged HRD-His. As an example, our analysis showed that in the DOCI state, DFG-Asp is more protonated when A-loop is open as compared to when it is closed. Thus, our work suggests a model comprising three states, DFG-in(Asp−), DFG-out(AspH), and DFG-out(Asp−), which differs from the model of Shan et. al.,9 which includes DFG-in(AspH) instead of DFG-out(Asp−). The existence of both DFG-out(AspH) and DFG-out(Asp−) and the thermally accessible barrier between DFG-out and DFG-in states (at most 3 kcal/mol in our replica-exchange simulations, Fig. 2a) may explain why the DFG flip to the DFG-out state was observed with a deprotonated Asp in a recent MD simulation.17
It was initially puzzling that the active-state population decreases as pH increases above pH 5. Our analysis revealed that without a DFG flip that involves concerted motion of the backbone of several residues, DFG-Asp can move into a back pocket and form a salt bridge with the catalytic Lys, resulting in a novel DOCO type of intermediate, which is further stabilized by a salt bridge between αC-Glu and Arg409. Interactions of αC-Glu with Arg409 and Arg385 have been known to stabilize the inactive αC position, based on crystal structures41 and MD studies.10,13 To our best knowledge, it is the first time that their impact on the DFG flip was revealed, further explaining the coupling between the DFG and αC-helix motions.
The current work is subject to several caveats. Although the pH dependence is robust, the relative conformational populations are likely not accurate due to the short sampling time and the presence of four states. The peak of the active pH range might be too low by 1–1.5 units, resulting from over-stabilization of the deprotonated carboxylates, e.g., DFG-Asp, αC-Glu, and catalytic Asp. We also note that while Cys, Tyr, and Lys sidechains were allowed to titrate, they did not change protonation states, i.e., Cys and Tyr remained neutral and Lys remained charged in the entire simulation pH range. Although this may well be true, there is a chance that the default (solution) protonation states were overly favored, as the pKa shifts for Cys, Tyr, and Lys using the hybrid-solvent CpHMD method have not been extensively validated. Our recent study employing an improved GB model demonstrated that the catalytic Lys can be neutral and some Cys can be charged in the DOCO conformations of some kinases.19
Our work does not consider ATP and magnesium binding, which is known to impact the conformational dynamics and catalytic activities of kinases. Phosphates and magnesium ions form an electrostatic network with key residues such as the catalytic Lys, DFG-Asp, and catalytic Asp,42 and stabilize the kinase core structure in the active state.1 The adenine ring of ATP forms multiple contacts with the protein, among which a β7 hydrophobic residue, and results in the assembly of the so-called catalytic spine required for catalysis.8,43 Additionally, our work does not consider phosphorylation, which has been demonstrated to stabilize the important kinase structural features in the active conformation for the cAMP-dependent protein kinase A38,39 and MAP kinase ERK2.44 Nonetheless, our data suggesting that the active conformation is not the predominant state for the unphosphorylated c-Src kinase at physiological pH is consistent with the above experimental studies and a previous computational work based on free energy simulations.31 Similar to inhibitor binding,45,46 phosphorylation likely induces a population shift, making the active state the most stable one.7
Notwithstanding the caveats, by exploring the proton-coupled conformational dynamics of the c-Src kinase, our simulations provided an intimate view of how key structural elements are electrostatically coupled to enable a remarkable conformational plasticity. Proton titration at peak activation pH enables the breakage and formation of electrostatic contacts at the heart of the kinase catalytic machinery and allows assembly and disassembly of the structural features required for kinase functions. In contrast, at lower pH protonated DFG-Asp and αC-Glu disfavor the active DFG and αC conformations, while at higher pH a DOCO intermediate is stabilized by the salt bridges involving the deprotonated DFG-Asp and αC-Glu. The novel DOCO intermediate found in this work can be tested in selective inhibitor design. Perhaps the most significant finding is that key residues change protonation states to facilitate the conformational transitions. The current work represents the beginning of a journey to understand the role of proton titration in kinase conformational dynamics. Many questions remain to be answered. For example, what are the electrostatic roles of HRD-Asp, HRD-His, and catalytic Lys? How does phosphorylation and inhibitor binding perturb the electrostatic networks and conformational landscape? Understanding proton-coupled conformational dynamics may hold a key to further innovation in kinase drug discovery. The current work also demonstrated the predictive capability of replica-exchange CpHMD to generate various conformational states for structure-based kinase inhibitor design.
METHODS and PROTOCOLS
System preparation.
The kinase domain structure of the chicken Src protein was retrieved from the Protein Data Bank (PDB ID 5K9I27 with resolution 2.5 Å). The crystal structure has two subunits; the backbone root-mean-square-deviation between the two subunits is 1.0 Å and the orientations of titratable sidechains are highly similar. Subunit B was used as the starting structure, as it has fewer unresolved residues (411–423), which we added using MODELLER (version 9.11).47 100 structures were generated and the one with the lowest MODELLER objective function score was used. The small molecule was removed. Crystallographic water molecules within 3.5 Å of any protein heavy atoms were kept. The N-terminus was acetylated and the C-terminus was amidated. Hydrogen atoms were added using the HBUILD facility48 in the CHARMM package (version c37a2).49 Dummy hydrogen atoms were added to Asp and Glu sidechains and placed in the syn positions, following the original continuous constant pH molecular dynamics protocol by Khandogin and Brooks.50
The protein was then placed in a truncated octahedral water box with a minimum distance of 10 Å between the protein and edges of the water box, resulting in a unit cell with a lattice parameter of 81.7 Å. Water molecules within 2.6 Å of any existing heavy atoms were deleted, as in the standard CHARMM protocol,49 which has been used in the previous kinase simulations by Roux and coworkers.12,17,31 The solvated system contained a total of 40447 atoms. No explicit ions were added, as the system net charge is dependent on the pH and as such cannot be compensated by a fixed number of ions. In lieu of explicit counterions, the particle-mesh (PME) Ewald calculation includes a neutralizing plasma to maintain charge neutrality. Note, as the net charge of the system is very small at pH 7 (−2 according to the estimate by the PropKa calculation51). Our previous simulations showed that the neglect of explicit ions has a negligible effect on the protonation-state calculations.20 To account for the salt effects on protonation and deprotonation, th physiological ionic strength of 0.15 M was taken into account via an approximated Debye-Hückel form52 in the generalized Born calculations for the hybrid-solvent CpHMD, consistent with our previous work on aspartyl proteases.23,53,54 To relax the hydrogen positions, the system was minimized by the steepest descent method followed by the adopted basis Newton-Raphson method with the protein heavy atoms fixed and subsequently harmonically restrained with a force constant of 5.0 kcal/mol·Å2.
Molecular dynamics protocol.
Molecular dynamics simulations were carried out using the hybrid-solvent continuous constant pH molecular dynamics (CpHMD) method and the pH-based replica-exchange protocol20 in the CHARMM package (version c37a2).49 The all-atom CHARMM22/CMAP55,56 and the CHARMM-style TIP3P water model57 were used to represent the protein and water, respectively. In the hybrid-solvent CpHMD simulations, the conformational degrees of freedom are propagated using explicit solvent, while the λ particles, which represent the protonation degrees of freedom, are propagated in the GBSW generalized Born model58 with the default setting in the PHMD module.20 All ionizable sidechains, including 10 Asp, 27 Glu, 3 His, 6 Cys and 13 Tyr, 15 Lys, and 17 Arg, were allowed to titrate in the CpHMD simulations.
The Leap-frog integrator was used to propagate the spatial coordinates. The SHAKE algorithm59 was applied to bonds involving hydrogen atoms to allow for a 2-fs time step. Simulations were conducted under the periodic boundary conditions and constant NPT ensemble at ambient temperature (300 K) and pressure (1 atm), controlled by the Nóse-Hoover thermostat60,61 and the Langevin piston pressure-coupling algorithm,62 respectively. Switching function was applied to the van der Waals potentials from 10 to 12 Å. Electrostatic potentials were computed using the particle–mesh Ewald method63,64 with a real-space cutoff of 12 Å and a sixth-order interpolation with approximately 1-Å grid spacing. The cutoffs for generating the nonbonded and image lists were 14 and 16 Å, respectively. The nonbond neighbor and image lists were heuristically updated during the simulations. The λ dynamics were governed by Langevin dynamics with a collision frequency of 5 ps−1 and updated every 10 MD steps to allow for water relaxation.20 The mass of the λ particles was set to 10 atomic mass units (default setting).
The energy minimized system was heated from 100 to 300 K within 20 ps, with the protein heavy atoms harmonically restrained with a force constant of 5 kcal/mol Å2. Following heating, the system was equilibrated for 30 ps under harmonic restraints, where the force constant was gradually reduced from 5 (10 ps), to 2 (10 ps), and 1 kcal/mol Å2 (10 ps). In the heating and equilibration stages, CpHMD was turned on at the crystallization pH 6.1.27 In the production run, the pH-based replica exchange protocol was applied. 48 replicas were placed in the pH range 3–9.5, with 0.25 or 0.125 pH intervals. The exchanges of conformational states between neighboring pH replicas were attempted every 500 MD steps or 1 ps. Each replica was subject to NPT molecular dynamics for 62 ns, resulting in an aggregate sampling time of 1,824 μs. Data was collected after every exchange attempt (500 MD steps or 1 ps). Unless otherwise specified, the last 20 ns simulation for each replica was used for analysis.
Supplementary Material
Acknowledgement
We acknowledge financial support provided by the National Institutes of Health (GM098818).
Footnotes
Supporting Information Available
Supporting Information contains supplementary tables and figures.
References
- (1).Kornev AP; Taylor SS Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci 2015, 40, 628–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ferguson FM; Gray NS Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov 2018, 17, 353–376. [DOI] [PubMed] [Google Scholar]
- (3).Klaeger S; Heinzlmeir S; Wilhelm M; Polzer H; Vick B; Koenig P-A; Reinecke M; Ruprecht B; Petzoldt S; Meng C; Zecha J; Reiter K; Qiao H; Helm D; Koch H; Schoof M; Canevari G; Casale E; Depaolini SR; Feuchtinger A; Wu Z; Schmidt T; Rueckert L; Becker W; Huenges J; Garz A-K; Gohlke B-O; Zolg DP; Kayser G; Vooder T; Preissner R; Hahne H; Tõnisson N; Kramer K; Götze K; Bassermann F; Schlegl J; Ehrlich H-C; Aiche S; Walch A; Greif PA; Schneider S; Felder ER; Ruland J; Médard G; Jeremias I; Spieker-mann K; Kuster B The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Huse M; Kuriyan J The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [DOI] [PubMed] [Google Scholar]
- (5).Müller S; Chaikuad A; Gray NS; Knapp S The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol 2015, 11, 818–821. [DOI] [PubMed] [Google Scholar]
- (6).Nolen B; Taylor S; Ghosh G Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell 2004, 15, 661–675. [DOI] [PubMed] [Google Scholar]
- (7).Tong M; Seeliger MA Targeting Conformational Plasticity of Protein Kinases. ACS Chem. Biol. 2015, 10, 190–200. [DOI] [PubMed] [Google Scholar]
- (8).Kornev AP; Haste NM; Taylor SS; Eyck LFT Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shan Y; Seeliger MA; Eastwood MP; Frank F; Xu H; Jensen MO; Dror RO; Kuriyan J; Shaw DE A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yang S; Banavali NK; Roux B Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. USA 2009, 106, 3776–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lovera S; Sutto L; Boubeva R; Scapozza L; Dölker N; Gervasio FL The Different Flexibility of c-Src and c-Abl Kinases Regulates the Accessibility of a Druggable Inactive Conformation. J. Am. Chem. Soc 2012, 134, 2496–2499. [DOI] [PubMed] [Google Scholar]
- (12).Meng Y; lin Lin Y; Roux B Computational Study of the “DFG-Flip” Conformational Transition in c-Abl and c-Src Tyrosine Kinases.J. Phys. Chem. B 2015, 119, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shukla D; Meng Y; Roux B; Pande VS Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun 2014, 3, 3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Foda ZH; Shan Y; Kim ET; Shaw DE; Seeliger MA A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat. Commun 2015, 6, 5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sala GL; Riccardi L; Gaspari R; Cavalli A; Hantschel O; Vivo MD HRD Motif as the Central Hub of the Signaling Network for Activation Loop Autophosphorylation in Abl Kinase. J. Chem. Theory Comput 2016, 12, 5563–5574. [DOI] [PubMed] [Google Scholar]
- (16).Sultan MM; Kiss G; Pande VS Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem 2018, 10, 903–909. [DOI] [PubMed] [Google Scholar]
- (17).Meng Y; Gao C; Clawson DK; Atwell S; Russell M; Vieth M; Roux B Predicting the Conformational Variability of Abl Tyrosine Kinase using Molecular Dynamics Simulations and Markov State Models. J. Chem. Theory Comput 2018, 14, 2721–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hanson SM; Georghiou G; Thakur MK; Miller WT; Rest JS; Chodera JD; Seeliger MA What Makes a Kinase Promiscuous for Inhibitors? Cell Chem. Biol 2019, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu R; Yue Z; Tsai C-C; Shen J Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc 2019, 141, 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wallace JA; Shen JK Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yue Z; Shen J pH-Dependent cooperativity and existence of a dry molten globule in the folding of a miniprotein BBL. Phys. Chem. Chem. Phys 2018, 20, 3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ellis CR; Shen J pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc 2015, 137, 9543–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Harris RC; Tsai C-C; Ellis CR; Shen J Proton-Coupled Conformational Allostery Modulates the Inhibitor Selectivity for β-Secretase. J. Phys. Chem. Lett 2017, 8, 4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huang Y; Chen W; Dotson DL; Beckstein O; Shen J Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA. Nat. Commun 2016, 7, 12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen W; Huang Y; Shen J Conformational Activation of a Transmembrane Proton Channel from Constant pH Molecular Dynamics.J. Phys. Chem. Lett 2016, 7, 3961–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yue Z; Chen W; Zgurskaya HI; Shen J Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J. Chem. Theory Comput 2017, 13, 6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc 2017, 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Möbitz H The ABC of protein kinase conformations. Biochim. Biophys. Acta 2015, 1854, 1555–1566. [DOI] [PubMed] [Google Scholar]
- (29).van Linden OPJ; Kooistra AJ; Leurs R; de Esch IJP; de Graaf C KLIFS: A Knowledge-Based Structural Database To Navigate Kinase–Ligand Interaction Space. J. Med. Chem 2014, 57, 249–277. [DOI] [PubMed] [Google Scholar]
- (30).Levinson NM; Kuchment O; Shen K; Young MA; Koldobskiy M; Karplus M; Cole PA; Kuriyan J A Src-Like Inactive Conformation in the Abl Tyrosine Kinase Domain. PLoS Biol 2006, 4, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Meng Y; Roux B Locking the Active Conformation of c-Src Kinase through the Phosphorylation of the Activation Loop. J. Mol. Biol 2014, 426, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ellis CR; Tsai C-C; Lin F-Y; Shen J Conformational dynamics of cathepsin D and binding to a small-molecule BACE1 inhibitor. J. Comput. Chem 2017, 38, 1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nagar B; Hantschel O; Young MA; Scheffzek K; Veach D; Bornmann W; Clarkson B; Superti-Furga G; Kuriyan J Structural Basis for the Autoinhibition of c-Abl Tyrosine Kinase. Cell 2003, 112, 859–871. [DOI] [PubMed] [Google Scholar]
- (34).Adams JA Activation Loop Phosphorylation and Catalysis in Protein Kinases: Is There Functional Evidence for the Autoinhibitor Model? Biochemistry 2003, 42, 601–607. [DOI] [PubMed] [Google Scholar]
- (35).Huang H; Zhao R; Dickson BM; Skeel RD; Post CB αC Helix as a Switch in the Conformational Transition of Src/CDK-like Kinase Domains. J. Phys. Chem. B 2012, 116, 4465–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lovera S; Morando M; Pucheta-Martinez E; MartinezTorrecuadrada JL; Saladino G; Gervasio FL Towards a Molecular Understanding of the Link between Imatinib Resistance and Kinase Conformational Dynamics. PLoS Comput. Biol 2015, 11, e1004578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Haldane A; Flynn WF; He P; Vijayan R; Levy RM Structural propensities of kinase family proteins from a Potts model of residue co-variation. Protein Sci 2016, 25, 1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Steichen JM; Iyer GH; Li S; Saldanha SA; Deal MS; Virgil L Woods, J.; Taylor, S. S. Global Consequences of Activation Loop Phosphorylation on Protein Kinase A. J. Biol. Chem 2010, 285, 3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Steichen JM; Kuchinskas M; Keshwani MM; Yang J; Adams JA; Taylor SS Structural Basis for the Regulation of Protein Kinase A by Activation Loop Phosphorylation. J. Biol. Chem 2012, 287, 14672–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Liao JJ-L Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J. Med. Chem 2007, 50, 409–424. [DOI] [PubMed] [Google Scholar]
- (41).Xu W; Doshi A; Lei M; Eck MJ; Harrison SC Crystal Structures of c-Src Reveal Features of Its Autoinhibitory Mechanism. Mol. Cell 1999, 3, 629–638. [DOI] [PubMed] [Google Scholar]
- (42).Gerlits O; Weiss KL; Blakeley MP; Veglia G; Taylor SS; Kovalevsky A Zooming in on protons: Neutron structure of protein kinase A trapped in a product complex. Sci. Adv 2019, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kim J; Ahuja LG; Chao F-A; Xia Y; McClendon CL; Kornev AP; Taylor SS; Veglia G A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv 2017, 3, e1600663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xiao Y; Lee T; Latham MP; Warner LR; Tanimoto A; Pardi A; Ahn NG Phosphorylation releases constraints to domain motion in ERK2. Proc. Natl. Acad. Sci. USA 2014, 111, 2506–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Vogtherr M; Saxena K; Swen Hoelder SG; Betz M; Schieborr U; Pescatore B; Robin M; Delarbre L; Langer T; Wendt KU; Schwalbe H NMR Characterization of Kinase p38 Dynamics in Free and Ligand-Bound Forms. Angew. Chem 2006, 45, 993–997. [DOI] [PubMed] [Google Scholar]
- (46).Kar G; Keskin O; Gursoy A; Nussinov R Allostery and population shift in drug discovery. Curr. Opin. Pharmacol 2010, 10, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Fiser A; Do RKG; Săli A Modeling of loops in protein structures. Protein Sci 2000, 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brünger AT; Karplus M Polar hydrogen positions in proteins: Empirical energy placement and neutron diffraction comparison. Proteins 1988, 4, 148–156. [DOI] [PubMed] [Google Scholar]
- (49).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Khandogin J; Brooks III, Constant CL pH molecular dynamics with proton tautomerism. Biophys. J 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Søndergaard CR; Mats HM Olsson MR; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- (52).Khandogin J; Brooks III, Toward CL the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 2006, 45, 9363–9373. [DOI] [PubMed] [Google Scholar]
- (53).Ellis CR; Tsai C-C; Hou X; Shen J Constant pH Molecular Dynamics Reveals pH-Modulated Binding of Two Small-Molecule BACE1 Inhibitors. J. Phys. Chem. Lett 2016, 7, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Henderson JA; Harris RC; Tsai C-C; Shen J How Ligand Protonation State Controls Water in Protein-Ligand Binding. J. Phys. Chem. Lett 2018, 9, 5440–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL Jr.; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prod-hom B; Reiher WE III; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiórkiewicz-Kuczera J; Yin D; Karplus M All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (56).MacKerell AD Jr.; Feig M; Brooks CL III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (57).Durell SR; Brooks BR; Ben-Naim A Solvent-Induced Forces between Two Hydrophilic Groups. J. Phys. Chem 1994, 98, 2198–2202. [Google Scholar]
- (58).Im W; Lee MS; Brooks III, Generalized CL Born model with a simple smoothing function. J. Comput. Chem 2003, 24, 1691–1702. [DOI] [PubMed] [Google Scholar]
- (59).Ryckaert JP; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (60).Nosé S A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys 1984, 52, 255–268. [Google Scholar]
- (61).Hoover WG Canonical dynamics: Equilibration phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- (62).Feller SE; Zhang Y; Pastor RW; Brooks BR Constant pressure molecular dynamics simulation: The Langevin piston method.J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- (63).Darden T; York D; Pedersen L Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (64).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald mesthod. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.