

Abstract
Protein domains constitute regions of distinct structural properties and molecular functions that are retained when removed from the rest of the protein. However, due to the lack of tertiary structure, the identification of domains has been largely neglected for long (>50 residues) intrinsically disordered regions. Here we present a sequence‐based approach to assess and visualize domain organization in long intrinsically disordered regions based on compositional sequence biases. An online tool to find putative intrinsically disordered domains (IDDomainSpotter) in any protein sequence or sequence alignment using any particular sequence trait is available at http://www.bio.ku.dk/sbinlab/IDDomainSpotter. Using this tool, we have identified a putative domain enriched in hydrophilic and disorder‐promoting residues (Pro, Ser, and Thr) and depleted in positive charges (Arg and Lys) bordering the folded DNA‐binding domains of several transcription factors (p53, GCR, NAC46, MYB28, and MYB29). This domain, from two different MYB transcription factors, was characterized biophysically to determine its properties. Our analyses show the domain to be extended, dynamic and highly disordered. It connects the DNA‐binding domain to other disordered domains and is present and conserved in several transcription factors from different families and domains of life. This example illustrates the potential of IDDomainSpotter to predict, from sequence alone, putative domains of functional interest in otherwise uncharacterized disordered proteins.
Keywords: compositional bias, DNA‐binding domain, domain, IDDomainSpotter, IDPs, low‐complexity regions, NMR, p53, plant MYB protein, transactivation domain, transcription factor
1. INTRODUCTION
For many proteins, molecular function is tightly coupled to the three‐dimensional structures of globular domains existing in the context of larger proteins. In contrast, intrinsically disordered regions (IDRs) lack fixed three‐dimensional structure1 instead populating dynamic ensembles of conformations that may display transient structural elements of functional relevance. In doing so, there are various ways in which IDRs enable fast, sensitive, dynamic, and graded regulation of protein activity, for example, by physical interactions with other proteins or molecules,2, 3, 4 through post‐translational modifications (PTMs),5, 6, 7, 8 or via allostery.9, 10
Protein domains have been defined as segments that may function, evolve, and fold independently of the rest of the protein.11, 12, 13 Thus, even when removed from its original sequence context and expressed independently or when fused to other polypeptide chains, a domain retains its fold and function. This definition does not exclude disordered protein segments that have conserved sequences, conserved disorder, and/or conserved functions independent of the rest of the polypeptide chain.14, 15, 16 Globular domains are predicted based on, for example, sequence similarity, predicted secondary structure content, patterns of hydrophobic residues, and relative solvent accessibility. These criteria fail to identify domains in IDRs, which usually evolve faster17 and do not fold into stable structures with a hydrophobic core. Thus, while multidomain globular proteins are frequent, domains within IDRs have generally not been considered, perhaps because domains have historically been viewed as primarily being linked to structure. Nonetheless, distinct regions within IDRs may have independent functional, chemical, and structural properties as well as evolve independently of the rest of the protein and may therefore similarly be considered as domains existing within a larger disordered context.
Compared to globular domains, IDRs are often compositionally biased with higher frequencies of Pro, Glu, Ser, Gln, Lys, Ala, and Gly.18 However, different biases and residue distributions within an IDR may lead to widely different chemical and structural properties giving rise to the plethora of diverse molecular functions IDRs confer.19 IDRs thus act as entropic chains linking other domains,20 as chaperones that assist folding of RNA or other proteins,21 as assemblers via short interaction motifs that allow building of higher order complexes in signal transduction,22 or displaying PTM sites for regulating protein activity.23 As an example, some IDRs enriched in polar and Gly residues with regularly spaced aromatic residues promote liquid–liquid phase separation to form biomolecular condensates under certain physiological conditions.24, 25, 26, 27 Conversely, for IDRs with a high fraction of charged residues, the distribution of opposite charges (quantified by the κ‐value) controls chain compaction.28 These cases exemplify how different sequence biases and distributions within IDRs can lead to markedly different chemical and structural properties and hence may allow for domain identification, especially in cases where those biases are evolutionarily conserved among homologs.
Many proteins involved in signaling and regulation utilize the advantages of intrinsic disorder when integrating information about cellular and organismal status.1 One such group of proteins are transcription factors (TFs) that integrate internal and external cues to fine‐tune gene expression and thus modulate the output of biological processes to make decisions of vital importance to the organism. Consistently, the transactivation domains (TADs) and other regulatory domains of TFs are predicted to contain extensive disordered regions.29, 30, 31 These IDRs are often of significant size, ranging up to several hundred residues uninterrupted by any ordered domains. Hence, this group of proteins constitute a suitable model system for analyzing the presence of disordered domains identified from compositional biases.
In this work we have used a compositional bias approach to spot domains in long IDRs from TFs by calculating fractions of residues within sliding windows. Our results suggest the presence of different disordered domains in TFs conserved across species and show that transitions and peaks on the sliding window profiles are linked to known structural and functional modules, supporting the validity of our method. Notably, several DNA‐binding domains (DBDs) of TFs are bordered by a segment enriched in Pro, Ser, and Thr and depleted in Arg and Lys compared to surrounding regions. We find this putative +PST‐RK domain to be evolutionarily conserved among homologs, indicating functional importance. In the predicted long IDRs of Arabidopsis thaliana MYB28 and MYB29,32 the +PST‐RK domain connects the folded N‐terminal DBD to the disordered C‐terminal part of the protein. Results from several complementary biophysical techniques revealed that the +PST‐RK domain lacks stable structure, being in a flexible and extended conformation, providing the first experimental evidence of intrinsic disorder in the plant R2R3 MYB TF family. We propose that the disordered +PST‐RK domain provides conformational flexibility between the connected domains as well as possibly chaperoning against undesired electrostatic attraction. Our findings illustrate the potential of IDDomainSpotter to identify disordered domains by exploiting compositional biases, enabling the decoding of long IDRs into structural and functional units, accelerating their characterization.
2. RESULTS
2.1. Sliding window profiles divide disordered regions into structural and functional domains
IDDomainSpotter calculates the fractions of residue types in either single protein sequences or sequence alignments in FASTA format. Fractions are calculated in sliding windows with a default size of 15 residues, chosen to achieve a balance between sensitivity to average characteristics of the sequence and resolution. Further, as the majority of short interaction motifs in IDRs are between 3 and 10 residues,33 this window size allows for the identification of such motifs that are biased in their composition relative to their neighboring regions. For each residue, k, the fraction of residues between k − 7 and k + 7 matching specific criteria is returned, and in the case of alignments, the result is averaged across the individual sequences. All criteria and combinations are possible, and the user can add as many features as desired, as well as choose a custom window size relevant to the user's study. Each feature consists of a specific combination of amino acids yielding a positive contribution and a combination of amino acids yielding a negative contribution. For each feature any combination of proteinogenic amino acids (or X for unusual or unnatural amino acids) can be used, making the approach highly flexible and customizable depending on the protein examined and the properties of choice.
In order to capture the diversity of long IDRs, we assembled a set of TFs from different families and different organisms, and with varying preexisting information on structure, function, and domain organization (Figures 1, 2, 3). We further gathered homolog sequences and constructed alignments to assess the conservation of the features we observed from analyses of the individual sequences. We then applied IDDomainSpotter to assess compositional bias throughout the IDRs and identify regions that may constitute segregated disordered domains.
Figure 1.
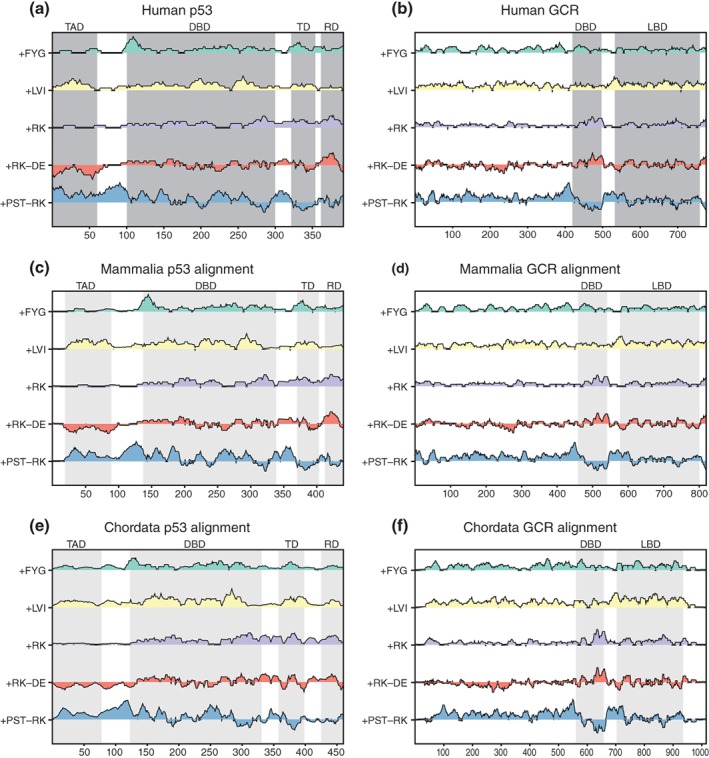
IDDomainSpotter profiles of p53 (a, c, e) and GCR (b, d, f). Profiles of human p53 (a), profiles of aligned mammalian p53 sequences (c), and of aligned chordate p53 sequences (e) shown above each other for comparison. Profiles of human GCR (b), profiles of aligned mammalian GCR sequences (d), and of aligned chordate GCR sequences (f) shown above each other for comparison. Gray boxes indicate domains, conserved regions, and motifs as named above the graph. Light gray boxes indicate the same information transferred to the aligned sequence profiles by locating the corresponding positions in the alignment. Profiles display scores for Phe + Tyr + Gly (+FYG), Leu + Val + Ile (+LVI), Arg + Lys (+RK), Arg + Lys‐Asp‐Glu (+RK−DE), and Pro+Ser + Thr‐Arg‐Lys (+PST−RK) calculated over 15 residues windows. See Tables S1–S4 for sequences used in alignments. DBD, DNA‐binding domain; LBD, ligand‐binding domain; RD, regulatory domain; TAD, transactivation domain; TD, tetramerization domain
Figure 2.
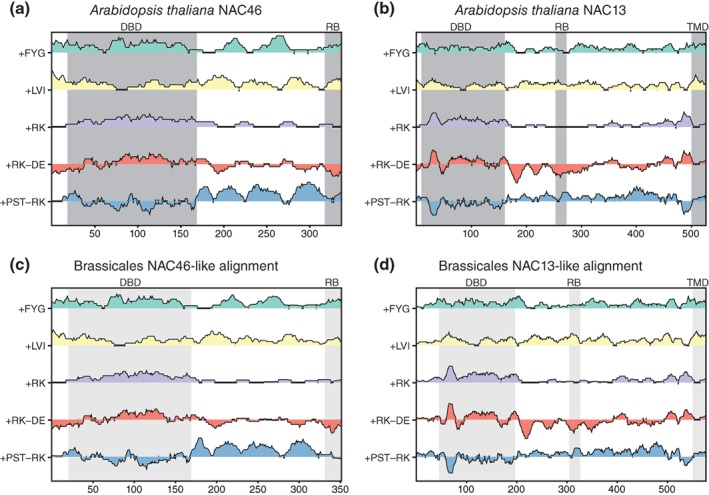
IDDomainSpotter profiles of NAC46 (a, c) and NAC13 (b, d). Profiles of A. thaliana NAC46 (a) and of aligned Brassicales NAC46 sequences (c) shown above each other for comparison. Profiles of A. thaliana NAC13 (b) and of aligned Brassicales NAC13 sequences (d) shown above each other for comparison. Gray boxes indicate domains, conserved regions and motifs as named above the graph. Light gray boxes indicate the same information transferred to the aligned sequence profiles by locating the corresponding positions in the alignment. Profiles display scores for Phe + Tyr + Gly (+FYG), Leu + Val + Ile (+LVI), Arg + Lys (+RK), Arg + Lys‐Asp‐Glu (+RK−DE), and Pro + Ser + Thr‐Arg‐Lys (+PST−RK) calculated over 15 residues windows. See Tables S5 and S6 for sequences used in alignments. DBD, DNA‐binding domain; RB, RCD1 binding region; TMD, transmembrane domain
Figure 3.
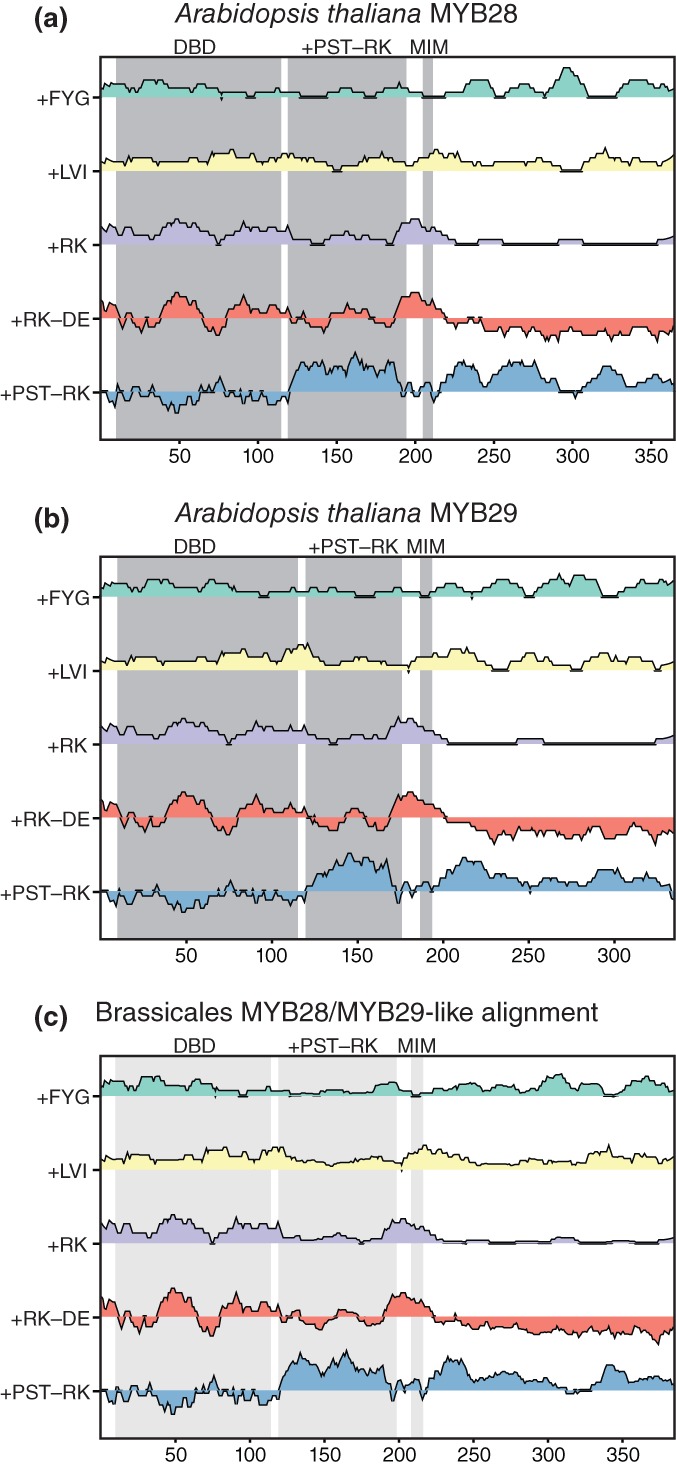
IDDomainSpotter profiles of A. thaliana MYB28 (a), A. thaliana MYB29 (b), and of aligned Brassicales MYB28/MYB29‐like sequences (c) shown above each other for comparison. Gray boxes indicate domains, conserved regions, and motifs as named above the graph. Light gray boxes indicate the same information transferred to the aligned sequence profiles by locating the corresponding positions in the alignment. Profiles display scores for Phe + Tyr + Gly (+FYG), Leu + Val + Ile (+LVI), Arg + Lys (+RK), Arg + Lys‐Asp‐Glu (+RK−DE), and Pro + Ser + Thr‐Arg‐Lys (+PST−RK) calculated over 15 residues windows. See Table S7 for sequences used in alignments. DBD, DNA‐binding domain; MIM, MYC‐interaction motif; +PST−RK, region investigated in this study
To analyze our assembled TF set, we chose several features. These included Phe, Tyr, and Gly (+FYG) as IDRs enriched in these residues may be involved in the promotion of liquid–liquid phase separation, and Leu, Val and Ile (+LVI) since hydrophobic residues are generally underrepresented in disordered regions. The DBDs of TFs are rich in positively charged residues gearing them to interact with the negative charges on the DNA phosphate backbone, while the activation domains are often negatively charged.34, 35, 36 We therefore included Arg and Lys (+RK) and net charge (+RK−DE), to specifically discriminate these regions. To look for highly disordered regions outside the DBD, we included a feature that located regions enriched in hydrophilic and disorder promoting residues (Pro, Ser, and Thr), and depleted in Arg and Lys (+PST−RK). For example, for the feature “+PST−RK”, residue k counted as +1 if the position was occupied by a Pro, Ser, or Thr, −1 if occupied by Arg or Lys, and 0 if occupied by any other residue. The sliding window profiles based on these features allowed us to visualize segments of the proteins which contained high fractions of residues with certain chemical properties, such as hydrophobicity or charge, or with certain structural properties, such as prolines (Figures 1, 2, 3). Differently biased regions could then be related to each other, and to regions of the proteins known to have distinct structural and/or functional properties.
We applied IDDomainSpotter to a previously structurally characterized TF to test whether it can identify functional domains. Human p53 contains a central DBD, a tetramerization domain (TD), a C‐terminal regulatory domain (RD), and an N‐terminal IDR, in which the TAD is embedded.37, 38 These domains have originally been assigned from structural and functional insight. Applying IDDomainSpotter, we observed that the borders between the original domains were associated with peaks and dips on the sliding window profiles (Figure 1a). Notably, within the N‐terminal IDR, the TAD stands out from the net charge (+RK−DE) and +LVI features. When comparing with the IDDomainSpotter profiles of an alignment of p53 sequences from mammals (without any primate p53 sequences) or an alignment of p53 sequences from chordates (without any mammal p53 sequences), these compositional biases were largely conserved (Figure 1c,e). Thus, IDDomainSpotter can detect previously defined domains, as well as suggest that other, disordered domains can meaningfully be defined within the disordered regions.
2.2. The DBDs are flanked by disordered +PST−RK domains
Notably, relative to the surrounding residues, the p53 DBD was flanked by segments enriched in Pro, Ser, and Thr but depleted in Arg and Lys (high +PST−RK score) (Figure 1a,c,e). This bias was found both N‐ and C‐terminally to the DBD.
To evaluate if the putative +PST−RK domain identified in p53 is a commonly occurring domain in TFs, we applied IDDomainSpotter to other TFs with known IDRs. The human glucocorticoid receptor (GCR) contains a central DBD, a C‐terminal ligand‐binding domain (LBD) and a large N‐terminal IDR.10, 39 Using IDDomainSpotter, we saw that the DBD was similarly surrounded by +PST−RK domains (Figure 1b). Further, when comparing the human GCR IDDomainSpotter profiles with those of an alignment of GCR sequences from mammals (without any primate GCR sequences) or an alignment of GCR sequences from chordates (without any mammal GCR sequences), these +PST−RK domains were highly conserved (Figure 1d,f).
Plants have highly expanded TF families.40 NAC46 and NAC13 from A. thaliana are both members of the NAC TF family. Apart from their DBDs, they contain long, largely disordered transcriptional regulatory domains41, 42, 43 including a region responsible for RCD1 binding (RB).44, 45 Similar to the observations for p53 and GCR, IDDomainSpotter showed that the DBD of NAC46 was bordered C‐terminally by a +PST‐RK domain and contained several notable other putative domains with different compositional biases throughout its C‐terminal (Figure 2a); features conserved among other Brassicales NAC46‐like proteins (Figure 2c). In contrast, the profile of NAC13 was markedly different in the DBD border region, being instead highly enriched in negatively charged residues (Figure 2b). This feature was also observed among other Brassicales NAC13‐like proteins (Figure 2d). Why the DBD is not linked by a +PST‐RK domain, is not clear. NAC13 belongs to a class of NAC TFs containing a transmembrane domain (TMD) tethering it to the membrane until cleaved and released by proteolytic activation.46, 47, 48 The location of this TMD was identified by a switch in the composition from enriched in +RK to being enriched in +LVI (Figure 2b,d).
MYB28 and MYB29 are also TFs from A. thaliana but belong to a different TF family. They both contain an N‐terminal DBD and a predicted disordered C‐terminal region with a MYC interaction motif (MIM).49 Throughout the disordered regions, IDDomainSpotter uncovered several putative domains with different compositional biases (Figure 3a,b). These included domains enriched in +PST‐RK, in +RK, in +FYG, or in +LVI, relative to their surroundings, and domains of net negative charge. A large putative domain (~80 residues for MYB28 and 60 residues for MYB29) immediately in extension of the DBDs was found to be enriched in +PST‐RK, similar to what we observed for several other TFs in our analysis. This +PST‐RK domain was also observed in the IDDomainSpotter profile of an alignment of MYB28‐ and MYB29‐like sequences from Brassicales (Figure 3c), indicating that such a compositional bias bordering the DBD is conserved.
The +PST‐RK domain, bordering the DBD, appeared to be shared by several TFs across different TF families and conserved among their homologs. The specific compositional bias of enrichment in hydrophilic and disorder‐promoting residues and depletion of positive charges, suggested a highly disordered domain with unique structural properties. To address these properties, we proceeded by investigating the +PST‐RK domains identified in MYB28 and MYB29.
2.3. The +PST‐RK domain lacks stable secondary and tertiary structure
To acquire empirical data describing the structural properties of the putative +PST‐RK domains of MYB28 and MYB29, we purified Escherichia coli expressed GST‐fused +PST‐RK domains covering M117‐R197 of MYB28 and G120‐R178 of MYB29. Following removal of the GST‐tag, only a residual Gly‐Ser dipeptide remained at the N‐terminals (Figure 4a). Far‐UV circular dichroism (CD) spectra acquired of MYB28117–197 (Figure 4b) and MYB29120–178 (Figure 4c) showed that the spectrum of both proteins exhibited a pronounced negative band around 200 nm, and hardly any negative ellipticity around 220 nm, characteristic of an unstructured chain without substantially populated secondary structures.50 In concurrence, the 15N‐1H heteronuclear single quantum coherence (HSQC) spectra recorded on 15N‐labeled MYB28117–197 (Figure 4d) and MYB29120–178 (Figure 4e), had a narrow peak distribution in the proton dimension, as seen for disordered proteins that lack tertiary structure.51, 52
Figure 4.
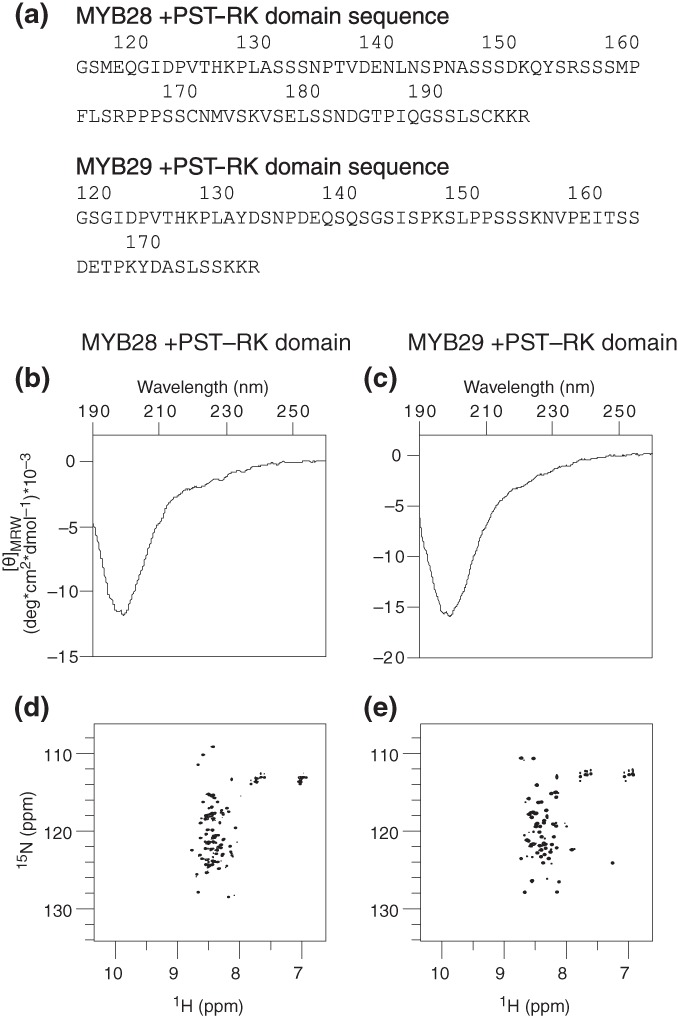
Structure propensities of the MYB28 (M117‐R197) and MYB29 (G120‐R178) putative +PST‐RK domains. (a) Amino acid sequences of MYB28117–197 and MYB29120–178 following removal of the N‐terminal GST‐tag. CD spectra of the MYB28 (b) and MYB29 (c) +PST‐RK domains. 15N‐1H HSQC spectra of the MYB28 (d) and MYB29 (e) +PST‐RK domains. CD, circular dichroism
Secondary chemical shift analysis based on sequence‐corrected random coil values53, 54 revealed that neither MYB28117–197 (Figure 5a) nor MYB29120–178 (Figure 5b) contained stable secondary structures, but both had smaller stretches of lowly populated, transient extended structures and putative turns in the C‐terminal. For comparison, p531–93 (chemical shift values from BMRB Entry 1776055) harbors transient α‐helices within the N‐terminal half of the IDR, corresponding to the TAD (Figure 5c). For p53, the second part of the IDR that corresponds to the +PST‐RK domain bordering the DBD, contained smaller stretches of lowly populated, transient extended structures, similar to MYB28117–197 and MYB29120–178. These results suggest a lack of secondary structure and a highly disordered nature of +PST‐RK domains bordering the DBDs of MYB28, MYB29, and p53.
Figure 5.
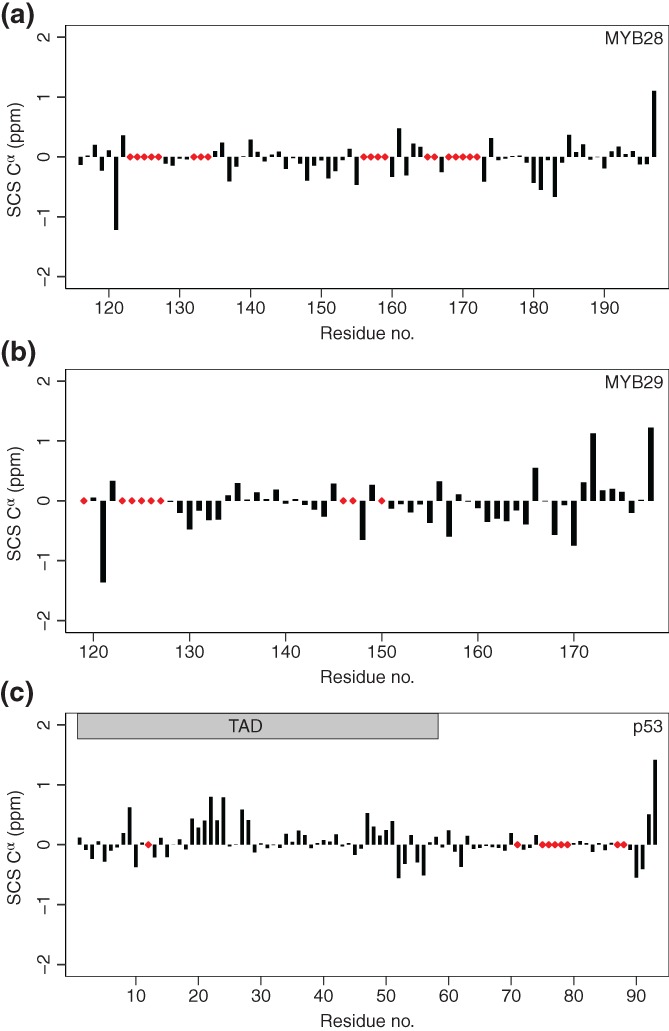
Secondary chemical shift analysis of the MYB28 putative +PST‐RK domain (a), the MYB29 putative +PST‐RK domain (b) and the human p53 N‐terminal IDR (c). The applied sequence‐corrected random coil Cα shifts were calculated with correction for temperature and pH.53, 54 p53 Cα shifts were obtained from BMRB Entry 17760.55 Red diamonds indicate unassigned residues. IDR, intrinsically disordered region
2.4. The MYB28 + PST‐RK domain is dynamic and extended
To further characterize the putative +PST‐RK domain, we quantified the structural dynamics occurring on the ps‐ns timescale. Nuclear magnetic resonance (NMR) relaxation experiments of MYB28117–197 revealed an average R 2/R 1 value of 2.2 ± 0.5 (Figure 6). Overall, heteronuclear nuclear Overhauser effect (NOE) values were also low, with most values ranging between 0.2 and 0.4. Due to repetitive prolines and compositional bias we were unable to assign some smaller segments (123PVTHKP128, 155RSSSMP160, and 165PPPSSCN171). The first of these (123PVTHKP128) is flanked by residues having higher R 2/R 1 values, suggesting a potential motional restriction, whereas this was not the case for the two other regions. R 2 relaxation rates were similar to fitted random coil values using the model of segmental motion56, 57 (line in Figure 6). In summary, the heteronuclear NOE values and relaxation rates are consistent with the MYB28 + PST‐RK domain being highly flexible and dynamic.
Figure 6.
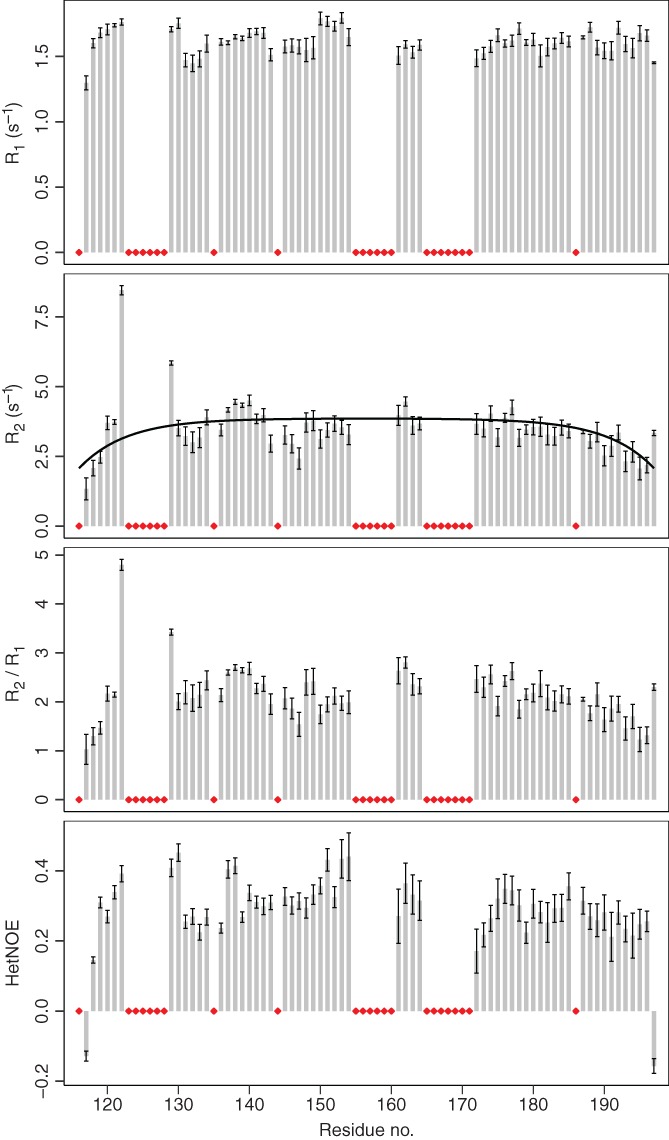
R 1, R 2, R 2/R 1, and heteronuclear NOE values from 15N‐relaxation NMR experiments of the MYB28 putative +PST‐RK domain. Bars represent fitted values of data from consecutive acquisitions ± error of the fits. Black line: R 2 relaxation rates fitted with random coil values.56, 57 Red diamonds indicate unassigned residues or prolines. NMR, nuclear magnetic resonance
Small‐angle X‐ray scattering (SAXS) analysis of MYB28117–197 (Figure 7a) resulted in a continuously rising Kratky plot (Figure 7b) as observed for unfolded chains58 and consistent with the CD‐ and NMR experiments. The R g of MYB28117–197 as estimated from the SAXS data was 23.8 ± 0.5 Å (Figure 7). Globular proteins of this size (51–100 residues) usually have R g values of 12–14 Å, and reach R g values in excess of 20 Å only when containing 250 residues or more.59 Scaling laws60, 61 based on length (83 residues) estimated the R g to ~12 Å for a folded protein or ~27 Å for a chemically unfolded protein (Table 1). This suggested that the MYB28 putative +PST‐RK domain is extended. Measurement of the hydrodynamic radius (R h) of MYB28117–197 using 15N‐filtered DOSY‐NMR resulted in an R h = 22.7 ± 0.02 Å (Table 1). Scaling laws62, 63 based on length estimated the R h to ~17 Å for a folded protein, ~24 Å for an intrinsically disordered protein, and ~27 Å for a chemically unfolded protein (Table 1). This was consistent with a disordered conformation of the MYB28 + PST‐RK domain that was slightly less extended than a chemically denatured protein.
Figure 7.
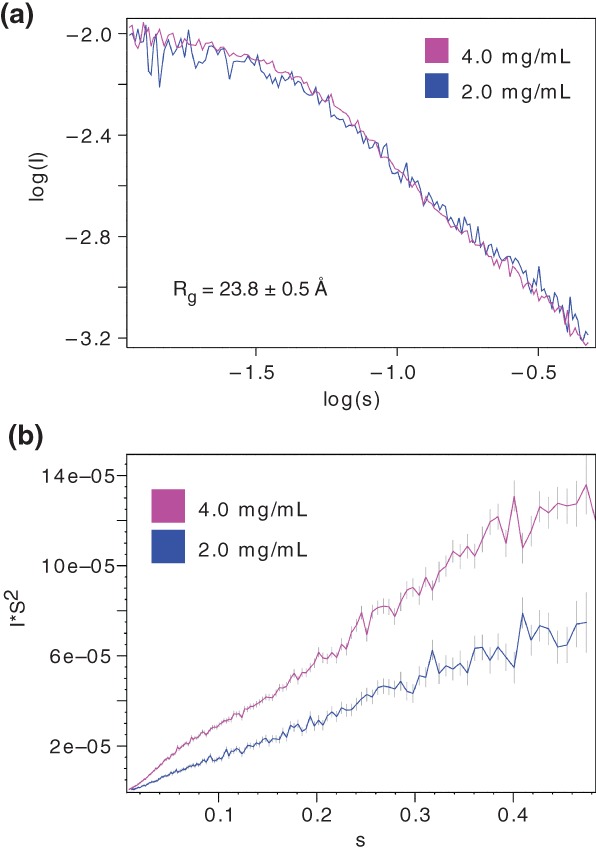
SAXS analysis of MYB28117–197. (a) SAXS scaled log versus log plot at 2.0 and 4.0 mg/ml. The radius of gyration (R g) calculated from the scattering data of the 4.0 mg/ml sample is shown. (b) Kratky plot at 2.0 and 4.0 mg/ml. SAXS, small‐angle X‐ray scattering
Table 1.
MYB28117–197 R g and R h values experimentally determined and calculated from previously determined scaling laws
| R x | Method/State | R x (Å) | References |
|---|---|---|---|
| R g | Experimental (SAXS) | 23.8 ± 0.5 | This study |
| R g | Folded | 11.8 | 60 |
| R g | Chemically unfolded | 27.1 | 61 |
| R h | Experimental (NMR) | 22.7 ± 0.02a | This study |
| R h | Folded | 17.3 | 63 |
| R h | Chemically unfolded | 26.4 | 63 |
| R h | Intrinsically disordered (original method) | 23.6 | 63 |
| R h | Intrinsically disordered (improved method) | 23.8 | 63 |
| R h | Folded | 17.1 | 62 |
| R h | Chemically unfolded | 27.4 | 62 |
Abbreviations: NMR, nuclear magnetic resonance; SAXS, small‐angle X‐ray scattering.
The error of the experimental R h value accounts for the error in the estimated diffusion coefficient but does not account for errors in the estimated viscosity or temperature.
The ratio between R g and R h (R g/R h) of an object yields information about its shape.64 A solid sphere has a R g/R h = √3/5 ≈ 0.77, while objects that are elongated may have R g/R h values in excess of 1, increasing toward ~1.4 for a denatured protein.65 From our experimentally determined R g and R h, we estimated a R g/R h = 1.1 for MYB28117–197. Using a method developed by Nygaard et al.66 to estimate the R g/R h value for unfolded proteins based on length (83 residues) and the experimentally determined R g (23.8 Å), we estimated the R g/R h = 0.96 Å for MYB28117–197. Comparing this to the R g/R h value derived from our experiments, MYB28117–197 was more extended than expected. Overall, the putative +PST‐RK domains lack stable secondary‐ and tertiary structure, and do not display globular, compact conformations, but can be characterized as fully disordered, highly dynamic, and extended.
3. DISCUSSION
3.1. IDRs contain disordered domains with distinct functional and structural properties
IDDomainSpotter revealed several putative domains within large IDRs with distinct compositional biases (Figures 1, 2, 3). These compositional biases were conserved among homologs, suggesting functional importance of the candidate domains. As the differently biased domains are linked to different chemical and structural properties, and therefore likely contribute uniquely to the overall function of the protein, we consider these to be a basis for domain discrimination within IDRs (Figure 8). The IDRs of MYB28 and MYB29 contain candidate domains enriched in +PST‐RK, +RK, +LVI, and +FYG, and negatively charged regions. These likely have different structural properties in terms of transient structure content, compactness, flexibility, and potential for participating in physical interactions (including protein–protein or protein–nucleic acid interactions). In this work, we structurally characterized one of these domains that was found bordering the DBDs of TFs from different families.
Figure 8.
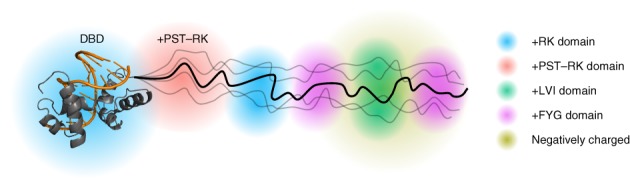
Domain model of MYB TFs. R2R3 MYB DNA‐binding domain in complex with DNA (PDB ID: http://firstglance.jmol.org/fg.htm?mol=1MSE), with the protein colored gray and DNA orange. Domains within the IDR have different sequence properties (i.e., enriched or depleted in side chains increasing hydrophilicity, flexibility, [net] charge, etc.) and thus distinct structural properties of relevance to differentiated molecular functions. The +PST‐RK domain, bordering the DNA‐binding domain is indicated. IDR, intrinsically disordered region
3.2. Decomposition of disordered domains by properties
Relating sequence information to conformational preferences and molecular functions is a central task in biology. Certain sequence attributes may in some cases directly correlate to conformation or function and can thus facilitate protein characterization. Such attributes can be global or local and consider either composition or patterning. CIDER is one approach that considers global sequence composition and global sequence patterning and relates these to conformational preferences.67, 68 Sequence patterning has been shown to be important for predicting the compactness of IDPs rich in positive and negative charges.28 Conversely, sequence patterning has minor influence on the cryoprotective function of intrinsically disordered dehydrin proteins, which is instead governed by size and sequence composition.69, 70 IDDomainSpotter aims at considering local differences in composition not limited to particular features (such as charge or proline content) but with the possibility to explore compositional biases among all amino acid combinations.
Similar to other predictors, IDDomainSpotter does not consider PTMs, although IDRs are often enriched in phosphorylation sites.71 Multisite (or even hyper‐)phosphorylation of IDRs may induce folding or modulate the conformational preferences, for example, by changing the charge patterning and affecting chain expansion.5, 6, 67 Thus, for the +PST‐RK domains discussed here, which we propose to be disordered spacers connecting the DBDs of many TFs to regulatory regions, phosphorylations may directly modulate activity by influencing the overall conformational preferences of the protein.
3.3. Optimal use of IDDomainSpotter
The intent of IDDomainSpotter is to dissect long IDRs into distinct structural and functional units, that is, putative domains. IDDomainSpotter does not predict disorder, and the user is referred to other tools for assessing disorder content, see for instance Reference 72, 73. Further, little information can be gained by applying IDDomainSpotter to short (<40) IDRs, as the objective is to look for differences in compositional biases along the length of an IDR. The user also must carefully consider the contributions of each amino acid when analyzing composite features. As an example, in the case of the +PST‐RK feature, many sequences with different combinations of Pro, Ser, Thr, Arg, and Lys may result in a similar output. Therefore, it is useful to observe the profiles of the individual amino acids when interpreting the composite features (see Figure S1). Finally, special care has to be taken when interpreting IDDomainSpotter profiles from sequence alignments. A genuine conserved signal may be hidden from the user if, for example, several sequences in the alignment contain a gap at this position, diluting the signal. Conversely, a strong signal could be an artifact of a phylogenetically unbalanced alignment. For example, if an alignment contains many primate sequences and few sequences from other vertebrates, the primate sequences dominate and many of the signals observed may not be conserved beyond primates. This is why we excluded primate and mammal sequences when considering conservation of the compositional biases in p53 and GCR among mammals and chordates, respectively.
3.4. An intrinsically disordered +PST‐RK domain in MYB28 and MYB29 links the DBD to the C‐terminal
The non‐MYB regions of MYB TFs are associated with numerous regulatory functions.32 Depending on how these tasks are carried out on a mechanistic level (e.g., through physical interaction), and what specific part of the protein that mediates it (such as a binding site), it can be of critical importance to control the spatial distance between the DBD and that region of the protein. Although MYB28 does not require interactions with other TFs to bind DNA in vitro,74 most TFs function in a combinatorial manner by interacting with other TFs.75 For these types of interactions, the spatial distance between the binding site and the DBD limits how far in space the TF can search for interactors while bound to DNA, and potentially even the distance between cis elements for TFs that act in combination. Characterizing conformations and compactness of the TFs involved and narrowing down the TF interaction sites are necessary before these questions can be explored in more detail.
3.5. Decoupling of intrachain binding avidity by highly disordered spacers?
The putative +PST‐RK domains in MYB28 and MYB29 are ~80 and 60 residues long, respectively, and highly disordered, flexible, and extended. Similar domains bordering the DBDs are conserved in several TFs from other protein families. DBDs are rich in positive charges to interact with the negative charges of the DNA phosphate backbone, while TADs are often enriched in negative charges,34, 35, 36 related to their ability to recruit the mediator complex and activate transcription.76 Thus, when not DNA‐bound, there is a possibility that intramolecular electrostatic attraction may occur between the two domains. When domains participating in intramolecular interactions are connected by a spacer, the length, conformation, and flexibility of the spacer are chief determinants of interaction kinetics.77, 78 Thus, we suggest that the +PST‐RK domain might serve to control inhibitory interactions between the DBD and the TAD. Indeed, further research examining additional functional and mechanistic properties of these +PSR‐RK domains is needed, although the fact that they are conserved in the IDDomainSpotter profiles of alignments of both close and distant homolog sequences (Figures 1, 2, 3), suggests that they confer molecular functions or conformational properties that are under evolutionary constraints.
Within several TFs including MYB28 and MYB29, IDDomainSpotter also identified other putative domains enriched in, for example, different charges or containing higher fractions of aliphatic or aromatic residues. These other domains could exhibit lower structural flexibility and affect overall protein conformation, mediate regulatory interactions through linear motifs, or be involved in the transactivation capability of the TF through mechanisms uncovered recently.35, 36, 76 Especially in long disordered regions, often associated with signaling and DNA homeostasis, integration of a multitude of cues as well as scaffolding of multicomponent complexes, pose the need for segregation of function. One way to help achieve this is via organization in domains of disorder with differentially encoded properties. Such domains are now possible to dissect using IDDomainSpotter.
4. CONCLUSIONS
In this work, we used a sequence‐based approach to explore the presence of distinct domains within long disordered regions in proteins. By applying the approach to proteins with characterized structural domains and functional regions, we demonstrated that compositional sliding window profiles can be used to identify putative domains in IDRs from sequence alone. An online tool for performing this analysis for any protein sequence or sequence alignment is available at http://www.bio.ku.dk/sbinlab/IDDomainSpotter. IDDomainSpotter allows the user to visually inspect compositional biases in long IDRs to predict candidate domains, and thus advances segregation of long, multifunctional IDRs into constituent domains. We identified a uniquely biased disordered putative domain conserved in several TFs from different families, hypothesized to be acting as disordered spacers separating the DBDs from the TADs and other regulatory regions. Through biophysical experiments we showed that this +PST‐RK domain from AtMYB28 and AtMYB29, hypothesized to form a domain separator, is highly extended, dynamic, and disordered in solution, without stable or noteworthily populated tertiary‐ or secondary structures. The presence of these compositionally distinct regions in several unrelated TFs suggests general roles in conformational decoupling of functional domains.
5. MATERIALS AND METHODS
5.1. IDDomainSpotter
IDDomainSpotter is a sliding window procedure which calculates position specific scores for a given specification of amino acid composition within a window over the sequence. The size of the window is 15 residues per default, but can be changed by the user. The composition specification consists of a list of amino acids that are allowed (associated with positive unit score) and a list of amino acids that are disallowed (associated with a negative unit score), with the remaining amino acids considered as neutral. When alignments are provided as input, the script will consider a window over the entire alignment, which corresponds to averaging over the signals in the individual sequences. The score is calculated as an average over the window size. For the special case of the first and last residues in a sequence, where the sliding window lies partially outside the sequence, we calculate the average only over the valid residues, which corresponds to using shorter window sizes in these boundary regions. The script is implemented in Javascript using the d3 library and runs in any modern browser.
5.2. Cloning
The coding sequences covering A. thaliana MYB28117–197 and MYB29120–178 were amplified by PCR from in‐house plasmids to add compatible overhangs for cloning into the pGEX‐4T‐1 plasmid (GE Healthcare) using the BamHI and SalI restriction sites. The constructs were verified by Sanger sequencing (Macrogen Europe).
5.3. Protein expression
E. coli BL21(DE3) were transformed with the resulting plasmid for protein expression. Liquid LB media with ampicillin (100 μg/ml) were inoculated from fresh transformation plates, and the cultures were grown overnight (37°C, 200 rpm). The next day, an OD600 = 0.2 culture in 800 ml LB + ampicillin (100 μg/ml) was started by dilution of the overnight culture, and grown for 3 hr (37°C, 200 rpm). The bacteria were pelleted by centrifugation (4,000g, 20 min), and washed with 400 ml M9 salts (6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NaCl, pH 7.0). After washing, the cells were resuspended in 400 ml M9 media (M9 salts +4 g/L glucose (for double labeling 13C glucose) + 1 g/L NH4Cl [for single or double labeling 15NH4Cl) + 1 mM MgSO4 + M2 trace element solution45 + ampicillin (100 μg/ml)]. The cultures were grown for 1 hr (37°C, 200 rpm), before induction of protein expression by addition of IPTG to a final concentration of 1 mM, and the cultures grew for further 4 hr (37°C, 200 rpm), before the cells were harvested by centrifugation (4,000g, 20 min). If not used immediately, the bacterial pellets were stored at −20°C.
5.4. Protein purification
Cells were lysed by resuspending the bacterial pellet in a total of 2.5 ml BugBuster Master Mix (Merck Millipore) (supplemented with 1x EDTA‐free cOmplete protease inhibitor cocktail [Roche] and 1 mM DTT) per 50 ml M9 media culture. The lysis reaction proceeded by end‐over‐end rotation at room temperature for 30 min, before clearing the lysate by centrifugation (20,000g, 20 min, 4°C). The supernatant was filtered through a 0.2 μm filter (Sarstedt) using a syringe, and added to pre‐equilibrated glutathione sepharose 4B (GE Healthcare) (0.5 ml slurry per 100 ml M9 media culture). The slurry was incubated with the cleared lysate for 60 min at room temperature and with end‐over‐end rotation. The slurry was pelleted by centrifugation (500g, 5 min), and washed six times (each with 5 ml PBS + 1 mM DTT per ml of slurry). PBS containing 1 mM DTT and thrombin (40 units per ml slurry) (GE Healthcare) was added, and cleavage proceeded overnight (>12 hr) at room temperature and with end‐over‐end rotation. The slurry was pelleted and the supernatant was filtered using a 30 kDa cut‐off centrifugal filter (Merck Millipore). The flow‐through was concentrated and the buffer changed to 10 mM sodium phosphate pH 7.0, or 50 mM sodium phosphate pH 7.0, with 100 μM TCEP using 3 kDa cut‐off PES membrane ultrafiltration centrifugal tubes (Thermo Scientific™ Pierce). Purity was confirmed by SDS‐PAGE, and concentration was estimated using the Lambert–Beer law based on absorbance at 280 nm.
5.5. CD spectroscopy
Each protein was diluted to 10 μM with 10 mM sodium phosphate buffer pH 7.0 containing 100 μM TCEP. Far‐UV CD spectra were recorded from 260 to 190 nm on a Jasco 810 spectropolarimeter (Jasco) at room temperature equipped with a Peltier temperature control and with 0.1 nm data pitch, 1 nm bandwidth, 2 s response time, and 50 nm/min scan speed. A total of 50 scans were accumulated for each sample. The spectra of the buffer were recorded immediately following acquisition of protein sample spectra using identical settings and subtracted before analysis.
5.6. NMR spectroscopy
Samples for NMR spectroscopy were prepared with 15N or 13C,15N‐labelled protein, 10% (vol/vol) D2O, 1 mM DSS, 100 μM TCEP, and 0.03% (vol/vol) NaN3 in 50 mM sodium phosphate buffer (pH 7.0), and all spectra were recorded at 5°C. Free induction decays were transformed and visualized in NMRPipe79 or Topspin (Bruker Biospin) and analyzed using CcpNmr Analysis software.80 Proton chemical shifts were referenced internally to DSS at 0.00 ppm, with heteronuclei referenced by relative gyromagnetic ratios.
Assignments of MYB29120–178 were performed manually from analysis of 15N‐1H HSQC‐, HNCA‐, HN(CA)CO‐, HNCB‐, HNCO‐, HN(CO)CA‐, and HN(CO)CACB spectra recorded on a 225 μM sample on a Bruker 800 MHz spectrometer at the Swedish NMR Centre, University of Gothenburg. Assignments of MYB28117–197 (350 μM) were performed manually from analysis of 15N‐1H HSQC, HNCACB, HNCOCACB, HN(CO)CA, HNCO, and HN(CA)NNH spectra acquired with nonuniform sampling81 using standard pulse sequences on a Bruker AVANCE III 750 MHz (1H) spectrometer equipped with a cryogenic probe. The content of transient structure in MYB28, MYB29, and p53 was evaluated from secondary Cα‐chemical shifts using a random coil reference set for intrinsically disordered proteins.53, 54
T1‐ and T2 15N‐relaxation times of MYB28117–197 (200 μM) were determined from two series of 15N‐1H HSQC spectra with varying relaxation delays recorded at 750 MHz (1H), using 10 (10, 20, 60, 100, 200, 400, 600, 800, 900, and 1,200 ms) and 10 (16, 32, 64, 80, 96, 112, 128, 160, 192, and 224 ms) different relaxation delays for T1 and T2, respectively, plus triplicate measurements. The relaxation decays were fitted to single exponentials and relaxation times determined using CcpNmr Analysis software.80 For calculation of the R 2 random coil values of MYB28117–197 the following equation was used56, 57
where R int is the intrinsic relaxation rate (R int = 0.2889 to adjust for the slower tumbling and increased R 2 at 5°C compared to 25°C used in the original Reference 57), N is the number of residues, and λ 0 is the persistence length of the chain (λ 0 = 6.67).
For DOSY‐NMR on MYB28117–197, 15N‐1H HSQC and 15N‐filtered DOSY spectra were recorded on a 150 μM sample (5°C for the HSQC and 25°C for the DOSY) on a Bruker AVANCE III 600 MHz spectrometer equipped with a cryogenic probe. Signal amplitudes were extracted from spectra recorded at varying gradient strengths, and the translational diffusion coefficients obtained by fitting the signal decay to the Stejskal–Tanner equation
where g is the gradient strength, γ is the gyromagnetic ratio, δ is the length of the gradient, Δ is the diffusion time, and D is the translational diffusion coefficients. R h was then calculated from the Stokes–Einstein relation
where k B is the Boltzmann constant, T the temperature, and η the solvent viscosity.
5.7. Small‐angle X‐ray scattering
SAXS data on MYB28117–197 (50 mM sodium phosphate pH 7.0, with 100 μM TCEP, pH 7.0) were collected at the PETRA III, P12 beamline (DESY synchrotron, Hamburg),82 at 25°C and following standard procedures. Two different concentrations of MYB28117–197 were measured, at 2‐ and 4 mg/ml. The two scattering curves, with each curve being an average of 20 frames, were recorded in succession flanked by recordings of the buffer alone. Data were processed and analyzed using the ATSAS program.83 For each data set, the two flanking background scattering curves were averaged and subtracted from the sample scattering curves.
6. DATA DEPOSITION
The chemical shifts of MYB28117–197 and MYB29120–178 have been deposited in the BMRB under the accession numbers 27992 and 27993, respectively.
Supporting information
Figure S1 IDDomainSpotter profiles of human p53. Grey boxes indicate domains, conserved regions and motifs as named above the graph. DBD: DNA‐binding domain, TAD: transactivation domain, TD: tetramer‐ization domain, RD: regulatory domain. Profiles display scores for Pro+Ser + Thr‐Arg‐Lys (+PST‐RK), Phe + Tyr + Gly (+FYG) and individual residues, calculated over 15 residues windows
Figure S2. IDDomainSpotter profiles of human p53. Grey boxes indicate domains, conserved regions and motifs as named above the graph. DBD: DNA‐binding domain, TAD: transactivation domain, TD: tetramerization domain, RD: regulatory domain. Profiles display scores for Pro+Ser + Thr‐Arg‐Lys (+PST‐RK), calculated with various window sizes (WS)
Table S1. Sequences used in Mammalia p53 alignment (Figure 1C)
Table S2. Sequences used in Chordata p53 alignment (Figure 1E)
Table S3. Sequences used in Mammalia GCR alignment (Figure 1D)
Table S4. Sequences used in Chordata GCR alignment (Figure 1F)
Table S5. Sequences used in Brassicales NAC46 alignment (Figure 2C)
Table S6. Sequences used in Brassicales NAC13 alignment (Figure 2D)
Table S7. Sequences used in Brassicales MYB28/MYB29‐like alignment (Figure 3C).
ACKNOWLEDGMENTS
We thank Andreas Prestel for assistance with NMR spectroscopy on the MYB28 + PST‐RK domain and analysis of the DOSY spectra. We thank Søren Roi Midtgaard for assistence with SAXS. We gratefully acknowledge beamtime at the DESY/EMBL P12 beamline at PETRA III, Hamburg. We thank Maxim Mayzel at the Swedish NMR Centre for recording the NMR spectra on the MYB29 + PST‐RK domain. The work was supported by grants from The Danish National Research Foundation, DNRF grant 99 (PSM, MB), Villumfonden, project no. 13169 (PSM, MB), the Department of Plant and Environmental Sciences, University of Copenhagen (PSM), the Danish Research Councils (to BBK: 12‐128803 and to BBK and WB: 4181‐00344), and the Novo Nordisk Foundation Challenge program—REPIN (BBK).
Millard PS, Bugge K, Marabini R, Boomsma W, Burow M, Kragelund BB. IDDomainSpotter: Compositional bias reveals domains in long disordered protein regions—Insights from transcription factors. Protein Science. 2020;29:169–183. 10.1002/pro.3754
Funding information Danmarks Grundforskningsfond, Grant/Award Number: grant 99; Novo Nordisk Fonden, Grant/Award Number: Challenge program ‐ REPIN; Statens Naturvidenskabelige Forskningsrad, Grant/Award Numbers: 12‐128803, 4181‐00344; Villum Fonden, Grant/Award Number: project no. 13169
Contributor Information
Wouter Boomsma, Email: wb@di.ku.dk.
Birthe B. Kragelund, Email: bbk@bio.ku.dk.
REFERENCES
- 1. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borcherds W, Theillet F‐X, Katzer A, et al. Disorder and residual helicity alter p53‐Mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10:1000–1002. [DOI] [PubMed] [Google Scholar]
- 3. Arai M, Sugase K, Dyson HJ, Wright PE. Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc Natl Acad Sci USA. 2015;112:9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clark S, Myers JB, King A, et al. Multivalency regulates activity in an intrinsically disordered transcription factor. Elife. 2018;7:e36258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kulkarni P, Jolly MK, Jia D, et al. Phosphorylation‐induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity. Proc Natl Acad Sci USA. 2017;114:E2644–E2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bah A, Vernon RM, Siddiqui Z, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. [DOI] [PubMed] [Google Scholar]
- 7. Lee CW, Ferreon JC, Ferreon ACM, Arai M, Wright PE. Graded enhancement of p53 binding to CREB‐binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci USA. 2010;107:19290–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dahal L, Shammas SL, Clarke J. Phosphorylation of the IDP KID modulates affinity for KIX by increasing the lifetime of the complex. Biophys J. 2017;113:2706–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J, White JT, Saavedra H, et al. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. Elife. 2017;6:e30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wetlaufer DB. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc Natl Acad Sci USA. 1973;70:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richardson JS. The anatomy and taxonomy of protein structure Advances in protein chemistry. Volume 34 Elsevier, 1981; p. 167–339. 10.1016/S0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 13. Bork P. Shuffled domains in extracellular proteins. FEBS Lett. 1991;286:47–54. [DOI] [PubMed] [Google Scholar]
- 14. Yegambaram K, Bulloch EMM, Kingston RL. Protein domain definition should allow for conditional disorder. Protein Sci. 2013;22:1502–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou J, Oldfield CJ, Yan W, Shen B, Dunker AK. Intrinsically disordered domains: Sequence ➔ disorder ➔ function relationships. Protein Sci. 2019;28:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays. 2009;31:328–335. [DOI] [PubMed] [Google Scholar]
- 17. Brown CJ, Takayama S, Campen AM, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. [DOI] [PubMed] [Google Scholar]
- 18. Theillet F‐X, Kalmar L, Tompa P, et al. The alphabet of intrinsic disorder. Intrinsically Disord Proteins. 2013;1:e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Lee R, Buljan M, Lang B, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daughdrill GW, Narayanaswami P, Gilmore SH, Belczyk A, Brown CJ. Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J Mol Evol. 2007;65:277–288. [DOI] [PubMed] [Google Scholar]
- 21. Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004;18:1169–1175. [DOI] [PubMed] [Google Scholar]
- 22. Wu H. Higher‐order assemblies in a new paradigm of signal transduction. Cell. 2013;153:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bah A, Forman‐Kay JD. Modulation of intrinsically disordered protein function by post‐translational modifications. J Biol Chem. 2016;291:6696–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin EW, Mittag T. Relationship of sequence and phase separation in protein low‐complexity regions. Biochemistry. 2018;57:2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin Y, Currie SL, Rosen MK. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J Biol Chem. 2017;292:19110–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brangwynne C, Tompa P, Pappu R. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11:899–904. [Google Scholar]
- 27. Kato M, Han TW, Xie S, et al. Cell‐free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das RK, Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci USA. 2013;110:13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shammas SL. Mechanistic roles of protein disorder within transcription. Curr Opin Struct Biol. 2017;42:155–161. [DOI] [PubMed] [Google Scholar]
- 31. Staby L, O'Shea C, Willemoës M, Theisen F, Kragelund BB, Skriver K. Eukaryotic transcription factors: Paradigms of protein intrinsic disorder. Biochem J. 2017;474:2509–2532. [DOI] [PubMed] [Google Scholar]
- 32. Millard PS, Kragelund BB, Burow M. R2R3 MYB transcription factors – Functions outside the DNA‐binding domain. Trends Plant Sci. 2019;24:934–946. [DOI] [PubMed] [Google Scholar]
- 33. Davey NE, Shields DC, Edwards RJ. SLiMDisc: Short, linear motif discovery, correcting for common evolutionary descent. Nucleic Acids Res. 2006;34:3546–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sigler PB. Transcriptional activation. Acid blobs and negative noodles. Nature. 1988;333:210–212. [DOI] [PubMed] [Google Scholar]
- 35. Ravarani CN, Erkina TY, De Baets G, Dudman DC, Erkine AM, Babu MM. High‐throughput discovery of functional disordered regions: Investigation of transactivation domains. Mol Syst Biol. 2018;14:e8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erkine AM. ‘Nonlinear’ biochemistry of nucleosome detergents. Trends Biochem Sci. 2018;43:951–959. [DOI] [PubMed] [Google Scholar]
- 37. Wells M, Tidow H, Rutherford TJ, et al. Structure of tumor suppressor p53 and its intrinsically disordered N‐terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Itahana Y, Ke H, Zhang Y. p53 Oligomerization is essential for its C‐terminal lysine acetylation. J Biol Chem. 2009;284:5158–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hilser VJ, Thompson EB. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J Biol Chem. 2011;286:39675–39682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shiu S‐H, Shih M‐C, Li W‐H. Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol. 2005;139:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kjaersgaard T, Jensen MK, Christiansen MW, Gregersen P, Kragelund BB, Skriver K. Senescence‐associated barley NAC (NAM, ATAF1,2, CUC) transcription factor interacts with radical‐induced cell death 1 through a disordered regulatory domain. J Biol Chem. 2011;286:35418–35429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Shea C, Kryger M, Stender EGP, Kragelund BB, Willemoës M, Skriver K. Protein intrinsic disorder in Arabidopsis NAC transcription factors: Transcriptional activation by ANAC013 and ANAC046 and their interactions with RCD1. Biochem J. 2015;465:281–294. [DOI] [PubMed] [Google Scholar]
- 43. Stender EG, O'Shea C, Skriver K. Subgroup‐specific intrinsic disorder profiles of Arabidopsis NAC transcription factors: Identification of functional hotspots. Plant Signal Behav. 2015;10:e1010967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Shea C, Staby L, Bendsen SK, et al. Structures and short linear motif of disordered transcription factor regions provide clues to the interactome of the cellular hub protein radical‐induced cell death. J Biol Chem. 2017;292:512–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bugge K, Staby L, Kemplen KR, et al. Structure of radical‐induced cell death1 hub domain reveals a common αα‐scaffold for disorder in transcriptional networks. Structure. 2018;26:734–746. [DOI] [PubMed] [Google Scholar]
- 46. Kim Y‐S, Kim S‐G, Park J‐E, et al. A membrane‐bound NAC transcription factor regulates cell division in Arabidopsis. Plant Cell. 2006;18:3132–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim S‐Y, Kim S‐G, Kim Y‐S, et al. Exploring membrane‐associated NAC transcription factors in Arabidopsis: Implications for membrane biology in genome regulation. Nucleic Acids Res. 2007;35:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Clercq I, Vermeirssen V, Van Aken O, et al. The membrane‐bound NAC transcription factor ANAC013 functions in mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. Plant Cell. 2013;25:3472–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Millard PS, Weber K, Kragelund BB, Burow M. Specificity of MYB interactions relies on motifs in ordered and disordered contexts. Nucleic Acids Res. 2019;47:9592–9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chemes LB, Alonso LG, Noval MG, de Prat‐Gay G. Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol Biol. 2012;895:387–404. [DOI] [PubMed] [Google Scholar]
- 51. Kosol S, Contreras‐Martos S, Cedeño C, Tompa P. Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules. 2013;18:10802–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yao J, Dyson HJ, Wright PE. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997;419:285–289. [DOI] [PubMed] [Google Scholar]
- 53. Kjaergaard M, Brander S, Poulsen FM. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J Biomol NMR. 2011;49:139–149. [DOI] [PubMed] [Google Scholar]
- 54. Kjaergaard M, Poulsen FM. Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J Biomol NMR. 2011;50:157–165. [DOI] [PubMed] [Google Scholar]
- 55. Wong TS, Rajagopalan S, Freund SM, et al. Biophysical characterizations of human mitochondrial transcription factor a and its binding to tumor suppressor p53. Nucleic Acids Res. 2009;37:6765–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwalbe H, Fiebig KM, Buck M, et al. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry. 1997;36:8977–8991. [DOI] [PubMed] [Google Scholar]
- 57. Wirmer J, Peti W, Schwalbe H. Motional properties of unfolded ubiquitin: A model for a random coil protein. J Biomol NMR. 2006;35:175–186. [DOI] [PubMed] [Google Scholar]
- 58. Kikhney AG, Svergun DI. A practical guide to small angle X‐ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015;589:2570–2577. [DOI] [PubMed] [Google Scholar]
- 59. Lobanov MY, Bogatyreva NS, Galzitskaya OV. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42:623–628. [PubMed] [Google Scholar]
- 60. Skolnick J, Kolinski A, Ortiz AR. MONSSTER: A method for folding globular proteins with a small number of distance restraints. J Mol Biol. 1997;265:217–241. [DOI] [PubMed] [Google Scholar]
- 61. Kohn JE, Millett IS, Jacob J, et al. Random‐coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci USA. 2004;101:12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques? Biochemistry. 1999;38:16424–16431. [DOI] [PubMed] [Google Scholar]
- 63. Marsh JA, Forman‐Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. 2010;98:2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tande BM, Wagner NJ, Mackay ME, Hawker CJ, Jeong M. Viscosimetric, hydrodynamic, and conformational properties of dendrimers and dendrons. Macromolecules. 2001;34:8580–8585. [Google Scholar]
- 65. Receveur‐Brechot V, Durand D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr Protein Pept Sci. 2012;13:55–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nygaard M, Kragelund BB, Papaleo E, Lindorff‐Larsen K. An efficient method for estimating the hydrodynamic radius of disordered protein conformations. Biophys J. 2017;113:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Holehouse AS, Ahad J, Das RK, Pappu RV. CIDER: Classification of intrinsically disordered ensemble regions. Biophys J. 2015;108:228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Das RK, Ruff KM, Pappu RV. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr Opin Struct Biol. 2015;32:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hughes SL, Schart V, Malcolmson J, et al. The importance of size and disorder in the cryoprotective effects of dehydrins. Plant Physiol. 2013;163:1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Palmer SR, De Villa R, Graether SP. Sequence composition versus sequence order in the cryoprotective function of an intrinsically disordered stress‐response protein. Protein Sci. 2019;28:1448–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Iakoucheva LM, Radivojac P, Brown CJ, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Oates ME, Romero P, Ishida T, et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nielsen JT, Mulder FAA. Quality and bias of protein disorder predictors. Sci Rep. 2019;9:5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aarabi F, Kusajima M, Tohge T, et al. Sulfur deficiency‐induced repressor proteins optimize glucosinolate biosynthesis in plants. Sci Adv. 2016;2:e1601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bemer M, van Dijk ADJ, Immink RGH, Angenent GC. Cross‐family transcription factor interactions: An additional layer of gene regulation. Trends Plant Sci. 2017;22:66–80. [DOI] [PubMed] [Google Scholar]
- 76. Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell. 2018;175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sørensen CS, Kjaergaard M. Disordered protein linkers: Predicting effective concentrations using polymer physics. Biophys J. 2018;114:368a–369a.29401434 [Google Scholar]
- 78. Sørensen CS, Kjaergaard M. Effective concentrations enforced by intrinsically disordered linkers are governed by polymer physics. BioRxiv. 2019. 10.1101/577536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 80. Vranken WF, Boucher W, Stevens TJ, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins. 2005;59:687–696. [DOI] [PubMed] [Google Scholar]
- 81. Orekhov VY, Jaravine VA. Analysis of non‐uniformly sampled spectra with multi‐dimensional decomposition. Prog Nucl Magn Reson Spectrosc. 2011;59:271–292. [DOI] [PubMed] [Google Scholar]
- 82. Blanchet CE, Spilotros A, Schwemmer F, et al. Versatile sample environments and automation for biological solution X‐ray scattering experiments at the P12 beamline (PETRA III, DESY). J Appl Cryst. 2015;48:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Petoukhov MV, Franke D, Shkumatov AV, et al. New developments in the ATSAS program package for small‐angle scattering data analysis. J Appl Cryst. 2012;45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 IDDomainSpotter profiles of human p53. Grey boxes indicate domains, conserved regions and motifs as named above the graph. DBD: DNA‐binding domain, TAD: transactivation domain, TD: tetramer‐ization domain, RD: regulatory domain. Profiles display scores for Pro+Ser + Thr‐Arg‐Lys (+PST‐RK), Phe + Tyr + Gly (+FYG) and individual residues, calculated over 15 residues windows
Figure S2. IDDomainSpotter profiles of human p53. Grey boxes indicate domains, conserved regions and motifs as named above the graph. DBD: DNA‐binding domain, TAD: transactivation domain, TD: tetramerization domain, RD: regulatory domain. Profiles display scores for Pro+Ser + Thr‐Arg‐Lys (+PST‐RK), calculated with various window sizes (WS)
Table S1. Sequences used in Mammalia p53 alignment (Figure 1C)
Table S2. Sequences used in Chordata p53 alignment (Figure 1E)
Table S3. Sequences used in Mammalia GCR alignment (Figure 1D)
Table S4. Sequences used in Chordata GCR alignment (Figure 1F)
Table S5. Sequences used in Brassicales NAC46 alignment (Figure 2C)
Table S6. Sequences used in Brassicales NAC13 alignment (Figure 2D)
Table S7. Sequences used in Brassicales MYB28/MYB29‐like alignment (Figure 3C).