

Abstract
KEGG is a reference knowledge base for biological interpretation of large‐scale molecular datasets, such as genome and metagenome sequences. It accumulates experimental knowledge about high‐level functions of the cell and the organism represented in terms of KEGG molecular networks, including KEGG pathway maps, BRITE hierarchies, and KEGG modules. By the process called KEGG mapping, a set of protein coding genes in the genome, for example, can be converted to KEGG molecular networks enabling interpretation of cellular functions and other high‐level features. Here we report a new version of KEGG Mapper, a suite of KEGG mapping tools available at the KEGG website (https://www.kegg.jp/ or https://www.genome.jp/kegg/), together with the KOALA family tools for automatic assignment of KO (KEGG Orthology) identifiers used in the mapping.
Keywords: genome annotation, KEGG, KEGG Mapper, KEGG module, KEGG Orthology, pathway analysis
1. INTRODUCTION
The KEGG database resource has been developed for the purpose of uncovering cellular and organism‐level functions from large‐scale molecular‐level datasets, especially gene sets in the complete genomes.1 The three databases, PATHWAY, BRITE, and MODULE, contain experimental knowledge of such high‐level functions captured from published literature and represented in terms of KEGG pathway maps, BRITE hierarchies, and KEGG modules, respectively. The KEGG pathway map is a manually drawn graphical diagram showing metabolic, signaling, and other molecular interaction/reaction networks. The BRITE hierarchy is a classification system for various biological objects including genes and proteins. The KEGG module is a manually defined functional unit in the metabolic and other networks represented by the logical expression for automatic evaluation of its presence or absence. Among the three, the PATHWAY database is the most widely used as a reference knowledge base for biological interpretation of users' datasets through KEGG pathway mapping, a type of gene set enrichment analysis.
KEGG PATHWAY can be compared with Gene Ontology (GO),2 a key database for gene set enrichment analysis. GO is a collection of controlled vocabularies for gene functions organized in three ontologies: biological process, cellular component, and molecular function. Many genomes are annotated with GO terms by community efforts, so that GO enrichment analysis can be performed on various gene sets. For example, an enrichment analysis finds GO terms that are over‐represented for a given set of human genes, enabling its biological interpretation. In contrast, KEGG pathway mapping presents additional information about how genes or gene products interact in pathways, but the coverage of genes is less comprehensive than GO.
The BRITE database, which is an ontology database, was introduced to supplement PATHWAY and to expand the coverage of genes for KEGG mapping. The MODULE database was added as an attempt to automate functional interpretation. Unfortunately, however, these two databases have not been well utilized. In the new version of KEGG Mapper, the pathway mapping tools are redesigned to search by default the three databases, PATHWAY, BRITE, and MODULE, as well as other databases for human gene sets. Here we report these new developments.
2. KEGG IDENTIFIERS
KEGG is an integrated database consisting of 18 databases in four categories, systems, genomic, chemical, and health information categories. Excluding the computationally generated SSDB database, which is used internally for genome annotation, Table 1 shows various biological objects accumulated in 17 databases. They are KEGG original databases, except the three databases, GENES, ENZYME, and VARIANT, whose data are taken from outside sources and given KEGG annotations.
Table 1.
KEGG identifiers
| Category | Database | Object | DB | Entry prefix | Example |
|---|---|---|---|---|---|
| Systems information | PATHWAY | KEGG pathway map | path | map, ko, ec, rn, <org> |
map00300 (Lysine biosynthesis pathway) hsa04010 (Human MAPK signaling pathway) |
| BRITE | BRITE hierarchy or table (classification system) | br | br, jp, ko, <org> | ko02000 (Transporter classification) | |
| MODULE | KEGG module | md | M, <org>_M | M00433 (Lysine biosynthesis module) | |
| Genomic information | KO | Functional ortholog | ko | K | K01655 (Homocitrate synthase) |
| GENOME | KEGG organism (complete genome) | gn | T |
T01001 (Homo sapiens) gn:hsa (Homo sapiens) |
|
| GENES | Gene and protein | <org> | – | hsa:1956 (EGFR) | |
| Chemical information | COMPOUND | Small molecule | cpd | C | C01290 (Lactosylceramide) |
| GLYCAN | Glycan | gl | G | G00092 (Lactosylceramide) | |
| REACTION | Biochemical reaction | rn | R | R03355 | |
| RCLASS | Reaction class | rc | RC | RC00049 | |
| ENZYME | Enzyme nomenclature | ec | – | ec:3.2.1.23 (beta‐Galactosidase) | |
| Health information | NETWORK |
Network variation map Network element |
ne |
nt N |
nt06210 (ERK signaling) N00014 (Mutation‐activated EGFR signaling) |
| VARIANT | Human gene variant | hsa_var | – | hsa_var:1956v2 (EGFR mutation) | |
| DISEASE | Human disease | ds | H | H00014 (Non‐small cell lung cancer) | |
| DRUG | Drug | dr | D | D01977 (Gefitinib) | |
| DGROUP | Drug group | dg | DG | DG01917 (Receptor tyrosine kinase inhibitor) | |
| ENVIRON | Health‐related substance | ev | E | E00017 (Opium) |
<org>: organism code such as hsa for Homo sapiens.
Each database entry is uniquely identified by specifying the database name and the entry identifier in the form of “db:entry,” but the database name may be omitted in the KEGG original databases, because the entry identifier takes the form of a database‐dependent prefix followed by a five‐digit number. The KEGG identifier refers to any of these prefix‐type identifiers or the db:entry type identifiers. Table 1 shows examples of KEGG identifiers: K01655 as a simplified form of ko:K01655, C01290 as a simplified form of cpd:C01290, and so forth. The prefix‐type identifier is often called K number, C number, and so forth. The actual content of each KEGG object (database entry) may be obtained by entering the KEGG identifier in the search box of the KEGG website top page.
3. KEGG MOLECULAR NETWORKS
The three databases in the systems information category (Table 1) are used as target databases in KEGG mapping. They contain KEGG pathway maps, BRITE hierarchies, and KEGG modules, collectively called KEGG molecular networks, as they represent certain aspects of molecular interaction, reaction, and relation networks. The KEGG molecular networks are developed in a generic way, namely, in terms of functional orthologs, called KO (KEGG Orthology) groups, rather than individual genes or proteins, so that experimental evidence in specific organisms can be extended to other organisms.3 Two types of KEGG mapping may be distinguished. One is KO‐based mapping, where genes and proteins are first assigned KO identifiers (also called K numbers) to generate organism‐specific versions of KEGG pathways, BRITE hierarchies, and KEGG modules, as implemented for over 6,000 complete genomes in KEGG. The other is direct mapping, where various molecular objects are directly mapped to molecular network objects, such as human genes mapped to nodes of human pathways and chemical substances mapped to metabolites of metabolic pathways.
The PATHWAY database contains pathway maps for metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, and human diseases. Figure 1a is an example, a part of human MAPK signaling pathway map. The BRITE database plays a role of classifying various objects shown in Table 1: genes and proteins, chemical compounds and reactions, drugs, diseases, and organisms. Its basic representation is the tree‐like structure implemented as a hierarchy file, such as shown in Figure 1b for transporter classification. An additional representation is the excel‐like structure implemented as an html table file, which is useful for comparison of various attributes associated with the objects.
Figure 1.
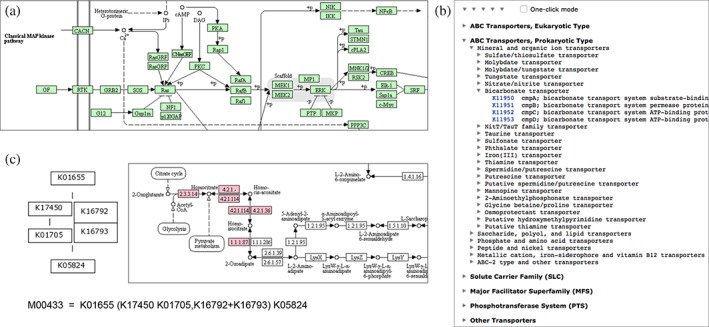
Knowledge representation of molecular interaction, reaction, and relation networks in KEGG. (a) KEGG pathway map for human MAPK signaling pathway (hsa04010). (b) BRITE hierarchy for transporter classification (ko02000). (c) KEGG module for lysine biosynthesis, 2‐oxoglutarate = > 2‐oxoadipate (M00433), corresponding to the highlighted part in lysine biosynthesis pathway (map00300). The logical expression of K numbers is also represented by the graphical diagram
The MODULE database is a collection of KEGG modules. Each module is a functional unit of genes (or proteins) characterizing metabolism and other high‐level features such as pathogenicity and antimicrobial resistance. The module is identified by the M number and defined by the logical expression of K numbers. Figure 1c shows an example, a lysine biosynthesis unit in fungi. When evaluating the logical expression, a space or a plus sign, representing a connection in the pathway or the molecular complex, is treated as the AND operator and a comma, used for alternatives, is treated as the OR operator. The logical expression allows automatic evaluation of whether the gene set is complete, that is, the module is present, in a given genome or metagenome.
4. FROM GENES TO PATHWAYS THROUGH KOS
KOs represent functional orthologs in the context of KEGG pathway maps and BRITE hierarchies and are defined by extending experimental knowledge in specific organisms to other organisms.3 Among 23,000 KOs currently defined, 87% contain published references reporting functional characterizations and 71% contain sequence data used in the original experiments. From such sequence data, a sequence similarity group is manually defined and then computationally expanded with the help of the KOALA tool described below. The similarity threshold is dependent on how far experimental evidence in a specific organism can be generalized to other organisms. Certain paralogs of human genes may be used to define finely classified KOs to represent, for example, mammalian specific cellular processes. There are so‐called tight KOs for distinguishing, for example, antimicrobial resistance due to beta‐lactamase variants, which tend to form tight clusters of high sequence similarity among more broadly distributed lower similarity groups.4
As of June 2019, the GENES database contains 30 million genes from 6,000 completely sequenced genomes. KOALA (Kegg Orthology And Links Annotation) is the annotation tool used internally for both manually defining the starting KO groups and automatically propagating manual KO assignments to the entire GENES database.3 KOALA is based on the SSDB database, a computationally generated database containing the results of SSEARCH5 comparison scores for all gene pairs and best‐hit relations for all genome pairs in the GENES database. Currently, KOs are given to 49% of 28 million protein‐coding genes and 24% of over one million RNA‐coding genes.
For each of the 6,000 organisms KEGG pathway maps and other molecular networks are automatically reconstructed using assigned KOs. Prefixes of KEGG identifiers (Table 1) are changed to distinguish organism‐specific pathways. For example, the human pathway hsa00010 is generated from the reference pathway map00010 and the KO assignment for Homo sapiens whose organism code is “hsa,” thus changing the prefix from “map” to “hsa.”
5. KOALA FAMILY TOOLS
At the KEGG/GenomeNet website, four servers are available for the KO assignment and subsequent KEGG mapping of the user's amino acid sequence data: KAAS6 released in 2005, BlastKOALA and GhostKOALA7 released in 2015, and KofamKOALA8 released in 2019. All the servers utilize e‐mail based job submission systems. It is no longer recommended to use KAAS, because it searches a small fraction of the GENES database and does not incorporate non‐genome categories of addendum genes (ag) and viral genes (vg). KAAS is the only option, though, to use nucleotide sequences rather than amino acid sequences as query data.
The internal KOALA tool for KO assignment examines the GFIT tables7 created from the SSDB database using the weighted scoring scheme that includes SW (Smith‐Waterman) score, best‐best flag, overlap of alignment, ratio of query and DB sequences, taxonomic category and Pfam domains. The web servers of BlastKOALA using the BLASTP9 program and GhostKOALA using the GHOSTX10 program search nonredundant pangenome datasets, which are generated by removing similar sequences in similar organisms at the species, genus, or family level,3, 7 and create GFIT‐like tables. BlastKOALA uses the weighted scoring scheme similar to internal KOALA, while GhostKOALA uses its own scores only. In contrast to these sequence similarity based tools, KofamKOALA searches the HMM profile database named KOfam and uses its bit scores with a precomputed adaptive score threshold for each KO group.8 The nonredundant pangenome datasets are updated weekly, while the KOfam database is updated every 1–2 months.
There are advantages and disadvantages of these three KOALA family tools. A comparison was made by using each tool to assign KOs to eight newly added genomes. After the internal annotation procedure was complete, the accuracy of reproducing the internal KO assignment was computed for each tool and for each genome. The accuracy is defined by (TP + TN)/(TP + FP + FN + TN), where TP and TN mean that the assigned KO and no assignment, respectively, matched the internal KOALA assignment, and FP and FN mean that they did not match (see Supplementary Material). The result is shown in Table 2. Sequence comparison based BlastKOALA and GhostKOALA performed better than profile based KofamKOALA, and BlastKOALA was slightly better than GhostKOALA. The main advantage of KofamKOALA is that the HMM database and the search program are downloadable and can be installed locally. The execution time was highly dependent on the server loads, but roughly speaking, GhostKOALA and KofamKOALA are comparable and BlastKOALA takes up to 100 times longer (see Supplementary Material).
Table 2.
Comparison of KOALA family tools
| Genome | Org code | Organism name | Number of genes | Assigned KO (%) | Accuracy | ||
|---|---|---|---|---|---|---|---|
| Blast | Ghost | Kofam | |||||
| T05867 | pcw | Phascolarctos cinereus (koala) | 19,945 | 72.2 | 0.972 | 0.968 | 0.878 |
| T05881 | pcan | Pomacea canaliculata (golden apple snail) | 21,144 | 43.1 | 0.913 | 0.873 | 0.828 |
| T05868 | qsu | Quercus suber (cork oak) | 49,388 | 37.4 | 0.961 | 0.956 | 0.864 |
| T05852 | lsd | Litorilituus sediminis (Gammaproteobacteria) | 3,652 | 53.6 | 0.976 | 0.964 | 0.89 |
| T05862 | rpod | Roseitalea porphyridii (Alphaproteobacteria) | 3,371 | 55.7 | 0.98 | 0.973 | 0.873 |
| T05864 | tvu | Thermoactinomyces vulgaris (Firmicutes) | 2,498 | 58.8 | 0.972 | 0.956 | 0.857 |
| T05863 | strr | Streptomonospora sp. M2 (Actinobacteria) | 5,057 | 37.9 | 0.976 | 0.977 | 0.871 |
| T05849 | ney | Neochlamydia sp. S13 (Chlamydiae) | 2,174 | 36.3 | 0.971 | 0.949 | 0.869 |
There is a simplified version of BlastKOALA, named “Annotate Sequence” and linked from the KEGG Mapper page, which can be used interactively to search a single genus or family only. This tool may be useful when similar genomes are already annotated in KEGG.
6. KEGG MAPPER TOOLS
KEGG Mapper is a collection of tools for KEGG mapping against PATHWAY, BRITE, and MODULE databases. Previously the tools were made available separately for separate databases, but in the new version released in July 2019, they are merged into three general mapping tools shown in Table 3. Each of them allows mapping against multiple databases at a time, and the result is displayed in multiple tabs in the result page.
Table 3.
General KEGG mapping tools in KEGG Mapper
| Tool | Search mode | Target database | Query data (KEGG identifier) |
|---|---|---|---|
| Reconstruct Pathway | Reference |
Pathway Brite hierarchy and table Module |
K number |
| Search Pathway | Reference |
Pathway Brite hierarchy and table Module |
K/R/EC number C/G/D/E number |
| Human‐specific |
Pathway Brite hierarchy Module Network Disease |
Human gene identifier C/G/D number |
|
| Other organism‐specific |
Pathway Brite hierarchy Module |
Gene identifier K/R/RC/EC number C/G/D number |
|
| Search & Color Pathway | Reference |
Pathway Brite hierarchy Module |
K/R/EC number C/G/D/E number |
| Organism‐specific |
Pathway Brite hierarchy Module |
Gene identifier K/R/RC/EC number C/G/D number Outside gene identifier |
“Reconstruct Pathway” is the tool for KO‐based mapping against KEGG pathway maps, BRITE hierarchies, BRITE tables, and KEGG modules. It is linked from the KOALA family annotation tools and useful for analyzing genome and metagenome sequences. The analysis can be done for a single genome or multiple genomes, for example, to examine host‐endosymbiont relationships. Here an example is shown for the combination of human genome (T01001) and a gut metagenome sample (T30003).11 As shown in Figure 2a, the mapping results are displayed in different tabs with the number of matched pathway maps, and so forth, in parentheses. Figure 2b is one of the matched pathways, the combined global map of metabolic pathways (map01100), in which the coloring of green, red, and blue indicates human only, gut microbiome only, and both, respectively, possibly revealing how microbiome may help human to metabolize chemical substances. Figure 2c is an example of complete module indicating that human and gut microbiome use different gene sets for this coenzyme A biosynthesis module.
Figure 2.
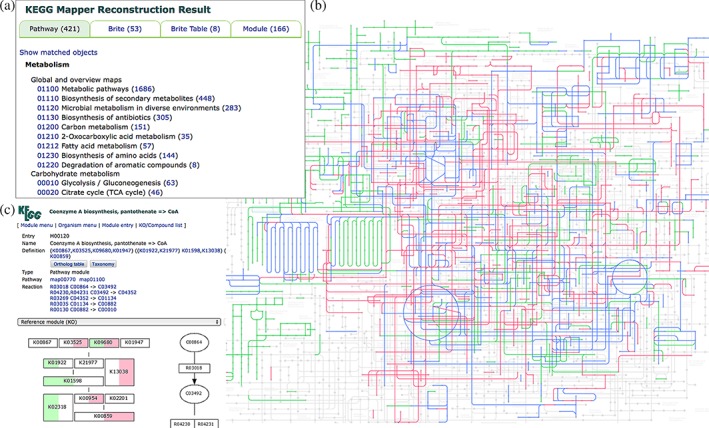
The result of Reconstruct Pathway from the combined gene set of Homo sapiens (T01001) and a gut metagenome sample (T30003). (a) the result page of KEGG pathway mapping, which shows that 421 matching pathway maps are found. (b) Reconstructed global map of metabolic pathways (map01100), where human specific pathways are shown in green, microbiome specific pathways in red, and shared pathways in blue. (c) the result in (a) shows that 166 complete modules are found. An example is coenzyme A biosynthesis (M00120), in which different gene sets, green for human and pink for microbiome, make this module complete
“Search Pathway” and “Search & Color Pathway” are traditional tools, which existed from the beginning of the KEGG project and now include expanded target databases. They are for direct mapping of objects, including genes, proteins, compounds, glycans, reactions and drugs, as they appear in KEGG pathway maps, BRITE hierarchies, and KEGG modules. Therefore, these tools can be used not only for genomes and metagenomes, but also for transcriptomes, proteomes, metabolomes, glycomes, and many other datasets. When the query data is a set of human genes in Search Pathway, additional mapping is performed against network variation maps in the NETWORK database12 and disease entries in the DISEASE database. Mapped objects are marked in red in Search Pathway, while they can be marked in any background and foreground colors in Search & Color Pathway.
In addition to these general mapping tools, two specialized tools are available. One of them is “Color Pathway” for mapping of genes associated with either color specification or numerical values, such as for gene expression data. Numerical values are displayed in color gradation or 3D bar graph. Multiple datasets may be given to display, for example, time‐series gene expression data. More details about individual tools and how to prepare query data sets can be found in the KEGG Mapper website (http://www.kegg.jp/kegg/mapper.html).
7. DISEASE GENES AND DRUG TARGETS
KEGG Mapper is used mostly for the analysis of users' data, but it can also be used to capture global and integrated views of different types of objects within KEGG, such as involving diseases and drugs. The KEGG DISEASE and DRUG databases contain disease genes and drug targets, respectively, with human gene identifiers linked to the GENES database and K numbers linked to the KO database. Here they are used to create the lists of H number (disease identifier) to K number relations and D number (drug identifier) to human gene identifier relations. Note that drug targets of nonhuman proteins, such as pathogen proteins, are not considered here.
The first list, containing 5,390 relations between H numbers and K numbers, is given to the Reconstruct Pathway tool in KEGG Mapper. Figure 3a shows a part of the mapping result for KEGG modules, indicating that 40 complete modules, as well as additional 34 almost complete (one block missing) modules, are generated from the entire list of disease genes in the DISEASE database. One example is the heme biosynthesis module M00868, which corresponds to the pink‐marked portion of the human pathway map for porphyrin and chlorophyll metabolism (hsa00860). Figure 3a also indicates that all of the eight genes that constitute M00868 are disease‐associated genes in H01763, which is porphyria, and two other H numbers for more finely classified disease names. Thus, so‐called single gene disorders are grouped by a functionally correlated gene set, and this set (module) is associated with a broadly categorized disease. There are many other similar examples of module‐disease associations, but they are currently limited to metabolic disorders, for the module dataset is highly biased to metabolism. Other types of KEGG mapping may also have potentials to reveal functional links among disease genes.
Figure 3.
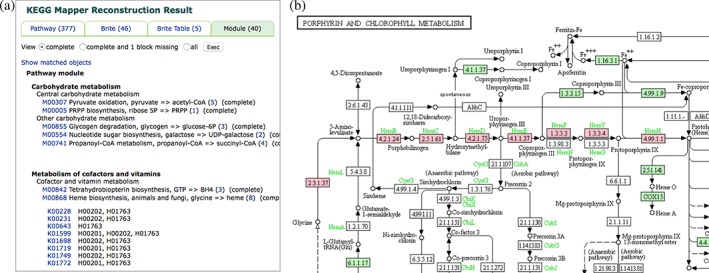
The result of Reconstruct Pathway from the entire set of disease genes represented by KOs in the KEGG DISEASE database. (a) the result page of mapping against KEGG modules, which shows that 40 complete modules are reconstructed. One of them is M00868 for heme biosynthesis, where all genes are associated with H01763 for porphyria. (b) the pathway module M00868 is shown in pink in the pathway map hsa00860 for porphyrin and chlorophyll metabolism. Eight genes in this module correspond to eight disease‐associated genes in H01763
The second list of 12,842 relations between D numbers and human gene identifiers contains 1,025 unique human genes. When the drug type is limited to monoclonal antibodies, the list is reduced to 295 relations containing 146 unique human genes. The full and reduced lists of human genes are given to the Search Pathway tool in KEGG Mapper. Figure 4 shows the mapping results against BRITE hierarchies: human proteins targeted by (A) all drugs or (B) monoclonal antibodies only. The results indicate which protein classes are targeted by drugs currently in use and for which protein classes monoclonal antibodies have been developed. Excluding the “KEGG Orthology (KO)” file (hsa00001) containing all genes/proteins with assigned KOs, the “Enzymes” file and the “CD molecule” file were enriched most by the full set and the monoclonal antibody set of drug target genes, respectively.
Figure 4.
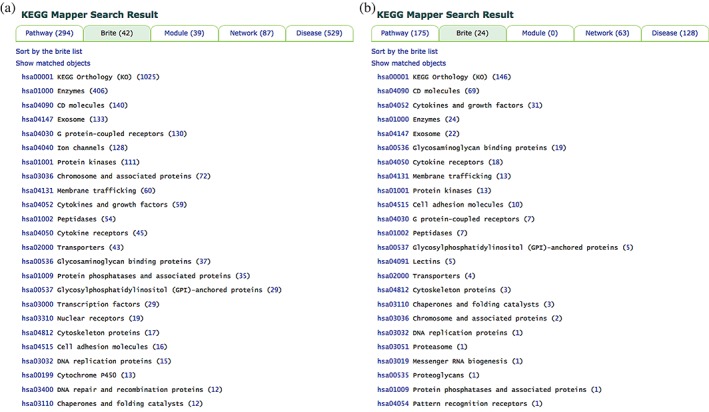
The result of Search Pathway from the human protein set of drug targets in the KEGG DRUG database. (a) the result page of mapping against BRITE hierarchies for the full set of drug targets, which shows that 42 matching hierarchy files are found with “enzymes” at the top, excluding the KEGG Orthology (KO) file. (b) the result page of mapping against BRITE hierarchies for the targets of monoclonal antibodies, which shows that 24 matching hierarchy files are found with “CD molecules” at the top
KEGG mapping against BRITE hierarchies is an operation similar to GO enrichment analysis. KEGG BRITE currently contains 53 classification systems (hierarchy files) for genes and proteins, in contrast to only three in GO. BRITE hierarchy files are created separately to represent specific knowledge in specific domains. Currently, 14,500 KOs are linked to BRITE hierarchies in comparison to 12,500 KOs linked to KEGG pathway maps, and over 20,000 KOs are linked to either or both of them. The BRITE enrichment analysis significantly increases the number of genes that can be mapped to the KEGG resource assisting functional interpretation.
8. CONCLUSION
The pathway mapping tools in KEGG Mapper have been widely used for biological interpretation of genome sequences and other high‐throughput data. The new release of KEGG Mapper integrates KEGG pathway mapping with BRITE enrichment analysis and module completeness check for genes and proteins. Furthermore, the integrated resource of KEGG pathway mapping and BRITE enrichment analysis will facilitate biological interpretation of other types of data, especially metabolites and drugs.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
We thank Kanae Morishima for helping to evaluate the KOALA family tools. The KEGG project is partially supported by the National Bioscience Database Center of the Japan Science and Technology Agency. Computational resources were provided by the Bioinformatics Center, Institute for Chemical Research, Kyoto University.
Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Science. 2020;29:28–35. 10.1002/pro.3711
Funding information Institute for Chemical Research, Kyoto University; National Bioscience Database Center, Japan Science and Technology Agency
REFERENCES
- 1. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanehisa M, Sato Y, Kawashima K, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanehisa M. Inferring antimicrobial resistance from pathogen genomes in KEGG. Methods Mol Biol. 2018;1807:225–239. [DOI] [PubMed] [Google Scholar]
- 5. Pearson WR. Searching protein sequence libraries: Comparison of the sensitivity and selectivity of the Smith‐Waterman and FASTA algorithms. Genomics. 1991;11:635–650. [DOI] [PubMed] [Google Scholar]
- 6. Moriya Y, Itoh M, Okuda S, Yoshizawa A, Kanehisa M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–731. [DOI] [PubMed] [Google Scholar]
- 8. Aramaki T, Blanc‐Mathieu R, Endo H, et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. bioRxiv. 2019. 10.1101/602110. [DOI] [PMC free article] [PubMed]
- 9. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki S, Kakuta M, Ishida T, Akiyama Y. GHOSTX: An improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS One. 2014;9:e103833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurokawa K, Itoh T, Kuwahara T, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2015;14:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47:D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information