

Abstract
N‐acetylglucosamine (NAG) belongs to the eight essential saccharides that are required to maintain the optimal health and precise functioning of systems ranging from bacteria to human. In the present study, we have developed a method, NAGbinder, which predicts the NAG‐interacting residues in a protein from its primary sequence information. We extracted 231 NAG‐interacting nonredundant protein chains from Protein Data Bank, where no two sequences share more than 40% sequence identity. All prediction models were trained, validated, and evaluated on these 231 protein chains. At first, prediction models were developed on balanced data consisting of 1,335 NAG‐interacting and noninteracting residues, using various window size. The model developed by implementing Random Forest using binary profiles as the main principle for identifying NAG‐interacting residue with window size 9, performed best among other models. It achieved highest Matthews Correlation Coefficient (MCC) of 0.31 and 0.25, and Area Under Receiver Operating Curve (AUROC) of 0.73 and 0.70 on training and validation data set, respectively. We also developed prediction models on realistic data set (1,335 NAG‐interacting and 47,198 noninteracting residues) using the same principle, where the model achieved MCC of 0.26 and 0.27, and AUROC of 0.70 and 0.71, on training and validation data set, respectively. The success of our method can be appraised by the fact that, if a sequence of 1,000 amino acids is analyzed with our approach, 10 residues will be predicted as NAG‐interacting, out of which five are correct. Best models were incorporated in the standalone version and in the webserver available at https://webs.iiitd.edu.in/raghava/nagbinder/
Keywords: Binary profile, Machine learning techniques, N‐acetylglucosamine, NAG, PSSM profile
Abbreviations
- Acc
Accuracy
- AUROC
Area Under Receiver Operating Characteristics
- ETree
ExtraTree classifier
- KNN
K‐Nearest Neighbor
- LPC
Ligand Protein Contact
- MCC
Matthews Correlation Coefficient
- MLP
Multilayer Perceptron
- NAG
N‐acetylglucosamine
- PDB
Protein Data Bank
- PSI‐BLAST
Position‐Specific Iterative Basic Local Alignment Search Tool
- PSSM
Position Specific Scoring Matrix
- RFC
Random Forest classifier
- Sen
Sensitivity
- Spc
Specificity
- SVC
Support Vector Classifier
1. INTRODUCTION
One of the primary hurdles in today's world of science is the annotation of a protein at structural as well as functional level. Due to the swift headway in sequencing innovations, the number of protein sequences are increasing at an exponential rate in the respective databases, but they lack annotation, and this gap is increasing every moment.1 Therefore, there is a pressing necessity for the development of computational methods, which can determine the function of the proteins at the residue level. The interaction between the proteins and their ligands is crucial for the well‐being of an organism.2 In the last few decades, substantial efforts have been made toward the identification of the ligand‐binding residues in a protein as shown in the review Sousa et al.3 At first, nonspecific methods were developed to predict the binding sites or pockets in the proteins, irrespective of their ligands,4, 5 but soon it was realized that each ligand possesses different physical and chemical properties. Therefore, new computational methods specific to the ligands came into the picture,6, 7, 8, 9, 10 which performed better as compared to nonspecific methods.11, 12
Broadly, computational methods can be divided into two categories, structure‐based and sequence‐based.13, 14 In the case of structure‐based methods, ligand binding to the protein structure can be studied using docking techniques.15, 16 These methods fail if the structure of a protein is not available. To overcome this limitation, sequence‐based methods have been developed in the past for predicting the protein residues that interact with specific ligands such as ATP,6, 17 GTP,18 NAD,19 and SAM.10 To the best of our knowledge, the method described here is the first for predicting NAG interacting residues in a protein from its amino acid sequence.
The monosaccharide N‐acetylglucosamine (NAG) is ubiquitous in the environment. It is known for playing essential structural roles at the cell surface ranging from bacteria to human.20 It is the principal component of the bacterial cell wall peptidoglycan,21 and of chitin in the fungal cell wall.22 Glycosaminoglycans are also present on the extracellular matrix in animal cells.23, 24 It is also involved in processes such as cell signaling in fungi and bacteria,23 and regulation of gene expressions.22 Plants and animal cells also use NAG for cell signaling and act as the sensors for the status of the nutrition that lead to the modification of the protein by the attachment of O‐GlcNAc.20 In recent years, NAG is suggested as a treatment for autoimmune disorders.25 In the case of human, NAG signaling facilitates the coexistence of the extensive range of bacteria, fungi, and human cells in the gut.26
In this study, we made a methodical attempt to predict the NAG interacting residues in the given protein sequence. We believe this study will be advantageous to the researchers working in the field of drug discovery. In order to facilitate the scientific community, a web server and standalone software has been developed for predicting the NAG interacting residues in a protein.
2. RESULTS
2.1. Compositional analysis
In this study, we analyzed NAG interacting and noninteracting residues to understand NAG interactions in protein. A residue is assigned as NAG interacting if any of its atoms are within the 4Å distance of bound NAG. The contact between residue and NAG was computed using Ligand Protein Contact (LPC) software and this is standard protocol commonly used in most of the previous studies.6, 18, 19 We also analyzed the amino acid composition of NAG interacting and noninteracting residues in the NAG binding proteins. As shown in Figure S1, certain types of residues like N, Q, R, T, W, Y, and H were more abundant among the NAG interacting residues than the non‐NAG interacting ones. It has been shown that residues like N, Y, and W are preferred in the NAG interacting sites,27 which corresponds with our analysis.
2.2. Propensity analysis
The propensities of N and W are higher in the NAG interacting sites in comparison to other amino acids as shown in Figure S2.
2.3. Physiochemical properties analysis
Small, polar, and aromatic amino acids had higher prevalence in the NAG‐interacting sites, whereas NAG noninteracting sites were rich in nonpolar and aliphatic amino acids (Figure S3).
2.4. Two sample logo
As shown in the past, the properties of a given residue can be influenced by its neighbor residues.28 The sample logo (Figure S4) shows that the NAG interacting residues are different than the NAG noninteracting ones.
2.5. Performance of machine learning models on balanced data set using binary profiles
Binary profiles show the composition as well as positional information of the residues present in the sequence.18, 29 We generated binary profiles for window sizes ranging from 5–23 amino acids in length and used to develop various machine learning models based on the balanced data set, where balanced data set consists of equal number of NAG interacting and noninteracting residues, which is 1985 in number. The best results for each window size is shown in Table 1 and AUROC plot was created for the best performing model for training and validation data set as exhibited in Figure 1a and 1b, respectively. On analyzing the performance of each prediction model, we determined that the Random Forest (RF)‐based model performed best among all the other models for window size 9. The model gained the Acc of 65.58%, MCC of 0.31, and AUROC of 0.73 on training data set and Acc of 62.69%, MCC of 0.25, and AUROC of 0.70 on the validation data set. Detailed performance achieved on various machine learning techniques on each window size is provided in Tables S1‐S10.
Table 1.
Performance of the machine learning classifiers using binary profile on balanced data set for various window sizes
| Pattern (classifier) | Training data set | Validation data set | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sen | Spc | Acc | MCC | AUROC | Sen | Spc | Acc | MCC | AUROC | |
| Pat5(SVC) | 67.42 | 60.90 | 64.16 | 0.28 | 0.71 | 67.18 | 55.69 | 61.43 | 0.23 | 0.68 |
| Pat7(SVC) | 66.52 | 64.12 | 65.32 | 0.31 | 0.72 | 66.26 | 60.00 | 63.13 | 0.26 | 0.70 |
| Pat9(RF) | 65.39 | 65.77 | 65.58 | 0.31 | 0.73 | 65.69 | 59.69 | 62.69 | 0.25 | 0.70 |
| Pat11(RF) | 66.07 | 65.17 | 65.62 | 0.31 | 0.72 | 69.08 | 60.62 | 64.85 | 0.30 | 0.71 |
| Pat13(RF) | 65.62 | 65.77 | 65.69 | 0.31 | 0.72 | 69.69 | 62.31 | 66.00 | 0.32 | 0.71 |
| Pat15(RF) | 66.52 | 65.24 | 65.88 | 0.32 | 0.72 | 68.00 | 59.23 | 63.62 | 0.27 | 0.71 |
| Pat17(RF) | 67.64 | 61.12 | 64.38 | 0.29 | 0.71 | 68.15 | 58.92 | 63.54 | 0.27 | 0.69 |
| Pat19(RF) | 66.37 | 62.47 | 64.42 | 0.29 | 0.71 | 67.69 | 59.54 | 63.62 | 0.27 | 0.70 |
| Pat21(RF) | 67.87 | 61.57 | 64.72 | 0.29 | 0.71 | 67.38 | 60.15 | 63.77 | 0.28 | 0.70 |
| Pat23(RF) | 67.57 | 62.02 | 64.79 | 0.30 | 0.71 | 66.00 | 59.85 | 62.92 | 0.26 | 0.69 |
Note: Various classifiers were used for building models and the performance obtained by the best classifier (mentioned in the bracket) for each window size has been reported.
Figure 1.
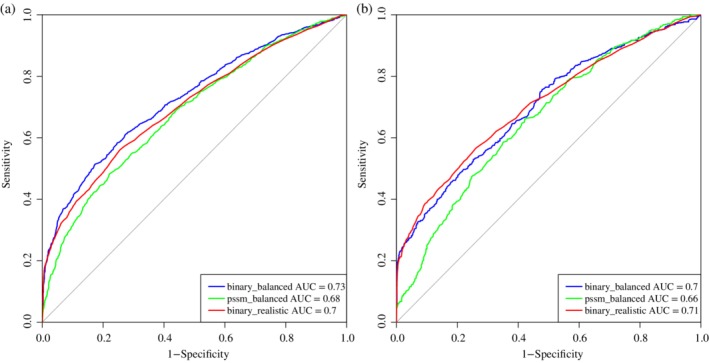
AUROC plots obtained for window length 9 developed using, binary profiles on balanced data set (binary_balanced), PSSM profiles on balanced data set (pssm_balanced), binary profiles on realistic dataset (binary_realistic) for (a) training data set and (b) validation data set
2.6. Performance of machine learning models on balanced data set using PSSM profile
As shown in the literature, evolutionary information provides more information about a protein than a single sequences.30, 31 In this study, evolution information is represented in the form of PSSM profiles. PSSM profiles were generated for each window size, to develop different machine learning models on the balanced data set. The best results for each window size are reported in Table 2, and AUROC plot was created for the best performing model for training and validation data set as exhibited in Figure 1a and 1b, respectively. On evaluating the performance of each model, we determined that the prediction model for RF on window size 9 performed best. This model obtained an Acc of 62.17%, MCC of 0.24, and AUROC of 0.69 on training data set and Acc of 61.15%, MCC of 0.22, and AUROC of 0.66 on the validation data set. In previous studies it has been shown that PSSM profile based model performs better than the binary profile‐based model.19, 32 However, in our case we observed that the binary profile‐based model performed better than the evolutionary profile (or PSSM profile) based model. One possible reason for this could be the nonconservation among residues present in the NAG binding proteins. Therefore, we randomly selected few proteins and analyzed the multiple sequence alignment file of those proteins obtained during PSSM profile generation. We observed that the sequence similarity of these proteins with the other proteins present in the nonredundant database was very low, due to which the PSSM profile generated is not suitable for developing the model to predict the NAG interacting residue in new protein. The multiple sequence alignment file of the selected proteins along with its sequence similarity is provided in Table S11.
Table 2.
The performance of the machine learning classifiers developed using PSSM profile on balanced data set for various window sizes
| Pattern (classifier) | Training data set | Validation data set | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sen | Spc | Acc | MCC | AUROC | Sen | Spc | Acc | MCC | AUROC | |
| Pat5(RF) | 61.27 | 61.57 | 61.42 | 0.23 | 0.67 | 52.00 | 64.46 | 58.23 | 0.17 | 0.64 |
| Pat7(RF) | 61.27 | 61.87 | 61.57 | 0.23 | 0.68 | 56.46 | 66.92 | 61.69 | 0.24 | 0.66 |
| Pat9(RF) | 62.47 | 61.87 | 62.17 | 0.24 | 0.69 | 55.38 | 66.92 | 61.15 | 0.22 | 0.66 |
| Pat11(RF) | 62.92 | 62.55 | 62.73 | 0.25 | 0.68 | 56.15 | 66.31 | 61.23 | 0.23 | 0.66 |
| Pat13(RF) | 64.27 | 62.17 | 63.22 | 0.26 | 0.68 | 56.92 | 66.46 | 61.69 | 0.23 | 0.66 |
| Pat15(RF) | 62.47 | 62.17 | 62.32 | 0.25 | 0.68 | 56.62 | 64.92 | 60.77 | 0.22 | 0.66 |
| Pat17(RF) | 63.67 | 61.8 | 62.73 | 0.25 | 0.68 | 54.15 | 63.23 | 58.69 | 0.17 | 0.65 |
| Pat19(ETree) | 64.04 | 62.77 | 63.41 | 0.27 | 0.68 | 53.85 | 65.38 | 59.62 | 0.19 | 0.65 |
| Pat21(ETree) | 65.02 | 62.25 | 63.63 | 0.27 | 0.69 | 54.31 | 65.69 | 60.00 | 0.20 | 0.66 |
| Pat23(ETree) | 63.45 | 63.00 | 63.22 | 0.26 | 0.68 | 54.62 | 66.77 | 60.69 | 0.22 | 0.65 |
Note: Various classifiers were used for building models and the performance obtained by the best classifier (mentioned in the bracket) for each window size has been reported.
The performance achieved by different classifiers on each window size is provided in the Tables S12‐S21.
2.7. Performance of machine learning models on balanced data set using hybrid feature
The hybrid feature is obtained by the elementwise addition of binary profile matrix and PSSM profile and then used as the feature to develop the prediction model. Table S22 holds the best results for each window size and Figures S5a and S5b exhibit the AUROC plots for training and validation data set respectively. The best results were obtained for Extratree classifier (ET) on window size 13 with an accuracy of 65.51%, MCC of 0.31, and AUROC of 0.73, where in case of the validation data set accuracy of 64.46%, MCC of 0.29, and AUROC of 0.70 was obtained. The performance reached by different classifiers on each window size is provided in the Tables S23‐S32.
2.8. Performance of the machine learning models on the realistic data set
On analyzing the results for various features, we found that the window size 9 is the optimum window size, as models developed using binary profiles and PSSM profiles exhibiting the consent and model developed using binary profiles performed best among them. Hence, we used window size 9 for developing the prediction models for the realistic dataset using binary profiles as the input feature. Maximum MCC 0.26 was achieved on RF classifier for the training data set and 0.27 for validation data set as shown in Table 3. The AUROC obtained corresponding to the training and validation data set are 0.70 and 0.71 respectively, as shown in Figure 1.
Table 3.
The performance of the various machine learning classifiers developed using binary profile on realistic dataset for window size 9
| Classifier | Main data set | Validation data set | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sen | Spc | Acc | MCC | AUROC | Sen | Spc | Acc | MCC | AUROC | |
| SVC | 14.91 | 99.47 | 97.15 | 0.25 | 0.71 | 18.95 | 99.43 | 97.59 | 0.28 | 0.72 |
| RF | 16.70 | 99.41 | 97.14 | 0.26 | 0.70 | 19.69 | 99.35 | 97.53 | 0.27 | 0.71 |
| ETree | 17.30 | 99.22 | 96.97 | 0.25 | 0.70 | 19.69 | 99.26 | 97.44 | 0.26 | 0.70 |
| KNN | 08.61 | 98.88 | 96.40 | 0.11 | 0.61 | 10.92 | 98.99 | 96.97 | 0.13 | 0.63 |
| MLP | 13.78 | 98.94 | 96.60 | 0.18 | 0.71 | 17.85 | 98.78 | 96.92 | 0.20 | 0.72 |
| Ridge | 13.11 | 99.11 | 96.74 | 0.18 | 0.70 | 16.62 | 99.2 | 97.31 | 0.22 | 0.71 |
2.9. Model implementation in the web server
To serve the scientific community, we have developed the web server “NAGbinder”; for predicting the NAG interacting residues, we have implemented our best models in the server. The web server consists of various modules such as “Sequence,” “PSSM Profile,” “Standalone,” and “Download.” The detailed description of each module is as follows.
2.9.1. Sequence
This module allows users to predict the NAG interacting residues in an uncharacterized protein from its sequence. The user can provide the sequence in FASTA format, can select the desired probability threshold and machine learning techniques. The user can provide either single or multiple sequences; on the other hand, the user can upload the sequence file in the FASTA format. In the output page, the NAG interacting residues are highlighted in red color with bigger font size, in the protein sequence. The output is downloadable in different formats such as pdf, txt, and png.
2.9.2. PSSM profile
This module generates the PSSM profile of all the provided sequences in FASTA format and predicts the NAG interacting residues. As the server permits users to select the threshold, we suggest a higher value for high specificity and lower value for higher sensitivity. The output would show NAG interacting residues highlighted in red color with bigger font size, in the protein sequence. The output is downloadable in pdf, txt, and png format.
2.9.3. Standalone
This module allows the user to predict the NAG interacting residues in a protein, even if the Internet is not present. The standalone has the compatibility with macOS, Linux, and Windows. Our best models are implemented in the back‐end, which takes FASTA sequence of proteins as input and provides the results that are comparable to the online server. This software is equipped with all the required files in the zip format and can download from the “Standalone” module of the online server “NAGbinder.” Moreover, this module also guides the user to use the standalone using docker technology.
2.9.4. Download
This module allow the users to download the data sets that we have used in this study. The training and validation data sets are provided separately and each data sets are comprises of NAG‐interacting and noninteracting chains. The data sets are provided in three different types, as first type contains annotated protein chains, type two and three comprises of binary and PSSM profiles for window size 9, respectively. These data sets are freely downloadable from the “Download” module of the online server of NAGbinder.
2.10. Standalone
The NAGbinder standalone is Python‐based and available on GitHub. The user can access it from the URL: https://github.com/raghavagps/nagbinder/. The standalone version of NAGbinder is also executed in the docker technology, its complete usage, and implementation is provided in the manual of “GPSRdocker” that can be downloaded from https://webs.iiitd.edu.in/gpsrdocker/.
3. DISCUSSION
NAG is ubiquitous in the environment, hence showing its importance in the maintenance and coordination in various systems ranging from bacteria to humans.20 For understanding the mechanisms behind the interactions, the determination of the structure is prerequisite, which is a very intricate process. The determination of the protein structure is a highly complex process, and moreover, due to the shortcomings of the present technology, a sequence‐based computational method to predict the NAG interacting residues in a protein, is the need of the current time. We investigated various properties of the NAG interacting protein chains such as compositional analysis, propensity, and physiochemical properties and developed various prediction models using machine learning techniques to predict the NAG interacting residues in the uncharacterized proteins using their sequence information. Initially, the models were developed on balanced data using different window sizes. We found that the prediction model developed on binary profiles using window size 9 performed best among all the models, and the performances of the models were validated on the independent data set. To serve the scientific community, we have provided the standalone version as well as “NAGbinder” web server, in the hope that it will allow the biologists in the identification of NAG binding proteins and their interacting residues for the purpose of annotation and functional analysis. The server is freely available on https://webs.iiitd.edu.in/raghava/nagbinder. The comprehensive workflow of NAGbinder is exhibited in Figure 2.
Figure 2.
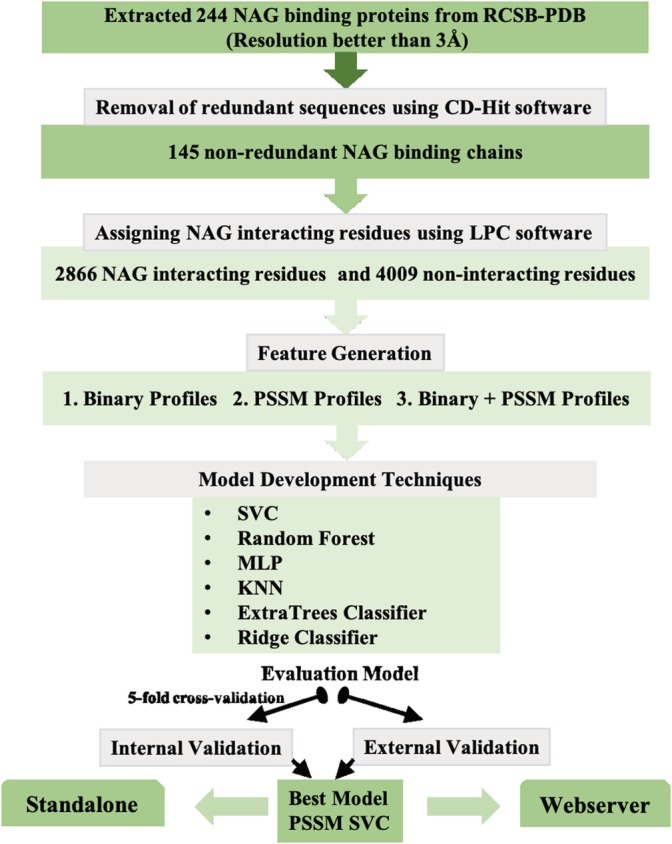
Architecture of NAGbinder
4. MATERIALS AND METHODS
4.1. Data set creation
Initially, we extracted 5,736 NAG binding proteins’ PDB IDs from the PDB April 2019 release, which resulted in 15,349 protein chains. Next, using CD‐HIT software33 further filtration of the sequences was done by applying the criteria of 40% sequence identity and obtained 1,279 protein chains having sequences bearing identity up to 40%. As demonstrated in the past, the quality of the protein structure is one of the principal components for reliable annotation.18 Hence, we have put the threshold of 3Å for resolution; therefore including only those chains having a resolution equal to or less than 3 Å. We were left with a total of 231 protein chains. Finally, we ran the LPC34 software on these chains and extracted the contact information of NAG interacting residues present in the protein chains with threshold cut off of 4 Å that is a standard criteria followed in many previous studies.18, 35 In total we obtained 1985 NAG interacting and 74,931 noninteracting residues from the 231 protein chains and average number of NAG‐interacting residues was found to be 3.75 residues per NAG binding site.
4.2. Internal and external validation
Following the findings of previous studies, the data sets are created at the protein sequence level, rather than pattern or residue level, as previous studies proposed that the data sets generated at the pattern level are biased and leads to higher performance.1 By pattern level we mean, generating patterns of a given window length using all proteins and then creating five folds. The data set (231 protein chains) was divided into two parts (a) training data set, which is comprised of 80% (186 protein chains) and (b) validation data set, which includes the remaining 20% (45 protein chains) of the data set. In total, 1,335 NAG interacting and 47,198 noninteracting residues were present in internal validation data set and 650 NAG interacting and 27,733 noninteracting residues were present in external validation data set.
Two types of data sets were generated for model development and analysis studies that is, (a) realistic data set, which consists of the original NAG interacting and noninteracting residues (i.e., 1985 NAG interacting and 74,931 NAG noninteracting) obtained initially and (b) balanced data set, in which an equal number of NAG interacting and noninteracting residues were present (i.e., 1985 NAG interacting and 1985 NAG noninteracting). As the noninteracting data were several fold higher, an equal number of noninteracting residues was randomly selected in order to avoid bias.
4.3. Five fold cross‐validation
The five fold cross‐validation technique was implemented to evaluate the performance of the various prediction models. In this technique, all the instances were divided into five different sets. Out of these five sets, four sets are kept for training, and last set is used for testing. The process is repeated five times such that each set gets the chance to be used for testing once. The overall performance is the average of performances of all these five sets.36
4.4. Window/pattern size
The overlapping patterns of each sequence were created with varying window size, where the length of the window ranges from five to 23 amino acids. The central residue of the generated pattern is the representative of the sequence segment. If this central residue is NAG interacting then the segment is designated as positive pattern; if not, then negative. For the terminus residues, (k‐1)/2 dummy residues as “X” are added at both termini of the protein chain (where k signifies the length of the pattern).
4.5. Percent amino acid composition
The realistic data set was used to compute percent amino acid composition of the NAG interacting and noninteracting residues using Equation 1.
| (1) |
where C i is the percent composition of residue of type i. A i and N are number of residues of type i, and the total number of residues, respectively.
4.6. Residue propensity
Preference or nonpreference of residue in NAG binding site is very crucial. Therefore, to address this issue, we computed residue propensity using our realistic data set. The propensity for each type of residue is computed using Equation 2 as used in previous studies.37
| (2) |
where P i is the propensity score for residue of type i. A i and N i are number of residues of type i, and total the number of residues (interacting and noninteracting) of type i, respectively.
4.7. Percent composition based on physiochemical property
Each amino acid possesses the unique physiochemical property that exhibits its functionality. We have considered important eight physiochemical properties that is, aromatic, aliphatic, polar, nonpolar, charged, small, acidic, and basic amino acids. We utilize the realistic data set to compute the percent composition of each residues of these eight properties (or composition of physicochemical property) using Equation 3.
| (3) |
where PC i is the percent composition of a physiochemical property type i. A i and N are number of residues possessing physiochemical property of type i, and total the number of residues, respectively.
4.8. Binary profile
The binary profiles are generated for each pattern, by providing the binary values to each amino acid in the fixed length pattern. A vector of size 21 designates each amino acid in the pattern, and hence a vector of size N*21 will be generated for pattern having the length equal to N. For example, Alanine residue was represented by [1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0]; which comprises 20 amino acids and one dummy amino acid “X,” where X was designated as [0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0].29
4.9. Evolutionary profile (PSSM)
The evolutionary profile was used as the second input feature, which is represented by the position‐specific scoring matrix (PSSM) generated for patterns.11 This was consolidated by using the PSSM generated during the PSI‐BLAST38 by searching against the SwissProt database.39 Three iterations were performed with the cutoff for e‐value was 0.001 for each sequence. Then, all the values were normalized between 0 and 1 using Equation 4, following calculation of the position‐specific score. The final matrix has the dimensions of N X 21, where N is the length of the pattern.
| (4) |
where val refers to PSSM score and PSSMnorm is the normalized value.
4.10. Machine learning techniques
We have used python‐based machine learning libraries/modules contained in the package SCIKIT‐learn,40 to develop the prediction models. Before developing the prediction models, the Grid Search module of scikit‐learn was used to optimize the parameters on the internal training data set. We have executed Random Forest classifier (RF), ExtraTree classifier (ET), Support Vector Classifier (SVC), MultiLayer Perceptron (MLP), K‐Nearest Neighbor (KNN), and Ridge classifier to develop the prediction models.
4.11. Performance evaluation
The performance of the prediction models is evaluated on various threshold‐dependent and threshold‐independent parameters. In this study, Sensitivity (Sen), Specificity (Spc), Accuracy (Acc), and Matthews Correlation Coefficient (MCC) are used as the threshold‐dependent parameters. Performance of the model were evaluated in terms of Sensitivity (Sen), which is the percentage of correctly predicted NAG interacting residues; Specificity (Spc), which is the percentage of correctly predicted noninteracting residues; Accuracy (Acc), defined as percentage of correct prediction (NAG interacting and noninteracting residues); Matthews Correlation Coefficient (MCC), which is the correlation between observed and predicted values. Area Under Receiver Operating Characteristics (AUROC) is used as the threshold‐independent parameter, which is the plot between Sen (True Positive Rate) and 1‐Spc (False Positive Rate), where Sen is on y‐axis and 1‐Spc is on x‐axis. It is the measure of separability, it signifies that how well the model is capable of distinguishing between the classes. AUROC was computed using the “pROC” package of R.41 The equations for the parameters are as follows:
| (5) |
| (6) |
| (7) |
| (8) |
TP refers to true positive; FN refers to false negative; TN refers to true negative; FP refers to false positive.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
SP collected, compiled, and curated the data sets. SP, PA, and GM have performed the experiments and formal analysis. SP, VK, RK, AD, and GPSR developed the web interface. SP and PA developed the standalone software. SP, PA, AD, RK, and GPSR wrote the manuscript. GPSR conceived the idea and supervised the project. All authors read and approved the final manuscript.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENT
Authors are thankful to J.C. Bose National Fellowship, Department of Science and Technology (DST), Government of India, and Department of Biotechnology (DBT) and Department of Science and Technology (DST‐INSPIRE) and Council of Scientific and Industrial Research (CSIR), University Grants Commission (UGC), Govt. of India for fellowships and the financial support.
Patiyal S, Agrawal P, Kumar V, et al. NAGbinder: An approach for identifying N‐acetylglucosamine interacting residues of a protein from its primary sequence. Protein Science. 2020;29:201–210. 10.1002/pro.3761
REFERENCES
- 1. Yu D‐J, Hu J, Yan H, Yang X‐B, Yang J‐Y, Shen H‐B. Enhancing protein‐vitamin binding residues prediction by multiple heterogeneous subspace SVMs ensemble. BMC Bioinformatics. 2014;15:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agrawal P, Singh H, Srivastava HK, Singh S, Kishore G, Raghava GPS. Benchmarking of different molecular docking methods for protein‐peptide docking. BMC Bioinformatics. 2019;19:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sousa SF, Fernandes PA, Ramos MJ. Protein‐ligand docking: Current status and future challenges. Proteins. 2006;65:15–26. [DOI] [PubMed] [Google Scholar]
- 4. Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chauhan JS, Mishra NK, Raghava GP. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics. 2009;10:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu D‐J, Hu J, Huang Y, et al. TargetATPsite: A template‐free method for ATP‐binding sites prediction with residue evolution image sparse representation and classifier ensemble. J Comput Chem. 2013;34:974–985. [DOI] [PubMed] [Google Scholar]
- 8. Hu X, Dong Q, Yang J, Zhang Y. Recognizing metal and acid radical ion‐binding sites by integrating ab initio modeling with template‐based transferals. Bioinformatics. 2016;32:3260–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu J, Li Y, Zhang Y, Yu D‐J. ATPbind: Accurate protein‐ATP binding site prediction by combining sequence‐profiling and structure‐based comparisons. J Chem Inf Model. 2018;58:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agrawal P, Mishra G, Raghava GPS. SAMbinder: A web server for predicting SAM binding residues of a protein from its amino acid sequence. bioRxiv. 2019;625806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen K, Mizianty MJ, Kurgan L. Prediction and analysis of nucleotide‐binding residues using sequence and sequence‐derived structural descriptors. Bioinformatics. 2012;28:331–341. [DOI] [PubMed] [Google Scholar]
- 12. Yu D‐J, Hu J, Yang J, Shen H‐B, Tang J, Yang J‐Y. Designing template‐free predictor for targeting protein‐ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Trans Comput Biol Bioinforma. 2013;10:994–1008. [DOI] [PubMed] [Google Scholar]
- 13. Dukka BK. Structure‐based methods for computational protein functional site prediction. Comput Struct Biotechnol J. 2013;8:e201308005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agrawal P, Raghav PK, Bhalla S, Sharma N, Raghava GPS. Overview of free software developed for designing drugs based on protein‐small molecules interaction. Curr Top Med Chem. 2018;18:1146–1167. [DOI] [PubMed] [Google Scholar]
- 15. Fukunishi Y, Nakamura H. Prediction of ligand‐binding sites of proteins by molecular docking calculation for a random ligand library. Protein Sci. 2011;20:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heo L, Shin W‐H, Lee MS, Seok C. GalaxySite: Ligand‐binding‐site prediction by using molecular docking. Nucleic Acids Res. 2014;42:W210–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen K, Mizianty MJ, Kurgan L. ATPsite: Sequence‐based prediction of ATP‐binding residues. Proteome Sci. 2011;9(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chauhan JS, Mishra NK, Raghava GP. Prediction of GTP interacting residues, dipeptides and tripeptides in a protein from its evolutionary information. BMC Bioinformatics. 2010;11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ansari HR, Raghava GP. Identification of NAD interacting residues in proteins. BMC Bioinformatics. 2010;11:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naseem S, Parrino SM, Buenten DM, Konopka JB. Novel roles for GlcNAc in cell signaling. Commun Integr Biol. 2012;5:156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev. 2008;72:211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gunasekera A, Alvarez FJ, Douglas LM, Wang HX, Rosebrock AP, Konopka JB. Identification of GIG1, a GlcNAc‐induced gene in Candida albicans needed for normal sensitivity to the chitin synthase inhibitor nikkomycin Z. Eukaryot Cell. 2010;9:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konopka JB. N‐acetylglucosamine (GlcNAc) functions in cell signaling. Scientifica. 2012;2012:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moussian B. The role of GlcNAc in formation and function of extracellular matrices. Comp Biochem Physiol B Biochem Mol Biol. 2008;149:215–226. [DOI] [PubMed] [Google Scholar]
- 25.Anon Sugar supplement may treat immune disease | New Scientist. Available from: https://www.newscientist.com/article/mg19426074-500-sugar-supplement-may-treat-immune-disease/
- 26. Nicholson JK, Holmes E, Kinross J, et al. Host‐gut microbiota metabolic interactions. Science. 2012;336:1262–1267. [DOI] [PubMed] [Google Scholar]
- 27. Ramakrishnan B, Boeggeman E, Qasba PK. Binding of N‐acetylglucosamine (GlcNAc) β1‐6‐branched oligosaccharide acceptors to β4‐galactosyltransferase I reveals a new ligand binding mode. J Biol Chem. 2012;287:28666–28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schweitzer‐Stenner R, Toal SE. Anticooperative nearest‐neighbor interactions between residues in unfolded peptides and proteins. Biophys J. 2018;114:1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agrawal P, Raghava GPS. Prediction of antimicrobial potential of a chemically modified peptide from its tertiary structure. Front Microbiol. 2018;9:2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuznetsov IB, Gou Z, Li R, Hwang S. Using evolutionary and structural information to predict DNA‐binding sites on DNA‐binding proteins. Proteins Struct Funct Bioinforma. 2006;64:19–27. [DOI] [PubMed] [Google Scholar]
- 31. Kaur H, Raghava GPS. Prediction of beta‐turns in proteins from multiple alignment using neural network. Protein Sci. 2003;12:627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liou Y‐F, Charoenkwan P, Srinivasulu Y, et al. SCMHBP: Prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinformatics. 2014;15(Suppl 16):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Y, Niu B, Gao Y, Fu L, Li W. CD‐HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. [DOI] [PubMed] [Google Scholar]
- 35. Mishra NK, Raghava GPS. Prediction of FAD interacting residues in a protein from its primary sequence using evolutionary information. BMC Bioinformatics. 2010;11(Suppl 1):S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagpal G, Chaudhary K, Agrawal P, Raghava GPS. Computer‐aided prediction of antigen presenting cell modulators for designing peptide‐based vaccine adjuvants. J Transl Med. 2018;16:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh H, Srivastava HK, Raghava GPS. A web server for analysis, comparison and prediction of protein ligand binding sites. Biol Direct. 2016;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bairoch A, Apweiler R. The SWISS‐PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit‐learn: Machine learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 41. Sachs MC. plotROC: A tool for plotting ROC curves. J Stat Softw. 2017;79:1–19. Available from:. http://www.jstatsoft.org/v79/c02/. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information