

Abstract
Engineered recombinant antibody‐based reagents are rapidly supplanting traditionally derived antibodies in many cell biological applications. A particularly powerful aspect of these engineered reagents is that other modules having myriad functions can be attached to them either chemically or through molecular fusions. However, these processes can be cumbersome and do not lend themselves to high throughput applications. Consequently, we have endeavored to develop a platform that can introduce multiple functionalities into a class of Fab‐based affinity reagents in a “plug and play” fashion. This platform exploits the ultra‐tight binding interaction between affinity matured variants of a Fab scaffold (FabS) and a domain of an immunoglobulin binding protein, protein G (GA1). GA1 is easily genetically manipulatable facilitating the ability to link these modules together like beads on a string with adjustable spacing to produce multivalent and bi‐specific entities. GA1 can also be fused to other proteins or be chemically modified to engage other types of functional components. To demonstrate the utility for the Fab‐GA1 platform, we applied it to a detection proximity assay based on the β‐lactamase (BL) split enzyme system. We also show the bi‐specific capabilities of the module by using it in context of a Bi‐specific T‐cell engager (BiTE), which is a therapeutic assemblage that induces cell killing by crosslinking T‐cells to cancer cells. We show that GA1‐Fab modules are easily engineered into potent cell‐killing BiTE‐like assemblages and have the advantage of interchanging Fabs directed against different cell surface cancer‐related targets in a plug and play fashion.
Keywords: Phage display, affinity maturation, engineered Fab, Protein G, proximity assay, bi‐specific T‐cell engagers, split enzyme
1. INTRODUCTION
Affinity reagents are the cornerstone of cell biology. They come in many manifestations, but antibodies are by far the most widely used format. Traditionally, antibodies were produced using animal immunization methodologies.1 While this approach is still in broad use, recombinant display technologies have now assumed the leading role in producing antibody‐based affinity reagents.2, 3, 4 Recombinant reagents have manifold advantages over traditionally produced monoclonal antibodies; for instance, economic and scalable production and permanent archiving are notable advantages.5, 6 While monoclonal antibodies can be reproduced, the maintenance and large‐scale culture of hybridoma cells can be cumbersome and expensive. However, the most compelling advantage of the recombinant approach is that display methods can be used to customize the affinity binders in ways not accessible by monoclonal antibodies.7, 8, 9 For instance, selection conditions to produce affinity reagents can be tuned to direct binders to target particular conformation states or bind a specific epitope.10, 11 Thus, the user has much more control over the characteristics of the affinity reagent being produced.
Over the last decade, we have developed a high throughput pipeline for the rapid production of antibody Fab‐based affinity reagents using phage display mutagenesis.6, 12 The Fab CDR libraries we use are based on the Herceptin Fab scaffold, which has been engineered for stability and expression. The phage display biopanning protocols we employ allow exquisite control of the properties of the Fabs being selected for and generally multiple high‐quality binders can be obtained for a given target.6, 8, 13 These binders have been utilized in a number of cell biological and biochemical applications.14, 15, 16 Furthermore, they have been exploited as powerful crystallization chaperones and fiducial marks for single‐particle cryo‐EM structural studies.17, 18, 19, 20, 21
While a principal reason for utilization of Fabs in many standard applications is due to their ease of production, it is their engineerability that makes them candidates for more sophisticated types of applications. In particular, Fabs are stable modules that are easily adapted to fusing or chemically linking other molecular entities to them for imaging and numerous biochemical manipulations.22 One challenge faced by antibody engineers has been to develop user‐friendly ways to endow these modules with multivalent or multi‐specificity properties. The key is to design simplified systems that can be used by cell biologists or biochemists that do not require significant expertise in protein engineering. A goal would be to combine Fabs as modules like lego blocks, pieces of which could be pre‐fabricated as a unit and then combined with other Fabs of other specificities to generate a variety of bi‐specific or multivalent entities in a plug and play fashion. Towards this end, we have developed a Fab binding module based on Protein G (PG) that can be fused onto many different molecular components. As such, they can be assembled in a variety of different formats in a straightforward way, allowing the researcher to design highly customized affinity reagents (Figure 1).
Figure 1.
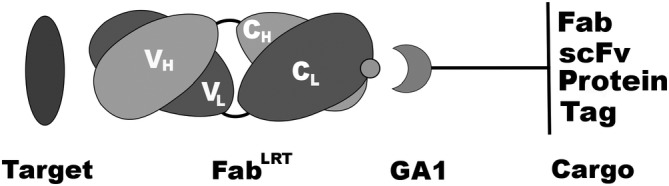
Basic Fab–GA1 construct. Fab can be coupled to a variety of GA1 fusions. The fusions can contain another Fab or scFv to generate a bi‐specific assemblage or another protein or protein fragment. Tags or chemical moieties can to attach to GA1 to further functionalize the fusion
Herein, we describe a platform that uses engineered Fab‐based modules to perform a series of complex tasks outside the capabilities of traditional antibodies. A key component of the modules is an affinity matured variant (GA1) derived from the immunoglobulin binding domain, PG. Importantly, GA1 binds to an epitope on the constant domain of the Fab far removed from its antigen‐binding loops. We had previously shown that Fabs can bind to GA1 domains that have been linked together to form multivalent entities.23 However, the initial GA1–Fab affinity was ~50 nM, which we deemed insufficient for the types of functions we envisioned for the modules described here. Therefore, using a stepwise phage display mutagenesis approach, we produced variants of the Fab scaffold (FabLRT) that form a complex with GA1 endowed with an ultra‐high affinity (100 pM). Exploiting the high affinity between Fabs and GA1, modules were designed and tested that facilitate high throughput sandwich assays, proximity complementary assays (PCA) and fabrication of potent bi‐specific T‐cell engagers (BiTES). The modules are designed to have interchangeable parts allowing a broad range of combinations that can be evaluated in a multiplexed fashion (Figure 1). Importantly, the work presents a blueprint to guide other protein engineers for how to expand the system for myriad applications.
2. RESULTS
2.1. Initial engineering of the GA1–Fab interface
PG is an immunoglobulin binding protein that has been used for antibody purification by virtue of its affinity to the Fc portion of the molecule. PG also has a weak affinity to the Fab framework (~3 μM). We had previously engineered an affinity improved PG variant (GA1) that bound specifically to a Herceptin Fab scaffold variant (E123S mutation in Fab constant light chain [Lc]) that could be utilized for applications that involved linking multiple copies of GA1 together to make multi‐valent and bi‐specific assemblages.23Thus, we considered the possibility of further developing the platform to facilitate building higher level modules that could incorporate multiple interchangeable Fabs in a plug‐and‐play fashion.
An important element of initial design was that the affinity matured GA1 would only bind to the specific E123S Fab Lc variants (FabS) that were designated parts of the assemblages, not any natural wild‐type Fabs (Table 1) that were contained in endogenous IgGs or other sources in the experimental milieu. However, although the GA1–FabS complex had a 60‐fold improved Kd of ~50 nM compared to the wild‐type (wt) PG domain, the binding is still characterized by fast dissociation kinetics that are not optimal for the desired non‐equilibrium applications. Therefore, we sought to further engineer the Herceptin variant scaffold (FabS) to have a significantly elevated affinity to GA1 accompanied with slow off‐rate kinetics.
Table 1.
Protein GA1‐binding affinity of different Fab Lc variants
| FAB LC scaffold (aa 123–127) | KD (nM) |
|---|---|
| SQLKS | 50 |
| LRT | 0.1 |
| GSLRS (selected) | 2.5 |
| SMLRS (selected) | 7.5 |
| ΔΔLKS (mutated) | 12 |
| SQLRT (mutated) | 8 |
| EQLKS (Hkappa) Fabκ | NB |
| EELQA (Hlambda) | NB |
| EQLTS (MKappa) | NB |
| EELET (Mlambda) | NB |
The initial affinity maturation of PG to produce GA1 involved phage display selections focusing on two points of contact with the FabS scaffold (Figure 2). The first region was through the formation of complementary β‐strands from PG (β2, residues 16–22) and residues 209–216 of the last β‐strand of the heavy chain (Hc) of the constant domain of the Fab. The engineered interface includes all of the main chain hydrogen bonds observed in the original structures.24 Although, β‐2 of GA1 contains several mutations that bury significant surface area at the protein interface, the overall affinity gain of these interactions is limited. The more noteworthy changes within GA1 accounting for the major affinity improvement occur at the C‐terminal cap of the α‐helix (Figure 2), where the original residues 40–43 (40NDNG43) were substituted for 40YVHE43 in the engineered GA1 variant. The helical cap of the engineered variant provides improved shape complementarity to interdigitate with the α‐helix residues SQLKS (residues 123–127) connecting β‐strands of the Lc domain of the Fab.
Figure 2.
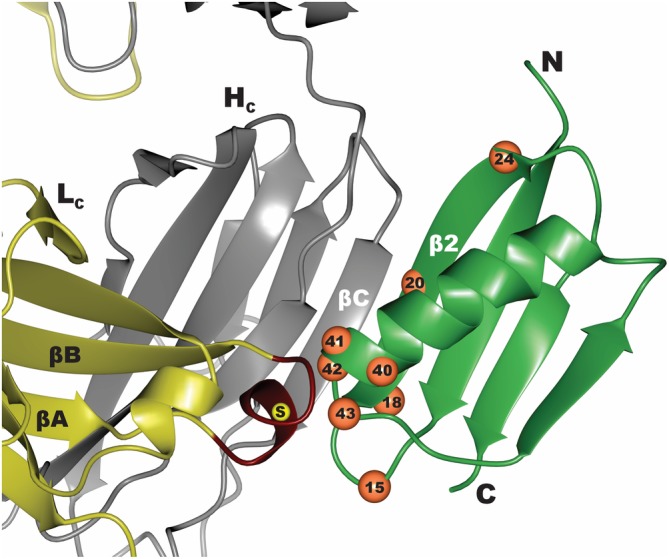
Interface between Protein G (green) and Fab (yellow–gray). Residues that were diversified in the phage display mutagenesis library to convert Protein G to GA1 are numbered and marked as orange spheres. Region in the Lc of FabS that was diversified to generate the affinity matured FabLRT involved residues 123–127 marked in red. The position of E123S mutation that differentiates between the FabH and FabS scaffolds is marked by a yellow sphere
2.2. Affinity maturation of the binding interface of FabS Lc to Protein GA1
While compared to wt‐ PG, GA1 produced a significant affinity boost in binding FabS, it still was not optimal for the engineered modules we envisioned. To further improve the affinity and the off‐rate kinetics, we hypothesized that a stepwise phage display approach was the best way to further increase the FabS–GA1 affinity. That is, as described above, GA1 was produced from phage display selections against the original FabS scaffold. In the stepwise selection scheme, the process is reversed; FabS is affinity matured against GA1. To perform phage display on the complementary surfaces on the FabS scaffold, we designed our phage display library focusing on residues 123–127 of the FabS Lc, since it formed the most extensive contact with GA1, as described above (Figure 2). Different levels of diversity were introduced into each of these positions depending on the amino acid character of the parent residue (see Section 4). This library had a theoretical diversity of roughly 107 Lc‐scaffold variants. A protocol for target immobilization through a cleavable SNAP‐tag was applied to all Fab selections as has been described.25 Using this approach, the C‐terminally SNAP‐tagged GA1 was biotinylated through the SNAP self‐modifying activity using commercially available SNAP‐Biotin enabling capture by streptavidin‐coated magnetic beads during selection.
Five rounds of selection were performed using the FabS Lc library. To introduce additional binding stringency, the concentration of the antigen was systematically reduced with each round of selection starting at 200 nM during the first round and ending with 1 nM in round 5. Phage enzyme‐linked immunosorbent assay (ELISA) was performed on 96 clones resulting in identifying six unique FabS variants. Notably, sequencing revealed that all six of the variants contained a K126R substitution (Figure S1). Two of the variants resulted in a 5–10‐fold improved GA1 binding affinity as determined by surface plasmon resonance (SPR) (GSLRS and SMLRS, Table 1). Importantly, a single variant containing a serendipitous two amino acid deletion, ΔΔLRT (FabLRT), produced significantly superior binding characteristics. The replacement of the original SQLKS sequence of FabS with ΔΔLRT produced a Kd of 100 pM and a slow dissociation rate. Overall, compared to the FabS, the FabLRT improved the affinity to GA1 by ~500‐fold (Figure 3C). This deletion mutation did not affect Fab stability or expression. We speculate that the deletion may have occurred during the synthesis of randomizing DNA oligonucleotides.
Figure 3.
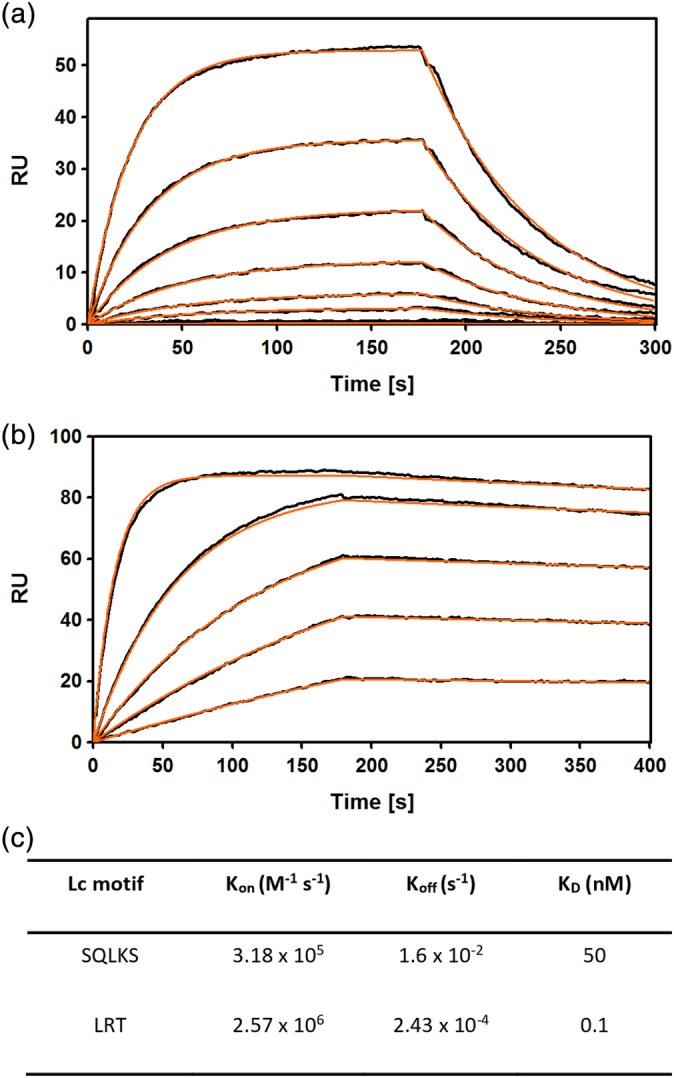
Affinity maturation of the Fab‐Protein G interface. (a). SPR sensograms showing fast on‐fast off binding kinetics between FabS and the affinity matured GA1. The concentration of Fab was serially diluted twofold for each run starting at 100 nM (b). Sensogram showing slow—dissociation kinetics for FabLRT binding to GA1. Initial concentration of Fab was 12 nM and serially diluted as in (a). (c). SPR kinetics for GA1 binding to FabS and FabLRT. Curve fit is shown in red. Chi2 value for the fit are 0.41 and 0.67, respectively
To evaluate the relative importance of the conservative mutations K126R and S127T relative to the two amino acid deletion at positions 123–124, two variants were constructed. The first included the deletion, but replaced the Arg with the wild type Lys (ΔΔLKT). The second one contained the wild type residues at positions 123–124 followed by LRT (SQLRT). SPR analysis determined that both these variants had intermediate affinities (12 and 8 nM, respectively) in the range between GA1 (50 nM) and ΔΔLRT (0.1 nM) (Table 1). These data, together with the fact that all selected scaffolds contained affinity‐improving K–R substitution, suggest direct and significant involvement of the Arg in the enhanced interactions with GA1.
2.3. The structure of GA1–FabLRT
The crystal structure of the FabLRT–GA1 complex was determined to gain structural insights into how the ΔΔLRT mutation enhances the binding affinity between the Fab and GA1 to the extent that it does. The complex crystallized in space group P3221 with two FabLRT–GA1 complexes in the asymmetric unit. The average root‐mean‐square deviation (RMSD) between the two Fab–GA1 complexes is ~0.2 Å (over 211, 220, and 56 Cα atoms of the Fab Lc, Fab Hc and GA1, respectively). The GA1–FabLRT interface is formed through two sets of contacts that bury ~560 and 160 Å2 of the Fab's Hc and Lc, respectively. The first contact is through the formation of an antiparallel β‐strand configuration that includes main chain H‐bonds between residues 16–22 of GA1 β2 and residues 221–227 of Fab Hc βC. A similar H‐bonding arrangement was reported in the structure of a wild‐type PG‐Fab complex.24 A second and more extensive set of contacts involves the C‐terminal α‐helical cap of GA1 and Fab residues comprising 137–140 of the Hc and 123–127 in the Lc, which includes the ΔΔLRT motif (Figure 2). Notably, the ΔΔLRT motif mates with the residues of GA1 (40YVHE43) that were involved in GA1's affinity maturation from PG to GA1. The structure shows that the loop containing the deleted residues in the ΔΔLRT motif induces a conformational change that positions the guanidium group of R126 to pack against the aromatic ring of Y40 of GA1 resulting in the formation a cation‐π interaction (Figure 4). Furthermore, the guanidinium group forms an H‐bond with the carbonyl of Y40. V41 of GA1 forms hydrophobic interactions with F139 of Fab Hc βA. Additionally, H42 of GA1 is buried at the Hc Fab interface, where its Nε2 nitrogen forms an H‐bond to the main chain nitrogen of the V129. The H‐bonding potential at this position appears to be conserved, as all phage display variants isolated have either His, Asn or Gln at this position. E43 is exposed to the solvent and protrudes into the cavity created by the two deletions at the ΔΔLRT motif.
Figure 4.
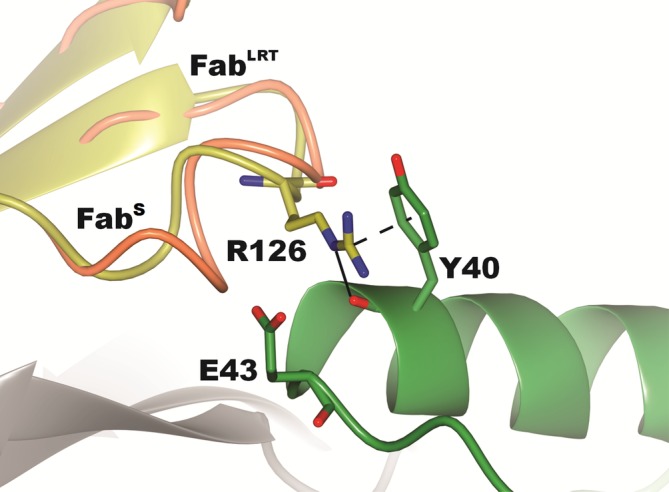
Interface contacts of FabLRT with GA1 showing the interactions of the key R126 side chain. The guanidinium portion of the side chain forms a cation‐π interaction with the ring of Y40 and also an H‐bond with that group's main chain carbonyl. There is also a significant rearrangement of the main chain 123–127 presumably induced by the deletion of two residues in the loop
2.4. GA1–FabLRT protein complementation assays: Principle and components
As the model system for the proof of principal of the plug‐and‐play GA1–FabLRT concept, we devised a protein complementation assay (PCA) based on the well‐established proximity‐driven refolding/reactivation of the TEM1 β‐lactamase (BL) split enzyme system.26 In this assay, the two separate fragments of the BL enzyme are attached through a linker to the two different targets that are to be evaluated for proximity. If the targets are in close vicinity, then the fragments can associate to form an active enzyme state. This can be evaluated readily by introducing a fluorogenic BL substrate that provides a distinct readout. The format generally requires that the individual complementary fragments be genetically fused by means of a linker to one or the other of the potential interaction partners. The linker lengths can be adjusted to fine tune the complementary efficiency. However, this requires multiple genetic fusions that can be cumbersome and time consuming.
To circumvent the issues involving serial genetic fusions, we developed a system that exploits the high affinity of a FabLRT to GA1. Our test case involves using complementation in the form of a canonical sandwich assay. The strategy is to express and purify two GA1 fusions with one or the other of the two complementary BL fragments (BLF): N‐terminal fragment‐ residues 26–196 and C‐terminal fragment—residues 198–220. In order to bring the BL fragments in proximity allowing for BL association and refolding at low concentrations in this type of antigen‐detection assay, the GA1 modules of complementary BLF fusions are associated separately with two LRT scaffold Fabs that bind the antigen at different epitopes. Then, upon addition of the antigen, simultaneous antigen‐binding of these Fabs results in BL refolding and activation (Figure 5). The induced BL activity is detected by the increase in fluorescence signal upon addition of Fluorocillin Green, a fluorogenic BL substrate. Notably, the Fab‐binding GA1 module genetically fused to BLFs could serve as a potent non‐covalent linker between the BLFs and any number of interchangeable FabLRT molecules, laying the basis for plug‐and‐play opportunities.
Figure 5.
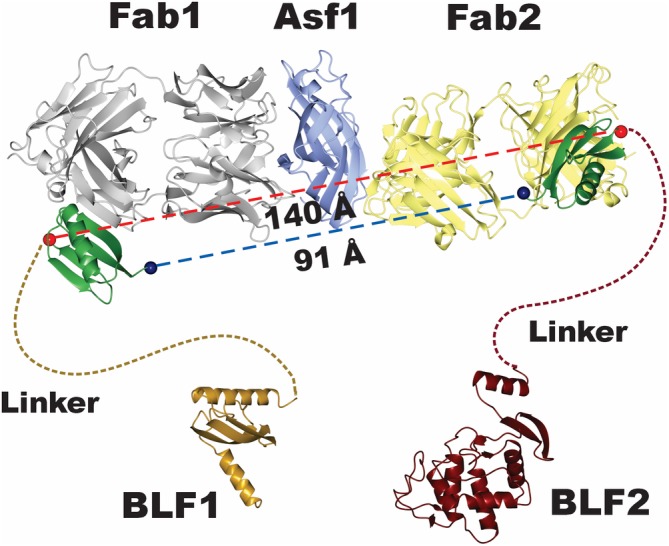
Model for components in the complementation proximity assay showing the potential fusion points between the Fabs and the linker‐BL fragments. The structure of the Asf1 Fab 1‐Fab 2 complex shows that the Fabs bind to the opposite faces of Asf1. In those positions, it is possible to measure the direct distances between the N‐ and C‐terminal fusion points the BL fragments on GA1 bound to its respective Fab. The direct distances range from ~90 to 140 Å. A 30‐residue linker was thought to have enough reach that it would be effective in all possible combinations
To optimize the system and explore the different options, we constructed and produced four fusions of combinations of the N‐terminal (BLF1) and C‐terminal (BLF2) fragments of BL connected to GA1 by a Gly–Ser linker of about 30 residues. Next, we demonstrated that BLF–GA1 fusion constructs in the absence of antigen were capable of BL reconstitution by testing them at 1 μM concentration in the β‐lactamase assay with its complementation partner (Figure 6a). When mixed together at different concentrations, the pair: 1 and 4 (BLF1 fused to the C‐term of GA1 and BLF2 fused to the N‐term of GA1) showed the lowest spontaneous activity level at 1 μM (Figure 6a). This pair was then used to establish the background level at concentrations between 2 μM and 15 nM. This showed that BL activity in the absence of antigen was triggered at concentrations above 500 nM. Thus, we chose a concentration of 250 nM (Figure 6b) that was well below this threshold as the baseline for the antigen‐detection conditions, since it was the highest concentration that displayed minimal background activity in the absence of antigen.
Figure 6.
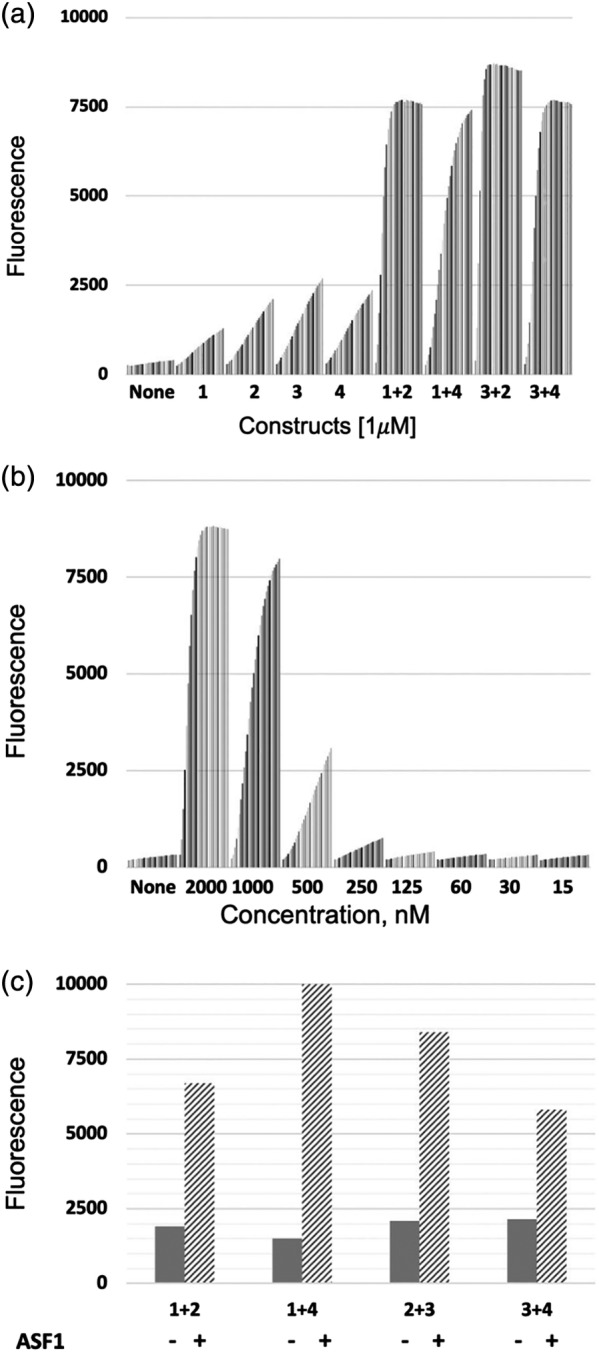
Establishing background levels of Beta Lactamase (BL) activity readouts. (a). Different BL fragments were mixed at 1 μM concentration. Fluorescent readings were taken every 2 mins over 20 time points. No activity was observed when the individual fusion components were mixed without their complementary pair. Activity was seen at this high concentration when the component pairs were mixed together. Although at the last time points activities are similar, the 1 + 4 pair, shows a distinct difference from the others over the time course. B). Background activity for the complementation pair 1‐4 (GA1‐BLF1(1) and BLF2‐GA1(4)) when mixed at varying concentrations. Readings were taken at 2 min intervals over a 1 hr incubation time frame. Data show that the signal is at background at 250 nM concentration of the pair. (c). Asf1 antigen detection using different BFL combinations. Fabs 11E and 12E were mixed with BLF fragments at 250 nM concentration. Then, 250 nM of Asf1 was added. (−) is the signal prior to Asf1 addition, (+) after addition of antigen. BL activity was measured after 20 mins incubation at RT
As an initial model for the sandwich assay development, we chose as the antigen a small, 158 amino acid histone chaperone protein, Asf1. Two Fabs (11E and 12E), shown to be binding to orthogonal epitopes of the protein, had previously been generated.27 A crystal structure of the Asf1 with these two Fabs indicated that they bound on opposite faces of the protein.20 To establish possible linker lengths that might work in this system, further examination of the superimposed crystal structure model of the tripartite ASF1:12E:11E complex with GA1 bound to each Fab, indicated a ~100–150 Å distance range between the termini of the two GA1 molecules (Figure 5). Two competing criteria were considered in selecting effective linker lengths. First, the length should not be too long as to diminish the local concentration effect. However, perhaps more importantly, effective linker lengths cannot be estimated by measuring directly between points A and B. There has to be built‐in excess to take into consideration their inherent flexibility and the fact that the Ramachandran plot has to be adhered to in the process.
Taking these issues into account, we surmised that a 30 amino acid Gly–Ser linker was a reasonable compromise between these requirements. Indeed, in pilot experiments where each of the pairs of complementary BLF–GA1 fusions were pre‐mixed separately with 11E or 12E Fabs having LRT mutations, a significant increase (up to 10‐fold) in the fluorescent signal was produced upon addition of an equimolar amount of Asf1 (Figure 6c). Although all combinations worked, we found as before that the 1–4 pairs reproducibly produced the best signal‐to‐noise ratio.
2.5. Dual epitope Fabs against NPCT EBOV and MT ZIKV
To further develop the GA1 fusion platform and apply it to antigen detection in the systems with unknown structural organization, we chose two viral protein antigens where previously generated Fabs were available. The first was the 98 residue C‐terminal domain of the Zaire strain of Ebola virus nucleoprotein (EBOV NTCT). The second was the 261 residue, N‐terminal methyltransferase domain of the Zika virus bifunctional NS5 enzyme (MT ZIKV). From the pool of Fabs selected against NTCT from five known major Ebola virus strains (Table S1), epitope binning revealed two distinct epitopes. The major epitope was highly dominant, while only a single Fab (MJ6) was found that bound to a second independent epitope. From the group of major epitope Fabs, MJ20 was selected as a representative binder and was used in subsequent studies. Using a dot blot analysis, Fab pairs, MJ20 (major epitope) and MJ6 (minor epitope), were shown to bind simultaneously to EBOV NTCT. The binding kinetics of the pair were subsequently determined by SPR analysis indicating affinities of 0.7 nM (MJ6) and 3.4 nM (MJ20), with dissociation rates of 1.0 ×10−3 s−1 and 6.1 × 10−4s−1, respectively (Figure 7a). Furthermore, it was shown by SPR that consecutive injections of Fabs MJ6 and MJ20 resulted in an approximately twofold increase of the mass signal compared to single injections of either of them or two consecutive injections of the same Fab (Figure 7b). This confirmed that MJ6 and MJ20 are capable of binding simultaneous to non‐overlapping epitopes of EBOV NTCT without affecting the affinities of either Fab.
Figure 7.
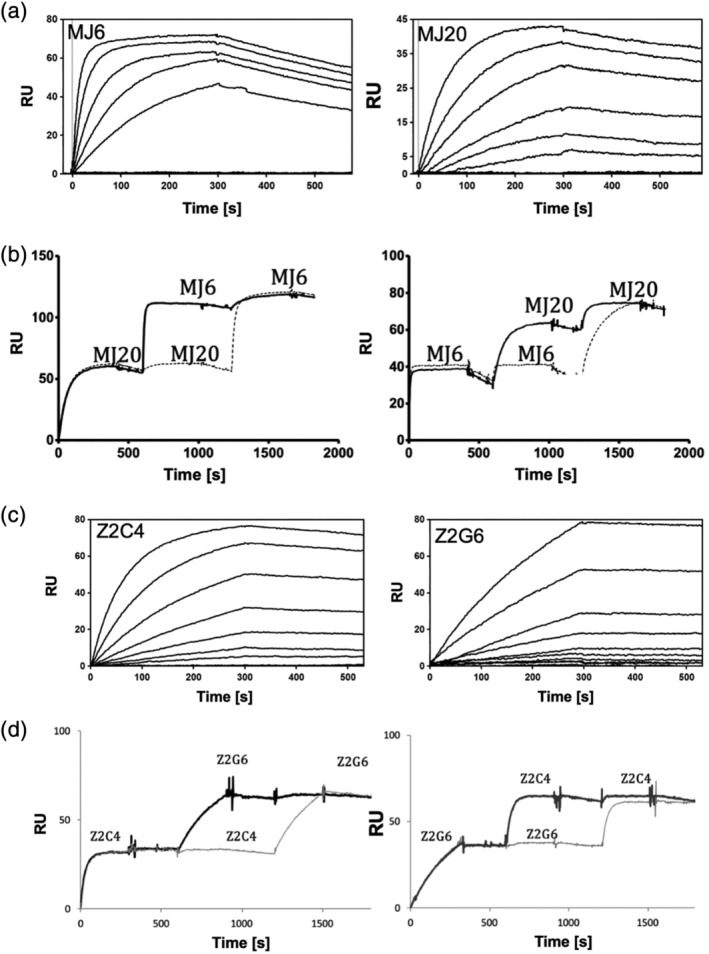
Analysis of binding and epitope binning using SPR. (a) SPR senograms used for kinetic analysis of Mj6 and Mj20 binding to EBOV NPCT. Initial concentrations (MJ6–50 nM; MJ20–100 nM) were serially diluted by 50% between each run. (b). Epitope binning experiment of Mj6 and Mj20 against EBOV NPCT showing the Fabs have non‐overlapping epitopes. Fab Mj20 (or Mj6) was injected as an analyte first, followed by a second injection of the other Fab. Substantial increase in RUs upon the second injection indicates the two Fabs bind simultaneously. c). Binding sensograms for Z2C4 and Z2G6 binding to ZIKV MT. (d). Epitope binning of the two Fabs, as described in B. Initial concentrations: Z2C4‐ 40 nM; Z2G6‐ 75 nM)
Among Fabs selected against MT ZIKV, using the procedures described above, a pair of Fabs, Z2C4 and Z2G6, was found to bind to non‐overlapping epitopes (Figure 7c,d). These Fabs exhibited KDs of 0.7 and 1.7 nM and dissociation rates of 2.8 × 104 s1 and 9.2 × 10−5 s−1, respectively (Figure 6e). Thus, we confirmed that pairs of Fabs for both systems (EBOV NTCT: MJ6 and MJ20; MT ZIKV: Z2C4 and Z2G6) possessed the desirable antigen‐binding characteristics for our GA1‐BL detection system (high affinity, slow dissociation rate, independent binding to the antigen molecule) and could be introduced into formats to test their abilities in the plug and play proximity assays.
2.6. Detection of viral proteins: NPCT EBOV Zaire and MT ZIKV
A challenge for the EBOV and ZIKV systems was the absence of information about the position of the epitopes of the Fabs that were being employed in the proximity assay. Only the crystal structure of EBOV NTCT with one Fab, MJ20, had been solved.25 As with the Asf1 system, we employed a 30 residue Gly–Ser linker to connect GA1 to the BLFs. To test this system in the context of the FabLRT components (MJ6 and MJ20) and the complementary fusions between protein GA1 and the BLFs, we individually premixed the Fabs with each of the complementary fusions at a final concentration of 250 nM. The BL activity induced upon addition of equimolar 250 nM NPCT revealed a preferential performance of the GA1_BLF1 + MJ6 and BLF2_GA1 + MJ20 premix out of the two possible active combinations (Figure S2). Notably, reversing the format, that is, matching BLF2 with MJ6 and BLF1 with MJ20, reduced the activity by about 40%, suggesting some sensitivity between the matched pairs. Negative‐control mixtures, containing the same BLF‐fusion or the same Fab component in the pre‐mixtures, did not show any significant activation upon antigen addition (Figure S2). Titration of NPCT into 250 nM of the combination of GA1_BLF1 + MJ6 and BLF2_GA1 + MJ20, resulted in a detectable fluorescent signal at 15 nM NPCT, which increased linearly over the range from 15 to 125 nM with a distinct maximum at 250 nM (Figure 8a). A reduction of the signal observed at NPCT excess, most likely was caused by a breakdown of the stoichiometry at high antigen concentration.
Figure 8.
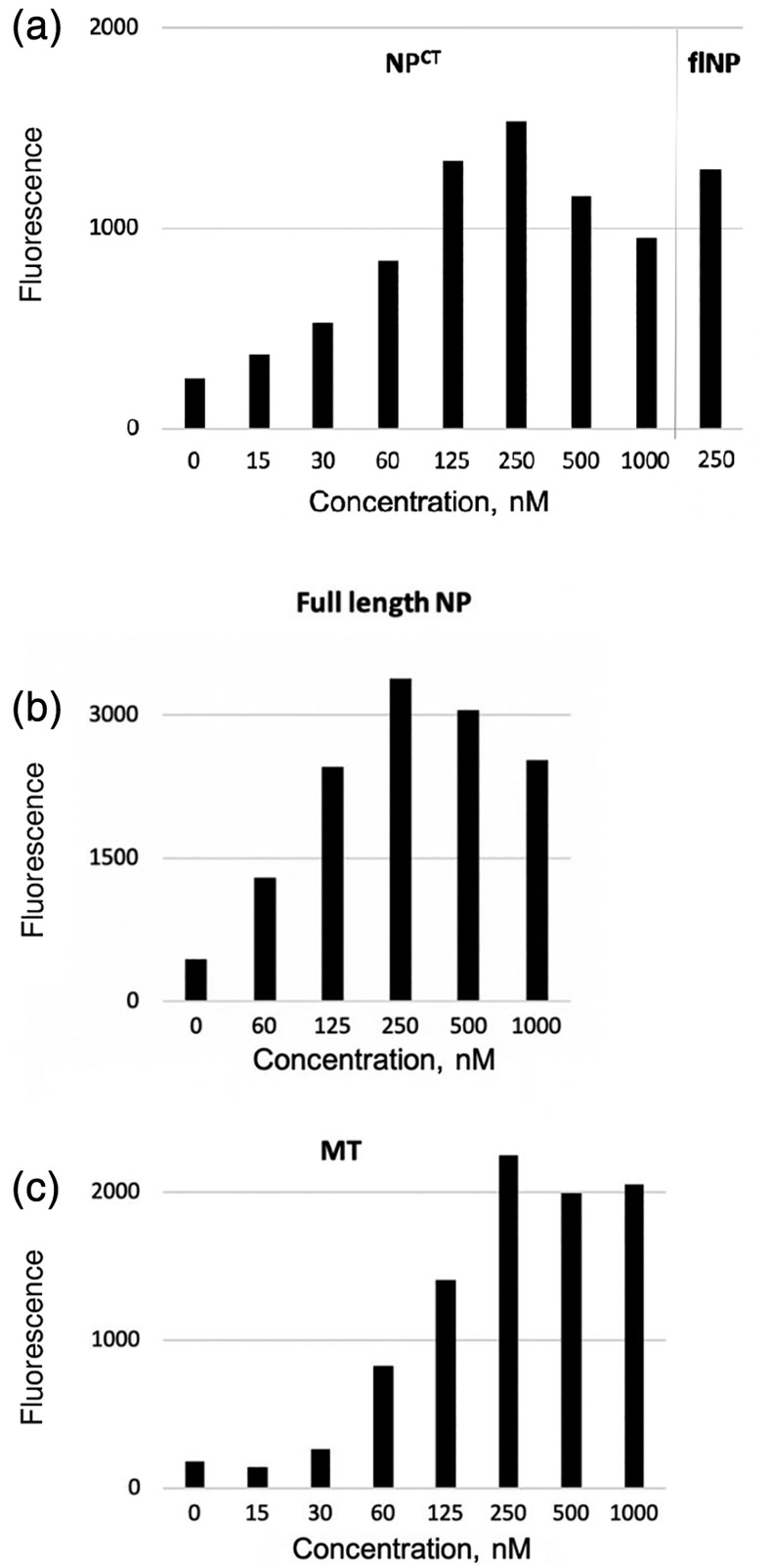
BL proximity assay results. (a). Detection of EBOV NPCT at different concentrations using complementary pairs: GA1(C‐term)‐BLF1/Mj6 and BLF2‐GA1(N‐term)/Mj20. Detectable signal was observed starting at 15 nM and peaking at 250 nM. Last bar shows that NPCT is readily detected in the context of the full length EBOV NP at 250 nM. (b). Concentration dependence of detection of full length EBOV NP. (c). Concentration dependence of ZIKV MT detection using complementary pairs: GA1(C‐term)‐BLF1/Z2C4 and BLF2‐GA1(N‐term)/Z2G6. In all experiments, reactions were incubated for 20 mins at RT; a background of 200 units of substrate fluorescence was subtracted
Next, we asked whether NPCT in context of the full‐length EBOV NP Zaire could be detected by the above system with comparable efficiency, since the additional N‐terminal NP domain might create a steric hindrance for Fab binding or BL refolding. However, the NPCT domain contained in the full‐length NP Zaire protein was readily detected, as measured by an increase in BL activity similar to the NPCT antigen alone making this assay applicable to full‐length NP and potentially to EBOV detection in biological samples containing the lysed virus (Figure 8a,b).
To further demonstrate the plug‐and‐play capabilities of the platform, we applied the same BLF‐GA1‐fusion constructs to detect MT ZIKV using the FABLRT format with Z2C4 and Z2G6. The results obtained for MT ZIKV were congruent with the findings of the EBOV detection system (Figure 8c). At a 250 nM concentration of the GA1_BLF1 + Z2C4 and BLF2_GA1 + Z2G6 combinations, the limit of antigen detection for both systems appeared to be roughly 30 nM, which falls within the range published for laboratory‐performed Ebola‐detection ELISA assay28 and the maximum of the signal was achieved at the equimolar 250 nM concentration of the BLFs and the antigen.
2.7. A novel plug‐and‐play Bi‐specific T‐cell engager immuno‐reagent
Bi‐specific T‐cell engagers (BiTEs) have recently emerged as an important class of immuno‐therapeutic assembly.29 BiTEs are molecules that are engineered to engage an activated T‐cell through one binding arm and to attach it to a cell surface target on an antigen‐presenting cancer cell (APC) through its second arm.30 This engagement leads to T‐cell dependent cell death of the cancer cell. BiTEs using several formats have been developed and successfully deployed.31, 32, 33, 34 The most prevalent formats to induce engagement between the two cells are: (a) bispecific antibody where one arm recognizes the T‐cell and the other the APC, and (b) two cell‐directed single‐chain Fvs attached by a flexible linker. Each of these formats has its strengths and weaknesses, but neither has the versatility provided by GA1‐FabLRT constructs described below.
The designed bi‐Fab constructions are based on a GA1‐FabLRT concept and are bi‐specific with adjustable linker lengths between the two antigen‐binding modules (Figure 9). A number of such fusion constructs were engineered with different linker lengths (from 3 to 73 aa long) between GA1 and the C‐terminus of the Hc of the Herceptin Fab scaffold with a specificity directed at one of the target antigens. The Herceptin scaffold differs from FabS by a single amino acid in that it has the wt kappa Lc with Glu at position 123, rather than Ser, as is the case for FabS. This scaffold is referred to as FabH. A fusion construct with 13 residue linker (GGSGSAGSGGAGA) was used for the proof of principal described below. The concept is that a Fab(1)H‐linker‐GA1 fusion that binds to antigen target 1 can be combined with a FabLRT that binds antigen target 2 (Fab(2)LRT) (Figure 9). This forms a noncovalent entity consisting of Fab(1)H—linker‐GA1–Fab(2)LRT. We refer to these modules as “bi‐Fab” BiTEs. Such constructs allow easy cloning of any desired Fab CDRs into the Fab scaffold (in this example, FabH), the resulting GA1–Fab(target 1) fusions can be efficiently produced through Escherichia coli periplasmic expression. Importantly, GA1 binds preferentially to Fabs containing the LRT motif with the FabS scaffold and does not bind to Fabs with wt kappa Lcs (FabH) (Table 1), therefore, there is no “self” association within the FabH‐linker‐GA1 component of the module.
Figure 9.
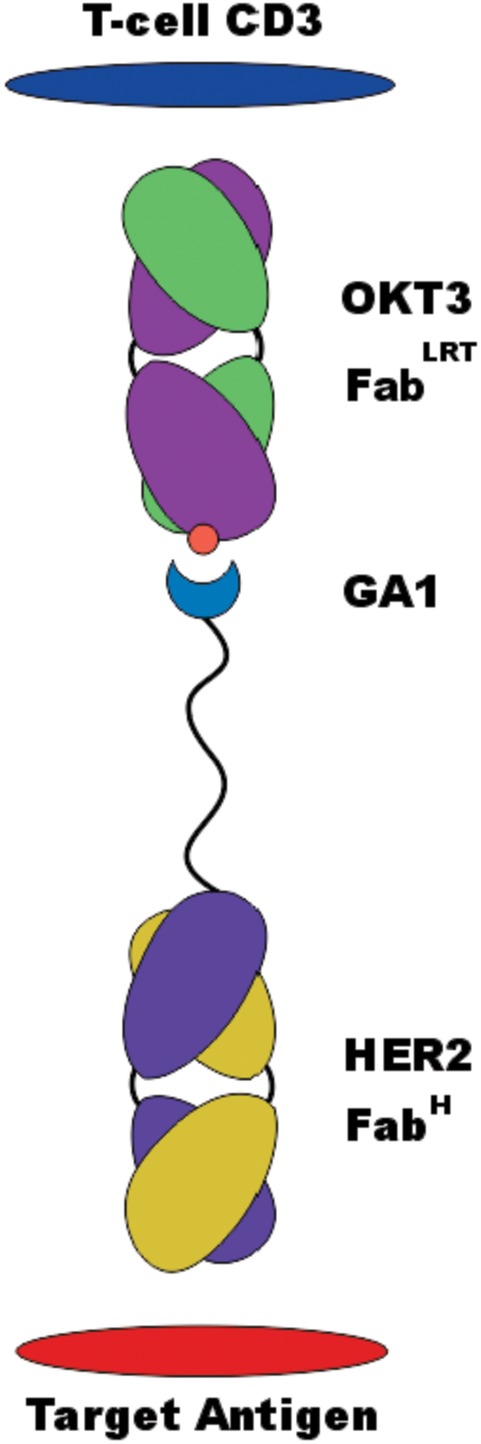
BiTE construct. FabH recognizes Her2 extracellular domain on the antigen‐presenting cells (APC). The Fab is attached by a 13 residue linker to GA1 via a fusion to the C‐term of its Lc. FabLRT component binds to CD3 of the T‐cell receptor. This Fab contains the CDRs of either OKT3 or UTCH1
To test the bi‐Fab module in a biological application, we chose to construct a BiTE that would induce engagement between a cell that had an overexpressed cell surface cancer marker through one arm and a cytotoxic T‐cell through the other. Thus, for the first arm (FabH), we chose to target the specific APC marker, Her2, which is highly over‐expressed on the surface of many breast cancer cell lines. For the Fab (target 2)LRT arm, we chose a humanized Fab version of an antibody that binds the CD3 component of the T‐cell receptor complex and activates it.37, 38 We hypothesized that the tight noncovalent link between T‐cells and tumor cells created by these bi‐Fab immuno‐reagents would induce robust immunological‐synapse formation, leading to T‐cell activation and secretion of cytokines and cytotoxic granules resulting in lysis of the tumor cell.
FabS of the bi‐Fab was derived from the α‐Her2 trastuzumab antibody. The Fab(2)LRT component was based on introducing the CDRs of either of the widely used CD3 antibodies, OKT3 or UCHT1 into the LRT engineered Fab scaffold. Thus, either CD3 Fab can be interchangeably plugged into the GA1 unit. The full bi‐Fab module was assembled and assessed for activity in a redirected tumor‐cell killing assay. The assay has three readouts: (a) the activity of a cytoplasmic enzyme, lactate dehydrogenase (LDH), released into the medium upon cell lysis, (b) interleukin IL2 and (c) interferon γ production by T‐helper cells. As the source of Effector T‐cells, we used isolated human PBMCs. The target cells were from Her2‐positive SKBR3 human breast‐cancer cells. Addition of bi‐Fabs in several different active combinations to PMBC‐SKBR3 co‐cultures at the optimal 50 nM, corresponding to early saturation concentration point, resulted in robust cell killing (up to 70%). Furthermore, these conditions led to prominent IL2 and IFNγ release (Figure 10). Notably, these levels somewhat surpassed those of the positive‐control bi‐specific antibody, representing hOKT3 Fab‐ hHer2 scFv genetic fusion (Figure 9). Importantly, the functional readouts were similar when the format was switched; that is, when the anti‐CD3 Fab is introduced into the construct as the fusion with GA1 and the Her2 is the FabLRT component (Figure 9). Thus, the activity of the bi‐Fab is independent of organization of the Fab components. All activities were abolished upon introduction of CDR mutations eliminating CD3 binding within the CD3 FabLRT element. As expected, the assembly of the functional non‐covalent bi‐Fab was dependent upon the genetic fusion of GA1 to FabH, as no detectable activity was observed when the proteins (FabH, FabLRT, GA1) were added as three separate unlinked entities. These results demonstrate the utility of the high affinity GA1–FabLRT binding pair for the facile construction of bi‐specific immuno‐reagents. Such a strategy should prove especially useful when large numbers of antibodies need to be screened in combination, streamlining the time and resource‐intensive expression and purification of bispecific molecules.
Figure 10.
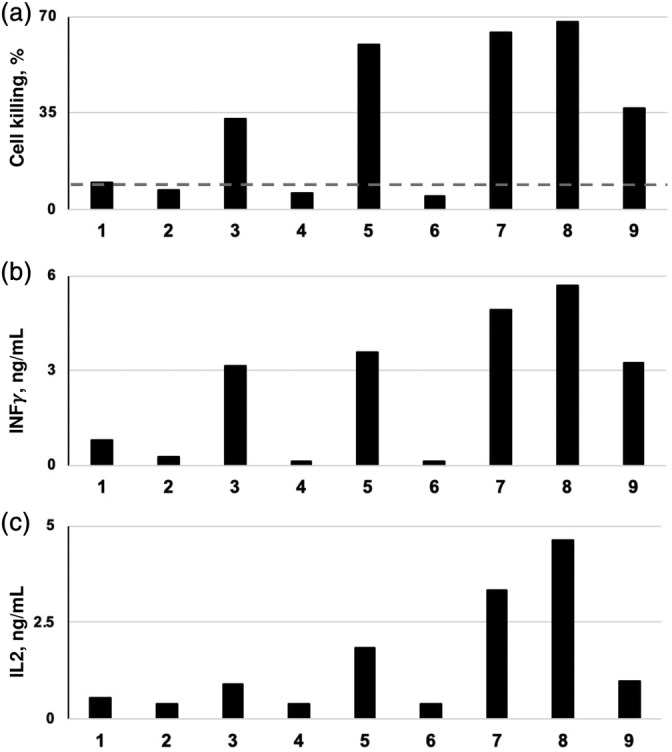
The effects of the FabH (Her2)‐linker‐GA1‐FabLRT(OKT3/UTCH1) BiTE on PBMC/SKBR3 (10:1) co‐cultures. To test the effect of the BiTE, 20K SKBR3 cells were cultured on a plate overnight. 200K of PBMCs were mixed with 50 nM of the BiTE and added on the SKBR3 cells. Cell killing effect measured by LDH activity (a) and cytokine release upon T cell activation (b, c) were measured after 24 hr incubation. As a control, all the individual components of the BiTE reagents (lanes 1 and 2) and with mutant CD3 FabLRT, deficient in CD3 binding (lanes 4 and 6) were tested and showed practically no effect on LDH or cytokine levels (dashed line). The CD3 activation and cell killing was observed only when both active components of the BiTE were present (lanes 3, 4, 7, and 8) and with genetically linked bi‐specific molecule used as a positive control for the immunological synapse formation 9. Results of representative experiments out of three (or more) are shown. Contents of lanes: 1 (GA1+ FabH(Her2) + FabLRT(OKT3); 2 (GA1+ FabH(Her2) + FabLRT(UTCH1), 3 (FabH(Her2) + GA1 + FabLRT(OKT3); 4 (FabH(Her2) + GA1 + mutFabLRT(OKT3)); 5 (FabH(Her2) + GA1 + FabLRT(UTCH1); 6 (FabH(Her2) + GA1+ mutFabLRT(UTCH1); 7 (FabH(OKT3) + GA1+ FabLRT(Her2); 8 FabH(UTCH1) + GA1+ FabLRT(Her2); 9 “BiTE control”: FabH(OKT3) fused to Her2 scFV
3. DISCUSSION
We have described the development of a platform that facilitates the coupling of Fab‐based affinity reagents in multi‐valent and multi‐specific formats. The core of the technology is a module of Protein‐G (GA1) that had been affinity matured by phage display mutagenesis to bind tightly to variant Herceptin Fab (FabS) scaffold.23 The interaction between FabS and GA1 was further enhanced by a subsequent affinity maturation of the FabS scaffold against GA1. Interestingly, the highest affinity FabS variant (FabLRT) contained a serendipitous two amino acid deletion within the region of five amino acids that were diversified in the phage display library. Together, this tandem mutagenesis approach resulted in an affinity of 100 pM between FabLRT and GA1, which was over 500‐fold tighter than that between the starting FabS and the wild‐type PG. While this is not a covalent interaction, our results indicate it is clearly of sufficient affinity for the applications that we investigated.
The impetus for developing this platform was to overcome myriad limitations of traditional antibody‐based affinity reagents. Antibodies have evolved structures and specificities optimized for in vivo immune recognition, not as tools for cell biologists. Recombinant technology has allowed for engineering scaffolds and optimizing specificities through relatively straightforward processes.2 This has enabled generating versatile assemblages with bi‐specific capabilities, allowing simultaneous recognition of multiple antigens. Nevertheless, these assemblages are generally constructed with particular pairs of targets in mind. If one or both of the targets change, then a new construct has to be designed, built and optimized. Thus, we endeavored to simplify the process by developing a tool kit that allows facile exchange of affinity modules in a plug and play fashion utilizing the ultra‐high affinity of the FabLRT–GA1 pair.
The first system we investigated involved the use of an enzyme complementation format to evaluate the properties of the GA1 fusions in the context of a sandwich assay. This requires two non‐overlapping epitopes on the antigen so that two independent Fabs can bind simultaneous. Several constructs were made; the first comprised the N‐terminal fragment of beta‐lactamase (BLF1) fused to a 30 amino acid linker with GA1, the second was identical except that the C‐terminal fragment (BLF2) was attached through a similar linker to a GA1 module (Figure 5). Furthermore, the formats were expanded whereby linkers were fused to either the N‐ or C‐terminal ends of GA1, introducing additional spatial variation. Each of these fusions was then mixed with one of the Fabs in a variety of combinations. A key concern of ours initially was that since the FabLRT–GA1 interaction is not covalent there might be exchange between components that might compromise the efficiency of the assay. However, we determined that with the 100 pM affinity between the pair, no measurable interchange occurs within the timeframe of the experiment.
Furthermore, it might be assumed that since this complementation assay was being performed using a small protein, Asf1, it does not offer a challenging system because the Fab binding sites are close together. However, the converse is actually true. The two epitopes on Asf1 are on directly opposing faces of the protein, effectively orienting the GA1 binding sites on the Fab scaffold such that the Gly–Ser linkers point in opposite directions (Figure 5). Anyone who has built a molecule model realizes that it is difficult to “turn a corner” in an efficient way while still adhering to reasonable conformational energies. That is why we added the option of fusing the linker to either the N‐ or C‐terminal end of GA1 to increase the potential spatial disposition of the BL fragments. Indeed, it was found that in the case of the Asf1 sandwich assay, there was some bias with regard to how the fragments were hooked up, but this did not carry over to other systems where pairings did not appear to matter. While no attempt was made to optimize the system, the general overall success of the complementation‐sandwich assay demonstrated that the system had enough inherent flexibility to suggest it could be broadly utilized for not only sandwich assays, but also other types of proximity assays. One can imagine a scenario where Fabs could be made to multiple cell surface targets. The Fabs could then be hooked up and profiled in high throughput to identify proximal cell surface neighbors.
A different format for the GA1 fusion was used in the development of the bi‐Fab BiTE construct.30, 31, 34 The concept of BiTEs has been developed to connect and bring together two different cell types, one being a cytotoxic T‐cell and the other a tumor cell.32, 33 A BiTE can take several different forms, but the basic construct is comprised of two linked antibody‐based moieties, one targeting a component of the T‐cell receptor on the T‐cell and the other targeting an over‐expressed surface antigen on the tumor cell (APC). Adding the BiTE initiates extensive crosslinking of the cells leading to T‐cell activation and subsequent tumor cell death. The effectiveness of the construct depends on multiple factors ranging from target density and binding potency to their linker length.35, 36 Importantly, simply linking a T‐cell to an APC does not induce cell death, the binding component of the T‐cell has to target certain components of the T‐cell receptor, most notably CD3.37, 38 We designed a bi‐Fab BiTE that could function as a cassette that allows facile interchange of Fabs directed at different cell surface targets. The basic component was a Fab–GA1 fusion that was directed at the HER2 antigen that is overexpressed on SKBR3 cells, a breast cancer cell line. Connecting GA1 to the Fab (FabH) is accomplished by fusing it by means of a 13 residue linker to the C‐term of the Hc of the Fab. Adjusting the linker length is straightforward and it does not affect the expression or stability of the basic cassette. Adding a FabLRT to this modality results in formation of the full‐length BiTE (FabH‐linker‐GA1–FabLRT). In our test case, the FabLRT was a humanized Fab version of either OKT3 or UTCH1, which are highly validated antibodies that activate T‐cells through their binding to CD3.37, 38 Either of these CD3 binding LRT Fabs could be interchangeably introduced into the HER2 FabH–GA1 module. Importantly, these constructs are highly soluble. Adding the assembled BiTEs to a mixture of PBMCs (which contain cytotoxic CD8 and CD4 cells) and SKBR3 breast cancer cells elicited readouts that verified induction of cancer cell death.
Given the ease of interchanging FabLRT in the BiTE module, one might envision a high throughput campaign to profile BiTE efficiencies targeting many different cancer‐specific cell surface antigens. To do this most efficiently, the format described above should be reversed. We showed that the “polarity” of the bi‐Fab makes no difference to its effectiveness; that is, which target Fab is included in the fusion with GA1 or as the FabLRT component (Figure 10). The Fab–GA1 fusion would be constructed to contain the CD3 binding Fab, OKT3 or UTCH1; this is an easy cloning step. Then, the FabLRT could be a Fab targeting any number of cell surface cancer markers. In this format, there is no need to make constructions for each bispecific pair, each BiTE can be rapidly assembled in a plug and play fashion. Furthermore, it is easy to change the linker lengths and even to put multiple GA1 modalities on the linker to exploit possible avidity effects.
To extend the system even further, one can imagine that by using the tandem phage display approach new sets of distinct high‐affinity Fab–GA1 interactions could be engineered. This could expand the distinct specificity of the fusion modules past being bi‐specific to tri‐specific or even tetra‐specific. While this remains a future goal, one can imagine the unique types of experiments that would be in reach with this technology in hand.
4. MATERIALS AND METHODS
4.1. Protein cloning, expression and purification
The sequences of all the constructs used are provided in Table S3. The open‐reading frames (ORFs) encoding the C‐terminal domain of Nucleoprotein (NPCT) from Zaire (EBOV), Reston and 3 other strains of Ebola virus, full‐size EBOV NP and Zika virus methyltransferase (MT ZIKV) optimized for bacterial expression, were gifted by Dr Z. Derewenda, University of Virginia. To serve as targets for phage selections, these viral ORFs as well as ORFs coding for yeast histone chaperone protein—Asf127 and protein GA1, an engineered high‐affinity Fab‐binding variant of PG domain C3,23 were cloned using Sma1 site into pEKD40 with the cleavable N‐terminal SNAP‐tag and the C‐terminal 6× His tag. pEKD40 is a derivative of pSNAP‐tag (T7)‐2 vector (NEB) that was modified with the thrombin‐cleavage site at the C‐terminus of the SNAP‐tag followed by Sma1 site and a C‐terminal 6× His tag added for enabling of protein purification. For the β‐lactamase split enzyme proximity applications, the viral proteins and Asf1 were cloned without SNAP‐tag using Xho1‐BamH1 sites of the pHFT2 version containing TEV‐cleavable N‐terminal 10× His tag.39 The same strategy using pHFT2 vector was applied for cloning of four of the BLF_GA1 fusion constructs comprised of one of two TEM‐1 β‐lactamase (BL) complementation fragments: BLF1, aa 26–196 bearing a M182T mutation26 or BLF2, aa 198–290, connected to the N‐ or C‐termini of GA1 by roughly 30 aa‐long GS linkers. Selected Fabs and Fab‐scaffold variants from phage clones were cloned into Sph1 sites of pSFV4 expression vector (https://www.thesgc.org/sites/default/files/toronto_vectors/pSFV4.pdf) using InFusion HD cloning kit (Clontech) as recommended.
To obtained Her2, OKT3 and UCHT1 Fabs (Table S3), their humanized CDR‐containing regions (synthesized as gBlocks by IDT) were cloned into pSFV4 using Nco1 and SgrA1 sites. To improve the bacterial expression of the OKT3 Fab, the Cys in CDR H3 of OKT3 was substituted with Ser. Genetic fusion of GA1 to the C‐terminus of FabH (Lc S123E) variants was achieved by cloning of GA1 containing an N‐terminal 13 aa long linker (GGSGSAGSGGAGA) into SgrA1 of pSFV4. FabLRT (∆∆LRT) and FabH (S123E) distinguishing mutations were grafted into FabS Lc at aa positions 123‐127 (SQLKS) using quick change site‐directed mutagenesis.
Expression of 6× and 10× His‐tagged proteins was induced by addition of 1 mM IPTG to 1L E. coli BL21 (DE3) cell cultures grown in 2xYT medium to a mid‐log phase (0.4–0.6 OD600). After the overnight incubation at 18°C (250 rpm), harvested cells were sonicated in buffer A: 50 mM Tris‐HCl, pH 8.0, 150 mM NaCl and centrifuged. Then, either native or denaturation purification protocols were applied for protein purification depending on their solubility. Soluble 6× and 10× His‐tagged proteins were purified from the cleared supernatants by TALON (Clontech) Immobilized metal affinity chromatography (IMAC) using a standard native‐condition procedure and elution by 100 mM imidazole in buffer A. The insoluble 10× His‐tagged BLF_GA1 fusion proteins were extracted from the pellets by 6M Gua‐HCl in buffer A with 0.3 mM TCEP and purified on TALON resin using a denaturation‐condition protocol and on‐column renaturation achieved by six washes of the column by the sequential twofold 6M Gua‐HCl dilutions in buffer A and a final buffer A wash. The renaturated BLF_GA1 fusion proteins eluted with 100 mM imidazole in buffer A were immediately diluted with buffer A in order to lower their concentration to 0.5 mg/ml (or less) to prevent them from precipitation. These fusion proteins were never frozen and were stored on ice.
Fabs and Fab_GA1 fusion proteins were expressed in the periplasm of E. coli BL21 cells for 4–5 hr at 37°C after induction by 1 mM IPTG at 0.8–1.2 OD600. The cells were harvested by centrifugation and sonicated in 50 mM phosphate buffer, pH 7.4, 500 mM NaCl. The centrifugation‐cleared sonicates were applied to one or the other affinity column depending on the LC scaffold of the Fab: FabS and FabLRT variants (possessing high affinity toward protein GA1) were purified on ProteinGA1 resin created in the lab as described23 using SulfoLink Coupling Resin (Thermo Scientific), while FabH_GA1 fusions lacking GA1‐bindnig affinity were purified using ProteinA resin (Genscript). In both cases, Fab variants and Fab_GA1 fusions were eluted from the column by 0.1 M glycine, pH 2.6, and neutralized with aliquots of 1 M Tris‐HCl pH 8.5. For short‐term storage, Fabs and Fab fusions were kept at 1 mg/ml on ice.
4.2. Phage display selection protocol
4.2.1. SNAP‐tagged target protein immobilization
The selection strategy for Fab generation was previously described in23, 27, 40 Purified SNAP‐tagged target proteins were SNAP‐biotinylated at 20% excess of SNAP‐Biotin (NEB) in the presence of 0.3 mM TCEP for 15 min at 37°C, followed by the binding onto streptavidin‐coated paramagnetic beads (Dynabeads M‐270, Invitrogen) using a standard protocol. 1–3 selections were performed with each of the SNAP‐biotinylated target proteins.
4.2.2. Phage display libraries
The Phage M13 Fab library, containing CDRs randomized at a diversity of >1010 in a variant of the human FabH scaffold, FabS, featuring a single aa substitution in Lc, E123S, Hc, C‐terminally fused to the M13 minor coat protein pIII, was used for sAB selection against various antigens. Another phage library was created for selection of Fab Lc scaffold variants against SNAP_GA1 as a target protein, using the strategy previously published.41 To that end, five residues in FabS light‐chain scaffold that interact with GA1 were chosen for hard randomization (Figure S4): DNA encoding aa 123–127 (SQLKS) in Lc MJ20 phagemid was replaced for NNK NNK NNT NNK NNK (K standing for G or T: NNK covers 32 codons for all 20 aa and TAG Stop codon) using Kunkel mutagenesis protocol.40
4.2.3. Library sorting procedure
Three to five rounds of phage sorting (depending on the selected phage specificity achieved) were performed at room temperature as described earlier23, 27 with some modifications. For the first round, 200 μl (original volume) streptavidin‐coated paramagnetic beads (Promega) with immobilized SNAP‐biotinylated target proteins were incubated in 1 ml phage library (1011 cfu) at 200 nM final target‐protein concentration for one hour at RT. The beads were washed manually two times using a magnetic stand and added to log phase E. coli XL1‐blue (Stratagene) for 20 min. Then, M13K07 helper phage was added to final concentration of 1,010 pfu/ml for the overnight phage amplification. For all subsequent rounds, the amplified phage was precipitated twice in 20% PEG/2.5 M NaCl, and placed at 1–2 OD286 /well into an automated Magnetic Particle processor (KingFisher 700, Thermo Scientific). The phage was captured from 100 μl well solution containing target‐coated beads (2 μl original bead volume/well) in the presence of 1 mM O6‐Benzylguanine‐blocked SNAP protein as a competitor. The final concentration of the antigen bound to the beads was dropped gradually from 200 to 1 nM from the first to the fifth round. After phage binding, the beads were subjected to five washing rounds and the phage particles bound to the target protein were eluted by 5 min incubation in 100 μl of 1 U/ml thrombin (1.3 U/μl, Novagen). Then, the phage eluate was used for E. coli infection and phage amplification as described above. After 103 and higher specificity enrichment of phage was achieved, the infected cells (without the helper phage) were directly plated on ampicillin agar for the overnight growth at 37°C and sets of 96 colonies were picked to produce phage clones for single‐point phage ELISA assays.27 The promising clones demonstrating high specific and low non‐specific binding were sequenced and reformatted into a pSFV4 vector as described above for Fab expression and purification.
4.3. FabLRT–GA1 purification and crystallization
Recombinant FabLRT11M42 and protein GA1 containing 10× His tag and the TEV‐cleavage site at the N‐terminus were produced as described above. Prior to the complex formation, 10× His tag on GA1 was removed using TEV protease. To obtain the FabLRT–GA1 complex, FabLRT11M was incubated with GA1 at 1:1 molar ratio on ice for 3 hr and the complex was purified by size‐exclusion chromatography on a Superdex 200 Increase 10/300 GL (GE Healthcare Life Sciences) column equilibrated with 20 mM HEPES, 150 mM sodium chloride, pH 7.5. The purity of the complex was confirmed by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE).
Initial crystallization trials of the complex were set up at room temperature using the hanging‐drop vapor‐diffusion method utilizing the Mosquito Crystal robot (TTP Labtech). FabLRT–GA1 complex at 17 mg/ml was crystallized by mixing 100 nl of protein complex solution with 100 nL of a Protein Complex Suite (QIAGEN) screen solution. The most promising crystals of FabLRT–GA1 were observed in 0.1 M magnesium chloride, 0.1 M sodium acetate pH 5.0, and 15% (w/v) PEG 4000 at 19°C. To improve crystal quality, the initial crystallization condition was optimized. Hanging‐drop crystallization trials were set up at room temperature by mixing 1 μl of complex solution with 1 μl of reservoir solution. Good quality crystals were obtained by the seeding technique43 in 0.1 M magnesium chloride, 0.1 M sodium acetate pH 5.0, and 20% (w/v) PEG 4000 at 19°C. The resulting crystals of FabLRT–GA1 were soaked in mother liquor containing 20% glycerol and flash‐frozen in liquid nitrogen for data collection.
4.4. Data collection, structure determination and refinement
X‐ray diffraction experiments were carried out at 100°K at beam line 23‐ID‐D at the General Medical Sciences and Cancer Institute Structural Biology Facility (GM/CA), Argonne National Laboratory (Argonne, Illinois). Data were indexed and integrated with XDS 44 and scaled using AIMLESS 45 integrated into the CCP4 program suite.46 Initially, the data set was processed in P6222 space group. However, molecular replacement failed to find a structure solution. Evaluation of images showed that there were split reflections at high resolution, which suggested twinning. To explore the possibility of twinning, data were reprocessed in P61, P321 and P312 point group symmetries. The best structure solution with R factor = 33.5% and R free = 37.8% was obtained in the P3221 space group by molecular replacement method using BALBES.47 A starting PDB model for the FabLRT–PGA1 complex structure was generated by BALBES based on proteins sequence similarity. The analysis by phenix.xtriage48 indicated crystal twinning with one twin operator (‐h,‐k, l) and estimated a twin fraction of 0.49. The structure was refined in PHENIX48 using obtained twin law to R work = 19.2% and R free = 25% compared to R work = 29.4% and R free = 37.1% with no twin refinement. Manual structure corrections were performed in Coot.49, 50 Atom contacts and structure validation were determined in MolProbity.51, 52 The data collection and refinement statistics are summarized in Table S2. The surface accessible solvent area between FabLRT and PGA1 was calculated in AREAIMOL.53 Structure alignment was performed using CCP4 support program LSKAB.46 Structural figures were created with CCP4mg.54 Coordinates and structure factors have been deposited in the Protein Data Bank under entry 6U8C.55
4.5. Surface plasmon resonance analysis
For Surface plasmon resonance (SPR) analysis, all the protein components were dialyzed into EB buffer. The experiments were performed on a BIAcore‐3000 (Biacore AB, Uppsala, Sweden). The target was immobilized via a 10× or 6× His tag to a Ni‐NTA chip (GE Healthcare), while Fab variants in twofold dilutions were run as analytes in EB buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 50 μM EDTA, 0.005% Tween20) at 30 μl/min flow rate, 20°C. Senogram traces were corrected through double referencing and fit using Scrubber (BioLogic Software, Campbell, Australia) to a 1:1 binding model.
4.6. PCA β‐lactamase assay
PCA reaction components: BLF–GA1 fusions and the antigen to be detected (viral proteins or Asf1) were combined on ice in 100 μl PBS containing 2 μM fluorogenic BL substrate, Fluorocillin Green 495/525 (Life Technologies), in a well of black FluoroNunc 96‐well microplate (Nunc) and the fluorescent signal was monitored at room temperature using Safire2 Tecan Plate Reader (483 nm excitation, 525 nm emission). The results were reproduced at least three times and the data from a representative experiment are shown.
4.7. T‐cell redirection cell‐culture assays
Human breast cancer cell line SKBR3 (ATCC), overexpressing Her2 gene product on the cell surface was cultured according to ATCC protocols. CD3+ PMBC cells were isolated from patient blood56, 57 and stored frozen in liquid nitrogen.
The day before the experiment, SKBR3 cells were seeded into a 96‐well plate (20K SKBR3 cells in 100 μl per well), while defrosted PBMC were placed into a suspension culture (2 ml cell/ml). After 16–24 hr incubation, PBMC cells were washed, transferred to the medium‐aspired SKBR3 wells at 10:1 Effector cell to tget cell ratio and then the bi‐specific components were added at 50 nM, unless otherwise stated, in the final volume of 100 μl/well. After 24 hr of co‐culturing, the medium in each plate was analyzed using commercially available kits: for LDH presence (CytoTox96, Promega #G1781, positive control – complete cancer‐cell lysis), and cytokine release (INFg, Cisbio #62HIFNGPEG) and (IL2, Cisbio #62HIL02PEG)—the values were normalized using protocols and standards provided in the kits. The results were reproduced at least three times and the data of a representative experiment are shown.
Supporting information
Table S1. CDR aa sequences for Fabs selected against NPCT of 5 Ebola virus strains
Table S2. X‐ray data collection and refinement statistics
Table S3. Constructs Results of LC scaffold GA1‐affinity maturation. (a) The weblogo and the list of 6 selected motifs for Lc aa 123–127 (note the deletions in position 123–124). (b). Phage ELISA results for the selected Fab clones. Blue and red—10 nM pGA1 well coating, Lilac – 1 mM SNAP well coating, red—100 nM pGA1 soluble GA1 competitor.
Figure S2. Antigen‐dependent BL activity of different combinations of GA1‐BLF fusions and FabLRTs. The chart represents BL activity measured by the fluorescent signal in reaction mixtures 1 to 12 after 20 min at RT. Bars for reactions detecting EBOV NPCT or ZIKV MT are shown in solid black or in black stripes, respectively. The presence of the antigen is indicated on the top. The components of each reaction mixtures are shown in the table below; the numbers for the active combinations are in red. Each component was present in the reaction at 250 nM.
Figure S3. Tumor‐cell killing by bi‐Fab BiTES: The effect of Her2_GA1+hUCHT1 concentration on LDH release in PMBC‐SKBR3 co‐cultures. 50 nM concentration corresponding to 70% killing was chosen for the further experimentation.
Figure S4. Strategy schematics for Kunkel‐based library generation for Fab LC scaffold affinity maturation. Phagemid containing Fab MJ20 with the stop codon introduced into Lc aa position 125 was subjected to Kunkel mutagenesis using NNK NNK NNT NNK NNK randomization primer for Lc aa positions 123–127. The library of 109 clones was produced, while the theoretical diversity for this library is approximately 1.7 × 107 variants.
ACKNOWLEDGMENTS
We acknowledge the contributions of A. Rohaim for valuable crystallography advice and T. Uchanski for developing the SNAP‐tag phage display format. We appreciate the use of the crystallization robot in J. Piccirilli's lab. The X‐ray data collection was performed at the GM/CA@APS Sector 23 at the Advanced Photon Source, a U.S. Department of Energy (DOE), Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB‐12002) and the National Institute of General Medical Sciences (AGM‐12006). The work was supported by NIH GM117372 to AAK. Support was also provided by the Searle Foundation under the auspices of the Chicago Biomedical Consortium.
Slezak T, Bailey LJ, Jaskolowski M, et al. An engineered ultra‐high affinity Fab‐Protein G pair enables a modular antibody platform with multifunctional capability. Protein Science. 2020;29:141–156. 10.1002/pro.3751
Present address Mateusz Jaskolowski, Institute of Molecular Biology and Biophysics, ETH Zurich, 8093 Zurich, Switzerland.
Dominik A. Nahotko, Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
Anthony Kossiakoff is the winner of the 2019 Christian B. Anfinsen Award.
Funding information Center for Scientific Review, Grant/Award Number: GM117372; Chicago Biomedical Consortium
REFERENCES
- 1. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. [DOI] [PubMed] [Google Scholar]
- 2. Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: The power of in vitro display technologies. Nat Biotechnol. 2011;29:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Groff K, Brown J, Clippinger AJ. Modern affinity reagents: Recombinant antibodies and aptamers. Biotechnol Adv. 2015;33:1787–1798. [DOI] [PubMed] [Google Scholar]
- 4. Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. [DOI] [PubMed] [Google Scholar]
- 5. Bradbury A, Pluckthun A. Reproducibility: Standardize antibodies used in research. Nature. 2015;518:27–29. [DOI] [PubMed] [Google Scholar]
- 6. Hornsby M, Paduch M, Miersch S, et al. A high through‐put platform for recombinant antibodies to folded proteins. Mol Cell Proteomics. 2015;14:2833–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bass S, Greene R, Wells JA. Hormone phage: An enrichment method for variant proteins with altered binding properties. Proteins. 1990;8:309–314. [DOI] [PubMed] [Google Scholar]
- 8. Paduch M, Koide A, Uysal S, Rizk SS, Koide S, Kossiakoff AA. Generating conformation‐specific synthetic antibodies to trap proteins in selected functional states. Methods. 2013;60:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ye JD, Tereshko V, Frederiksen JK, et al. Synthetic antibodies for specific recognition and crystallization of structured rna. Proc Natl Acad Sci USA. 2008;105:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paduch M, Kossiakoff AA. Generating conformation and complex‐specific synthetic antibodies. Methods Mol Biol. 2017;1575:93–119. [DOI] [PubMed] [Google Scholar]
- 11. Gao J, Sidhu SS, Wells JA. Two‐state selection of conformation‐specific antibodies. Proc Natl Acad Sci USA. 2009;106:3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miersch S, Li Z, Hanna R, et al. Scalable high throughput selection from phage‐displayed synthetic antibody libraries. J Vis Exp. 2015;95:51492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fellouse FA, Esaki K, Birtalan S, et al. High‐throughput generation of synthetic antibodies from highly functional minimalist phage‐displayed libraries. J Mol Biol. 2007;373:924–940. [DOI] [PubMed] [Google Scholar]
- 14. Rizk SS, Kouadio JL, Szymborska A, et al. Engineering synthetic antibody binders for allosteric inhibition of prolactin receptor signaling. Cell Commun Signal. 2015;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rizk SS, Paduch M, Heithaus JH, Duguid EM, Sandstrom A, Kossiakoff AA. Allosteric control of ligand‐binding affinity using engineered conformation‐specific effector proteins. Nat Struct Mol Biol. 2011;18:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marcon E, Jain H, Bhattacharya A, et al. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat Methods. 2015;12:725–731. [DOI] [PubMed] [Google Scholar]
- 17. Zhang Z, Liang WG, Bailey LJ, et al. Ensemble cryoem elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme. Elife. 2018;7:e33572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang Y, Kuybeda O, de Waal PW, et al. Cryo‐em structure of human rhodopsin bound to an inhibitory g protein. Nature. 2018;558:553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dominik PK, Borowska MT, Dalmas O, et al. Conformational chaperones for structural studies of membrane proteins using antibody phage display with nanodiscs. Structure. 2016;24:300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bailey LJ, Sheehy KM, Dominik PK, et al. Locking the elbow: Improved antibody fab fragments as chaperones for structure determination. J Mol Biol. 2018;430:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bukowska MA, Grutter MG. New concepts and aids to facilitate crystallization. Curr Opin Struct Biol. 2013;23:409–416. [DOI] [PubMed] [Google Scholar]
- 22. Rizk SS, Luchniak A, Uysal S, Brawley CM, Rock RS, Kossiakoff AA. An engineered substance p variant for receptor‐mediated delivery of synthetic antibodies into tumor cells. Proc Natl Acad Sci USA. 2009;106:11011–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bailey LJ, Sheehy KM, Hoey RJ, Schaefer ZP, Ura M, Kossiakoff AA. Applications for an engineered protein‐g variant with a ph controllable affinity to antibody fragments. J Immunol Methods. 2014;415:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Derrick JP, Wigley DB. Crystal structure of a streptococcal protein g domain bound to an fab fragment. Nature. 1992;359:752–754. [DOI] [PubMed] [Google Scholar]
- 25. Radwanska MJ, Jaskolowski M, Davydova E, et al. The structure of the c‐terminal domain of the nucleoprotein from the bundibugyo strain of the ebola virus in complex with a pan‐specific synthetic fab. Acta Cryst. 2018;D74:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta‐lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol. 2002;20:619–622. [DOI] [PubMed] [Google Scholar]
- 27. Schaefer ZP, Bailey LJ, Kossiakoff AA. A polar ring endows improved specificity to an antibody fragment. Protein Sci. 2016;25:1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu JS, Liao HX, Gerdon AE, et al. Detection of ebola virus envelope using monoclonal and polyclonal antibodies in elisa, surface plasmon resonance and a quartz crystal microbalance immunosensor. J Virol Methods. 2006;137:219–228. [DOI] [PubMed] [Google Scholar]
- 29. Chames P, Baty D. Bispecific antibodies for cancer therapy: The light at the end of the tunnel? MAbs. 2009;1:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen L, Flies DB. Molecular mechanisms of t cell co‐stimulation and co‐inhibition. Nat Rev Immunol. 2013;13:227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huehls AM, Coupet TA, Sentman CL. Bispecific t‐cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single‐chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA. 1995;92:7021–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bluemel C, Hausmann S, Fluhr P, et al. Epitope distance to the target cell membrane and antigen size determine the potency of t cell‐mediated lysis by bite antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jarantow SW, Bushey BS, Pardinas JR, et al. Impact of cell‐surface antigen expression on target engagement and function of an epidermal growth factor receptor x c‐met bispecific antibody. J Biol Chem. 2015;290:24689–24704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gebel HM, Lebeck LK, Jensik SC, Webster K, Bray RA. T cells from patients successfully treated with okt3 do not react with the t‐cell receptor antibody. Hum Immunol. 1989;26:123–129. [DOI] [PubMed] [Google Scholar]
- 38. Landegren U, Andersson J, Wigzell H. Mechanism of t lymphocyte activation by okt3 antibodies. A general model for t cell induction. Eur J Immunol. 1984;14:325–328. [DOI] [PubMed] [Google Scholar]
- 39. Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci USA. 2008;105:6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kunkel TA. Rapid and efficient site‐specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuh G, Sidhu SS. Efficient phage display of polypeptides fused to the carboxy‐terminus of the m13 gene‐3 minor coat protein. FEBS Lett. 2000;480:231–234. [DOI] [PubMed] [Google Scholar]
- 42. Mukherjee S, Griffin DH, Horn JR, et al. Engineered synthetic antibodies as probes to quantify the energetic contributions of ligand binding to conformational changes in proteins. J Biol Chem. 2018;293:2815–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luft JR, DeTitta GT. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Cryst. 1999;D55:988–993. [DOI] [PubMed] [Google Scholar]
- 44. Kabsch W. Integration, scaling, space‐group assignment and post‐refinement. Acta Cryst. 2010;D66:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Cryst. 2013;D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the ccp4 suite and current developments. Acta Cryst D. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Long F, Vagin AA, Young P, Murshudov GN. Balbes: A molecular‐replacement pipeline. Acta Cryst. 2008;D64:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.Refine. Acta Cryst. 2012;D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 50. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst. 2010;D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen VB, Arendall WB 3rd, Headd JJ, et al. Molprobity: All‐atom structure validation for macromolecular crystallography. Acta Cryst D. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davis IW, Leaver‐Fay A, Chen VB, et al. Molprobity: All‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55:379–400. [DOI] [PubMed] [Google Scholar]
- 54. McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: The ccp4mg molecular‐graphics software. Acta Cryst. 2011;D67:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ferrante A, Thong YH. Optimal conditions for simultaneous purification of mononuclear and polymorphonuclear leucocytes from human blood by the hypaque‐ficoll method. J Immunol Methods. 1980;36:109–117. [DOI] [PubMed] [Google Scholar]
- 57. Vissers MC, Jester SA, Fantone JC. Rapid purification of human peripheral blood monocytes by centrifugation through ficoll‐hypaque and sepracell‐mn. J Immunol Methods. 1988;110:203–207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. CDR aa sequences for Fabs selected against NPCT of 5 Ebola virus strains
Table S2. X‐ray data collection and refinement statistics
Table S3. Constructs Results of LC scaffold GA1‐affinity maturation. (a) The weblogo and the list of 6 selected motifs for Lc aa 123–127 (note the deletions in position 123–124). (b). Phage ELISA results for the selected Fab clones. Blue and red—10 nM pGA1 well coating, Lilac – 1 mM SNAP well coating, red—100 nM pGA1 soluble GA1 competitor.
Figure S2. Antigen‐dependent BL activity of different combinations of GA1‐BLF fusions and FabLRTs. The chart represents BL activity measured by the fluorescent signal in reaction mixtures 1 to 12 after 20 min at RT. Bars for reactions detecting EBOV NPCT or ZIKV MT are shown in solid black or in black stripes, respectively. The presence of the antigen is indicated on the top. The components of each reaction mixtures are shown in the table below; the numbers for the active combinations are in red. Each component was present in the reaction at 250 nM.
Figure S3. Tumor‐cell killing by bi‐Fab BiTES: The effect of Her2_GA1+hUCHT1 concentration on LDH release in PMBC‐SKBR3 co‐cultures. 50 nM concentration corresponding to 70% killing was chosen for the further experimentation.
Figure S4. Strategy schematics for Kunkel‐based library generation for Fab LC scaffold affinity maturation. Phagemid containing Fab MJ20 with the stop codon introduced into Lc aa position 125 was subjected to Kunkel mutagenesis using NNK NNK NNT NNK NNK randomization primer for Lc aa positions 123–127. The library of 109 clones was produced, while the theoretical diversity for this library is approximately 1.7 × 107 variants.