

Abstract
Next‐generation sequencing methods have not only allowed an understanding of genome sequence variation during the evolution of organisms but have also provided invaluable information about genetic variants in inherited disease and the emergence of resistance to drugs in cancers and infectious disease. A challenge is to distinguish mutations that are drivers of disease or drug resistance, from passengers that are neutral or even selectively advantageous to the organism. This requires an understanding of impacts of missense mutations in gene expression and regulation, and on the disruption of protein function by modulating protein stability or disturbing interactions with proteins, nucleic acids, small molecule ligands, and other biological molecules. Experimental approaches to understanding differences between wild‐type and mutant proteins are most accurate but are also time‐consuming and costly. Computational tools used to predict the impacts of mutations can provide useful information more quickly. Here, we focus on two widely used structure‐based approaches, originally developed in the Blundell lab: site‐directed mutator (SDM), a statistical approach to analyze amino acid substitutions, and mutation cutoff scanning matrix (mCSM), which uses graph‐based signatures to represent the wild‐type structural environment and machine learning to predict the effect of mutations on protein stability. Here, we describe DUET that uses machine learning to combine the two approaches. We discuss briefly the development of mCSM for understanding the impacts of mutations on interfaces with other proteins, nucleic acids, and ligands, and we exemplify the wide application of these approaches to understand human genetic disorders and drug resistance mutations relevant to cancer and mycobacterial infections.
Statement for a Broader Audience
Genetic or somatic changes in genes can lead to mutations in human proteins, which give rise to genetic disorders or cancer, or to genes of pathogens leading to drug resistance. Computer software described here, using statistical approaches or machine learning, uses the information from genome sequencing of humans and pathogens, together with experimental or modeled 3D structures of gene products, the proteins, to predict impacts of mutations in genetic disease, cancer and drug resistance.
Keywords: amino acid substitution probabilities, drug resistance, genetic disorders, machine learning, mutations, protein stability and interactions, protein structure
1. INTRODUCTION
Next‐generation sequencing methods have not only allowed an understanding of genome sequence variation during the evolution of organisms1 but have also provided invaluable information about human genetic disorders and cancer.2, 3 At the same time, genome sequences have facilitated our understanding of the emergence of resistance to drugs in tumors as well as antibiotics in infectious disease.4 A major challenge is to distinguish mutations that are “drivers” of disease or drug resistance from “passengers” that are neutral in these respects or even selectively advantageous to the organism. This requires understanding of the impacts of missense mutations in gene expression and regulation, and on protein function. Missense mutations can disrupt function not only by modulating protein stability but also through disturbing interactions with other biological molecules, including proteins, nucleic acids or polysaccharides, small molecule ligands, and metal ions.
Experimental approaches to understanding changes in stability and interactions between wild‐type and mutant proteins are clearly most accurate but are also time‐consuming and costly. Over the past three decades, this has encouraged the development of computational techniques, not only sequence‐based methods using support vector machines (INPS),5 neural networks,6 and decision trees (iPTREE‐STAB and MuStab)7, 8 but also structure‐based techniques using potential‐energy‐based approaches,9, 10, 11 machine learning algorithms,12, 13, 14, 15, 16, 17, 18, 19 or combinations of them.20 The development and validation of these computational methods have also been supported by databases documenting experimentally defined changes in free energy between the wild‐type and mutant proteins.21, 22, 23
Here, we briefly review various computational tools used to predict the impacts of mutations on protein function through impairment of stability and interactions with other molecules including proteins, nucleic acids, and small molecules. We then focus on two structure‐based approaches that were originally developed in Cambridge in the Blundell lab. The first of these, site‐directed mutator (SDM),24, 25 published in 1997, is based on conformationally constrained, environment‐specific substitution tables (ESSTs).26 The second more recent approach published in 2013, mCSM, uses graph‐based signatures to represent the wild‐type structural environment in order to use machine learning to predict the effect of mutations on stability. The mCSM family of software has been further developed by Douglas Pires and David Ascher after they left the Blundell laboratory;27 the current state of the mCSM software can be accessed at the Ascher Laboratory in Melbourne, Australia (https://biomedicalsciences.unimelb.edu.au/sbs-research-groups/biochemistry-and-molecular-biology-research/ascher-laboratory-structural-biology-and-bioinformatics).
In 2014, we brought together SDM and mCSM for predicting the impacts of mutations on protein stability as DUET.18 Here, we focus on developments that are more recent. These include elaboration of SDM to consider the effects of depth from the surface and the residue packing density on the impacts of mutations on protein stability28 and broadening the application of mCSM to consider impacts on protein function not only of residue mutations that impair interactions with other macromolecules19 but also small molecule ligands, which are critical for understanding drug resistance.16 We exemplify recent uses of these approaches to understand human genetic disease, mutations in cancer from the Cancer Gene Census of COSMIC,29 and resistance mutations occurring in mycobacterial and other infections.
2. SEQUENCE‐BASED PREDICTION OF IMPACT OF MUTATION ON PROTEIN FUNCTION
The availability of extensive exome sequencing data provides a wealth of previously unknown mutations that are either causal or lead to increased susceptibility to disease by affecting protein function, both outcomes providing challenges to human health and design of new medicines. Computational approaches to prediction of impacts of the mutations can be based on understanding sequence conservation, for example, SIFT,30, 31 PROVEAN,32 PANTHER‐PSEP,33 MutationAssessor,34 MutPred,35 LRT,36 FATHMM,37 and DEOGEN.38 More recently, machine learning has become more frequently used, and be trained on data describing annotation of functions, or sequence and structural features; for example, PolyPhen‐2,39 MutationTaster,40 SNAP,41 SNPs&GO,42 nsSNPAnalyzer,43 PhD‐SNP,44 SuSPect,45 DEOGEN2,46 and PMut.47 Various ensemble‐based methods, which exploit a gamut of available independent predictors, have also been developed. They include REVEL,48 MetaLR/support vector machine (SVM),49 Eigen,50 CADD,51 DANN,52 KGGSeq,53 Condel,54 and PON‐P.55 In addition, independent scoring functions based on sequence conservation have been developed to quantify the impact of mutations: they include PhyloP,56 SiPhy,57 GERP++,58 and GV‐GD.59
Detailed descriptions of different variant predictors have been reviewed elsewhere.60, 61, 62, 63, 64 Computational prediction tools rely on established databases like VariBench,65 VariSNP,66 dbSNP,67 ClinVar,68 UniProtKB,69 and PhenCode70 for variation benchmark data sets to develop, train, and validate the software.
3. STRUCTURE‐BASED PREDICTION OF THE IMPACT OF MUTATION ON PROTEIN STABILITY
Structure‐based methods use a combination of sequence and structural information. They can be grouped into statistical, physical, and empirical methods based on the potential energy functions. Statistical potentials include SDM,24, 25, 28 PoPMuSiC 2.0,9 CUPSAT,71 AUTO‐MUTE,72 I‐Mutant 2.0,13 DFIRE,73 Hoppe,74 PROTS,75 PROTS‐RF,76 Delaunay tessellation based four‐body statistical scoring function,77 MAESTRO,20 and Rosetta.11 Methods based on empirical energy functions include FoldX,78 ERIS,79 PEAT‐SA,80 and LIE.81 More accurate but computationally expensive physical‐energy‐based methods include CC/PBSA82 and EGAD.83 Machine‐learning approaches include mCSM15 described below, as well as Pro‐Maya, a mutant stability predictor that uses available data on mutations.84 iStable85 and DUET,18 described below, employ machine learning to provide a consensus prediction based on more than one independent predictor. A review with further information about the software tools is available.86 Databases that support the development and validation of the software include ProTherm,21 Platinum,22 and SKEMPI,23 which contain data on changes in protein stability, ligand affinity, and protein–protein interactions upon mutation, respectively.
4. SITE‐DIRECTED MUTATOR
We first focus on SDM,24, 25, 28 a statistical method developed in the Blundell lab that exploits protein sequence and structural data to predict the impacts of mutations on protein stability (Figure 1). SDM uses ESSTs derived from the analysis of amino acid substitutions that are tolerated within families of homologous proteins of known 3‐D structure.26 In a recent comparison study among 14 most efficient mutant prediction tools, SDM228 was ranked in the top five that are least biased toward stabilizing and destabilizing mutants.87 SDM2 is available at http://structure.bioc.cam.ac.uk/sdm2. Below we briefly describe the method.
Figure 1.
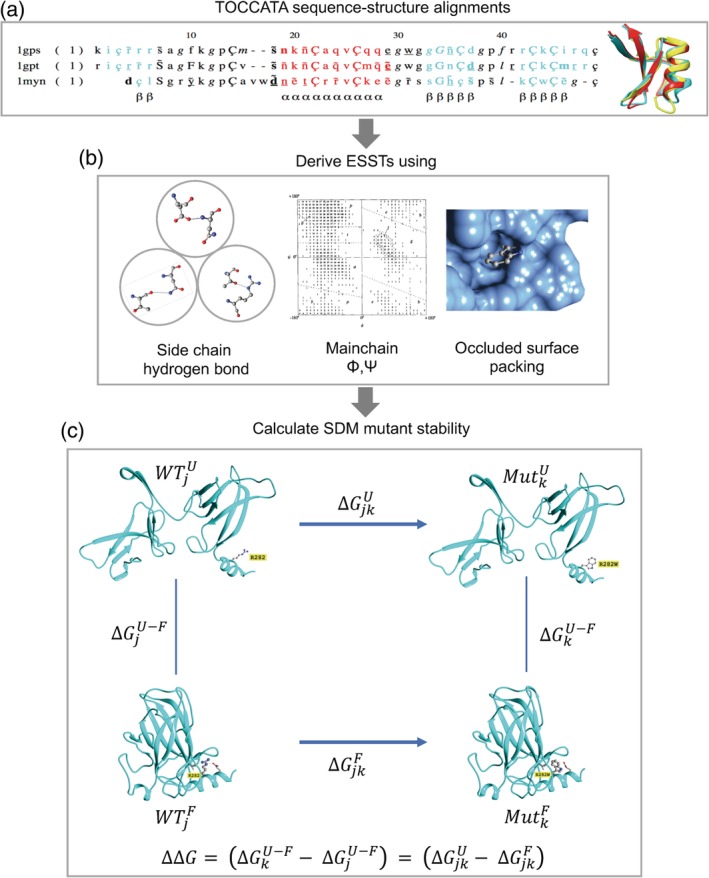
Site‐directed mutator (SDM) method. (a) Sequence‐structure alignments from TOCCATA for protein families are used to calculate the ESSTs. (b) 216 ESSTs are calculated using a combination of eight side‐chain hydrogen bonding patterns, nine main‐chain conformations based on ϕ and ψ dihedral angles, and three residue‐occluded surface packings. (c) The folding–unfolding free energy diagram represented as a thermodynamic cycle for site‐directed mutagenesis. The difference in free energy of unfolding of the wild‐type residue j, and mutant residue k, is related to the free energy changes associated with the mutation in the unfolded and folded states. Using ESSTs, the difference in stability score upon mutation [as shown in Equation (2)] is calculated analogously with Equation (1). As an example, the structure of p53 (PDB http://firstglance.jmol.org/fg.htm?mol=2OCJ) is used to illustrate the mutation at residue position 282 from arginine to tryptophan. Parts of the figure were derived from the graphical abstract taken from Ref. [27]
ESSTs are constructed using the TOCCATA database (Skwark, Torres, Ochoa‐Montano, and Blundell, manuscript in preparation) that contains 2,054 protein sequence‐structure alignments of homologous families taken from SCOP88 and CATH89 domain classification databases and represents a total of 12,038 structures (Figure 1a). In early versions, structural features including the main‐chain conformational angles, relative solvent accessibility (RSA), and hydrogen bonding patterns were used to define a set of local structural environments for the purpose of calculating ESSTs.26 These structural environments are characterized by distinct amino acid substitution propensities and have been successfully used to identify templates in homology modelling90 and finding key catalytic residues,91 as well as prediction of impacts on protein stability upon mutation.24 More recently, in SDM2,28 we introduced a combination of residue‐occluded surface packing (OSP)92, 93 and residue depth.94, 95 We have shown that the residue conservation progressively increases with residue depth and packing density and can be used to classify disease and non‐disease mutations.96 As default, SDM2 uses 216 ESSTs defined by the combination of nine main‐chain conformations, three residue occluded surface packings, and eight hydrogen bonding patterns [Figure 1(b)]. ESSTs defined using OSP showed an improvement in the quality of prediction compared to the previous version of SDM that used 54 ESSTs with RSA.25
The reversible folding and unfolding processes, represented as a thermodynamic cycle, are used to estimate the energetic contribution for a mutation of residue type j in the wild‐type protein to residue type k (Figure 1c). The relationship between the difference in free energy of unfolding of the wild type and mutant and the respective free energy changes associated with the mutation of residue j to k in the unfolded and folded states is expressed as:
| (1) |
As it is time‐consuming to use experiments to measure free energies of denaturation of mutant and wild‐type proteins , SDM uses ESSTs to calculate the difference in the stability scores (∆∆S) of the unfolded () and folded () state for the wild‐type and mutant protein structures, respectively, as:
| (2) |
where
| (3) |
where P(rk/Rj, εwt) is the conditional probability for replacement of residue type Rj in the wild‐type environment εwt by residue rk in an undefined environment and P(rj/Rk, εmut) is the conditional probability for replacement of residue type Rk in the mutant environment by residue rj in an undefined environment. Reference‐state probabilities P(rj/Rj, εwt) and P(rk/Rk, εmut) are introduced to normalize probabilities combined from different substitution tables. The difference in stability score of the unfolded state is calculated in a similar way using conformationally constrained substitution tables representing non‐hydrogen‐bonded, surface‐exposed amino acids whose main‐chain dihedral angles fall outside regular secondary structure regions of the Ramachandran phi–psi map. In addition to the previously defined disruption penalty term for buried residues (24), a cavity penalty term for the substitution of buried bulky hydrophobic residues (Phe, Leu, and Ile) by Ala or Val is added.28 All residues with RSA <17% are considered to be buried.
5. MACHINE LEARNING USING MUTATION CUTOFF SCANNING MATRIX
The family of mCSM computer programs uses the graph‐based approach based on Cutoff Scanning Matrix (CSM)97 to predict the impact of point mutations not only on protein stability but also on protein–protein, protein‐nucleic acid, and protein‐ligand affinities.15 Feature vectors, known as mCSM signatures, defined as inter‐atomic distance patterns around the mutated residue, are used to describe the structural environment in the wild‐type protein. Supervised machine learning is used to train predictive models using mCSM signatures (Figure 2). The mCSM predictive models for protein stability, protein–protein, and protein‐nucleic acid interactions are trained using thermodynamic data sets taken from ProTherm,21 SKEMPI,23 and ProNIT21 databases, respectively. mCSM is available at http://biosig.unimelb.edu.au/mcsm/.
Figure 2.
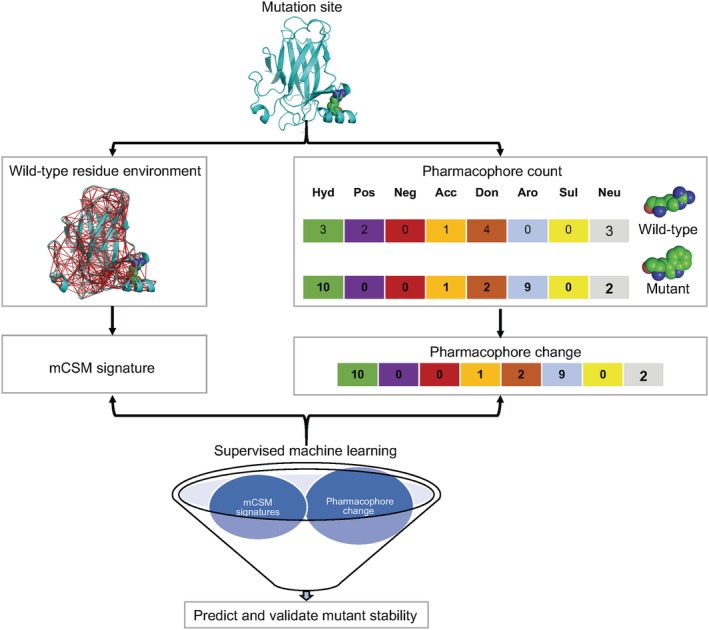
Mutation cutoff scanning matrix (mCSM) method. Starting from the mutant site of the wild‐type protein structure, the method calculates an atom contact graph based on a given distance criterion that defines the wild‐type residue environment. The cumulative distribution obtained from the distances of all pairwise atoms in the contact graph is used to construct the mCSM signature. Pharmacophore count vectors are used to describe the frequencies of atom classes found in the wild‐type and mutant residues. The pharmacophore change vector is used to quantify the difference between the mutant and wild‐type pharmacophore count vectors. mCSM signatures and the pharmacophore change vectors, along with their respective experimental thermodynamic stabilities and affinity data, were used to train predictive models using supervised machine learning techniques to predict the effects of mutations on stability and affinity
The residue environment is defined using graph‐based distance patterns, constructed by collecting atoms within a radial cutoff distance of 30 Å from the centroid of the wild‐type residue atoms. Atoms represent the nodes of the contact graph and edges that connect them are defined by the cutoff distance. The cumulative distribution obtained from the distances of all pairwise atoms in the contact graph forms the major component of mCSM signature that represents the environment of the wild‐type residue. The cumulative distributions were segmented using three types of atom classification as described previously.98
The mCSM signature captures the environment of the wild‐type residue only and, therefore, homology models of the mutants are not required as they are for SDM. mCSM uses a pharmacophore count vector to describe the frequencies of eight atom classes (hydrophobic, positive, negative, hydrogen acceptor, hydrogen donor, aromatic, sulfur, and neutral) based on PMapper program.99 The difference between the mutant and wild‐type pharmacophore count vectors is used to define the pharmacophore change vector, which is then appended to mCSM signature. Experimental conditions including pH, temperature, as well as the RSA of wild‐type residue are added to the mCSM signature.
Extensions to the mCSM method have been developed in Cambridge to predict the impact of mutation on protein–ligand (mCSM‐lig)16 and protein–protein interactions (mCSM‐PPI2),27 and more recently in Fiocruz, Brazil and in Melbourne, Australia by Douglas Pires and David Ascher for protein–nucleic acid (mCSM‐NA),19 antibody–antigen (mCSM‐AB)17 interactions and protein conformations and dynamics in combination based on normal mode dynamics (DynaMut).100
6. DUET
The use of ESSTs to define substitution probabilities in local structural environments and mCSM structural signatures to define long‐range interactions represent two independent complementary approaches to predict the impacts of mutations on protein stability. DUET18 was developed to leverage the individual strengths of SDM and mCSM into a consensus machine‐learning‐based predictor using a SVM101 trained with sequential minimal optimization. The SVM‐based supervised machine‐learning algorithm uses inputs as complementary features, including residue secondary structural annotation from SDM, pharmacophore change vector from mCSM, as well as the individual predictions from SDM and mCSM. The regression model tree is used prior to the machine learning in order to optimize SDM prediction results in combination with residue RSA. The method was trained on the test set of 2,297 mutant stability data taken from ProTherm database. The performance of the predictive model was tested on two independent blind sets that included thermodynamic stability data for 42 mutations in tumor suppressor p53 protein. In all cases, the method achieved higher performance compared to SDM and mCSM.18 DUET is available at http://structure.bioc.cam.ac.uk/duet.
We have recently performed a comparison of DUET and the latest version of the well‐established force‐field‐based mutant‐stability predictor FoldX 5.0.78 The comparison was done using the largest ProTherm benchmark containing 2,648 mutant stability data. DUET achieved a significantly higher Pearson's correlation coefficient (r) of 0.68 between the predicted and the experimental stability change compared to 0.51 obtained using FoldX (Figure 3). The root mean square error obtained for DUET and FoldX is 1.09 and 1.71, respectively.
Figure 3.
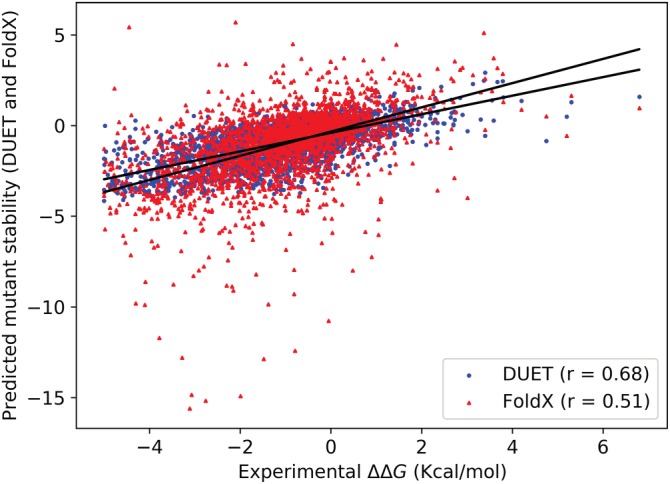
Scatterplot comparing DUET and FoldX stability prediction using the ProTherm benchmark containing 2,468 mutant thermodynamic stability data. Data points for DUET and FoldX are shown as dots (in blue) and triangles (in red), respectively. Pearson's correlation coefficient (r) for DUET and FoldX is shown at the lower right corner of the plot
7. EXAMPLES: UNDERSTANDING MUTATIONS IN GENETIC DISEASE, CANCER AND DRUG/ANTIMICROBIAL RESISTANCE
SDM2, mCSM, and DUET approaches have been used to understand the effects of mutations on human cancer‐related genes in the COSMIC database,102 inhibition of inosine‐5′‐monophosphate dehydrogenase in Mycobacterium tuberculosis,103 isoniazid and rifampicin resistance in Mycobacterium tuberculosis,104 phosphodiesterase somatic mutations implicated in cancer and retinitis pigmentosa,105 protein presenilin 2 linked to familial Alzheimer's disease,106 rifampin resistance in Mycobacterium leprae,107 ESX‐5 type VII‐secreted protein implicated in the transmission of Mycobacterium tuberculosis,108 carbapenem resistance in Acinetobacter baumannii,109 drug resistance in epidermal growth factor receptor,110 POT1 gene implicated in maintaining the telomere homeostasis,111 human Atg8 gene involved in autophagy,112 K13 propeller domain associated with artemisinin drug resistance,113 missense mutation in SLC6A1 associated with Lennox–Gastaut syndrome,114 and TN1 Gene mutations involved in coat plus syndrome.115
Some of these applications are ongoing, a most important example of which is COSMIC. Experimental structural data relevant to COSMIC are limited. Most genes in the Cancer Gene Census (the most frequently mutated cancer genes with mutations in COSMIC) are multidomain, but many of the reported structures for the gene products are single domain or functional substructures of the complete protein. We have already spent 2 years predicting models and the impacts of the 172,000 unique mutations reported for the 700 gene products of the Cancer Census (Alsulami, Torres, and Blundell, Unpublished). This has underlined the importance of full 3D models for the multidomain and multicomponent systems and software so that protein–protein, protein–nucleic acid, protein–ligand and other interactions can be considered. These factors are also important for understanding mutations in genetic disease and drug resistance in cancer therapeutics and antimicrobials.
An example of understanding genetic disease is the application of the SDM and mCSM software to understanding von Hippel–Lindau Syndrome, which leads to the development of clear cell renal carcinoma and is caused by mutations in the VHL gene that encodes protein pVHL (Figure 4). We earlier sought to identify a patient's risk of developing clear cell renal carcinoma from understanding the impacts of the mutations not only on pVHL protein stability but also on the affinity for the binding partners, HIF‐1α, and Elongin B and C.116, 117 At that time of the first paper,116 SDM was the only structure‐guided program available from our lab. We showed that the molecular mechanisms of renal cell carcinoma (RCC) and pheochromocytoma (PCC) in VHL disease appear to be decoupled: RCC can arise from disruption of HIF‐1α interactions or binding at the elongin B interface, while PCC is driven by mutations at binding site of elongin C. In the second publication when mCSM software117 was available, we used DUET18 to estimate impacts on stability and mCSM software to assess the impacts on protein–protein interactions. The observation of important mutations in protein–protein interactions has proved typical of many human Mendelian diseases, although mutations that affect stability are very common.118, 119, 120, 121
Figure 4.
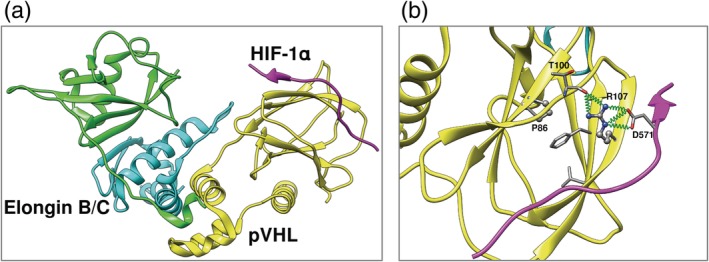
Clear cell renal carcinoma in von Hippel–Lindau disease. (a) Ternary complex of pVHL with Elongin B/C, critical for pVHL stability and function. (b) Protein–protein interactions mediated by arginine (R107), mutation of proline at position 86 to arginine alters complementarity of charge between subunits and destabilize protein–protein interactions, essential to function. Hydrogen bond interactions are shown as springs in green
The software SDM and mCSM can also be used to begin to understand the emergence of drug resistance. Looking at single‐point coding mutations in Mycobacterium tuberculosis, strong correlations have been observed between the structural features of mutations and their link to antibiotic sensitivity.4 This suggests that prediction of drug resistance mutations before they arise, using our software with genomic sequences and structures, could be useful in guiding drug development. This idea has been applied in our efforts to develop small‐molecule inhibitors of GuaB2 to treat Mycobacterium tuberculosis infections.122 Computational saturation mutagenesis on the crystal structure of GuaB2 with VCC234718 using SDM, mCSM, and mCSM‐lig suggested that a resistance mutation would likely occur at Y487, altering interactions with the inhibitor but not NAD, while not disrupting protein stability or the interactions between the homo‐tetramer units. In fact, Y487C was observed to be an important resistance mutation. Other molecules were identified that did not make these interactions and were active against the Y487C mutant. Further work is required to show that inhibitors that are active against Mtb in vitro through the inhibition of GuaB2 have therapeutic benefits in vivo.
These and many other analyses highlight the importance of considering the structural environment of a mutation in order to understand the molecular and biological consequences. In evolution, they may indicate new functions emerging. For genetic disease, they should help identify mechanisms of disease emergence, while for the emergence of drug resistance they may help identify molecules that are less likely to lead to the emergence of resistance.
The authors recognize that there are limitations of most computational approaches that attempt to predict mutational effects on the whole organism. As demonstrated above, we have moved in the direction of recognizing that mutations affect higher‐level interactions rather than just the stability of individual proteins, by considering the impacts of mutations on the interactions between various components of macromolecular assemblies. In cases of resistance to drugs and genetic disease, the correlations are evident at a whole organism level. However, the level of expression in vivo, how this exceeds the threshold required for function in the organism and how alterations in stability will affect protein levels and protein solubility are more difficult to assess. Furthermore, for diploid organisms, it is unclear whether a single mutation will show a phenotype. None of these factors is currently considered. Hence, while predictions of mutational effects on stability are undoubtedly important, translation of these to predict disease phenotypes will likely require individual disease‐specific training sets to calibrate predictions of programs like SDM2, mCSM, and DUET. Thus, studying the impact of mutations on in vivo protein expression, as well as within the context of recessive and dominant mutants must in future become central to the system‐level understanding for the whole organism.
ACKNOWLEDGMENTS
We thank our collaborators on the development of SDM (Drs. Chris Topham, N. Srinivasan, and Catherine Worth) and of mCSM and DUET (Drs. Douglas Pires and David Ascher) for their very significant contributions. We thank the Bill & Melinda Gates Foundation HIT‐TB (Grant Code: RG60453 to T.L.B.) and the EU MM4TB [Project ID: 260872 to A.P.P. and T.L.B.]; Wellcome Trust Investigator Award [200814/Z/16/Z to T.L.B.]; Collaboration of Medical Research Council (MRC) UK and Department of Biotechnology (DBT) India, Cambridge‐Chennai Centre Partnership on Antimicrobial Resistance (RG77600); Newton Fund RCUK‐CONFAP Grant awarded by The Medical Research Council (MRC) [MR/M026302/1 to T.L.B.]. A.P.P. thanks the MRC and BBSRC for support in the Laboratory of Professor Julian Gough at LMB, Cambridge.
Pandurangan AP, Blundell TL. Prediction of impacts of mutations on protein structure and interactions: SDM, a statistical approach, and mCSM, using machine learning. Protein Science. 2020;29:247–257. 10.1002/pro.3774
Funding information Bill & Melinda Gates Foundation HIT‐TB, Grant/Award Number: RG60453; Medical Research Council, Grant/Award Number: Antimicrobial Resistance / RG77600; Wellcome Trust, Grant/Award Number: Investigator Award / 200814/Z/16/Z
Contributor Information
Arun Prasad Pandurangan, Email: apandura@mrc-lmb.cam.ac.uk.
Tom L. Blundell, Email: tlb20@cam.ac.uk.
REFERENCES
- 1. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabbani B, Nakaoka H, Akhondzadeh S, Tekin M, Mahdieh N. Next generation sequencing: Implications in personalized medicine and pharmacogenomics. Mol Biosyst. 2016;12:1818–1830. [DOI] [PubMed] [Google Scholar]
- 3. Burgess DJ. Disease genetics: Network effects of disease mutations. Nat Rev Genet. 2015;16:317. [DOI] [PubMed] [Google Scholar]
- 4. Phelan J, Coll F, McNerney R, et al. Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti‐tuberculosis drug resistance. BMC Med. 2016;14:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fariselli P, Martelli PL, Savojardo C, Casadio R. INPS: Predicting the impact of non‐synonymous variations on protein stability from sequence. Bioinformatics. 2015;31:2816–2821. [DOI] [PubMed] [Google Scholar]
- 6. Capriotti E, Fariselli P, Casadio R. A neural‐network‐based method for predicting protein stability changes upon single point mutations. Bioinformatics. 2004;20(Suppl 1):i63–i68. [DOI] [PubMed] [Google Scholar]
- 7. Huang LT, Gromiha MM, Ho SY. iPTREE‐STAB: Interpretable decision tree based method for predicting protein stability changes upon mutations. Bioinformatics. 2007;23:1292–1293. [DOI] [PubMed] [Google Scholar]
- 8. Teng S, Srivastava AK, Wang L. Sequence feature‐based prediction of protein stability changes upon amino acid substitutions. BMC Genomics. 2010;11(Suppl 2):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC‐2.0. Bioinformatics. 2009;25:2537–2543. [DOI] [PubMed] [Google Scholar]
- 10. Bordner AJ, Abagyan RA. Large‐scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins. 2004;57:400–413. [DOI] [PubMed] [Google Scholar]
- 11. Kellogg EH, Leaver‐Fay A, Baker D. Role of conformational sampling in computing mutation‐induced changes in protein structure and stability. Proteins. 2011;79:830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masso M, Vaisman II. Accurate prediction of stability changes in protein mutants by combining machine learning with structure based computational mutagenesis. Bioinformatics. 2008;24:2002–2009. [DOI] [PubMed] [Google Scholar]
- 13. Capriotti E, Fariselli P, Casadio R. I‐Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins. 2006;62:1125–1132. [DOI] [PubMed] [Google Scholar]
- 15. Pires DE, Ascher DB, Blundell TL. mCSM: Predicting the effects of mutations in proteins using graph‐based signatures. Bioinformatics. 2014;30:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pires DE, Blundell TL, Ascher DB. mCSM‐lig: Quantifying the effects of mutations on protein‐small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep. 2016;6:29575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pires DE, Ascher DB. mCSM‐AB: A web server for predicting antibody‐antigen affinity changes upon mutation with graph‐based signatures. Nucleic Acids Res. 2016;44:W469–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pires DE, Ascher DB, Blundell TL. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42:W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pires DEV, Ascher DB. mCSM‐NA: Predicting the effects of mutations on protein‐nucleic acids interactions. Nucleic Acids Res. 2017;45:W241–W246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laimer J, Hofer H, Fritz M, Wegenkittl S, Lackner P. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinf. 2015;16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar MD, Bava KA, Gromiha MM, et al. ProTherm and ProNIT: Thermodynamic databases for proteins and protein‐nucleic acid interactions. Nucleic Acids Res. 2006;34:D204–D206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pires DE, Blundell TL, Ascher DB. Platinum: A database of experimentally measured effects of mutations on structurally defined protein‐ligand complexes. Nucleic Acids Res. 2015;43:D387–D391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jankauskaite J, Jimenez‐Garcia B, Dapkunas J, Fernandez‐Recio J, Moal IH. SKEMPI 2.0: An updated benchmark of changes in protein‐protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2019;35:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Topham CM, Srinivasan N, Blundell TL. Prediction of the stability of protein mutants based on structural environment‐dependent amino acid substitution and propensity tables. Protein Eng. 1997;10:7–21. [DOI] [PubMed] [Google Scholar]
- 25. Worth CL, Preissner R, Blundell TL. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Overington J, Johnson MS, Sali A, Blundell TL. Tertiary structural constraints on protein evolutionary diversity: Templates, key residues and structure prediction. Proc Biol Sci. 1990;241:132–145. [DOI] [PubMed] [Google Scholar]
- 27. Rodrigues CHM, Myung Y, Pires DEV, Ascher DB. mCSM‐PPI2: Predicting the effects of mutations on protein‐protein interactions. Nucleic Acids Res. 2019;47:W338–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pandurangan AP, Ochoa‐Montano B, Ascher DB, Blundell TL. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45:W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tate JG, Bamford S, Jubb HC, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- 32. Choi Y, Chan AP. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang H, Thomas PD. PANTHER‐PSEP: Predicting disease‐causing genetic variants using position‐specific evolutionary preservation. Bioinformatics. 2016;32:2230–2232. [DOI] [PubMed] [Google Scholar]
- 34. Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li B, Krishnan VG, Mort ME, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raimondi D, Gazzo AM, Rooman M, Lenaerts T, Vranken WF. Multilevel biological characterization of exomic variants at the protein level significantly improves the identification of their deleterious effects. Bioinformatics. 2016;32:1797–1804. [DOI] [PubMed] [Google Scholar]
- 39. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 41. Bromberg Y, Rost B. SNAP: Predict effect of non‐synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease‐related mutations in proteins. Hum Mutat. 2009;30:1237–1244. [DOI] [PubMed] [Google Scholar]
- 43. Bao L, Zhou M, Cui Y. nsSNPAnalyzer: Identifying disease‐associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005;33:W480–W482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. [DOI] [PubMed] [Google Scholar]
- 45. Yates CM, Filippis I, Kelley LA, Sternberg MJ. SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol. 2014;426:2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raimondi D, Tanyalcin I, Ferte J, et al. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017;45:W201–W206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lopez‐Ferrando V, Gazzo A, de la Cruz X, Orozco M, Gelpi JL. PMut: A web‐based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017;45:W222–W228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ionita‐Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quang D, Chen Y, Xie X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li MX, Kwan JS, Bao SY, et al. Predicting mendelian disease‐causing non‐synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gonzalez‐Perez A, Lopez‐Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Olatubosun A, Valiaho J, Harkonen J, Thusberg J, Vihinen M. PON‐P: Integrated predictor for pathogenicity of missense variants. Hum Mutat. 2012;33:1166–1174. [DOI] [PubMed] [Google Scholar]
- 56. Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009;25:i54–i62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tavtigian SV, Deffenbaugh AM, Yin L, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. [DOI] [PubMed] [Google Scholar]
- 61. Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: State‐of‐the‐art web‐based tools. Brief Bioinform. 2013;14:448–459. [DOI] [PubMed] [Google Scholar]
- 62. Ritchie GR, Flicek P. Computational approaches to interpreting genomic sequence variation. Genome Med. 2014;6:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Butkiewicz M, Bush WS. In silico functional annotation of genomic variation. Curr Protoc Hum Genet. 2016;88(6):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Niroula A, Vihinen M. Variation interpretation predictors: Principles, types, performance, and choice. Hum Mutat. 2016;37:579–597. [DOI] [PubMed] [Google Scholar]
- 65. Sasidharan Nair P, Vihinen M. VariBench: A benchmark database for variations. Hum Mutat. 2013;34:42–49. [DOI] [PubMed] [Google Scholar]
- 66. Schaafsma GC, Vihinen M. VariSNP, a benchmark database for variations from dbSNP. Hum Mutat. 2015;36:161–166. [DOI] [PubMed] [Google Scholar]
- 67. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat. 2018;39:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. UniProt Consortium . UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Giardine B, Riemer C, Hefferon T, et al. PhenCode: Connecting ENCODE data with mutations and phenotype. Hum Mutat. 2007;28:554–562. [DOI] [PubMed] [Google Scholar]
- 71. Parthiban V, Gromiha MM, Schomburg D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239–W242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Masso M, Vaisman II. AUTO‐MUTE: Web‐based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng Des Sel. 2010;23:683–687. [DOI] [PubMed] [Google Scholar]
- 73. Zhou H, Zhou Y. Distance‐scaled, finite ideal‐gas reference state improves structure‐derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hoppe C, Schomburg D. Prediction of protein thermostability with a direction‐ and distance‐dependent knowledge‐based potential. Protein Sci. 2005;14:2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li Y, Zhang J, Tai D, Middaugh CR, Zhang Y, Fang J. PROTS: A fragment based protein thermo‐stability potential. Proteins. 2012;80:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li Y, Fang J. PROTS‐RF: A robust model for predicting mutation‐induced protein stability changes. PLoS One. 2012;7:e47247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Deutsch C, Krishnamoorthy B. Four‐body scoring function for mutagenesis. Bioinformatics. 2007;23:3009–3015. [DOI] [PubMed] [Google Scholar]
- 78. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: An online force field. Nucleic Acids Res. 2005;33:W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yin S, Ding F, Dokholyan NV. Modeling backbone flexibility improves protein stability estimation. Structure. 2007;15:1567–1576. [DOI] [PubMed] [Google Scholar]
- 80. Johnston MA, Sondergaard CR, Nielsen JE. Integrated prediction of the effect of mutations on multiple protein characteristics. Proteins. 2011;79:165–178. [DOI] [PubMed] [Google Scholar]
- 81. Wickstrom L, Gallicchio E, Levy RM. The linear interaction energy method for the prediction of protein stability changes upon mutation. Proteins. 2012;80:111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural ensembles. Nat Methods. 2009;6:3–4. [DOI] [PubMed] [Google Scholar]
- 83. Pokala N, Handel TM. Energy functions for protein design: Adjustment with protein‐protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347:203–227. [DOI] [PubMed] [Google Scholar]
- 84. Wainreb G, Wolf L, Ashkenazy H, Dehouck Y, Ben‐Tal N. Protein stability: A single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics. 2011;27:3286–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen CW, Lin J, Chu YW. iStable: Off‐the‐shelf predictor integration for predicting protein stability changes. BMC Bioinf. 2013;14(Suppl 2):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kulshreshtha S, Chaudhary V, Goswami GK, Mathur N. Computational approaches for predicting mutant protein stability. J Comput Aided Mol Des. 2016;30:401–412. [DOI] [PubMed] [Google Scholar]
- 87. Pucci F, Bernaerts KV, Kwasigroch JM, Rooman M. Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics. 2018;34:3659–3665. [DOI] [PubMed] [Google Scholar]
- 88. Andreeva A, Howorth D, Chandonia JM, et al. Data growth and its impact on the SCOP database: New developments. Nucleic Acids Res. 2008;36:D419–D425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dawson NL, Lewis TE, Das S, et al. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017;45:D289–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Johnson MS, Overington JP, Blundell TL. Alignment and searching for common protein folds using a data bank of structural templates. J Mol Biol. 1993;231:735–752. [DOI] [PubMed] [Google Scholar]
- 91. Chelliah V, Chen L, Blundell TL, Lovell SC. Distinguishing structural and functional restraints in evolution in order to identify interaction sites. J Mol Biol. 2004;342:1487–1504. [DOI] [PubMed] [Google Scholar]
- 92. Pattabiraman N, Ward KB, Fleming PJ. Occluded molecular surface: Analysis of protein packing. J Mol Recognit. 1995;8:334–344. [DOI] [PubMed] [Google Scholar]
- 93. Fleming PJ, Richards FM. Protein packing: Dependence on protein size, secondary structure and amino acid composition. J Mol Biol. 2000;299:487–498. [DOI] [PubMed] [Google Scholar]
- 94. Chakravarty S, Varadarajan R. Residue depth: A novel parameter for the analysis of protein structure and stability. Structure. 1999;7:723–732. [DOI] [PubMed] [Google Scholar]
- 95. Tan KP, Nguyen TB, Patel S, Varadarajan R, Madhusudhan MS. Depth: A web server to compute depth, cavity sizes, detect potential small‐molecule ligand‐binding cavities and predict the pKa of ionizable residues in proteins. Nucleic Acids Res. 2013;41:W314–W321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pandurangan AP, Ascher DB, Thomas SE, Blundell TL. Genomes, structural biology and drug discovery: Combating the impacts of mutations in genetic disease and antibiotic resistance. Biochem Soc Trans. 2017;45:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pires DE, de Melo‐Minardi RC, dos Santos MA, da Silveira CH, Santoro MM, Meira W Jr. Cutoff scanning matrix (CSM): Structural classification and function prediction by protein inter‐residue distance patterns. BMC Genomics. 2011;12(Suppl 4):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pires DE, de Melo‐Minardi RC, da Silveira CH, Campos FF, Meira W Jr. aCSM: Noise‐free graph‐based signatures to large‐scale receptor‐based ligand prediction. Bioinformatics. 2013;29:855–861. [DOI] [PubMed] [Google Scholar]
- 99. Schneider G, Lee ML, Stahl M, Schneider P. De novo design of molecular architectures by evolutionary assembly of drug‐derived building blocks. J Comput Aided Mol Des. 2000;14:487–494. [DOI] [PubMed] [Google Scholar]
- 100. Rodrigues CH, Pires DE, Ascher DB. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shevade SK, Keerthi SS, Bhattacharyya C, Murthy KK. Improvements to the SMO algorithm for SVM regression. IEEE Trans Neural Netw. 2000;11:1188–1193. [DOI] [PubMed] [Google Scholar]
- 102. Malhotra S, Alsulami AF, Heiyun Y, et al. Understanding the impacts of missense mutations on structures and functions of human cancer‐related genes: A preliminary computational analysis of the COSMIC cancer gene census. PLoS One. 2019;14:e0219935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Singh V, Pacitto A, Donini S, et al. Synthesis and structure‐activity relationship of 1‐(5‐isoquinolinesulfonyl)piperazine analogues as inhibitors of Mycobacterium tuberculosis IMPDH. Eur J Med Chem. 2019;174:309–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Munir A, Kumar N, Ramalingam SB, et al. Identification and characterization of genetic determinants of isoniazid and rifampicin resistance in Mycobacterium tuberculosis in southern India. Sci Rep. 2019;9:10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Maryam A, Vedithi SC, Khalid RR, et al. The molecular organization of human cGMP specific phosphodiesterase 6 (PDE6): Structural implications of somatic mutations in cancer and retinitis pigmentosa. Comput Struct Biotechnol J. 2019;17:378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dehury B, Tang N, Kepp KP. Insights into membrane‐bound presenilin 2 from all‐atom molecular dynamics simulations. J Biomol Struct Dyn. 2019;1–15. 10.1080/07391102.2019.1655481. [DOI] [PubMed] [Google Scholar]
- 107. Vedithi SC, Malhotra S, Das M, et al. Structural implications of mutations conferring Rifampin resistance in mycobacterium leprae. Sci Rep. 2018;8:5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Holt KE, McAdam P, Thai PVK, et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat Genet. 2018;50:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hawkey J, Ascher DB, Judd LM, et al. Evolution of carbapenem resistance in Acinetobacter baumannii during a prolonged infection. Microb Genom. 2018;2018:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kannan S, Fox SJ, Verma CS. Exploring gatekeeper mutations in EGFR through computer simulations. J Chem Inf Model. 2019;59:2850–2858. [DOI] [PubMed] [Google Scholar]
- 111. Amir M, Kumar V, Mohammad T, et al. Investigation of deleterious effects of nsSNPs in the POT1 gene: A structural genomics‐based approach to understand the mechanism of cancer development. J Cell Biochem. 2019;120:10281–10294. [DOI] [PubMed] [Google Scholar]
- 112. Jatana N, Ascher DB, Pires DEV, Gokhale RS, Thukral L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. 2019;1–17. 10.1080/15548627.2019.1606636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. He Y, Campino S, Diez Benavente E, et al. Artemisinin resistance‐associated markers in Plasmodium falciparum parasites from the China‐Myanmar border: Predicted structural stability of K13 propeller variants detected in a low‐prevalence area. PLoS One. 2019;14:e0213686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cai K, Wang J, Eissman J, et al. A missense mutation in SLC6A1 associated with Lennox‐Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp Neurol. 2019;320:112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Amir M, Mohammad T, Kumar V, et al. Structural analysis and conformational dynamics of STN1 gene mutations involved in Coat Plus syndrome. Front Mol Biosci. 2019;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Forman JR, Worth CL, Bickerton GR, Eisen TG, Blundell TL. Structural bioinformatics mutation analysis reveals genotype‐phenotype correlations in von Hippel‐Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins. 2009;77:84–96. [DOI] [PubMed] [Google Scholar]
- 117. Pires DE, Chen J, Blundell TL, Ascher DB. In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci Rep. 2016;6:19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nemethova M, Radvanszky J, Kadasi L, et al. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: Focus on 'black bone disease’ in Italy. Eur J Hum Genet. 2016;24:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Usher JL, Ascher DB, Pires DE, Milan AM, Blundell TL, Ranganath LR. Analysis of HGD gene mutations in patients with Alkaptonuria from the United Kingdom: Identification of novel mutations. JIMD Rep. 2015;24:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jubb HC, Pandurangan AP, Turner MA, Ochoa‐Montano B, Blundell TL, Ascher DB. Mutations at protein‐protein interfaces: Small changes over big surfaces have large impacts on human health. Prog Biophys Mol Biol. 2017;128:3–13. [DOI] [PubMed] [Google Scholar]
- 121. Jafri M, Wake NC, Ascher DB, et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015;5:723–729. [DOI] [PubMed] [Google Scholar]
- 122. Singh V, Donini S, Pacitto A, et al. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect Dis. 2017;3:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]