

Abstract
Aims
Subjects with lipoprotein(a) [Lp(a)] elevation have increased arterial wall inflammation and cardiovascular risk. In patients at increased cardiovascular risk, arterial wall inflammation is reduced following lipid-lowering therapy by statin treatment or lipoprotein apheresis. However, it is unknown whether lipid-lowering treatment in elevated Lp(a) subjects alters arterial wall inflammation. We evaluated whether evolocumab, which lowers both low-density lipoprotein cholesterol (LDL-C) and Lp(a), attenuates arterial wall inflammation in patients with elevated Lp(a).
Methods and results
In this multicentre, randomized, double-blind, placebo-controlled study, 129 patients {median [interquartile range (IQR)]: age 60.0 [54.0–67.0] years, Lp(a) 200.0 [155.5–301.5] nmol/L [80.0 (62.5–121.0) mg/dL]; mean [standard deviation (SD)] LDL-C 3.7 [1.0] mmol/L [144.0 (39.7) mg/dL]; National Cholesterol Education Program high risk, 25.6%} were randomized to monthly subcutaneous evolocumab 420 mg or placebo. Compared with placebo, evolocumab reduced LDL-C by 60.7% [95% confidence interval (CI) 65.8–55.5] and Lp(a) by 13.9% (95% CI 19.3–8.5). Among evolocumab-treated patients, the Week 16 mean (SD) LDL-C level was 1.6 (0.7) mmol/L [60.1 (28.1) mg/dL], and the median (IQR) Lp(a) level was 188.0 (140.0–268.0) nmol/L [75.2 (56.0–107.2) mg/dL]. Arterial wall inflammation [most diseased segment target-to-background ratio (MDS TBR)] in the index vessel (left carotid, right carotid, or thoracic aorta) was assessed by 18F-fluoro-deoxyglucose positron-emission tomography/computed tomography. Week 16 index vessel MDS TBR was not significantly altered with evolocumab (−8.3%) vs. placebo (−5.3%) [treatment difference −3.0% (95% CI −7.4% to 1.4%); P = 0.18].
Conclusion
Evolocumab treatment in patients with median baseline Lp(a) 200.0 nmol/L led to a large reduction in LDL-C and a small reduction in Lp(a), resulting in persistent elevated Lp(a) levels. The latter may have contributed to the unaltered arterial wall inflammation.

Keywords: PCSK9 antibodies, Evolocumab, Lipoprotein(a), Arterial wall inflammation, Atherosclerosis
Introduction
Lipoprotein(a) [Lp(a)] is a potential independent and causal risk factor for cardiovascular disease (CVD).1,2 Lipoprotein(a) consists of an apolipoprotein(a) [apo(a)] tail covalently bound to a low-density lipoprotein cholesterol (LDL-C) core by a disulphide bridge. Lipoprotein(a)-mediated CVD risk is partly driven by pro-inflammatory oxidized phospholipids (OxPL), which are abundant on the apo(a) tail of Lp(a).2 Previously, we reported that Lp(a)-carried OxPL are crucial intermediates in the arterial wall inflammation process among patients with elevated Lp(a).3
Contrary to LDL-C, no large outcome studies on dedicated Lp(a) lowering are available, since potent Lp(a)-lowering therapies remain in development.4 The current European Society of Cardiology/European Atherosclerosis Society guideline therefore proposes altering other modifiable CVD risk factors such as LDL-C to lower CVD risk in patients with elevated Lp(a).5 Treatment with monoclonal antibodies directed against proprotein convertase subtilisin/kexin type 9 (PCSK9) may benefit patients with elevated Lp(a), as these agents produce a strong LDL-C reduction combined with a modest Lp(a) reduction. Evolocumab is an anti-PCSK9 monoclonal antibody that reduces LDL-C by a mean of 60% and Lp(a) by approximately 20–30% in patients without elevated Lp(a) levels.6 In post hoc analysis of the phase III FOURIER study, patients with higher baseline Lp(a) levels had a greater absolute CVD risk reduction after evolocumab treatment.7 It should be noted, however, that PCSK9 inhibition in patients with Lp(a) elevation induces a lesser percent Lp(a) reduction compared with the 20–30% reduction observed in patients with normal Lp(a) levels.8 Hence, PCSK9 inhibition fails to establish low Lp(a) levels in patients with baseline Lp(a) elevation. Moreover, agents offering modest Lp(a) reduction without LDL-C reduction have not been shown to reduce CVD risk.9–11
In ANITSCHKOW, we evaluated whether potent LDL-C lowering, combined with modest Lp(a) lowering with evolocumab, would attenuate arterial inflammation as a surrogate for CVD risk in patients with elevated Lp(a).
Methods
Study design
This study was a phase 3b, multicentre, randomized, double-blind, placebo-controlled trial. Eligible patients were randomized 1:1 to monthly subcutaneous injections of either evolocumab 420 mg or placebo for 16 weeks. Randomization was performed with an interactive voice or web response system.
The study was conducted according to the principles of the Declaration of Helsinki and the study protocol, including amendments, were approved by the ethic committees at all participating sites. All patients provided written informed consent prior to enrolment. Qualified researchers may request data from Amgen clinical studies. Complete details are available at http://www.amgen.com/datasharing. Clinical trial registration information is accessible at https://clinicaltrials.gov/ct2/show/NCT02729025.
Study population
Eligible patients were ≥50 years of age, had a fasting LDL-C of ≥2.6 mmol/L (100 mg/dL), an Lp(a) level of ≥125 nmol/L (50 mg/dL), and arterial wall inflammation as assessed by a most diseased segment target-to-background ratio (MDS TBR) of ≥1.6 in an index vessel measured with 18F-fluoro-deoxyglucose positron-emission tomography/computed tomography (18F-FDG PET/CT). For patients receiving lipid-lowering therapy, the treatment and dosage had to be stable for ≥8 weeks prior to screening. Key exclusion criteria included diabetes mellitus and a cardiovascular event within 3 months before randomization. The complete eligibility criteria are in the Supplementary material online.
Biochemical measurements
Patients fasted for ≥9 h before lipid samples were obtained. Total cholesterol, high-density lipoprotein (HDL)-C, triglycerides, and apolipoprotein B-100 (ApoB-100) were measured by commercially available kits at the Medpace core lab (Medpace Reference Laboratories; Leuven, Belgium). Low-density lipoprotein cholesterol was calculated using the Friedewald formula.12 For calculated LDL-C values <40 mg/dL or triglycerides >400 mg/dL, ultracentrifugation-determined LDL-C was measured and reported. Lipoprotein(a) levels were measured at baseline, Week 8, and Week 16 using an isoform-independent immunoturbidometric assay (Polymedco, Cortlandt Manor, NY, USA) and reported in nmol/L. A conversion factor of 2.5 was used to provide approximate values in mg/dL.
Positron-emission tomography/computed tomography imaging
Arterial inflammation was assessed using 18F-FDG PET/CT. Arterial 18F-FDG uptake is correlated with arterial macrophage content,13 and predicts cardiovascular events.14
18F-FDG PET/CT scans were performed on dedicated PET/CT scanners. Patients fasted for ≥6 h prior to infusion of 240 MBq of 18F-FDG; 90 min later, an ultra-low dose (20 mAs), non-contrast enhanced computed tomography of the carotid arteries and ascending thoracic aorta for attenuation correction and anatomic co-registration was performed, followed by PET. Images were analysed using a dedicated US Food and Drug Administration-approved analysis software package (OsiriX MD, Pixmeo SARL, Switzerland). An experienced radiologist blinded to all patient characteristics analysed the PET/CT images; 10% of all datasets were reanalysed by a separate analyst, and for a second time by the primary analyst, to assess inter- and intra-observer reproducibility.
Target-to-background ratio was calculated from the ratio of the standardized uptake value of the artery (left carotid, right carotid, or thoracic aorta) and the background venous activity according to previously reported methods.15
Endpoints
The primary endpoint was percentage change from baseline in MDS TBR of the index vessel (left carotid, right carotid, or thoracic aorta) measured by 18F-FDG PET/CT after 16 weeks of study drug treatment. Secondary endpoints were percentage change in Lp(a), LDL-C, and ApoB from baseline at Week 16. Adverse events were assessed during the study.
Statistical analysis
The planned sample size was 120 patients. This sample size, accounting for a 25% drop-out rate, provides >90% power for testing superiority of evolocumab over placebo in the percentage change in MDS TBR at Week 16, assuming an effect size of 14% reduction in the evolocumab arm compared with the placebo arm,13 and a common standard deviation of 20%.
Randomization was stratified by background statin therapy and by final screening Lp(a) (Supplementary material online). To estimate the treatment difference in the primary endpoint, a multivariate regression was used and modelled on the primary endpoint as well as three other response variables (percent change in Lp[a] at Weeks 8 and 16, baseline MDS TBR, and baseline Lp[a]). The primary endpoint was regressed on the treatment group and statin stratification factor; baseline MDS TBR and Lp(a) were regressed on the statin stratification factor, and percent changes in Lp(a) were regressed on the treatment group, statin stratification factor, visit, and treatment group by visit. Missing data for the primary endpoint were handled using the correlations of the error terms from the response variables in the model. Lipoprotein(a) in nmol/L is used for all statistical analyses.
Analysis of the secondary endpoints Lp(a), ApoB, and LDL-C was performed with a repeated measures linear mixed effects model including terms for treatment group, statin stratification, scheduled visit, and the interaction of treatment with scheduled visit. Low-density lipoprotein cholesterol levels corrected for Lp(a)-derived cholesterol were calculated using the Dahlen formula.16
Summary statistics for continuous variables were reported. For categorical variables, the frequency and percentage were reported. No adjustments for multiplicity were applied.
Results
A total of 240 patients were screened and 129 patients (evolocumab, n = 65; placebo, n = 64) were enrolled at 14 sites in the Netherlands, Canada, and United States between April 2016 and April 2018 (Supplementary material online, Figure). Baseline characteristics were generally comparable between groups (Table 1). Mean [standard deviation (SD)] LDL-C was 3.7 (1.0) mmol/L [144.0 (39.7) mg/dL] and median [interquartile range (IQR)] Lp(a) was 200.0 (155.5, 301.5) nmol/L [80.0 (62.5–121.0) mg/dL] in the total population. Mean (SD) LDL-C corrected for Lp(a)-derived cholesterol was 3.0 (1.1) mmol/L. Baseline statin use was present in 54.3% of patients. Baseline MDS TBR of the index vessel was comparable between groups [median (IQR) 2.2 (2.0–2.5), evolocumab vs. 2.2 (1.9–2.6), placebo].
Table 1.
Baseline characteristicsa
| Evolocumab (n = 65) | Placebo (n = 64) | |
|---|---|---|
| Age (years), median (IQR) | 59.0 (55.0–65.0) | 60.5 (54.0–68.0) |
| Male, n (%) | 26 (40.0) | 34 (53.1) |
| Caucasian, n (%) | 58 (89.2) | 58 (90.6) |
| BMI (kg/m2), median (IQR) | 26.7 (24.2–29.0) | 26.6 (24.0–28.9) |
| Cardiovascular disease, n (%) | ||
| Coronary artery disease | 9 (13.8) | 11 (17.2) |
| Peripheral artery disease | 2 (3.1) | 1 (1.6) |
| Cerebrovascular disease | 5 (7.7) | 0 |
| Hypertension | 26 (40.0) | 19 (29.7) |
| Current smoker, n (%) | 9 (13.8) | 5 (7.8) |
| NCEP high risk, n (%) | 18 (27.7) | 15 (23.4) |
| Statin use, n (%) | 36 (55.4) | 34 (53.1) |
| High-intensityb | 15 (23.1) | 14 (21.9) |
| Moderate-intensityb | 19 (29.2) | 18 (28.1) |
| Low-intensityb | 2 (3.1) | 2 (3.1) |
| Ezetimibe, n (%) | 10 (15.4) | 17 (26.6) |
| Lipidsc | ||
| Total cholesterol (mmol/L)d | 5.9 (1.3) | 5.8 (1.1) |
| HDL-cholesterol (mmol/L)d | 1.4 (0.4) | 1.5 (0.4) |
| LDL-cholesterol (mmol/L)d | 3.8 (1.1) | 3.7 (0.9) |
| Triglycerides (mmol/L)e | 1.4 (0.5) | 1.5 (1.0) |
| Lp(a) (nmol/L)f | 203.0 (162.5–301.5) | 198.0 (151.3–300.0) |
| ApoB (g/L) | 1.1 (0.2) | 1.1 (0.2) |
| hs-CRP (mg/L), median (IQR) | 1.1 (0.6–2.2) | 1.0 (0.6–1.6) |
| Glucose (mmol/L), median (IQR) | 5.1 (4.8–5.6) | 5.3 (5.2–5.6) |
| MDS TBR of index vessel,g median (IQR) | 2.2 (2.0–2.5) | 2.2 (1.9–2.6) |
ApoB, apolipoprotein B; BMI, body mass index; HDL, high-density lipoprotein; hs-CRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); MDS TBR, most diseased segment target-to-background ratio.
Baseline characteristics were generally comparable between groups. Numerical imbalances between groups are likely due to chance after randomization given the small sample size of each treatment group.
Intensity per American College of Cardiology/American Heart Association definition.39
Values are mean (SD) with the exception of Lp(a), which is median (IQR).
To convert to mg/dL, multiply by 38.7.
To convert to mg/dL, multiply by 88.6.
To convert to mg/dL, divide by 2.5.40
Mean of the maximum TBR in the MDS of the index vessel.
Lipid profile
Evolocumab significantly reduced LDL-C at Week 16 {mean [95% confidence interval (CI)] percent change treatment difference vs. placebo: −60.7% [−65.8 to −55.5]; P < 0.0001} (Table 2; Figure 1); total cholesterol and triglycerides were also reduced (Table 2). Mean (SD) LDL-C was 1.6 (0.7) mmol/L [60.1 (28.1) mg/dL] at Week 16 in the evolocumab group. The evolocumab-induced mean (SD) percent reduction in LDL-C corrected for Lp(a)-derived cholesterol was greater than the reduction in LDL-C [−74.9% (23.9%) vs. −59.0% (14.8%), respectively; Table 2]. Evolocumab resulted in a mean (95% CI) percent change treatment difference in Lp(a) vs. placebo of −13.9% (−19.3% to −8.5%; P < 0.0001) (Table 2; Figure 1). Median (IQR) absolute changes in Lp(a) were −28.0 (−56.5 to 9.0) nmol/L [−11.2 (−22.6 to 3.6) mg/dL] for evolocumab vs. 1.5 (−19.0 to 18.0) nmol/L [0.6 (−7.6 to 7.2) mg/dL] for placebo (Table 2). The median (IQR) Lp(a) was 188.0 (140.0–268.0) nmol/L [75.2 (56.0–107.2) mg/dL] in the evolocumab group at Week 16.
Table 2.
Absolute and percent change in plasma lipid levels from baseline at week 16
| Evolocumab (n = 65) | Placebo (n = 64) | |
|---|---|---|
| Lipid levels—absolute changea | ||
| Total cholesterol (mmol/L)b | −2.2 (0.8) | 0.0 (0.6) |
| HDL-cholesterol (mmol/L)b | 0.1 (0.2) | 0.0 (0.2) |
| LDL-cholesterol (mmol/L)b | −2.2 (0.8) | 0.0 (0.6) |
| LDL-cholesterol corrected for Lp(a) (mmol/L) | −2.1 (0.8) | 0.0 (0.5) |
| Triglycerides (mmol/L)c | −0.3 (0.4) | −0.0 (0.5) |
| Lp(a) (nmol/L)d | −28.0 (−56.5 to 9.0) | 1.5 (−19.0 to 18.0) |
| ApoB (g/L) | −0.5 (0.2) | 0.0 (0.1) |
| Lipid levels—LS mean (95% CI) percent change (%) | ||
| LDL-cholesterol | −59.0 (−62.6 to −55.4) | 1.6 (−2.0 to 5.3) |
| Treatment differencee | −60.7 (−65.8 to −55.5) | |
| LDL-cholesterol corrected for Lp(a) | −74.53 (−79.69 to −69.36) | 1.23 (−4.03 to 6.50) |
| Treatment differencee | −75.76 (−83.13 to −68.39) | |
| Lp(a) | −12.8 (−16.6 to −9.0) | 1.1 (−2.8 to 4.9) |
| Treatment differencee | −13.9 (−19.3 to −8.5) | |
| ApoB | −48.3 (−51.3 to −45.3) | 3.3 (0.3–6.3) |
| Treatment differencee | −51.6 (−55.9 to −47.3) | |
| Total cholesterol | −37.99 (−40.59 to −35.38) | 0.83 (−1.82 to 3.48) |
| Treatment differencee | −38.82 (−42.53 to −35.10) | |
| HDL-cholesterol | 9.31 (5.66–12.95) | 0.00 (−3.72 to 3.73) |
| Treatment differencee | 9.30 (4.09–14.52) | |
| Triglycerides | −16.45 (−22.67 to −10.22) | −0.06 (−6.43 to 6.30) |
| Treatment differencee | −16.38 (−25.29 to −7.48) | |
| hs-CRP (mg/L)— absolute changea | −1.2 (7.9) | −0.4 (4.5) |
ApoB, apolipoprotein B; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); LS, least squares.
Values are mean (SD) with the exception of Lp(a), which is median (IQR).
To convert to mg/dL, multiply by 38.7.
To convert to mg/dL, multiply by 88.6.
To convert to mg/dL, divide by 2.5.
P < 0.001 for evolocumab vs. placebo.
Figure 1.

Mean change from baseline in (A) low-density lipoprotein cholesterol and (B) lipoprotein(a) over time. Vertical bar indicates 95% confidence interval. QM, monthly.
Arterial wall inflammation
Least squares (LS) mean (95% CI) percentage change from baseline in MDS TBR of the index vessel was −8.3% (−11.6% to −5.0%) in the evolocumab group and −5.3% (−8.6% to −2.0%) in the placebo group [treatment difference −3.00 (−7.40 to 1.39) P = 0.18; Figure 2]. Among patients receiving evolocumab, percentage change from baseline in MDS TBR of the index vessel was similar regardless of baseline statin use [LS mean (95% CI) percent change −8.7% (−12.7% to −4.6%), statin; −7.7% (−12.9% to −2.5%), no statin]. The treatment difference in MDS TBR of the index vessel was −5.62 (95% CI −11.10 to −0.14; P = 0.045) in patients who received statins and 0.09 (95% CI −6.90 to 7.08; P = 0.98) in those who did not; the interaction P-value was 0.21. Also, no correlation was found between baseline Lp(a) and baseline MDS TBR [Pearson correlation coefficient (R) = −0.05; P = 0.61] or between baseline LDL-C levels and baseline MDS TBR (R = −0.06; P = 0.48). Absolute change in LDL-C or Lp(a) and change in MDS TBR were not correlated in patients receiving evolocumab (R = 0.01; P = 0.95, LDL-C and R = −0.16; P = 0.21, Lp[a]). Exploratory endpoints of percent change from baseline in the mean of the maximum TBR in the MDS of the whole index vessel, active slices of the index vessel, and the non-index vessel at week 16 are shown in Supplementary material online, Tables S1 and S2.
Figure 2.
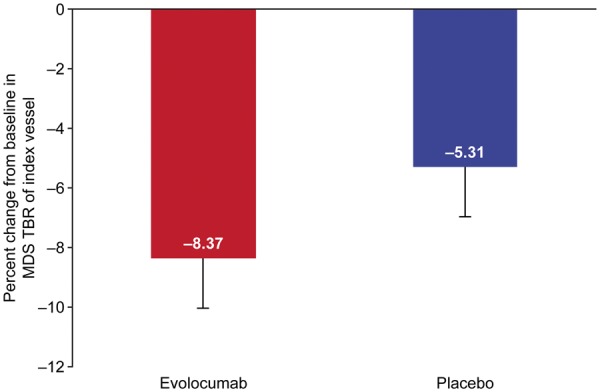
Least squares mean percentage change from baseline in most diseased segment target-to-background ratio of index vessel P = 0.18 for evolocumab vs. placebo). Vertical bar indicates 95% confidence interval.
Safety
Rates of total adverse events, serious adverse events, or adverse events leading to discontinuation of study drug were similar between the placebo and evolocumab groups (Supplementary material online).
Discussion
This placebo-controlled study evaluated the effect of evolocumab on lipids and arterial wall inflammation in patients with elevated Lp(a) [median 200 nmol/L (80 mg/dL)]. Sixteen weeks of evolocumab resulted in a 61% mean reduction in LDL-C and a 14% mean reduction in Lp(a) vs. placebo. However, evolocumab did not significantly alter arterial wall inflammation, assessed as MDS TBR of the index vessel, in patients with elevated Lp(a) (Take home figure). The current data imply that in patients with persistently elevated Lp(a) levels, 16 weeks of potent LDL-C reduction is unable to attenuate the pro-inflammatory state of the arterial wall.
Take home figure.
The combination of lowering low-density lipoprotein cholesterol by 61% compared with placebo to a mean of 1.6 mmol/L (60 mg/dL), and lipoprotein(a) by 14% compared with placebo to a median of 188 nmol/L (75 mg/dL) with PCSK9 inhibition does not reduce inflammatory activity in the arterial wall of patients with lipoprotein(a) elevation, highlighting an unmet need for potent lipoprotein(a)-lowering interventions in order to adequately test the benefit of such an intervention.
Lipoprotein(a) lowering by PCSK9 antibody depends on baseline lipoprotein(a) levels
The 61% LDL-C reduction with evolocumab resulted in a mean post-treatment LDL-C of 1.6 mmol/L (61.9 mg/dL), consistent with results reported in other patient groups.17–20 Notably, this study found only a 14% Lp(a) reduction compared with placebo in patients with elevated Lp(a), instead 20–30% as reported in previous studies in which the baseline Lp(a) values were much lower.6,18–20 The pathways contributing to Lp(a) clearance are incompletely understood and may depend on the concentration of circulating Lp(a) and possibly other lipoproteins.21 In vitro data and meta-analyses of human intervention trials suggest the involvement of the LDL-receptor (LDL-R) in mediating Lp(a) clearance, as Lp(a) was found to bind to the LDL-R and its reduction was closely associated with the degree of LDL-C reduction.6,21,22 In support, fractional catabolic rate of apo(a) is increased during PCSK9 antibody treatment,23 lending further support to a role for LDL-R in Lp(a) reduction. Conversely, PCSK9 antibody treatment also has LDL-R-independent effects, as it can lower Lp(a) particle production by 36%.24 The attenuated Lp(a) reduction in this study may relate to the fact that this is the first study that exclusively included patients with elevated Lp(a), with median levels of 200 nmol/L (80 mg/dL) vs. a median Lp(a) below 40 nmol/L (16 mg/dL) in previous studies.6,17,19,20 Patients with elevated Lp(a) have Lp(a) that is characterized by smaller apo(a) isoforms.25 The attenuated Lp(a) reduction could reflect less efficient clearance of smaller isoforms by the LDL-R. In support, a post hoc analysis of FOURIER also reported a −16% Lp(a) change among patients in the upper baseline Lp(a) quartile [median Lp[a] >165 nmol/L (66 mg/dL)].7 Similarly, LAPLACE demonstrated a −15% Lp(a) change following evolocumab in the highest baseline Lp(a) quartile.8
No impact of evolocumab on arterial wall inflammation in patients with high lipoprotein(a)
Previous studies substantiated that patients with Lp(a) elevation have marked pro-inflammatory changes in the arterial wall assessed using PET/CT,3 and arterial inflammation is a major risk factor for cardiovascular events.14,26 Intervention studies targeting LDL-C have reported a reduction in arterial wall inflammation in patients at increased cardiovascular risk, following either statin therapy or lipoprotein apheresis.13,27–29 In the absence of available Lp(a)-lowering strategies, intensive LDL-C reduction in patients with elevated Lp(a) appeared promising, as previous studies suggested that Lp(a) confers risk predominantly in conjunction with elevated LDL-C levels.30,31 In support, we previously substantiated that the risk associated with Lp(a) was attenuated at LDL-C levels lower than 2.5 mmol/L (96.8 mg/dL) in the primary prevention setting.32 However, despite robust LDL-C reduction combined with a 14% Lp(a) reduction with evolocumab compared with placebo, MDS TBR of the index vessel did not change significantly compared with placebo in this study. In comparison, previous LDL-C-lowering strategies reported a 2.1–3.2% reduction in MDS TBR of the arterial wall for every 10% reduction in LDL-C in patients with CVD.13,29 Several factors may have contributed to this discrepancy. First, we included patients with Lp(a) elevation.33 A direct consequence is that a small Lp(a) reduction following evolocumab still leads to elevated post-treatment levels [median 188.0 nmol/L (75.2 mg/dL)].5,33 In a prior study, Lp(a) levels in this range were associated with a pro-inflammatory effect on the arterial wall in untreated patients.3 Second, the absence of an effect of modest Lp(a) reduction on MDS TBR fits with recent data obtained from Mendelian randomization studies,34 estimating that an absolute Lp(a) reduction of 100 mg/dL (250 nmol/L) may be required to achieve a meaningful cardiovascular risk reduction. The need for substantial Lp(a) changes is further supported by the absence of clinical benefit by other compounds offering only modest Lp(a)-lowering potential.10,11 Third, our findings could relate to the absence of an anti-inflammatory effect specifically when targeting the PCSK9 pathway.35 Since no prior study has addressed arterial wall inflammation assessed with PET/CT after PCSK9-antibody-induced LDL-C lowering, this is currently being investigated in a separate study evaluating the impact of PCSK9 inhibition on arterial wall inflammation in participants at increased CV-risk but normal Lp(a) levels (EU Clinical Trials register 2016-004794-41). However, since multiple other modes of LDL-C lowering associate with MDS TBR lowering13,28,29 and PCSK9 inhibition was also found to attenuate cellular inflammation,36 it is less likely that the mode of LDL-C lowering is responsible. Prior studies as well as ours show that PCSK9 inhibitors do not reduce the inflammatory marker C-reactive protein (CRP). However, CRP is not a good biomarker for arterial wall inflammation, since CRP level is not correlated with MDS-TBR in patients with cardiovascular risk factors.37 Finally, a causal role of other inflammatory pathways, unresponsive to LDL-C lowering, may also contribute.
Clinical implications
In contrast to the anti-inflammatory effect of LDL-C lowering in previous studies, PCSK9 antibody treatment does not reduce arterial wall inflammation in patients with persistent Lp(a) elevation. In this environment, lowering other modifiable risk factors in patients with elevated Lp(a)5 may be less likely to fully mitigate the increased cardiovascular risk. Highly potent Lp(a)-lowering antisense can be expected to reduce pro-inflammatory changes, as 80% Lp(a) reduction was found to reduce cellular inflammatory responses.4
A strength of our trial is that it is a randomized, placebo-controlled study, and the largest lipid lowering-PET/CT trial focusing on arterial wall inflammation to date. A potential limitation of our study is that the 16-week timeframe may not be sufficient to observe a change in arterial wall inflammation. However, arterial wall inflammation measured by PET/CT is a dynamic functional parameter and 12 weeks of statin therapy or a single apheresis episode have been associated with significant reductions in arterial wall inflammation.13,29 A second limitation is the absence of a correlation between baseline Lp(a) and MDS TBR. However, in this study we deliberately excluded all patients with normal Lp(a) levels. This exclusion minimizes the chance of demonstrating a significant relation between (elevated) Lp(a) and MDS TBR. Finally, an additional limitation is that this study evaluated just one potential mechanism of the effect of Lp(a) on cardiovascular risk. The mechanism by which Lp(a) may mediate CVD risk is multifactorial, comprising arterial wall inflammation,3 pro-thrombogenic effects,38 and other pro-atherogenic effects.2 Hence, absence of a significant anti-inflammatory effect does not indicate absence of a potential plaque stabilizing effect of the lipid reduction observed in this study.
Conclusion
Sixteen weeks of PCSK9 inhibition with evolocumab 420 mg led to large percent reductions in LDL-C and modest percent reductions in Lp(a) plasma levels in patients with median baseline Lp(a) of 200 nmol/L (80 mg/dL), resulting in persistent Lp(a) elevation during evolocumab therapy. This persistent elevation may have contributed to the observation that lipid lowering by evolocumab did not lead to a reduction in arterial wall inflammation. These data support further evaluation of novel therapies with a potent Lp(a) lowering effect on both arterial wall inflammation and, eventually, cardiovascular outcome.
Supplementary Material
Acknowledgements
The authors thank Mahta Nili, PhD formerly of Amgen Inc., Tim Peoples of Amgen Inc., and Laura Evans, PharmD on behalf of Amgen Inc. for their editorial assistance.
Funding
This work was funded by Amgen Inc.
Conflict of interest: E.S.G.S. reports that his institution has received lecturing fees and advisory board fees from Amgen Inc., Regeneron, Sanofi, Akcea, Novartis and Athera. H.K., L.C., and S.M.W. are employees and stockholders of Amgen Inc. M.S.S. reports that his institution has received research grant support from Amgen Inc.; AstraZeneca; Daiichi-Sankyo; Eisai; GlaxoSmithKline; Intarcia; Janssen Research and Development; Medicines Company; MedImmune; Merck; Novartis; Pfizer; Poxel; Takeda (All >$10 000 per year); and he has consulted for Amgen; AstraZeneca, Bristol-Myers Squibb; CVS Caremark; Dyrnamix; Esperion; Intarcia; Janssen Research and Development; Medicines Company; MedImmune; Merck; Novartis (all ≤$10 000 per year except Amgen and Esperion). V.M. reports that his institution has received funding from Amgen Inc., Daiichi Sankyo, and Medlion Inc. Z.A.F. reports research funding from Amgen Inc. and Daiichi Sankyo. All other authors declared no conflict of interest.
See page 2782 for the editorial comment on this article (doi: 10.1093/eurheartj/ehz087)
References
- 1. Gencer B, Kronenberg F, Stroes ES, Mach F.. Lipoprotein(a): the revenant. Eur Heart J 2017;38:1553–1560. [DOI] [PubMed] [Google Scholar]
- 2. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol 2017;69:692–711. [DOI] [PubMed] [Google Scholar]
- 3. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ES.. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016;134:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S.. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 5.The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology and European Atherosclerosis Society. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016;37:2999–3058. [DOI] [PubMed] [Google Scholar]
- 6. Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Langslet G, Bays H, Blom D, Eriksson M, Dent R, Wasserman SM, Huang F, Xue A, Albizem M, Scott R, Stein EA.. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol 2014;63:1278–1288. [DOI] [PubMed] [Google Scholar]
- 7. O'Donoghue M, Giugliano R, Keech A, Kanevsky E, Im K, Lira Pineda A, Somaratne R, Sever P, Pederson T, Sabatine M.. Lipoprotein(a), PCSK9 inhibition and cardiovascular risk: insights from the FOURIER trial Presented at the European Atherosclerosis Society Congress, May 7, Lisbon, Portugal: 2018. https://services.aimgroup.eu/ASPClient/prgsci/search.asp (22 June 2018). [Google Scholar]
- 8. Desai NR, Kohli P, Giugliano RP, O'Donoghue ML, Somaratne R, Zhou J, Hoffman EB, Huang F, Rogers WJ, Wasserman SM, Scott R, Sabatine MS.. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation 2013;128:962–969. [DOI] [PubMed] [Google Scholar]
- 9. Albers JJ, Slee A, O'Brien KD, Robinson JG, Kashyap ML, Kwiterovich PO Jr, Xu P, Marcovina SM.. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol 2013;62:1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hps2 Thrive Collaborative G. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 2014;371:203–212. [DOI] [PubMed] [Google Scholar]
- 11. Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, Gibson CM, Granger C, Menon V, Montalescot G, Rader D, Tall AR, McErlean E, Wolski K, Ruotolo G, Vangerow B, Weerakkody G, Goodman SG, Conde D, McGuire DK, Nicolau JC, Leiva-Pons JL, Pesant Y, Li W, Kandath D, Kouz S, Tahirkheli N, Mason D, Nissen SE; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med 2017;376:1933–1942. [DOI] [PubMed] [Google Scholar]
- 12. Friedewald WT, Levy RI, Fredrickson DS.. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499–502. [PubMed] [Google Scholar]
- 13. Tawakol A, Fayad ZA, Mogg R, Alon A, Klimas MT, Dansky H, Subramanian SS, Abdelbaky A, Rudd JH, Farkouh ME, Nunes IO, Beals CR, Shankar SS.. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multicenter fluorodeoxyglucose-positron emission tomography/computed tomography feasibility study. J Am Coll Cardiol 2013;62:909–917. [DOI] [PubMed] [Google Scholar]
- 14. Figueroa AL, Abdelbaky A, Truong QA, Corsini E, MacNabb MH, Lavender ZR, Lawler MA, Grinspoon SK, Brady TJ, Nasir K, Hoffmann U, Tawakol A.. Measurement of arterial activity on routine FDG PET/CT images improves prediction of risk of future CV events. JACC Cardiovasc Imaging 2013;6:1250–1259. [DOI] [PubMed] [Google Scholar]
- 15. Gaztanaga J, Farkouh M, Rudd JH, Brotz TM, Rosenbaum D, Mani V, Kerwin TC, Taub R, Tardif JC, Tawakol A, Fayad ZA.. A phase 2 randomized, double-blind, placebo-controlled study of the effect of VIA-2291, a 5-lipoxygenase inhibitor, on vascular inflammation in patients after an acute coronary syndrome. Atherosclerosis 2015;240:53–60. [DOI] [PubMed] [Google Scholar]
- 16. Dahlen GH, Incidence of Lp(a) among populations In: Scanuu AM, ed. Lipoprotein(a). New York, NY: Academic Press; 1990, pp. 151–173. [Google Scholar]
- 17. Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H; MENDEL-2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab . J Am Coll Cardiol 2014;63:2531–2540. [DOI] [PubMed] [Google Scholar]
- 18. Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, Hovingh GK, Cariou B, Gouni-Berthold I, Somaratne R, Bridges I, Scott R, Wasserman SM, Gaudet D; RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:331–340. [DOI] [PubMed] [Google Scholar]
- 19. Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, Bruckert E, Cho L, Dent R, Knusel B, Xue A, Scott R, Wasserman SM, Rocco M; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014;63:2541–2548. [DOI] [PubMed] [Google Scholar]
- 20. Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, Somaratne R, Legg JC, Nelson P, Scott R, Wasserman SM, Weiss R; LAPLACE-2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014;311:1870–1882. [DOI] [PubMed] [Google Scholar]
- 21. Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Seidah NG, Honarpour N, Lira A, Xue A, Chiruvolu P, Jackson S, Di M, Peach M, Somaratne R, Wasserman SM, Scott R, Stein EA.. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor's role. J Lipid Res 2016;57:1086–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Havekes L, Vermeer BJ, Brugman T, Emeis J.. Binding of LP(a) to the low density lipoprotein receptor of human fibroblasts. FEBS Lett 1981;132:169–173. [DOI] [PubMed] [Google Scholar]
- 23. Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike J, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK, Ginsberg HN.. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation 2017;135:352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watts GF, Chan DC, Somaratne R, Wasserman SM, Scott R, Marcovina SM, Barrett PHR.. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur Heart J 2018;39:2577–2585. [DOI] [PubMed] [Google Scholar]
- 25. Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP.. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem 1995;41:246–255. [PubMed] [Google Scholar]
- 26. Rominger A, Saam T, Wolpers S, Cyran CC, Schmidt M, Foerster S, Nikolaou K, Reiser MF, Bartenstein P, Hacker M.. 18F-FDG PET/CT identifies patients at risk for future vascular events in an otherwise asymptomatic cohort with neoplastic disease. J Nucl Med 2009;50:1611–1620. [DOI] [PubMed] [Google Scholar]
- 27. Tahara N, Kai H, Ishibashi M, Nakaura H, Kaida H, Baba K, Hayabuchi N, Imaizumi T.. Simvastatin attenuates plaque inflammation: evaluation by fluorodeoxyglucose positron emission tomography. J Am Coll Cardiol 2006;48:1825–1831. [DOI] [PubMed] [Google Scholar]
- 28. van der Valk FM, Bernelot Moens SJ, Verweij SL, Strang AC, Nederveen AJ, Verberne HJ, Nurmohamed MT, Baeten DL, Stroes ES.. Increased arterial wall inflammation in patients with ankylosing spondylitis is reduced by statin therapy. Ann Rheum Dis 2016;75:1848–1851. [DOI] [PubMed] [Google Scholar]
- 29. van Wijk DF, Sjouke B, Figueroa A, Emami H, van der Valk FM, MacNabb MH, Hemphill LC, Schulte DM, Koopman MG, Lobatto ME, Verberne HJ, Fayad ZA, Kastelein JJ, Mulder WJ, Hovingh GK, Tawakol A, Stroes ES.. Nonpharmacological lipoprotein apheresis reduces arterial inflammation in familial hypercholesterolemia. J Am Coll Cardiol 2014;64:1418–1426. [DOI] [PubMed] [Google Scholar]
- 30. Afshar M, Pilote L, Dufresne L, Engert JC, Thanassoulis G.. Lipoprotein(a) interactions with low-density lipoprotein cholesterol and other cardiovascular risk factors in premature acute coronary syndrome (ACS). J Am Heart Assoc 2016;5e003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suk Danik J, Rifai N, Buring JE, Ridker PM.. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA 2006;296:1363–1370. [DOI] [PubMed] [Google Scholar]
- 32. Verbeek R, Hoogeveen RM, Langsted A, Stiekema LCA, Verweij SL, Hovingh GK, Wareham NJ, Khaw K-T, Boekholdt SM, Nordestgaard BG, Stroes ESG.. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J 2018;39:2589–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J 2010;31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, Bolton TR, Peters JE, Kamstrup PR, Tybjærg-Hansen A, Benn M, Langsted A, Schnohr P, Vedel-Krogh S, Kobylecki CJ, Ford I, Packard C, Trompet S, Jukema JW, Sattar N, Di Angelantonio E, Saleheen D, Howson JMM, Nordestgaard BG, Butterworth AS, Danesh J; European Prospective Investigation Into Cancer and Nutrition-Cardiovascular Disease Consortium. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bohula EA, Giugliano RP, Leiter LA, Verma S, Park JG, Sever PS, Lira Pineda A, Honarpour N, Wang H, Murphy SA, Keech A, Pedersen TR, Sabatine MS.. Inflammatory and cholesterol risk in the FOURIER trial. Circulation 2018;138:131–140. [DOI] [PubMed] [Google Scholar]
- 36. Bernelot Moens SJ, Neele AE, Kroon J, van der Valk FM, Van den Bossche J, Hoeksema MA, Hoogeveen RM, Schnitzler JG, Baccara-Dinet MT, Manvelian G, de Winther MPJ, Stroes ESG.. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J 2017;38:1584–1593. [DOI] [PubMed] [Google Scholar]
- 37. Verweij SL, Duivenvoorden R, Stiekema LCA, Nurmohamed NS, van der Valk FM, Versloot M, Verberne HJ, Stroes ESG, Nahrendorf M, Bekkering S, Bernelot Moens SJ.. CCR2 expression on circulating monocytes is associated with arterial wall inflammation assessed by 18F-FDG PET/CT in patients at risk for cardiovascular disease. Cardiovasc Res 2018;114:468–475. [DOI] [PubMed] [Google Scholar]
- 38. Langsted A, Kamstrup PR, Nordestgaard BG.. High lipoprotein(a) and low risk of major bleeding in brain and airways in the general population: a Mendelian randomization study. Clin Chem 2017;63:1714–1723. [DOI] [PubMed] [Google Scholar]
- 39. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC Jr, Watson K, Wilson PW.. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2889–2934. [DOI] [PubMed] [Google Scholar]
- 40. Brown WV, Ballantyne CM, Jones PH, Marcovina S.. Management of Lp(a). J Clin Lipidol 2010;4:240–247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.