

Abstract
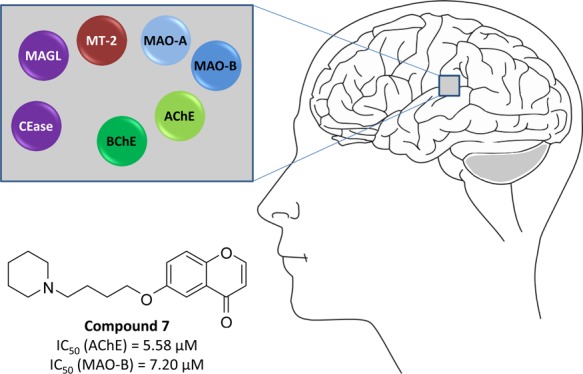
The complex nature of multifactorial diseases, such as Morbus Alzheimer, has produced a strong need to design multitarget-directed ligands to address the involved complementary pathways. We performed a purposive structural modification of a tetratarget small-molecule, that is contilisant, and generated a combinatorial library of 28 substituted chromen-4-ones. The compounds comprise a basic moiety which is linker-connected to the 6-position of the heterocyclic chromenone core. The syntheses were accomplished by Mitsunobu- or Williamson-type ether formations. The resulting library members were evaluated at a panel of seven human enzymes, all of which being involved in the pathophysiology of neurodegeneration. A concomitant inhibition of human acetylcholinesterase and human monoamine oxidase B, with IC50 values of 5.58 and 7.20 μM, respectively, was achieved with the dual-target 6-(4-(piperidin-1-yl)butoxy)-4H-chromen-4-one (7).
Introduction
The treatment of multifactorial diseases requires interactions with several biological targets. Multitarget compounds combine activity at more than a single biological target in one molecule. They offer two or more pharmacophores, which are either structurally overlapping or separated by a spacer. The design of single-molecule multitarget drugs has become an increasingly attractive option with a variety of advantages, compared to combination therapies.1,2 The concept and application of multitarget small molecules (MSMs) include the simultaneous modulation of receptors and the inhibition of enzymatic systems implemented in the progress of the disease.2−4 Neurodegenerative diseases are intensively investigated employing multitarget approaches. Alzheimer’s disease (AD) is a neurodegenerative process characterized by a progressive decline of memory and learning capabilities resulting from complex and multifactorial, but not completely identified, biological events. Among these, oxidative stress and a deficit of neurotransmitters as well as neuronal death play a key role.5 Even the most substantiated and investigated hypotheses of AD, beta-amyloid and tau, have not resulted in Alzheimer’s drug breakthrough yet.6
Several strategies are currently applied for AD therapy; acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), as well as monoamine oxidases (MAO) A and B, are investigated as MSM targets for AD treatment.2,4 From these, AChE still remains a valuable target with clinical success in AD treatment, as the selective loss of cholinergic neurons in AD results in a deficit of acetylcholine in specific brain regions that mediate cognitive functions. Inhibition of AChE increases the acetylcholine concentration in the synaptic cleft, thus enhancing central cholinergic neurotransmission. In addition, the noncholinergic actions of AChE might affect the aggregation of toxic amyloid peptides.2 MAO enzymes catalyze the deamination of neurotransmitters and thereby release reactive oxygen species. These enzymes contribute to the symptoms of dementia and dopamine degradation. MAO inhibitors possess neuroprotective effects, reduce oxidative stress, and are frequently administered in the therapy of neurodegenerative disorders such as AD and Parkinson’s disease.7,8 A variety of dual-targeting compounds has been developed combining the moieties for AChE and MAO inhibition in one molecule.2,4
A recent communication9 on the pharmacological profile of a highly advanced hit compound for AD (contilisant, Figure 1) showed it to be an antioxidant, permeable, strong neuroprotective agent, able to increase the level of neurotransmitters in the brain. This tetratarget compound inhibited human cholinesterases (AChE, IC50 = 0.53 μM; BChE, IC50 = 1.69 μM) and human monoamine oxidases (MAO-B, IC50 = 0.078 μM; MAO-A, IC50 = 0.145 μM) and specifically modulated the human histamine H3 (Ki = 10.8 nM, antagonist) and sigma 1 receptors (Ki = 65.2 nM, agonist) in a manner suitable for AD therapy. Being active in appropriate AD animal models, contilisant exceeded the efficacy of the marketed drug donepezil.9
Figure 1.

Design of chromenone-based MSMs.
The current research project was directed toward MSMs, which, in contrast to contilisant, might act as reversible MAO inhibitors. For this purpose, we planned to substitute the N-methyl propargylamine motif responsible for irreversible MAO inhibition by a typical pharmacophore for reversible inhibition, serving at the same time as the central heterocyclic core replacing the indole. This strategy can be realized with several building blocks such as 4H-3,1-benzothiazin-4-ones,10 3,4-dihydro-2(1H)-quinolinones,11 and coumarins.12−14 We decided to incorporate the chromen-4-one structure into our envisaged molecules (Figure 1), as chromen-4-ones constitute well-established reversible MAO inhibitors.14−18 Moreover, as potential targets for our MSMs, we considered further human enzymes which are involved in pathophysiological processes of neurodegenerative diseases (see below), that is, matriptase-2 (MT-2), monoacylglycerol lipase (MAGL), and cholesterol esterase (CEase).
Results and Discussion
The present versatile design allowed us to introduce linkers of various lengths attached to differently sized cyclic (or acyclic) tertiary amine moieties. The envisaged series 1–28 consists of groups of four compounds each with the same amino motif and n methylene units ranging from 2 to 5 (Table 1). In order to synthesize these compounds, the Laboratory of Medicinal Chemistry (IQOG, CSIC) established a collaboration with Eli Lilly’s Open Innovation Drug Discovery program, granting access to their Automated Synthesis Lab. This program featured a globally accessible, remote-controlled automated lab that can perform many different types of chemical transformations.19 All steps are controlled by a customized software and can be monitored in real time. The lab works continuously and is equipped with conventional and microwave heating, liquid-handling robots, work-up stations, liquid chromatography–mass spectrometry for reaction monitoring, solvent evaporation, and sample preparation for automated purification.
Table 1. Inhibition of Human Enzymes by Chromenones 1–28.
![]()


The method for the preparation of the compounds is given in parentheses.
n.i. (no inhibition) refers to IC50 > 100 μM (for AChE and BChE) and to <5% inhibitory activity@10 μM (for MAO-A, MT-2, MAGL, and CEase), respectively.
The products were synthesized through the formation of an aryl–alkyl ether bond, either by Mitsunobu- or Williamson-type etherification (Scheme 1, Supporting Information). Thus, the readily available 6-hydroxychromen-4-one20 and the respective amino alcohol were subjected to the Mitsunobu reaction using triphenylphosphine and di-tert-butyl azodicarboxylate (DBAD) in dichloromethane (Method A, Table 1).21 Combining triphenylphosphine and the dialkyl azodicarboxylate into one reagent, (cyanomethylene)tributylphosphorane (CMBP) has been shown to be a particularly effective phosphorane ylide for the Mitsunobu reaction.21,22 This reagent was applied and the reactions were performed in toluene at 100 °C for the preparation of additional final compounds (Method B, Table 1). A further subset of products was synthesized by following a two-step route, involving the O-alkylation of 6-hydroxychromen-4-one with an appropriate α,ω-dibromoalkane to produce an ether intermediate, which was used in situ, owing to its remaining alkyl bromide moiety, for the subsequent N-alkylation of the amino component (Method C, Table 1).23,24 In case of compounds 4, 24, and 27, we observed an unexpected cleavage of the vinylogous lactone, and the final compounds were therefore produced through an acid-promoted, microwave-assisted ring closure (see Supporting Information).
Scheme 1. Synthetic Entries to Chromenones 1–28.

Moreover, (R)-2-methylpyrrolidine was alkylated with 2-bromoethanol and 3-bromo-1-propanol, respectively, and the hydroxyl groups were converted to mesylates which were finally used for the ether bond formation providing 25 and 26 (Method D, Table 1). All compounds were isolated by a strong cation exchange (SCX) procedure, applying a benzenesulfonic acid-bonded sorbent. The basic products were liberated with ammoniacal methanol and obtained in moderate yields and excellent purity (see Supporting Information).
The in vitro measurements of cholinesterase activity were carried out following the method of Ellman, with 5,5′-dithio-bis-2-nitrobenzoic acid applied as a chromogenic reagent to detect the thiocholine liberated from acetylthiocholine and butyrylthiocholine, respectively.25 The majority of the chromenones showed inhibitory activity against AChE (Table 1), with the most advantageous amino moieties being pyrrolidine (1–4), piperidine (5–8), and azepane (21–24). More polar termini, that is, morpholine (9–12) and N-methylpiperazine (13–16), caused a reduction of potency. Derivatives 25–28 were weaker AChE inhibitors than their isomeric counterparts 5–8, probably because of the sterically more demanding 2-methylpyrrolidine moiety. Our data revealed a clear influence of the linker length on AChE inhibition, with four- and five-membered carbon-containing chains (n = 4 and 5) being more favorable than ethylene and trimethylene linkers (n = 2 and 3). This trend was observed throughout all the seven groups of tested chromenones.
AChE-catalyzed substrate cleavage follows an acyl transfer mechanism and is mediated by the catalytic triad Ser203–His447–Glu334. While the esteratic site is located at the bottom of a deep and narrow gorge, the peripheral anionic site (PAS) on the AChE surface is involved in ligand guiding and noncholinergic functions. It is formed by the residues of Tyr72, Asp74, Tyr124, Trp286, and Tyr341, which are clustered around the entrance of the active site gorge. It was expected from structurally related AChE inhibitors,13,18,24,26 including donepezil,27 that the accommodation of the active chromenones is stabilized by cation−π interactions between the inhibitor’s protonated tertiary amine and the aromatic residues of Trp86 and Phe338 at the midpoint of the gorge. However, based on our docking investigations with the prototypical compound 7, it aligned in an inversely oriented manner with the chromenone interacting with the catalytic anionic site (CAS) and the amine pointing toward the PAS (Figure 2A). Specifically, the chromenone system of 7 is assumed to undergo π–π stacking with the indole side chain of Trp86 and hydrophobic interactions with His447 of the catalytic triad. The piperidinium moiety of 7 is engaged in a π–cation formation with Tyr341 and an attractive charge interaction with Asp74. The analogous inhibitor 24 bearing a linkage elongated by one methylene group and an azepanium moiety showed a similar accommodation in the active site of AChE (see Supporting Information, Figure S1).
Figure 2.

Docking studies of compound 7 with AChE and MAO-B. (A) Proposed docked pose of 7 in the active site of AChE (PDB-ID: 1B41). Compound 7 is rendered with cyan sticks. The side-chain conformations of active site amino acids are shown, including the mobile residues of Trp86, Tyr72, Asp74, Tyr124, Trp286, Tyr337, and Tyr341. The catalytic triad is colored in green, the oxyanion hole in magenta, the CAS and the acyl-binding pocket in orange, and the PAS in light blue. (B) Binding mode prediction of compound 7 in the active site of MAO-B (PDB-ID: 2V5Z). Compound 7 is rendered with yellow sticks. The amino acid residues that are involved in inhibitor binding are shown. The FAD cofactor and the six water molecules as an integral part of the MAO-B structure model are displayed as orange sticks and light red balls, respectively. Intermolecular interactions are shown as dashed lines according to their type: green, hydrogen bond; blue, hydrogen bond to water; purple, π–π T-shaped interaction; pink, π–alkyl and alkyl–alkyl contacts.
Mixed-type inhibition is known for donepezil28 and several other AChE inhibitors.4,29,30 We chose active chromenone inhibitors out of each group, namely 3, 8, 11, 15, 20, 24, and 27 (Table 2), and carried out kinetic measurements with different substrate and inhibitor concentrations. The dissociation constants Ki and αKi were obtained, where Ki describes the competitive and αKi the uncompetitive contribution to AChE inhibition. A pronounced competitive mode was mainly determined (α = 2.1–3.3), with the exception of 11 (α = 0.7), where the affinity to the enzyme–substrate complex may arise from interactions between the inhibitor and regions not involved in substrate binding.
Table 2. Kinetic Parameters of Selected Chromenones.
![]()
| AChE |
MAO-B | MAO-A | ||
|---|---|---|---|---|
| compd | Ki ± SE (μM) | αKi ± SE (μM) | IC50 ± SE (μM) | IC50 ± SE (μM) |
| 3 | 1.15 ± 0.27 | 2.82 ± 0.16 | 19.6 ± 0.9 | n.d.a |
| 7 | n.d. | n.d. | 7.20 ± 0.41 | n.d. |
| 8 | 1.26 ± 0.28 | 3.80 ± 0.61 | n.d. | n.d. |
| 9 | n.d. | n.d. | 6.73 ± 0.80 | n.d. |
| 10 | n.d. | n.d. | 1.18 ± 0.04 | 1.70 ± 0.22 |
| 11 | 18.6 ± 12.4 | 13.5 ± 4.1 | n.d. | n.d. |
| 15 | 2.06 ± 0.56 | 5.36 ± 0.70 | n.d. | n.d. |
| 20 | 1.61 ± 1.44 | 5.23 ± 1.46 | n.d. | n.d. |
| 21 | n.d. | n.d. | 3.99 ± 0.42 | n.d. |
| 24b | 4.70 ± 1.46 | 13.1 ± 2.5 | 19.9 ± 2.6 | 30.1 ± 3.2 |
| 27 | 9.23 ± 5.01 | 19.0 ± 4.1 | n.d. | n.d. |
Not determined.
Kinetic parameters of 24 at BChE: Ki ± SE = 0.0887 ± 0.0470 μM; αKi ± SE = 0.446 ± 0.088 μM. A selectivity ratio Ki(AChE)/Ki(BChE) of 53 was determined for 24.
Both cholinesterases have widespread but different brain distributions. AChE represents the major therapeutic target of cholinesterase inhibitors, whereas BChE is considered to act as a coregulator of the neurotransmitter acetylcholine. In AD patients, a significant increase of cortical BChE levels was found. In contrast to AChE activity, that of BChE progressively increases with the advance of dementia. Hence, BChE inhibition may provide additional benefits in AD therapy.2,31−33 We identified just two chromenones, 24 and 25, with stronger activity against BChE than AChE (Table 1). The most potent BChE inhibitor of the series, that is 24, bears the most extended aliphatic substituent of the present chromenone series, which obviously facilitates a preferred interaction with the active site of BChE being larger and able to accommodate bulkier substrates, compared to AChE (for docking studies with 24 in the active site of BChE, see Supporting Information, Figure S2).34
Compound 24 was selected for a kinetic study at BChE in the presence of different substrate and inhibitor concentrations. The mixed-type inhibition as noted for AChE was also observed for BChE with a pronounced competitive portion and an α value of 5.0 for this dual cholinesterase inhibitor (Table 2).
The two MAO isozymes are involved in neurodegenerative diseases. Hyperactivity of MAO enzymes, decreasing the concentration of dopaminergic and serotoninergic neurotransmitters, has been observed in AD patients.4,7,35 The expression of MAO-B in neuronal tissues increases with aging, resulting in increased dopamine metabolism and higher levels of hydrogen peroxide and oxidative free radicals. MAO-B is therefore thought to play a pivotal role in the progression of AD.7,18 As MAO-A contributes to toxin-induced apoptosis and striatal damage of neuronal cells, this isoform also represents a target for the development of MSMs.4,18 In this study, chromenones 1–28 were investigated for the inhibition of MAO-B and MAO-A using human recombinant enzymes and applying the Amplex Red monoamine oxidase assay.12 At a single concentration of 10 μM, several chromenones exhibited MAO-B inhibitory activity, whereas only one compound, namely 10, was notably active against MAO-A (Table 1). The best four compounds at MAO-B (7, 9, 10, and 21), as well as two selected because of their anticholinesterase activity (3 and 24), were subjected to concentration-dependent analyses (Table 2). IC50 values between 1 and 7 μM were obtained for the most potent MAO-B inhibitors out of this series. The reversibility of MAO-B inhibition was exemplarily demonstrated for compound 7 (see Supporting Information, Figure S3). Chromenone 10 turned out to be equipotent against both MAO isoforms (Table 2).
For 3-substituted coumarins and chromen-4-ones, a plausible binding mode within the MAO-B active site has been proposed where the unsubstituted face of the heterobicyclic core is directed to the flavin adenine dinucleotide (FAD) cofactor, in close proximity to Tyr398 and Tyr435.14,17 A similar accommodation of our 6-substituted chromen-4-ones with the fused heteroaromatic ring oriented toward FAD was expected and confirmed by an exemplary docking approach with inhibitor 7 (Figure 2B).
Human MT-2, a type-2 transmembrane serine protease, is the main proteolytic regulator of iron homeostasis. MT-2 cleaves hemojuvelin, a coreceptor for bone morphogenic proteins, which induces hepcidin transcription through a Smad signaling pathway. The inhibition of MT-2, leading to reduced plasma iron levels, was postulated as a new therapeutic opportunity to treat iron overload diseases, such as hemochromatosis and β-thalassemia.36,37 MT-2 is mainly expressed in the liver, but low expression levels have also been found in the brain.38 As brain iron accumulation is a characteristic feature in several neurodegenerative disorders such as AD,39 we included MT-2 in our multitarget screening. MT-2 exhibits primary substrate specificity for basic amino acids, and these chromenones, all of them with a basic nitrogen within the side chain, were considered as possible candidates for MT-2 inhibition. As a source for MT-2, we used stably transfected human embryonic kidney (HEK) cells as wild-type HEK cells do not produce MT-2.36 An established fluorometric assay with the peptidic substrate Boc–Gln–Ala–Arg–AMC was applied, and the formation of 7-amino-4-methylcoumarin (AMC) was monitored.40 However, none of the chromenones caused a noteworthy MT-2 inhibition (Table 1).
MAGL and CEase also belong to the family of serine hydrolases. MAGL preferably degrades monoacylglycerols including the endocannabinoid 2-arachidonyl glycerol that is hydrolyzed to glycerol and the eicosanoid arachidonic acid, a precursor of prostaglandins. 2-Arachidonyl glycerol is the endogenous ligand of the G-protein-coupled cannabinoid receptors 1 and 2.41 Inhibition of MAGL was shown to have protective effects in AD progression by preventing the formation and accumulation of amyloid-β plaques and the expression of BACE 1, maintaining the integrity of hippocampal synaptic structure and function, thus improving spatial learning and memory in mice.42 Cholesterol is a regulator of lipid organization, the precursor of neurosteroid biosynthesis, and important in the development and progression of AD.43 It was recently reported that the inhibition of CEase reduced the concentrations of amyloid-β42 oligomers and increased the concentrations of the corresponding monomers, which protected neurons against synapse damage.44 Thus, MAGL and CEase seemed to be adequate enzymes for the multitarget approach together with cholinesterases and MAO-A and B. For the quantification of the MAGL and CEase activities, the chromogenic 4-nitrophenyl butyrate and the fluorogenic 4-methylumbelliferyl butyrate were used.45 However, the chromenones 1–28 exhibited neither MAGL nor CEase inhibitory activity (Table 1).
Conclusions
In this study, we have selected seven human enzymes which can be considered to constitute relevant drug targets for an MSM approach for neurodegenerative diseases. Our inhibitor design provided a systematically combined library of chromen-4-ones employing structural features for affinity toward cholinesterases and monoamine oxidases. Screening of this library revealed chromenone 7 as a potent dual-target small molecule with single-digit micromolar IC50 values at the particularly AD-relevant enzyme combination of AChE and MAO-B. Another library member, 24, targeted both cholinesterases, and chromenone 10 showed inhibitory activity against both MAO isozymes. We expanded our screening portfolio by further enzymes which have hitherto not been considered for a multitarget application against Morbus Alzheimer and related disorders. Although not successful with the present set of compounds, we will focus on elaborating such an extended set of enzymes in future multitarget studies.
Acknowledgments
The authors thank Christin Vielmuth, Sabine Terhart-Krabbe, and Martin Mangold for support.
Glossary
Abbreviations
- AChE
acetylcholinesterase
- AD
Alzheimer’s disease
- AMC
7-amino-4-methylcoumarin
- BChE
butyrylcholinesterase
- CEase
cholesterol esterase
- CAS
catalytic anionic site
- CMBP
(cyanomethylene)tributylphosphorane
- DBAD
di-tert-butyl azodicarboxylate
- FAD
flavin adenine dinucleotide
- HEK
human embryonic kidney
- MAGL
monoacylglycerol lipase
- MAO
monoamine oxidase
- MSM
multitarget small molecule
- MT-2
matriptase 2
- PAS
peripheral anionic site
- SCX
strong cation exchange
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03409.
Docking studies; enzymatic assays for AChE, BChE, MAO-B, MAO-A, MT-2, MAGL, and CEase; general synthetic procedures; and specific analytical data for chromenones 1–28 (PDF)
Author Contributions
J.M.C. and M.G. designed and supervised the study. C.L., J.C., J.Y., J.M.A., E.S., M.A.M.G., C.D.B., T.V., F.M.D., and I.I. performed experiments. All authors contributed to data analyses. C.L., P.W.E., J.M.C., and M.G. wrote the manuscript. All authors have given approval to the final version of the manuscript.
J.M.C. thanks MINECO (grant SAF2015-65586-R), UCJC (grants 2015-12, 2014-35, and 2015-21), and EU (COST Action CA15135) for support.
The authors declare no competing financial interest.
This paper published ASAP on December 11, 2019 with an error in Table 2. The paper was revised and reposted on December 16, 2019.
Supplementary Material
References
- Proschak E.; Stark H.; Merk D. Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2019, 62, 420–444. 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- León R.; Garcia A. G.; Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- Oset-Gasque M. J.; Marco-Contelles J. Alzheimer’s Disease, the “One-Molecule, One-Target” Paradigm, and the Multitarget Directed Ligand Approach. ACS Chem. Neurosci. 2018, 9, 401–403. 10.1021/acschemneuro.8b00069. [DOI] [PubMed] [Google Scholar]
- Knez D.; Sova M.; Košak U.; Gobec S. Dual inhibitors of cholinesterases and monoamine oxidases for Alzheimer’s disease. Future Med. Chem. 2017, 9, 811–832. 10.4155/fmc-2017-0036. [DOI] [PubMed] [Google Scholar]
- Citron M. Alzheimer’s disease: strategies for disease modification. Nat. Rev. Drug Discovery 2010, 9, 387–398. 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B.; Gupta V. P.; Kumar V. A perspective on monoamine oxidase enzyme as drug target: challenges and opportunities. Curr. Drug Targets 2016, 18, 87–97. 10.2174/1389450117666151209123402. [DOI] [PubMed] [Google Scholar]
- Kumar B.; Kumar M.; Dwivedi A. R.; Kumar V. Synthesis, biological evaluation and molecular modeling studies of propargyl-containing 2,4,6-trisubstituted pyrimidine derivatives as potential Anti-Parkinson agents. ChemMedChem 2018, 13, 705–712. 10.1002/cmdc.201700589. [DOI] [PubMed] [Google Scholar]
- Bautista-Aguilera Ó. M.; Budni J.; Mina F.; Medeiros E. B.; Deuther-Conrad W.; Entrena J. M.; Moraleda I.; Iriepa I.; López-Muñoz F.; Marco-Contelles J. Contilisant, a tetratarget small molecule for Alzheimer’s disease therapy combining cholinesterase, monoamine oxidase inhibition, and H3R antagonism with S1R agonism profile. J. Med. Chem. 2018, 61, 6937–6943. 10.1021/acs.jmedchem.8b00848. [DOI] [PubMed] [Google Scholar]
- Stößel A.; Schlenk M.; Hinz S.; Küppers P.; Heer J.; Gütschow M.; Müller C. E. Dual targeting of adenosine A(2A) receptors and monoamine oxidase B by 4H-3,1-benzothiazin-4-ones. J. Med. Chem. 2013, 56, 4580–4596. 10.1021/jm400336x. [DOI] [PubMed] [Google Scholar]
- Meiring L.; Petzer J. P.; Petzer A. Inhibition of monoamine oxidase by 3,4-dihydro-2(1H)-quinolinone derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 5498–5502. 10.1016/j.bmcl.2013.08.071. [DOI] [PubMed] [Google Scholar]
- Mertens M. D.; Hinz S.; Müller C. E.; Gütschow M. Alkynyl-coumarinyl ethers as MAO-B inhibitors. Bioorg. Med. Chem. 2014, 22, 1916–1928. 10.1016/j.bmc.2014.01.046. [DOI] [PubMed] [Google Scholar]
- Farina R.; Pisani L.; Catto M.; Nicolotti O.; Gadaleta D.; Denora N.; Soto-Otero R.; Mendez-Alvarez E.; Passos C. S.; Muncipinto G.; Altomare C. D.; Nurisso A.; Carrupt P.-A.; Carotti A. Structure-based design and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J. Med. Chem. 2015, 58, 5561–5578. 10.1021/acs.jmedchem.5b00599. [DOI] [PubMed] [Google Scholar]
- Fonseca A.; Reis J.; Silva T.; Matos M. J.; Bagetta D.; Ortuso F.; Alcaro S.; Uriarte E.; Borges F. Coumarin versus chromone monoamine oxidase B inhibitors: quo vadis?. J. Med. Chem. 2017, 60, 7206–7212. 10.1021/acs.jmedchem.7b00918. [DOI] [PubMed] [Google Scholar]
- Legoabe L. J.; Petzer A.; Petzer J. P. Selected C7-substituted chromone derivatives as monoamine oxidase inhibitors. Bioorg. Chem. 2012, 45, 1–11. 10.1016/j.bioorg.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Legoabe L. J.; Petzer A.; Petzer J. P. Inhibition of monoamine oxidase by selected C6-substituted chromone derivatives. Eur. J. Med. Chem. 2012, 49, 343–353. 10.1016/j.ejmech.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Reis J.; Cagide F.; Chavarria D.; Silva T.; Fernandes C.; Gaspar A.; Uriarte E.; Remião F.; Alcaro S.; Ortuso F.; Borges F. Discovery of new chemical entities for old targets: insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J. Med. Chem. 2016, 59, 5879–5893. 10.1021/acs.jmedchem.6b00527. [DOI] [PubMed] [Google Scholar]
- Reis J.; Cagide F.; Valencia M. E.; Teixeira J.; Bagetta D.; Pérez C.; Uriarte E.; Oliveira P. J.; Ortuso F.; Alcaro S.; Rodríguez-Franco M. I.; Borges F. Multi-target-directed ligands for Alzheimer’s disease: discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. 10.1016/j.ejmech.2018.07.056. [DOI] [PubMed] [Google Scholar]
- Godfrey A. G.; Masquelin T.; Hemmerle H. A remote-controlled adaptive medchem lab: an innovative approach to enable drug discovery in the 21st Century. Drug Discov. Today 2013, 18, 795–802. 10.1016/j.drudis.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Chand K.; Tiwari R. K.; Kumar S.; Shirazi A. N.; Sharma S.; Van der Eycken E. V.; Parmar V. S.; Parang K.; Sharma S. K. Synthesis, antiproliferative, and c-Src kinase inhibitory activities of 4-oxo-4H-1-benzopyran derivatives. J. Heterocycl. Chem. 2015, 52, 562–572. 10.1002/jhet.2106. [DOI] [Google Scholar]
- Swamy K. C. K.; Kumar N. N. B.; Balaraman E.; Kumar K. V. P. P. Mitsunobu and related reactions: advances and applications. Chem. Rev. 2009, 109, 2551–2651. 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- Tsunoda T.; Nagino C.; Oguri M.; Itô S. Mitsunobu-type alkylation with active methine compounds. Tetrahedron Lett. 1996, 37, 2459–2462. 10.1016/0040-4039(96)00318-8. [DOI] [Google Scholar]
- Nowakowska Z.; Kędzia B.; Schroeder G. Synthesis, physicochemical properties and antimicrobial evaluation of new (E)-chalcones. Eur. J. Med. Chem. 2008, 43, 707–713. 10.1016/j.ejmech.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Sun Y.; Guo Y.; Wang Z.; Huang L.; Li X. Dual functional cholinesterase and MAO inhibitors for the treatment of Alzheime’s disease: synthesis, pharmacological analysis and molecular modeling of homoisoflavonoid derivatives. J. Enzyme Inhib. Med. Chem. 2015, 31, 389–397. 10.3109/14756366.2015.1024675. [DOI] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V.; Featherstone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Bautista-Aguilera O. M.; Samadi A.; Chioua M.; Nikolic K.; Filipic S.; Agbaba D.; Soriano E.; de Andrés L.; Rodríguez-Franco M. I.; Alcaro S.; Ramsay R. R.; Ortuso F.; Yañez M.; Marco-Contelles J. N-Methyl-N-((1-methyl-5-(3-(1-(2-methylbenzyl)piperidin-4-yl)propoxy)-1H-indol-2-yl)methyl)prop-2-yn-1-amine, a new cholinesterase and monoamine oxidase dual inhibitor. J. Med. Chem. 2014, 57, 10455–10463. 10.1021/jm501501a. [DOI] [PubMed] [Google Scholar]
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Nochi S.; Asakawa N.; Sato T. Kinetic study on the inhibition of acetylcholinesterase by 1-benzyl-4-((5,6-dimethoxy-1-indanon)-2-yl)methylpiperidine hydrochloride (E2020). Biol. Pharm. Bull. 1995, 18, 1145–1147. 10.1248/bpb.18.1145. [DOI] [PubMed] [Google Scholar]
- Piazzi L.; Rampa A.; Bisi A.; Gobbi S.; Belluti F.; Cavalli A.; Bartolini M.; Andrisano V.; Valenti P.; Recanatini M. 3-(4-{[Benzyl(methyl)amino]methyl}phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: a dual function lead for Alzheimer’s disease therapy. J. Med. Chem. 2003, 46, 2279–2282. 10.1021/jm0340602. [DOI] [PubMed] [Google Scholar]
- Elsinghorst P. W.; Härtig W.; Goldhammer S.; Grosche J.; Gütschow M. A gorge-spanning, high-affinity cholinesterase inhibitor to explore β-amyloid plaques. Org. Biomol. Chem. 2009, 7, 3940–3946. 10.1039/b909612d. [DOI] [PubMed] [Google Scholar]
- Lane R. M.; Potkin S. G.; Enz A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- Montanari S.; Bartolini M.; Neviani P.; Belluti F.; Gobbi S.; Pruccoli L.; Tarozzi A.; Falchi F.; Andrisano V.; Miszta P.; Cavalli A.; Filipek S.; Bisi A.; Rampa A. Multitarget strategy to address Alzheimer’s disease: design, synthesis, biological evaluation, and computational studies of coumarin-based derivatives. ChemMedChem 2016, 11, 1296–1308. 10.1002/cmdc.201500392. [DOI] [PubMed] [Google Scholar]
- Montanari S.; Scalvini L.; Bartolini M.; Belluti F.; Gobbi S.; Andrisano V.; Ligresti A.; Di Marzo V.; Rivara S.; Mor M.; Bisi A.; Rampa A. Fatty acid amide hydrolase (FAAH), acetylcholinesterase (AChE), and butyrylcholinesterase (BuChE): networked targets for the development of carbamates as potential anti-Alzheimer’s disease agents. J. Med. Chem. 2016, 59, 6387–6406. 10.1021/acs.jmedchem.6b00609. [DOI] [PubMed] [Google Scholar]
- Knez D.; Brus B.; Coquelle N.; Sosič I.; Šink R.; Brazzolotto X.; Mravljak J.; Colletier J.-P.; Gobec S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23, 4442–4452. 10.1016/j.bmc.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Kumar B.; Dwivedi A. R.; Sarkar B.; Gupta S. K.; Krishnamurthy S.; Mantha A. K.; Parkash J.; Kumar V. 4,6-Diphenylpyrimidine derivatives as dual inhibitors of monoamine oxidase and acetylcholinesterase for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 252–265. 10.1021/acschemneuro.8b00220. [DOI] [PubMed] [Google Scholar]
- Stirnberg M.; Maurer E.; Horstmeyer A.; Kolp S.; Frank S.; Bald T.; Arenz K.; Janzer A.; Prager K.; Wunderlich P.; Walter J.; Gütschow M. Proteolytic processing of the serine protease matriptase-2: identification of the cleavage sites required for its autocatalytic release from the cell surface. Biochem. J. 2010, 430, 87–95. 10.1042/bj20091565. [DOI] [PubMed] [Google Scholar]
- Gitlin-Domagalska A.; Mangold M.; Dębowski D.; Ptaszyńska N.; Łęgowska A.; Gütschow M.; Rolka K. Matriptase-2: monitoring and inhibiting its proteolytic activity. Future Med. Chem. 2018, 10, 2745–2761. 10.4155/fmc-2018-0346. [DOI] [PubMed] [Google Scholar]
- Hooper J. D.; Campagnolo L.; Goodarzi G.; Truong T. N.; Stuhlmann H.; Quigley J. P. Mouse matriptase-2: identification, characterization and comparative mRNA expression analysis with mouse hepsin in adult and embryonic tissues. Biochem. J. 2003, 373, 689–702. 10.1042/bj20030390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaldan S.; Bush A. I.; Devos D.; Rolland A. S.; Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. 10.1016/j.freeradbiomed.2018.09.033. [DOI] [PubMed] [Google Scholar]
- Häußler D.; Mangold M.; Furtmann N.; Braune A.; Blaut M.; Bajorath J.; Stirnberg M.; Gütschow M. Phosphono bisbenzguanidines as irreversible dipeptidomimetic inhibitors and activity-based probes of matriptase-2. Chem.—Eur. J. 2016, 22, 8525–8535. 10.1002/chem.201600206. [DOI] [PubMed] [Google Scholar]
- Long J. Z.; Cravatt B. F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011, 111, 6022. 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.; Zhang J.; Wu Y.; Wang D.; Feng G.; Tang Y.-P.; Teng Z.; Chen C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329. 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibhatla R. M.; Hatcher J. F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008, 49, 241–268. 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale-Owen H.; Bate C. Cholesterol ester hydrolase inhibitors reduce the production of synaptotoxic amyloid-β oligomers. Biochim. Biophys. Acta, Mol. Basis Dis. 2018, 1864, 649. 10.1016/j.bbadis.2017.12.017. [DOI] [PubMed] [Google Scholar]
- Dato F. M.; Sheikh M.; Uhl R. Z.; Schüller A. W.; Steinkrüger M.; Koch P.; Neudörfl J.-M.; Gütschow M.; Goldfuss B.; Pietsch M. ω-Phthalimidoalkyl aryl ureas as potent and selective inhibitors of cholesterol esterase. ChemMedChem 2018, 13, 1833–1847. 10.1002/cmdc.201800388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.