

Abstract
Analyses of publicly available structural data reveal interesting insights into the impact of the three‐dimensional (3D) structures of protein targets important for discovery of new drugs (e.g., G‐protein‐coupled receptors, voltage‐gated ion channels, ligand‐gated ion channels, transporters, and E3 ubiquitin ligases). The Protein Data Bank (PDB) archive currently holds > 155,000 atomic‐level 3D structures of biomolecules experimentally determined using crystallography, nuclear magnetic resonance spectroscopy, and electron microscopy. The PDB was established in 1971 as the first open‐access, digital‐data resource in biology, and is now managed by the Worldwide PDB partnership (wwPDB; http://www.wwpdb.org). US PDB operations are the responsibility of the Research Collaboratory for Structural Bioinformatics PDB (RCSB PDB). The RCSB PDB serves millions of http://rcsb.org users worldwide by delivering PDB data integrated with ∼40 external biodata resources, providing rich structural views of fundamental biology, biomedicine, and energy sciences. Recently published work showed that the PDB archival holdings facilitated discovery of ∼90% of the 210 new drugs approved by the US Food and Drug Administration 2010–2016. We review user‐driven development of RCSB PDB services, examine growth of the PDB archive in terms of size and complexity, and present examples and opportunities for structure‐guided drug discovery for challenging targets (e.g., integral membrane proteins).
Keywords: GPCR, integral membrane proteins, ion channel, Protein Data Bank, protein structure and function, structural biology, structure‐guided drug discovery, transporter, ubiquitin ligase
1. OVERVIEW OF THE PDB ARCHIVE, THE WWPDB, AND THE RCSB PDB
The Protein Data Bank (PDB) archive was established in 1971 as the first open‐access digital‐data resource in the biological sciences with seven protein structures.1, 2 Current PDB archival holdings encompass >155,000 atomic‐level structures of proteins, DNA, and RNA, experimentally determined by macromolecular X‐ray crystallography (MX: ∼90%), nuclear magnetic resonance spectroscopy (NMR: ∼9%), and three‐dimensional electron microscopy (3DEM: ∼1%). Nearly three quarters (∼73%) of PDB structures also include one or more ligands (e.g., enzyme cofactors and inhibitors, US Food and Drug Administration (FDA) approved drugs, and metals), and ∼10% of PDB structures include one or more carbohydrate components. Virtually, all of these public‐domain structures were determined with the support of research funding from governments or private philanthropies, and the PDB archive is now widely regarded as an international public good. Replacement value of current PDB archival holdings is conservatively estimated at >15 billion US dollars.3
The PDB archive is jointly managed by the Worldwide PDB partnership (wwPDB; http://www.wwpdb.org),4 consisting of the Research Collaboratory for Structural Bioinformatics (RCSB) PDB,5, 6 PDB Japan (PDBj),7 PDB in Europe (PDBe),8 and BioMagResBank.9 The wwPDB operates under an international agreement (http://wwpdb.org/about/agreement). Adhering to the FAIR principles of findability, accessibility, interoperability, and reusability,10 under management by the wwPDB partners, the single global archive of macromolecular data is disseminated to the scientific community without charge or restrictions on usage.
US PDB operations are the responsibility of the RCSB PDB (RCSB PDB; http://rcsb.org) with financial support from the National Science Foundation, the National Institute of General Medical Sciences, the National Cancer Institute, the National Institute of Allergy and Infectious Disease, and Department of Energy. RCSB PDB team members are hosted by Rutgers, the State University of New Jersey, the San Diego Supercomputer Center at the University of California San Diego, and the University of California San Francisco. The RCSB PDB also serves as the global Archive Keeper, responsible for ensuring disaster recovery of PDB data and coordinating weekly archival updates among wwPDB partners in Europe and Asia.
RCSB PDB serves millions of users worldwide, primarily through the web portal at http://rcsb.org. The website, as described in Nucleic Acids Research,6 provides tools and services to access and explore PDB content. Each week, all PDB structural data are integrated with ∼40 external data resources to provide rich, up‐to‐date structural views of fundamental biology, biomedicine, and energy sciences. Data can be searched and explored through individual Structure Summary Pages, or as groups of structures displayed in tabular reports.
Since RCSB PDB users extend well beyond experts in structural biology,11, 12 our website features are designed to enable finding a variety of structures related to a particular topic using search tools (e.g., by sequence, sequence similarity, small molecule name). The website also offers alternatives to searching, such as the Browse by Annotation tool that organizes PDB structures into hierarchical trees based upon several different classifications, including Anatomical Therapeutic Chemical drug classification system developed by the World Health Organization Collaborating Centre for Drug Statistics Methodology (http://www.whocc.no/atc_ddd_index/); protein residue modifications in the PDB archive using the protein modification ontology from the Proteomics Standards Initiative13; and Biological Process, Cellular Component, and Molecular Function based upon descriptions from the Gene Ontology (GO) Consortium14 mapped to corresponding PDB structures by the SIFTS initiative.15
Different visualization options are available. Protein Feature View offers graphical summaries of full‐length UniProt16 protein sequences and how they correspond to PDB entries, together with annotations from external databases (such as Pfam),17 homology model information,18, 19 predicted regions of protein disorder, and hydrophobic regions. Rapid 3D visualization of structures large and small is possible using the NGL viewer,20 which includes specialized options for viewing ligand interactions and electron density maps. Many RCSB PDB features available on http://rcsb.org are also provided as Web Services supporting programmatic access by increasing numbers of users.
A separate website, http://pdb101.rcsb.org (“101,” as in an entry‐level course), hosts educational materials that encourage learning about proteins and nucleic acids in 3D. A main focus is the Molecule of the Month series,21 currently in its 20th year of “telling molecular stories” about structure and function. Other materials include molecular origami paper models, posters, animations, and curricular materials. A “Guide to Understanding PDB Data” is built around more PDB‐specific information: PDB data, visualizing structures, reading coordinate files, and potential challenges (including biological assembly versus asymmetric unit).
All RCSB PDB activities are supported by robust infrastructure that ensures 24/7/365 support of millions of PDB data depositors and users worldwide. A full description of RCSB PDB services has been published,6 along with various analyses of the impact of structural biologists, the PDB archive, and the RCSB PDB.12, 22, 23
2. GROWTH IN THE SIZE AND COMPLEXITY OF THE PDB ARCHIVE
Figure 1 illustrates the growth in the PDB archive since 2000. Atomic coordinates for >11,200 new structures together with experimental data/metadata (∼7.6% year‐on‐year growth) were made available in 2018. Most of these new structures were determined using MX (∼88.8%), with the remainder determined by 3DEM (∼7.6%), and NMR (∼3.5%). The number of 3DEM structures populating the archive has been growing rapidly since structural biologists ushered in the “resolution revolution”24 (Figure 1b). Starting in 2016, annual 3DEM structure depositions now exceed NMR structure depositions. Global data deposition statistics, maintained from 2000 onwards, are updated on a weekly basis (http://www.wwpdb.org/stats/deposition).
Figure 1.
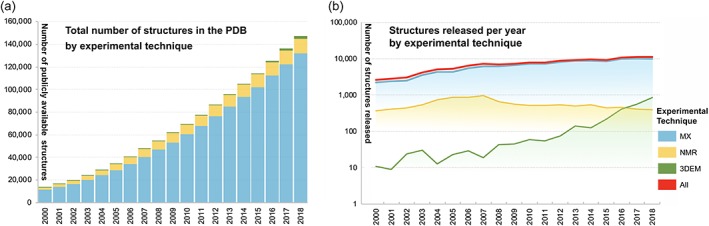
Growth of PDB archive, 2000–2018. (a) Total number of structures publicly available each year by experimental method and (b) new structures released annually by experimental method, shown using logarithmic scale
The global structural biology community has also been depositing increasingly more complex structures into the PDB archive. Figure 2a,b reflects the size and diversity of structures deposited to the PDB archive versus time. Growth in the number of distinct small‐molecule ligands represented in the PDB chemical component dictionary (CCD)25 is illustrated in Figure 2a (2,498 new ligands were added in 2018, corresponding to year‐on‐year growth of 7.7%). Entries in the PDB CCD include amino acids; nucleosides and nucleotides; carbohydrates; metals and other ions; crystallization and buffer solutes; enzyme cofactors, substrates, and products; prosthetic groups (e.g., heme); oligopeptides; small organic molecules; and pharmacologic agents. In parallel with the growth of the CCD, the average size of each PDB entry, as gauged by mean aggregate molecular weight of the biological assembly, is also growing (Figure 2b). Not surprisingly, 3DEM has contributed disproportionately to the growth in the number of larger PDB structures since early 2014 (Figure 2c).
Figure 2.
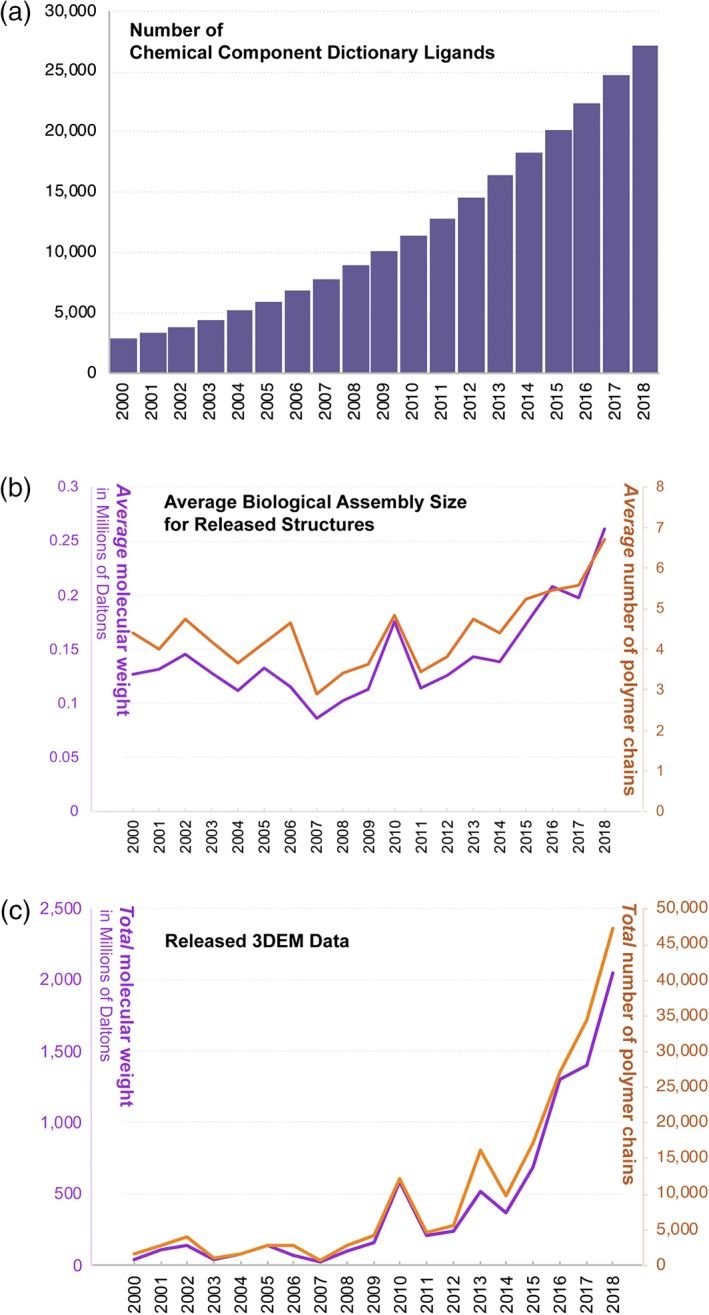
Growth in the complexity of PDB archival holdings 2000–2018. (a) Cumulative number of unique ligands maintained in the Chemical Component Dictionary each year. In 2018, 2,498 new entries were added. (b) Average molecular weight (solid purple line) and average number of polymer chains (solid orange line) of structures released each year. (c) Growth in available EM structure data, shown by annual accumulation of number of chains and molecular weight
3. DRUG TARGET STRUCTURES IN THE PDB
Structure‐guided drug discovery is a well‐established tool for large and small biopharmaceutical companies alike.26 3D structural studies frequently aid in optimizing small‐molecule ligand affinity and selectivity for target proteins (e.g., vemurafenib approved for treatment of the 50% of late‐stage metastatic melanoma patients with the Val600➔Glu mutation that activates the BRAF protein kinase, PDB structure 3og727). A recent RCSB PDB analysis23 documented that US FDA approval of 88% of 210 new molecular entities (NMEs or new drugs from 2010 to 2016) was facilitated by open access to ∼6,000 PDB structures containing the protein targeted by the NME and/or the new drug itself. More than half of these structures were described in the scientific literature and publicly released >10 years before final drug approval. Moreover, these structures were cited in a significant fraction of more than 2 million papers reporting publicly funded, precompetitive research on the drug targets that influenced drug company investment decisions, leading ultimately to the US FDA approvals and patient access to new life‐altering drugs. Finally, the impact of structural biologists and the PDB archive on US FDA new drug approvals was similar across all therapeutic areas.
4. INTEGRAL MEMBRANE PROTEIN STRUCTURES IN THE PDB
Contemporary successes enjoyed by structural biologists studying integral membrane proteins document that the PDB archive will represent an increasingly important source of precompetitive information supporting ongoing and future drug discovery campaigns directed at these challenging targets. More than 50% of the targets of current US FDA approved drugs are integral membrane proteins.28 The vast majority of these drug targets fall within four well‐studied protein families (G‐protein‐coupled receptors [GPCRs]: ∼30%; voltage‐gated ion channels [VGICs]: 8%; ligand‐gated ion channels [LGICs]: 7%; and transporters: 7%; examples of each are displayed in Figure 3). The following sections briefly review current PDB holdings and highlight opportunities for structure‐guided drug discovery for each of these major classes of target proteins.
Figure 3.
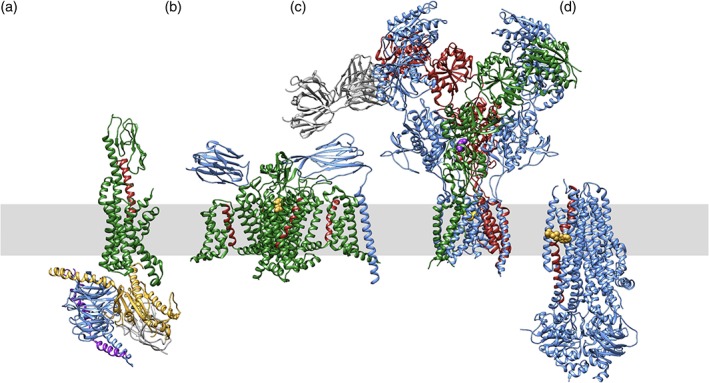
Ribbon drawings of exemplar structures from each of the four classes of membrane‐bound proteins of pharmacologic interest, viewed parallel to the lipid bilayer (shaded grey rectangle). (a) GPCR (PDB 5vai)36: GLP1‐R (glucagon‐like peptide‐1 receptor, active conformation in green) bound to GLP1 (red) and heterotrimeric G‐protein (blue, yellow, and magenta). (b) VGIC (PDB 6j8i)106: Nav1.7 (green), beta1 and beta2 (blue), bound to inhibitor tetrodotoxin (yellow). Voltage‐sensing helices are shown in red. (c) LGIC (PDB 5uow)72: NMDA receptor (blue, green, and red) bound to channel blocker MK‐801 (magenta). An antibody Fab (grey) was used in the structure determination. (d) Transporter (PDB 6o2p)85: CFTR (cystic fibrosis transmembrane conductance regulator (blue) bound to ivacaftor (yellow), which interacts with a long transmembrane helix involved in gating (red)
4.1. G‐protein‐coupled receptors
PDB archival holdings of GPCRs at the time of writing are summarized in Table 1. The landmark structure of bovine rhodopsin (PDB 1f88) gave the first view of the class in 2000,29 and initially, GPCR structure depositions to the PDB were restricted to the Rhodopsin subfamily, many of them crystallized using lipidic mesophases.30 Progress in this arena was accelerated by advances in protein engineering of the beta‐adrenergic receptor, creating chimeras with entire proteins or smaller protein domains inserted into extramembranous loops (e.g., T4 phage lysozyme31) that facilitate crystal lattice formation without perturbing the structure of the 7‐transmembrane helix domain.32 Currently, more than 300 GPCR structures from four of the five GPCR subfamilies have been determined and deposited in the archive, including A‐rhodopsin, B1‐secretin, C‐glutamate, and F‐frizzled/taste 2 (but not B2‐adhesion).33 The vast majority of these structures were determined using MX (∼91%), with a small number coming from NMR (∼2%), and a growing number coming from 3DEM (∼7%). At present, the PDB archive contains structures for more than 60 unique GPCRs (representing examples or orthologs of ∼15% of the entire complement of more than 800 GPCRs encoded by the human genome). GPCR structures have been elucidated in both active and inactive conformational states, some including bound small‐molecule ligands or drugs, bound peptide/protein ligands, bound heterotrimeric G proteins, and in some cases stabilizing Fab fragments and/or camelid‐nanobodies.34 Structure‐guided drug discovery for GPCRs (particularly Class A members) using MX is currently being pursued within many of the large biopharmaceutical companies, targeting both receptors represented within the PDB and novel receptors, exclusive to one or more companies.
Table 1.
G‐protein‐coupled receptors in the PDB archive
| All | Class A (rhodopsin) | Class B1 (secretin) | Class B2 (adhesion) | Class C (glutamate) | Class F (frizzled/taste 2) | |
|---|---|---|---|---|---|---|
| Structures | 339 | 295 | 23 | 0 | 8 | 13 |
| MX | 311 |
278 (7.7–1.7 Å) |
15 (3.3–1.9 Å) |
0 |
6 (3.1–2.2 Å) |
12 (3.9–2.4 Å) |
| NMRa | 6 | 6 | 0 | 0 | 0 | 0 |
|
3DEM (resolution) |
22 |
11 (4.5–3.0 Å) |
8 (4.1–3.0 Å) |
0 |
2 (4.0 Å) |
1 (3.8 Å) |
| Unique receptors | 62 | 52 | 6 | 0 | 2 | 2 |
One Solid State NMR entry (PDB 2lnl)35.
Note: Table generated in June 2019 using sequence searching with representative members of each class.
Abbreviations: 3DEM, three‐dimensional electron microscopy; MX, macromolecular X‐ray crystallography; NMR, nuclear magnetic resonance.
The first 3DEM structure of a GPCR to become publicly available (PDB 5vai)36 revealed the structure of glucagon‐like peptide 1 (GLP1) analog being recognized by the GLP1 receptor (GLP1‐R: active conformation) that was embedded in a detergent micelle and bound to a G‐protein heterotrimer (Figure 3a). Underscoring the power of cryo‐EM to enable structural studies of large/complex and very challenging samples, this Class B1 (secretin) GPCR was visualized at the atomic level in the act of recognizing its peptide hormone ligand, while engaging with a G‐protein heterotrimer.
GLP1‐R37 is the target of six oligopeptide agonists (exenatide [PDB 3c59, 3c9t],38 liraglutide [4apd],39 lixisenatide, albiglutide, dulaglutide, and semaglutide [4zgm]40) approved by the US FDA for treatment of type II diabetes mellitus. These biologic agents, the newest of which was approved in 2017, mimic endogenous GLP1 and slow gastric emptying/increase secretion of insulin by the patient's own pancreas in response to elevated blood glucose levels. The principal advantage of this treatment strategy versus older/cheaper small‐molecule insulin secretagogues is that it carries a lower risk of hypoglycemia. At present, there are no publicly available structures of any of the approved GLP1‐R agonists bound to full‐length GLP1‐R. With open access to PDB structure 5vai, detailed knowledge of how 5vai and related structures were determined, and recent acquisitions of state‐of‐the‐art 3DEM instrumentation by biopharmaceutical companies, the stage is now set for structure‐guided discovery of the next generation of GLP1‐R agonists with improved pharmacologic properties (i.e., longer half‐lives that will permit less frequent dosing and improve the likelihood of compliance). It also appears highly likely that 3DEM will shortly reveal one or more structures of Class B2 (Adhesion) GPCRs, some of which are drug discovery targets,41, 42 and all of which have thus far eluded 3D structure determination by any experimental method.
4.2. Voltage‐gated ion channels
VGICs open and close ion‐selective pores in response to small changes in membrane potential, playing central roles in nerve signal transmission. They form a large superfamily that includes voltage‐gated sodium (Nav), calcium (Cav), potassium (Kv), and other ion channels, encoded by at least 143 human genes,43, 44 making them the third largest family of signaling proteins after GPCRs and protein/lipid kinases. Rod MacKinnon's pioneering work on the homotetrameric potassium channel KcsA from S. lividans 45 and A. pernix KvAP46 revealed the mechanistic bases for ion selectivity and gating at the atomic level, but structural information for Nav and Cav channels, which lack the structural fourfold symmetry seen in KcsA, is relatively new to the archive.
Voltage‐gated sodium channels give rise to the rapid action potentials that mediate nerve transmission, making them targets for natural and designed toxins, inhibitors, and drugs.47, 48 Many examples are found in nature, including the potent and exquisitely selective tetrodotoxin, a neurotoxin found in puffer fish (and other organisms) with a lethal dose being less than a milligram. Many anticonvulsants, antiarrhythmics, and local anesthetics, such as lamotrigine, flecainide, and lidocaine, also act by blocking these channels.49 The PDB currently contains >750 VGIC structures (Table 2).
Table 2.
Voltage‐gated ion channels available from the PDB archive
| Voltage‐gated ion channels | Voltage‐gated potassium channel activity | High voltage‐gated calcium channel activity | Voltage‐gated proton channel activity | NMDA glutamate receptor activity | Voltage‐gated anion channel activity | Voltage‐gated ion channel activity involved in regulation of postsynaptic membrane potential | Voltage‐gated ion channel activity involved in regulation of presynaptic membrane potential | |
|---|---|---|---|---|---|---|---|---|
| Structures | 756 | 235 | 237 | 8 | 135 | 31 | 21 | 17 |
|
MX (resolution) |
494 (1.2–4.8 Å) |
156 (1.2–4.8 Å) |
132 (1.4–4.4 Å) |
7 (1.4–3.5 Å) |
94 (1.3–4.0 Å) |
11 (1.6–4.1 Å) |
16 (1.4–2.6 Å) |
8 (1.9–3.0 Å) |
| NMR | 64 | 30 | 25 | 1 | 2 | 2 | 5 | 7 |
|
3DEM (resolution) |
197 (2.9‐35 Å) |
48 (2.9‐10 Å) |
80 (3.0‐35 Å) |
0 |
39 (4.5–16.5 Å) |
18 (3.2–6.6 Å) |
0 |
2 (3.0–3.8 Å) |
| Hybrid | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Unique channels | 105 | 41 | 30 | 1 | 6 | 7 | 4 | 4 |
Note: Table generated in July 2019 based on gene ontology or GO #0005244.
Abbreviations: 3DEM, three‐dimensional electron microscopy; MX, macromolecular X‐ray crystallography; NMDA, N‐methyl‐D‐aspartate; NMR, nuclear magnetic resonance.
The human genome encodes nine voltage‐gated sodium channels (Nav, designated Nav1.1 to Nav1.9), which support a range of cellular and biological functions. Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are expressed primarily in the central nervous system; Nav1.4 is found in the skeletal muscle; Nav1.5 is found in cardiac muscle; and Nav1.7, Nav1.8, and Nav1.9 are typically found in the peripheral tissues. Much of the early structural work on Nav channels was performed using bacterial homologs, which have a simpler homotetrameric structure and proved relatively easy to express, purify, and crystallize. These first MX structures were reported in 2011 for A. butzleri NavAb (PDB 3rvy, 3rvz, and 3rw0).50 In contrast, the mammalian channel is composed of one long alpha chain, which forms four membrane‐spanning domains similar in arrangement to the four identical bacterial subunits. In addition, the alpha subunit associates with one or more copies of five beta‐subunits (beta1, beta1B, beta2, beta3, and beta4). 3DEM structures have been determined for human Nav1.4/beta2 (PDB 6agf),51 Nav1.2/beta2 with a conotoxin (PDB 6j8e),52 and Nav1.7/beta1/beta2 with tetrodotoxin and saxotoxin (PDB 6j8i and others, Figure 3b).52
Drug discovery efforts have focused considerable resources on Nav1.7.53 This work began in earnest following the 2004 discovery that a Nav1.7 gain‐of‐function mutation causes persistent pain.54 In 2006, a loss‐of‐function mutation was identified in several Pakistani street performers, who show no sensitivity to pain while walking on hot coals, and so forth.55 Selectivity remains an elusive challenge in this arena. Pair‐wise sequence identities among the nine human Nav VGICs exceed 70%. To complicate matters further, multiple functional‐binding sites for both large and small molecules are present on the solvent‐accessible surfaces of these integral membrane proteins. Prior to the availability of 3DEM structures of Nav VGICs, much of the early drug design work was performed using homology models based on distantly related bacterial proteins.
Today, medicinal chemists are sifting through multiple sites of action of natural toxins and poisons with the aim of finding druggable sites with potential for specificity, and then targeting them with small molecules, peptides, or antibodies. Notwithstanding insights from these new structures, serious challenges remain for structure‐guided drug discovery. Nav VGICs are conformationally dynamic, existing in multiple functional states (e.g., closed/resting, open, and closed/inactivated), each of which will need to be structurally characterized. Single‐particle 3DEM, however, offers a critical advantage versus MX in that multiple conformations of a macromolecular assembly can be accommodated via focused classification procedures56 to reveal multiple structural states on the EM grid.57
4.3. Ligand‐gated ion channels
LGICs mediate transmission of signals across nerve synapses in response to binding of neurotransmitters. There are three major structural classes of these channels (Table 3): pentameric “Cys‐loop” receptors, ionotropic glutamate receptors (iGluRs), and P2X receptors.58 The pentameric Cys‐loop receptors include excitatory cation‐selective channels, such as the nicotinic acetylcholine receptors and inhibitory anion‐selective channels (e.g., the GABAA receptor). In 2005, Nigel Unwin's ground‐breaking EM structure of the nicotinic acetylcholine receptor from the marbled electric ray (PDB 2bg9) revealed at the atomic‐level both ligand‐binding subunits and channel geometry.59 A large collection of toxins, poisons, and drugs act through these pentameric receptors, including the two well‐known poisons curare and strychnine60; anesthetics and alcohol61; benzodiazopine antidepressants62; and the antiparasitic agent ivermectin.63
Table 3.
Ligand‐gated ion channels in the PDB archive
| Alla | Cyclic nucleotide‐gated ion channel activity | Extracellular ligand‐gated ion channel activity | Intracellular ligand‐gated ion channel activity | Ligand‐gated anion channel activity | Ligand‐gated cation channel activity | Ligand‐gated ion channel activity involved in regulation of presynaptic membrane potential | |
|---|---|---|---|---|---|---|---|
| Structures | 968 | 38 | 647 | 241 | 88 | 865 | 159 |
|
MX (resolution) |
685 (1.15–7.4 Å) |
26 (1.65–3.28 Å) |
506 (1.15–4.79 Å) |
122 (1.21–7.4 Å) |
59 (1.55–3.8 Å) |
612 (1.15–7.4 Å) |
115 (1.24–3.96 Å) |
| NMR | 57 | 5 | 29 | 18 | 9 | 47 | 2 |
|
3DEM (resolution) |
223 (2.94‐50 Å) |
4 (3.4–3.51 Å) |
112 (2.95‐50 Å) |
98 (2.94–8.5 Å) |
20 (3.04–6.6 Å) |
203 (2.94‐50 Å) |
45 (3.8–16.5 Å) |
|
Electron crystallography (resolution) |
3 (3.54–3.8 Å) |
3 (3.54–3.8 Å) |
0 |
3 (3.54–3.8 Å) |
0 |
3 (3.54–3.8 Å) |
0 |
| Unique genes | 84 | 8 | 45 | 24 | 11 | 69 | 5 |
N.B.: PDB structures may appear in multiple LGIC classification categories.
Note: Table generated in July 2019 based on gene ontology or GO#0015276.
Abbreviations: 3DEM, three‐dimensional electron microscopy; MX, macromolecular X‐ray crystallography; NMR, nuclear magnetic resonance.
iGluRs fall into four main classes, based on their small‐molecule‐binding properties: AMPA receptors (GluA1‐4), kainate receptors (GluK1‐5), NMDA (N‐methyl‐D‐aspartate) receptors (GluN1, GluN2A‐D, and GluN3A‐B), and delta receptors (GluD1‐2).64, 65 These polypeptide chains can form both homo‐ and heterotetramers, and associate with a variety of modulatory auxiliary subunits. They are modular in structure. An N‐terminal domain (homologous to bacterial periplasmic‐binding proteins) mediates dimerization between subunits of the same iGluR class. The C‐terminal portion contains the extracellular agonist‐binding domain, which consists of two polypetide chain segments separated by the portion that forms the membrane‐spanning ion channel pore. Structures of extracellular fragments of iGluR proved instrumental in characterizing some functionally important properties of these channels.66 Beginning in 2009 with publication of the MX structure of GluA2 AMPA receptor (PDB 3kg2),67 work in this area has moved rapidly. Today, multiple 3DEM structures of iGluR and their complexes with ligands, toxins, and accessory proteins are also publicly available.68, 69
As seen for the VGICs, the iGluRs display multiple sites for binding of toxins and poisons, and many of these LGICs are currently the focus of structure‐guided drug discovery efforts (see the 2019 special issue of ACS Med. Chem. Lett. on Allosteric Modulation of iGluR).70 For example, memantine, an NMDA receptor channel blocker, has been approved for treatment of moderate‐to‐severe Alzheimer's patients.71 A 3DEM structure of the heterotrimeric GluN1/GluN2A/GluN2B NMDA receptor with a similar agent (MK‐801, PDB 5uow,72 Figure 3c) revealed the ligand‐binding site within a vestibule of the ion channel. Preclinical characterization of MK‐801 underscores both the promise and the challenges posed by targeting these receptors. Neuroprotection was observed in animal models of stroke, traumatic brain injury, and Parkinsonism, accompanied by side effects of induced psychotic behavior and neuronal degeneration. A subsequently determined 3.6 Å resolution MX structure of an N‐terminal truncated form of the receptor enabled molecular dynamics simulations of MK‐801 and memantine binding (PDB 5un1),73 further advancing structure‐guided drug design efforts aimed at improving side‐effect profiles.
4.4. Transporters
The transporters constitute a large, heterogenous class of membrane‐spanning proteins involved in trafficking of small cargo molecules across lipid bilayers. Sequence mapping of PDB structures to the Transporter Classification Database (http://www.tcdb.org)74 identified 9,834 transporter structures in the PDB archive (as of July 2019, Table 4). Membrane transporters play central roles in ADME (absorption, distribution, metabolism, and elimination) and pharmacodynamic properties of drugs.75 The human genome encodes >400 membrane transporters that fall into two superfamilies: ATP‐binding cassette (ABC) superfamily and solute carrier (SLC) family. All ABC transporters and many SLC transporters function as active transporters, using either ATP or electrochemical gradients to drive transport.
Table 4.
Transporter proteins in the PDB Archive
| All | Class 1: Channels/pores | Class 2: Electrochemical potential‐driven transporters | Class 3: Primary active transporters | Class 4: Group translocators | Class 5: Transmembrane electron carriers | Class 8: Accessory factors involved in transport | Class 9: Incompletely characterized transport systems | |
|---|---|---|---|---|---|---|---|---|
| Structures | 9,834 | 4,131 | 721 | 2,203 | 80 | 96 | 1,651 | 952 |
|
MX (resolution) |
8,207 |
3,364 (7.6–0.82 Å) |
669 (5.97–1.0 Å) |
1911 (7.78–0.88 Å) |
52 (3.91–1.45 Å) |
79 (3.7–0.99 Å) |
1,272 (7.81–0.73 Å) |
860 (7.0–0.85 Å) |
| NMR | 716 | 313 | 15 | 80 | 21 | 13 | 216 | 58 |
|
3DEM (resolution) |
911 |
454 (50.0–1.9 Å) |
37 (14.0–3.0 Å) |
212 (37.0–2.0 Å) |
7 (4.3–2.6 Å) |
4 (3.8–3.1 Å) |
163 (35.0–2.6 Å) |
34 (35.0–3.0 Å) |
| Unique | 3,429 | 1,318 | 214 | 810 | 46 | 43 | 655 | 343 |
Note: Table generated in July 2019 using sequences from the Transporter Classification Database.74
Abbreviations: 3DEM, three‐dimensional electron microscopy; MX, macromolecular X‐ray crystallography; NMR, nuclear magnetic resonance.
ABC transporters were first identified in bacterial nutrient import systems, bearing a characteristic ATP‐binding domain with a phosphate‐binding loop (commonly known as the P‐loop or Walker A motif) and a short “Leu‐Ser‐Gly‐Gly‐Gln” consensus sequence.76 Similar motifs were later found in the bacterial multidrug‐resistance export pump P‐glycoprotein (P‐gp). Subsequent studies revealed that 1–3% of bacterial genomes encode ABC transporters, which act as variously as importers or exporters. In all, the human genome encodes 48 ABC exporters, which fall into seven subfamilies (designated A–G). Multiple MX structures of bacterial/archaeal ABC transporters are available from the PDB archive, with some bound to periplasmic‐binding proteins responsible for delivering substrates to the transporter.77 These structures revealed various conformational states that cycle between inward‐ and outward‐facing configurations. Instructive examples include an early structure of the vitamin b12 transporter (BtuCD, PDB 1l7v),78 and three states of the E. coli maltose transporter MalEFGK2 (inward‐open, PDB 3fh679; pretranslocation, PDB 3pv080; and outward‐open, PDB 2r6g81).
ABC transporters are also relevant to human health and therapies. For example, P‐gp and breast cancer resistance protein, found on the luminal surfaces of cells in the gut, modulate oral bioavailability of drug, and are, therefore, key determinants of ADME properties.75, 82 For example, increased expression of P‐gp in cancer cells confers resistance to various chemotherapeutic agents. 3DEM structures, beginning with the complex of P‐gp with cyclic peptide inhibitors (PDB 3g61),83 are revealing the mechanism(s) of action of these transporters and modes of targeted inhibition.84 The cystic fibrosis transmembrane conductance regulator (CFTR) transporter (Figure 3d) is an ABC chloride ion transporter.85 More than 2000 CFTR gene variants have been identified in humans. Many of these differences are causative of cystic fibrosis, the most common autosomal recessive genetic disease affecting Caucasians. Inadequate chloride transport causes accumulation of mucus in lung and pancreas, leading to chronic pulmonary inflammation/infection and exocrine pancreatic insuffiency.86 The most common disease‐causing CFTR variants include deletion of Phe508, which accelerates protein degradation, and Arg117➔His and Gly551➔Asp, which yield transporters with gating defects. The US FDA‐approved drug ivacaftor acts as a potentiator of these gating variants, yielding increase chloride transmission.87 A 3DEM structure of human CFTR (PDB 6o2p)85 revealed that the drug binds at the protein‐lipid interface within the transmembrane region, at a hinge site known to be involved in gating. It has been hypothesized that ivacaftor, which was discovered via phenotypic screening, stabilizes the open configuration of the transporter. With an EM structure in hand and a druggable site identified, the door is now open to structure‐guided drug discovery of second‐generation drugs targeting a broader spectrum of mutations causative of cystic fibrosis in affected individuals with superior side‐effect profiles.
The SLC superfamily is highly heterogeneous, with 52 distinct human protein families that show little sequence or structural similarity, sharing simply their roles in intake and/or efflux of small molecules and inorganic ions across membranes.88 A 2017 review89 tabulates atomic‐level 3D structure determinations for members of 23 SLC families, largely prokaryotic proteins, such as the long‐awaited and much‐anticipated structure of lactose permease (PDB 1pv6).90 These structures revealed much diversity in the mechanism(s) of substrate recognition (as might have be expected), but also commonalities in the local conformational changes responsible for opening and closing “gates” on either side of the membrane to regulate transport. SLC proteins are only now being explored as drug discovery targets. A recent perspective issued a “call‐to‐arms” to explore this diverse and functionally important subset of transporters.91 Successes to date include various US FDA‐approved drugs, such as selective serotonin reuptake inhibitors (SSRIs) for treatment of depression, and sodium/glucose cotransporter inhibitors for treatment of type 2 diabetes.92 Recent structure‐guided development of SSRIs built on MX studies of a bacterial homolog LeuT,93 first determined in 2005 (PDB 2a65).94 Other structures of human serotonin transporters (PDB 5i6x and others)95 will almost certainly improve the impact of this approach for discovery and development of new pharmacologic agents targeting neuropsychiatric disorders.
5. E3 UBIQUITIN LIGASE STRUCTURES IN THE PDB
Successes enjoyed by structural biologists studying complex multiprotein assemblies show that the PDB archive will come to represent an increasingly important source of precompetitive information facilitating structure‐guided drug discovery of other challenging targets that are not integral membrane proteins. Some of the most exciting new drug targets among the large macromolecular machines can be found within the large family of E3 ubiquitin ligases. These enzymes confer substrate selectivity on the ubiquitin‐proteasome system (UPS) for degrading cytosolic proteins.96 The UPS pathway is regulated by sequential action of three classes of activating enzymes: E1 (two human enzymes), E2 (∼40 human enzymes), and E3 (>600 human enzymes) (Figure 4a).97 The end result of this combinatorial three‐step enzyme cascade is an ubiquitinated substrate that is in turn recognized and degraded by the 26S proteasome.
Figure 4.
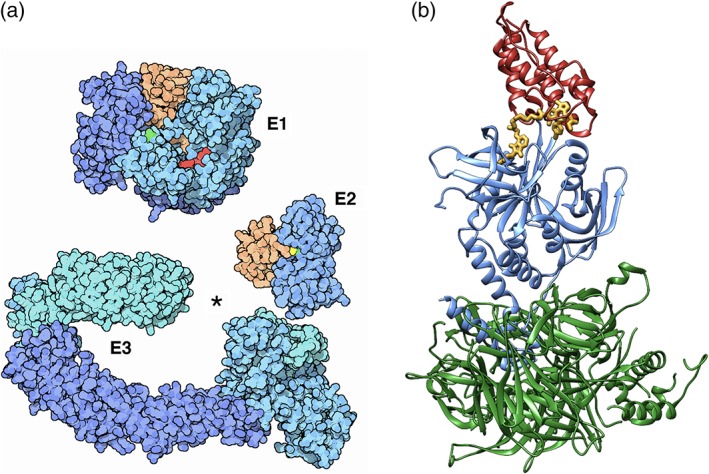
(a) Early structures of the components of the ubiquitin ligase system (E1,107 E2,108 and E3109, 110). Image from PDB‐101's Molecule of the Month.21 (b) Ribbon structure of the PROTAC (proteolysis targeting chimera) degrader MZ1 (yellow) linking cancer target Brd4 (red) to a ubiquitin ligase complex of pVHL:ElonginC:ElonginB (blue and green) from PDB 6bn7103
E3 ubiquitin ligases include components, respectively, responsible for catalysis (i.e., ubiquitination) and substrate recognition. In some cases, both functionalities are embedded within a single polypeptide chain. In many others, E3 is made up of multiple protein chains. As might be expected from the many types of substrate proteins that undergo targeted ubiquitination, E3 substrate‐recognition components are highly heterogenous, and variously recognize short substrate peptides called “degrons” or larger protein surface features. Ubiquitination machines have been classified into three major families: Really Interesting New Gene (RING), Homologous with E6‐associated protein C‐Terminus (HECT), and the family of hybrid RBR (RING‐IBR‐RING) E3s. Extensive structural studies (Table 5) have revealed the central role played by flexibility in influencing interactions of E3s with E2‐ubiquitin conjugates. This work has also explored the mechanisms by which multiple bacterial and viral proteins hijack the UPS.98
Table 5.
Ubiquitin‐like structures in the PDB archive
| Ubiquitin‐like protein ligase activity (alla) | NEDD8 ligase activity | SUMO ligase activity | Ubiquitin protein ligase activity | |
|---|---|---|---|---|
| Structures | 984 | 113 | 5 | 979 |
|
MX (resolution) |
804 (0.8–8.3 Å) |
104 (1.1–3.3 Å) |
3 (1.7–2.4 Å) |
801 (0.8–8.3 Å) |
| NMR | 138 | 9 | 2 | 136 |
|
3DEM (resolution) |
42 (2.9–16 Å) |
0 | 0 |
42 (2.9–16 Å) |
| Unique E3 ubiquitin ligases by gene names | 123 | 1 | 4 | 119 |
N.B.: PDB structures may appear in multiple ubiquitin‐like classification categories.
Note: Table generated in July 2019 based on gene ontology or GO#0061659, ubiquitin‐like protein ligase activity.
Abbreviations: 3DEM, three‐dimensional electron microscopy; MX, macromolecular X‐ray crystallography; NEDD8: neural precursor cell expressed, developmentally down‐regulated 8; NMR, nuclear magnetic resonance; SUMO: small ubiquitin like modifier.
An exciting new development in this arena is the prospect of targeting the UPS to proteins of pharmacologic interest. This work was inspired, at least in part, by natural products (e.g., auxin, a well‐characterized small‐molecule plant hormone) that stabilize interactions between an E3 ligase and its degradation target.99 Similarly, immunomodulatory imide drugs, such as the teratogen thalidomide, are being repurposed on the strength of their ability to stabilize binding of E3 ligases to several lymphoid transcription factors.100 A new strategy is also being explored to design bifunctional molecules, called proteolysis targeting chimera or degraders, that recognize a common site on the surface of an E3 and a specific‐binding site on the surface of a target protein, bringing them together to promote targeted degradation.101 Recently deposited PDB MX structures have revealed at the atomic level how such a degrader links the chromatin‐reader protein Brd4 (bromodomain‐containing protein 4), a target for cancer therapy, with ubiquitin ligase complexes, such as von Hippel–Lindau disease tumor suppressor:ElonginC:ElonginB (Figure 4b, PDB 5t35)102 and DDBI:CRBN (PDB 6bn7).103 These proof‐of‐concept structures open the door‐to‐structure‐guided discovery of similar degraders selective for other protein targets, via engineering of linkers based on structures of specific ligands bound to each of the partners.
6. CONCLUSION/PERSPECTIVE
The success of the discipline we have come to know as structural biology and the relentless growth of the open‐access PDB archive bode well for the continued impact of 3D biostructure data on basic and applied research across the biological, medical, and energy sciences. Of particular, importance looking ahead will be the explosive growth of 3DEM depositions to the archive. Since 2016, annual 3DEM depositions have exceeded those coming from NMR spectroscopy. Notwithstanding whispers to the contrary in some quarters, MX is actually “alive and well” and remains the mainstay experimental method for atomic‐level 3D structure determinations of macromolecules, accounting for ∼90% of 2018 PDB depositions. The precise role that 3DEM will play in structure‐guided drug discovery going forward remains to be determined. It is clear, however, from private communications received from biopharmaceutical company colleagues that they are benefiting from even lower‐resolution 3DEM structures of macromolecular machines wherein tool compounds can be visualized bound to druggable surface features, such as deep invaginations and protein–protein interface clefts. Knowledge of the functional groups presented by the amino acid sidechains comprising putative drug‐binding sites is particularly helpful for hypothesis generation during synthesis of early lead compounds. MX is likely to remain the method of choice for any drug‐discovery target that supports facile crystallization and production of higher‐resolution (i.e., better than 2.2 Å) cocrystal structures with pharmaceutically acceptable lead compounds and even drug candidates. Diffraction data in these cases are typically obtained at modern synchrotron radiation sources in <1 min of beam time, and refined structures therefrom can often be generated with automated scripts within 1 hr following the experiment. It is not unusual for structural biologists working in biopharmaceutical companies to deliver new, highly informative cocrystal structures with 1–2 weeks of compound synthesis. 3DEM will have to come a long way in terms of efficiency before it can rival the speed and relatively low cost of MX structure determination. The impact of X‐ray free electron lasers and serial femtosecond crystallography in time‐resolved studies of drug discovery targets remains an open question.104, 105 Whatever the outcome of this “horse race,” the open‐access PDB archive will continue to play central roles in research and education, facilitating discovery of new biomaterials, new drugs, and new diagnostic tools around the world.
ACKNOWLEDGMENTS
The RCSB PDB is jointly funded by the National Science Foundation (DBI‐1832184), the National Institutes of Health (R01GM133198), and the United States Department of Energy (DE‐SC0019749). We gratefully acknowledge contributions from current and past members of the RCSB PDB and our wwPDB partners.
Goodsell DS, Zardecki C, Di Costanzo L, et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Science. 2020;29:52–65. 10.1002/pro.3730
Present address Luigi Di Costanzo, Department of Agricultural Sciences, University of Naples Federico II, Portici (Napoli), 80055, Italy.
Funding information National Science Foundation, Grant/Award Number: 1832184; National Institutes of Health, Grant/Award Number: R01GM133198; U.S. Department of Energy, Grant/Award Number: DE‐SC0019749
REFERENCES
- 1. Protein Data Bank . Protein Data Bank. Nature New Biol. 1971;233:223.20480989 [Google Scholar]
- 2. Protein Data Bank . Cold spring harbor symposia on quantitative biology. Vol 36 New York, NY: Cold Spring Laboratory Press, 1972. [Google Scholar]
- 3. Sullivan KP, Brennan‐Tonetta P, Marxen LJ (2017) Economic impacts of the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank. doi: 10.2210/rcsb_pdb/pdb-econ-imp-2017. [DOI]
- 4. wwPDB consortium . Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019;47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burley SK, Berman HM, Bhikadiya C, et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47:D464–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kinjo AR, Bekker GJ, Wako H, et al. New tools and functions in data‐out activities at Protein Data Bank Japan (PDBj). Protein Sci. 2018;27:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mir S, Alhroub Y, Anyango S, et al. PDBe: Towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res. 2018;46:D486–D492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ulrich EL, Akutsu H, Doreleijers JF, et al. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2016;160018:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basner J (2017) Impact analysis of “Berman HM et al., (2000), The Protein Data Bank”. doi: 10.2210/rcsb_pdb/pdb-cit-anal-2017. [DOI]
- 12. Markosian C, Di Costanzo L, Sekharan M, Shao C, Burley SK, Zardecki C. Analysis of impact metrics for the Protein Data Bank. Sci Data. 2018;5:180212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao J, Prlic A, Bi C, et al. BioJava‐ModFinder: Identification of protein modifications in 3D structures from the Protein Data Bank. Bioinformatics. 2017;33:2047–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gene Ontology Consortium . Gene ontology consortium: Going forward. Nucleic Acids Res. 2015;43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Velankar S, Dana JM, Jacobsen J, et al. SIFTS: Structure integration with function, taxonomy and sequences resource. Nucleic Acids Res. 2013;41:D483–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. The UniProt Consortium . UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pieper U, Webb BM, Dong GQ, et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014;42:D336–D346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlić A, Rose PW. NGL viewer: Web‐based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodsell DS, Dutta S, Zardecki C, Voigt M, Berman HM, Burley SK. The RCSB PDB "Molecule of the Month": Inspiring a molecular view of biology. PLoS Biol. 2015;13:e1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burley SK, Berman HM, Christie C, et al. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018;27:316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Westbrook JD, Burley SK. How structural biologists and the Protein Data Bank contributed to recent FDA new drug approvals. Structure. 2018;27:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuhlbrandt W. Biochemistry. The resolution revolution. Science. 2014;343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 25. Westbrook JD, Shao C, Feng Z, Zhuravleva M, Velankar S, Young J. The chemical component dictionary: Complete descriptions of constituent molecules in experimentally determined 3D macromolecules in the Protein Data Bank. Bioinformatics. 2015;31:1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gilliland GL, Luo J, Vafa O, Almagro JC. Leveraging SBDD in protein therapeutic development: Antibody engineering. Methods Mol Biol. 2012;841:321–349. [DOI] [PubMed] [Google Scholar]
- 27. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF‐mutant melanoma. Nature. 2010;467:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: A G protein‐coupled receptor. Science. 2000;289:739–745. [DOI] [PubMed] [Google Scholar]
- 30. Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thorsen TS, Matt R, Weis WI, Kobilka BK. Modified T4 lysozyme fusion proteins facilitate G protein‐coupled receptor crystallogenesis. Structure. 2014;22:1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenbaum DM, Cherezov V, Hanson MA, et al. GPCR engineering yields high‐resolution structural insights into beta2‐adrenergic receptor function. Science. 2007;318:1266–1273. [DOI] [PubMed] [Google Scholar]
- 33. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. [DOI] [PubMed] [Google Scholar]
- 34. Katritch V, Cherezov V, Stevens RC. Structure‐function of the G protein‐coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park SH, Das BB, Casagrande F, et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Sun B, Feng D, et al. Cryo‐EM structure of the activated GLP‐1 receptor in complex with a G protein. Nature. 2017;546:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rajeev SP, Wilding J. GLP‐1 as a target for therapeutic intervention. Curr Opin Pharmacol. 2016;31:44–49. [DOI] [PubMed] [Google Scholar]
- 38. Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand‐bound glucagon‐like peptide‐1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. [DOI] [PubMed] [Google Scholar]
- 39. Ludvigsen S, Steensgaard DB, Thomsen JK, Strauss H, Normann M. Liraglutide. 2013. 10.2210/pdb2214APD/pdb. [DOI] [Google Scholar]
- 40. Lau J, Bloch P, Schaffer L, et al. Discovery of the once‐weekly glucagon‐like peptide‐1 (GLP‐1) analogue semaglutide. J Med Chem. 2015;58:7370–7380. [DOI] [PubMed] [Google Scholar]
- 41. Purcell RH, Hall RA. Adhesion G protein‐coupled receptors as drug targets. Annu Rev Pharmacol Toxicol. 2018;58:429–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Folts CJ, Giera S, Li T, Piao X. Adhesion G protein‐coupled receptors as drug targets for neurological diseases. Trends Pharmacol Sci. 2019;40:278–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu FH, Catterall WA (2004) The VGL‐chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004; re15. [DOI] [PubMed] [Google Scholar]
- 44. Zhang AH, Sharma G, Undheim EAB, Jia X, Mobli M. A complicated complex: Ion channels, voltage sensing, cell membranes and peptide inhibitors. Neurosci Lett. 2018;679:35–47. [DOI] [PubMed] [Google Scholar]
- 45. Jiang Y, Lee A, Chen J, et al. X‐ray structure of a voltage‐dependent K+ channel. Nature. 2003;423:33–41. [DOI] [PubMed] [Google Scholar]
- 46. Doyle DA, Morais Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. [DOI] [PubMed] [Google Scholar]
- 47. de Lera Ruiz M, Kraus RL. Voltage‐gated sodium channels: Structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58:7093–7118. [DOI] [PubMed] [Google Scholar]
- 48. Xu L, Ding X, Wang T, Mou S, Sun H, Hou T. Voltage‐gated sodium channels: Structures, functions, and molecular modeling. Drug Discov Today. 2019;24:1389–1397. [DOI] [PubMed] [Google Scholar]
- 49. Bagal SK, Marron BE, Owen RM, Storer RI, Swain NA. Voltage gated sodium channels as drug discovery targets. Channels. 2015;9:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage‐gated sodium channel. Nature. 2011;475:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pan X, Li Z, Zhou Q, et al. Structure of the human voltage‐gated sodium channel Nav1.4 in complex with beta1. Science. 2018;362:eaau2486. [DOI] [PubMed] [Google Scholar]
- 52. Pan X, Li Z, Huang X, et al. Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu‐conotoxin KIIIA. Science. 2019;363:1309–1313. [DOI] [PubMed] [Google Scholar]
- 53. Kingwell K. Nav1.7 withholds its pain potential. Nat Rev Drug Discov. 2019;18:321–323. [DOI] [PubMed] [Google Scholar]
- 54. Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang C, Cantara W, Jeon Y, Musier‐Forsyth K, Grigorieff N, Lyumkis D. Analysis of discrete local variability and structural covariance in macromolecular assemblies using Cryo‐EM and focused classification. Ultramicroscopy. 2019;203:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roh SH, Hryc CF, Jeong HH, et al. Subunit conformational variation within individual GroEL oligomers resolved by Cryo‐EM. Proc Natl Acad Sci U S A. 2017;114:8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand‐gated ion channels. Neuropharmacology. 2009;56:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol. 2005;346:967–989. [DOI] [PubMed] [Google Scholar]
- 60. Nasiripourdori A, Taly V, Grutter T, Taly A. From toxins targeting ligand gated ion channels to therapeutic molecules. Toxins. 2011;3:260–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Olsen RW, Li GD, Wallner M, et al. Structural models of ligand‐gated ion channels: Sites of action for anesthetics and ethanol. Alcohol Clin Exp Res. 2014;38:595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rammes G, Rupprecht R. Modulation of ligand‐gated ion channels by antidepressants and antipsychotics. Mol Neurobiol. 2007;35:160–174. [DOI] [PubMed] [Google Scholar]
- 63. Fox LM. Ivermectin: Uses and impact 20 years on. Curr Opin Infect Dis. 2006;19:588–593. [DOI] [PubMed] [Google Scholar]
- 64. Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462. [DOI] [PubMed] [Google Scholar]
- 65. Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kumar J, Mayer ML. Functional insights from glutamate receptor ion channel structures. Annu Rev Physiol. 2013;75:313–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sobolevsky AI, Rosconi MP, Gouaux E. X‐ray structure, symmetry and mechanism of an AMPA‐subtype glutamate receptor. Nature. 2009;462:745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang JX, Furukawa H. Dissecting diverse functions of NMDA receptors by structural biology. Curr Opin Struct Biol. 2019;54:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen S, Gouaux E. Structure and mechanism of AMPA receptor—auxiliary protein complexes. Curr Opin Struct Biol. 2019;54:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Carruthers NI, Lovenberg TW, Traynelis SF. Allosteric modulation of ionotropic glutamate receptors special issue. ACS Med Chem Lett. 2019;10:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Alam S, Lingenfelter KS, Bender AM, Lindsley CW. Classics in chemical neuroscience: Memantine. ACS Chem Nerosci. 2017;8:1823–1829. [DOI] [PubMed] [Google Scholar]
- 72. Lu W, Du J, Goehring A, Gouaux E. Cryo‐EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science. 2017;355:eaal3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Redziniak G, Leclerc M, Panijel J, Monsigny M. Separation of two different populations of axial organ cells of Asterias rubens by the use of lectins. Biochimie. 1978;60:525–527. [DOI] [PubMed] [Google Scholar]
- 74. Saier MH Jr, Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno‐Hagelsieb G. The Transporter Classification Database (TCDB): Recent advances. Nucleic Acids Res. 2016;44:D372–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Y. Overview of transporters in pharmacokinetics and drug discovery. Curr Protoc Pharmacol. 2018;82:e46. [DOI] [PubMed] [Google Scholar]
- 76. Wilkens S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Beis K. Structural basis for the mechanism of ABC transporters. Biochem Soc Trans. 2015;43:889–893. [DOI] [PubMed] [Google Scholar]
- 78. Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. [DOI] [PubMed] [Google Scholar]
- 79. Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Alternating access in maltose transporter mediated by rigid‐body rotations. Mol Cell. 2009;33:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Oldham ML, Chen J. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science. 2011;332:1202–1205. [DOI] [PubMed] [Google Scholar]
- 81. Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. [DOI] [PubMed] [Google Scholar]
- 82. Hennessy M, Spiers JP. A primer on the mechanics of P‐glycoprotein the multidrug transporter. Pharmacol Res. 2007;55:1–15. [DOI] [PubMed] [Google Scholar]
- 83. Aller SG, Yu J, Ward A, et al. Structure of P‐glycoprotein reveals a molecular basis for poly‐specific drug binding. Science. 2009;323:1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stoll F, Goller AH, Hillisch A. Utility of protein structures in overcoming ADMET‐related issues of drug‐like compounds. Drug Discov Today. 2011;16:530–538. [DOI] [PubMed] [Google Scholar]
- 85. Liu F, Zhang Z, Levit A, et al. Structural identification of a hotspot on CFTR for potentiation. Science. 2019;364:1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–2531. [DOI] [PubMed] [Google Scholar]
- 87. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hediger MA, Clemencon B, Burrier RE, Bruford EA. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol Aspects Med. 2013;34:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bai X, Moraes TF, Reithmeier RAF. Structural biology of solute carrier (SLC) membrane transport proteins. Mol Membr Biol. 2017;34:1–32. [DOI] [PubMed] [Google Scholar]
- 90. Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli . Science. 2003;301:610–615. [DOI] [PubMed] [Google Scholar]
- 91. Cesar‐Razquin A, Snijder B, Frappier‐Brinton T, et al. A call for systematic research on solute carriers. Cell. 2015;162:478–487. [DOI] [PubMed] [Google Scholar]
- 92. Rives ML, Javitch JA, Wickenden AD. Potentiating SLC transporter activity: Emerging drug discovery opportunities. Biochem Pharmacol. 2017;135:1–11. [DOI] [PubMed] [Google Scholar]
- 93. Immadisetty K, Geffert LM, Surratt CK, Madura JD. New design strategies for antidepressant drugs. Expert Opin Drug Discov. 2013;8:1399–1414. [DOI] [PubMed] [Google Scholar]
- 94. Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−‐dependent neurotransmitter transporters. Nature. 2005;437:215–223. [DOI] [PubMed] [Google Scholar]
- 95. Coleman JA, Green EM, Gouaux E. X‐ray structures and mechanism of the human serotonin transporter. Nature. 2016;532:334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bulatov E, Ciulli A. Targeting Cullin‐RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small‐molecule modulation. Biochem J. 2015;467:365–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–953. [DOI] [PubMed] [Google Scholar]
- 98. Zheng N, Shabek N. Ubiquitin ligases: Structure, function, and regulation. Annu Rev Biochem. 2017;86:129–157. [DOI] [PubMed] [Google Scholar]
- 99. Shabek N, Zheng N. Plant ubiquitin ligases as signaling hubs. Nat Struct Mol Biol. 2014;21:293–296. [DOI] [PubMed] [Google Scholar]
- 100. Asatsuma‐Okumura T, Ito T, Handa H. Molecular mechanisms of cereblon‐based drugs. Pharmacol Ther. 2019;202:132–139. 10.1016/j.pharmthera.2019.1006.1004. [DOI] [PubMed] [Google Scholar]
- 101. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1‐Cullin‐F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gadd MS, Testa A, Lucas X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nowak RP, DeAngelo SL, Buckley D, et al. Plasticity in binding confers selectivity in ligand‐induced protein degradation. Nat Chem Biol. 2018;14:706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mishin A, Gusach A, Luginina A, Marin E, Borshchevskiy V, Cherezov V. An outlook on using serial femtosecond crystallography in drug discovery. Expert Opin Drug Discov. 2019;14:933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Olmos JL Jr, Pandey S, Martin‐Garcia JM, et al. Enzyme intermediates captured “on the fly” by mix‐and‐inject serial crystallography. BMC Biol. 2018;16:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shen H, Liu D, Wu K, Lei J, Yan N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303–1308. [DOI] [PubMed] [Google Scholar]
- 107. Walden H, Podgorski MS, Huang DT, et al. The structure of the APPBP1‐UBA3‐NEDD8‐ATP complex reveals the basis for selective ubiquitin‐like protein activation by an E1. Mol Cell. 2003;12:1427–1437. [DOI] [PubMed] [Google Scholar]
- 108. Hamilton KS, Ellison MJ, Barber KR, et al. Structure of a conjugating enzyme‐ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure. 2001;9:897–904. [DOI] [PubMed] [Google Scholar]
- 109. Zheng N, Schulman BA, Song L, et al. Structure of the Cul1‐Rbx1‐Skp1‐F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. [DOI] [PubMed] [Google Scholar]
- 110. Schulman BA, Carrano AC, Jeffrey PD, et al. Insights into SCF ubiquitin ligases from the structure of the Skp1‐Skp2 complex. Nature. 2000;408:381–386. [DOI] [PubMed] [Google Scholar]