

Abstract
Background
The incidence of cholangiocarcinoma (CCA) has risen in recent years, and it has become a significant health burden worldwide. However, the mechanisms underlying tumorigenesis and progression of this disease remain largely unknown. An increasing number of studies have demonstrated crucial biological functions of epigenetic modifications, especially DNA methylation, in CCA. The present study aimed to identify and analyze methylation-regulated differentially expressed genes (MeDEGs) involved in CCA tumorigenesis and progression by bioinformatics analysis.
Methods
The gene expression profiling dataset (GSE119336) and gene methylation profiling dataset (GSE38860) were obtained from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) and differentially methylated genes (DMGs) were identified using the limma packages of R and GEO2R, respectively. The MeDEGs were obtained by overlapping the DEGs and DMGs. Functional enrichment analyses of these genes were then carried out. Protein–protein interaction (PPI) networks were constructed using STRING and visualized in Cytoscape to determine hub genes. Finally, the results were verified based on The Cancer Genome Atlas (TCGA) database.
Results
We identified 98 hypermethylated, downregulated genes and 93 hypomethylated, upregulated genes after overlapping the DEGs and DMGs. These genes were mainly enriched in the biological processes of the cell cycle, nuclear division, xenobiotic metabolism, drug catabolism, and negative regulation of proteolysis. The top nine hub genes of the PPI network were F2, AHSG, RRM2, AURKB, CCNA2, TOP2A, BIRC5, PLK1, and ASPM. Moreover, the expression and methylation status of the hub genes were significantly altered in TCGA.
Conclusions
Our study identified novel methylation-regulated differentially expressed genes (MeDEGs) and explored their related pathways and functions in CCA, which may provide novel insights into a further understanding of methylation-mediated regulatory mechanisms in CCA.
Keywords: Cholangiocarcinoma, Methylation, Bioinformatics
Background
Cholangiocarcinoma (CCA) is a fatal malignancy originating from the epithelial cells of the biliary tree [1]. According to its anatomical location, the most contemporary classification of CCA includes intrahepatic, perihilar, and distal subtypes [2]. Surgical resection remains the only potentially curative treatment for all subtypes [1]. Unfortunately, early diagnosis is rare due to a lack of specific clinical symptoms [3]. Only about one-third of CCA patients are eligible for surgery; most patients are often diagnosed with unresectable or metastatic disease [4]. Even so, the recurrence and progression to distant metastasis are common within 2 years of resection [5]. Overall survival for patients with unresectable CCA does not exceed 14 months, even with systemic chemotherapy [6]. In addition, the incidence of CCA seems to be on the rise, especially for intrahepatic tumors [6]. Therefore, cumulative cholangiocarcinoma mortality rates have also increased by 39% [2]. Thus, exploring the molecular mechanisms underlying the pathogenesis and development of CCA is urgently needed.
DNA methylation is a central epigenetic modification that plays a key role in cellular processes, such as genome regulation, organism development, and disease [7]. Importantly, the dysregulation of DNA methylation patterns has been increasingly recognized as an important cellular event during both the initiation and late stages of oncogenesis [8, 9]. To date, numerous studies have demonstrated a crucial role for both hypermethylation of tumor suppressor genes and global hypomethylation of oncogenes in cancer development and progression, including in CCA [8, 10]. For instance, Chen et al. [11] found that the O6-methylguanine-DNA methyltransferase (MGMT) promoter was highly methylated, and the expression level of MGMT was positively correlated with overall survival rates and histological grade in CCA. In addition, the aberrant methylation of GATA-5, ANGPTL4, and DLEC1 has been shown to participate in the initiation and progression of this disease [12–14]. Despite the identification of several individual genes with specific hypomethylation or hypermethylation in CCA, comprehensive network studies based on methylation profiles and related pathways for these genes have been largely inadequate.
Over the past decade, gene profiling and next-generation sequencing technology have emerged as indispensable tools for cancer studies because they allow the detection of cancer-related genetic and epigenetic alterations, such as mutations, copy number variations, and DNA methylation changes across more extensive genomic regions [15, 16]. The bioinformatics analysis of these data can provide valuable information for CCA research. For example, Kong et al. identified three differentially expressed genes (DEGs), UCA1, miR-122, and CLIC1, based on next-generation sequencing data in CCA [17]. Analysis of these dysregulated genes showed that they could promote CCA progression through the regulation of miR-122/CLIC1 and activation of the ERK/MAPK signaling pathway [17]. Furthermore, both Farshidfar et al. and Jusakul et al. identified many differentially methylated genes (DMGs) by methylation profiling or whole-genome sequencing that contributed to cholangiocarcinogenesis and might serve as methylation biomarkers in CCA [18, 19]. However, separate DEG and DMG analysis from a single study are limited, and multiple overlapping available datasets may provide more accurate and reliable clues through comprehensive bioinformatics analysis [20]. Thus far, there is still a lack of conjoint analysis involving both gene expression and methylation profiling microarray datasets in CCA.
In the present study, we performed an integrated bioinformatics analysis based on gene expression profiling by high-throughput sequencing (GSE119336) and gene methylation profiling microarray (GSE38860). The methylation-regulated differentially expressed genes (MeDEGs) were identified and then subjected to enrichment analysis. Moreover, we constructed a protein–protein interaction (PPI) network to identify hub genes to screen novel biomarkers and therapeutic targets in CCA for future research.
Methods
RNA-Seq and microarray data
We obtained the gene expression profiling dataset generated by high-throughput sequencing (GSE119336) and the microarray-based gene methylation profiling dataset (GSE38860) from the publicly available Gene Expression Omnibus database (GEO, https://www.ncbi.nlm.nih.gov/geo/). The expression dataset (GSE119336) contained 15 pairs of CCA tumors and matched non-tumor tissues, which was based on the GPL11154 platform (Illumina HiSeq 2000). The DNA methylation dataset (GSE38860), which was generated on the GPL8490 platform (Illumina HumanMethylation27 BeadChip), included a total of 28 primary CCA tissues and six matched adjacent normal tissues.
Identification of MeDEGs
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an online analysis tool that allows users to compare two or more groups of samples in a GEO Series to identify deregulated genes under specific experimental conditions. In this study, GEO2R was adopted to screen for differentially methylated genes (DMGs). |t| > 2 and P < 0.05 were considered statistically significant. In addition, differentially expressed genes (DEGs) were identified using the limma R package with a threshold of |log2FoldChange| > 2 and P < 0.05. Using the lookup function (VLOOKUP) of excel, we overlapped the GSE38860 and GSE119336 datasets. Finally, hypomethylation-high expression genes were obtained after superimposition of upregulated and hypomethylated genes, and hypermethylation-low expression genes were obtained after superimposition of downregulated and hypermethylated genes. The hypomethylation-high expression genes and hypermethylation-low expression genes were identified as methylation-regulated differentially expressed genes (MeDEGs).
Functional enrichment analysis
Gene Ontology (GO) annotation and pathway analysis of the MeDEGs were conducted using The Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/) [21], Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.kegg.jp/), and Reactome (https://reactome.org/). Statistically significant thresholds were set at a P < 0.05.
PPI network construction, module analysis, and identification of hub genes
The PPI network of hypomethylation-high expression genes and hypermethylation-low expression genes was constructed using the STRING online database. An interaction score of > 0.4 and P < 0.05 were regarded as statistically significant. Cytoscape (version 3.6.1; http://www.cytoscape.org/) software was utilized to visualize the network. Molecular Complex Detection (MCODE), an app in Cytoscape, was used to screen modules within the PPI network with a standard of combined scores > 3 and nodes ≥ 5. Hub genes were identified using the CytoHubba in Degree-ranked method with a selection criterion for hub genes set at a degree score of greater than 16.
Validation of hub genes
MEXPRESS (http://mexpress.be) is a straightforward and easy-to-use web tool for the integration and visualization of expression and DNA methylation based on The Cancer Genome Atlas (TCGA) database at a single-gene level [22, 23]. We used MEXPRESS to validate hypomethylation-high expression genes and hypermethylation-low expression genes. The cBioPortal (http://cbioportal.org) is an open-access resource for interactive exploration of multidimensional cancer genomics datasets that provides access to data for more than 5000 tumor samples from 20 cancer studies [24]. The cBioPortal was used to explore the genetic alterations of hub genes.
Results
Identification of MeDEGs in CCA
The limma packages of R and GEO2R were used to screen for DEGs and DMGs, respectively. For the gene expression dataset GSE119336, a total of 2338 DEGs were identified, comprising 936 upregulated genes and 1402 downregulated genes. For the DMGs in the gene methylation dataset (GSE38860), 1020 hypermethylated genes and 1210 hypomethylated genes were obtained. As shown in Fig. 1, we identified 98 hypermethylated, downregulated genes and 93 hypomethylated, upregulated genes after the overlapping of the DEGs and DMGs. The heat map of the top 50 MeDEGs is shown in Fig. 2.
Fig. 1.

Identification of methylation-regulated differentially expressed genes (MeDEGs)
Fig. 2.

Heat map of the top 50 MeDEGs. X-axis represents samples, Y-axis represents genes, red stands for upregulation, and green stands for downregulation
Functional enrichment analysis of MeDEGs
The results of the gene ontology enrichment analysis for the MeDEGs are shown in Table 1. In the biological process group, hypomethylated, upregulated genes were mainly enriched in the cell cycle, nuclear division, negative regulation of cell cycle process, cell cycle process, and negative regulation of mitotic nuclear division. The hypermethylated, downregulated genes were mainly enriched in xenobiotic metabolic process, drug catabolic process, negative regulation of proteolysis, acute-phase response, and monocarboxylic acid metabolic process. We also found that the hypermethylated, downregulated genes were related to endopeptidase inhibitor activity, enzyme inhibitor activity, and serine-type endopeptidase inhibitor activity in the molecular function group and extracellular region in the cellular component group. Pathway enrichment was also performed using KEGG and Reactome, and the results are presented in Table 2. We found that hypermethylated genes predominantly participated in metabolism-related pathways, including general metabolic pathways, drug or xenobiotic metabolism by cytochrome P450, and cholesterol metabolism. For hypomethylated genes, the most significantly enriched pathways involved the cell cycle (mitotic G1-G1/S phases; G0 and early G1), SLC-mediated transmembrane transport, and signaling by MST1.
Table 1.
Gene ontology enrichment analysis of MeDEGs
| Category | Term ID | Term description | Count | False discovery rate |
|---|---|---|---|---|
| Hypermethylated downregulated genes | ||||
| BP_FAT | GO:0019752 | Carboxylic acid metabolic process | 22 | 5.98E−07 |
| BP_FAT | GO:0071466 | Cellular response to xenobiotic stimulus | 9 | 8.39E−05 |
| BP_FAT | GO:0006805 | Xenobiotic metabolic process | 7 | 8.00E−04 |
| BP_FAT | GO:0042737 | Drug catabolic process | 7 | 8.00E−04 |
| BP_FAT | GO:0045861 | Negative regulation of proteolysis | 11 | 8.00E−04 |
| BP_FAT | GO:0006953 | Acute-phase response | 5 | 1.30E−03 |
| BP_FAT | GO:0010951 | Negative regulation of endopeptidase activity | 9 | 1.30E−03 |
| BP_FAT | GO:0032787 | Monocarboxylic acid metabolic process | 12 | 1.30E−03 |
| BP_FAT | GO:0001676 | Long-chain fatty acid metabolic process | 6 | 3.30E−03 |
| BP_FAT | GO:0044281 | Small molecule metabolic process | 23 | 3.30E−03 |
| CC_FAT | GO:0005576 | Extracellular region | 38 | 2.40E−08 |
| CC_FAT | GO:0005615 | Extracellular space | 25 | 2.40E−08 |
| CC_FAT | GO:0044421 | Extracellular region part | 28 | 2.40E−08 |
| CC_FAT | GO:0044432 | Endoplasmic reticulum part | 19 | 1.30E−03 |
| MF_FAT | GO:0004866 | Endopeptidase inhibitor activity | 9 | 1.20E−04 |
| MF_FAT | GO:0004857 | Enzyme inhibitor activity | 11 | 4.20E−04 |
| MF_FAT | GO:0004867 | Serine-type endopeptidase inhibitor activity | 6 | 7.80E−04 |
| MF_FAT | GO:0008395 | Steroid hydroxylase activity | 4 | 1.90E−03 |
| MF_FAT | GO:0015370 | Solute:sodium symporter activity | 5 | 3.60E−03 |
| MF_FAT | GO:0016491 | Oxidoreductase activity | 13 | 3.60E−03 |
| Hypomethylated upregulated genes | ||||
| BP_FAT | GO:0007049 | Cell cycle | 20 | 3.20E−03 |
| BP_FAT | GO:0000280 | Nuclear division | 8 | 1.61E−02 |
| BP_FAT | GO:0010948 | Negative regulation of cell cycle process | 8 | 1.61E−02 |
| BP_FAT | GO:0022402 | Cell cycle process | 15 | 1.61E−02 |
| BP_FAT | GO:0045839 | Negative regulation of mitotic nuclear division | 4 | 1.61E−02 |
| BP_FAT | GO:0045930 | Negative regulation of mitotic cell cycle | 8 | 1.61E−02 |
| BP_FAT | GO:0048519 | Negative regulation of biological process | 41 | 1.61E−02 |
| BP_FAT | GO:0051726 | Regulation of cell cycle | 17 | 1.61E−02 |
| BP_FAT | GO:1903047 | Mitotic cell cycle process | 12 | 1.61E−02 |
| BP_FAT | GO:0007093 | Mitotic cell cycle checkpoint | 6 | 1.66E−02 |
| BP_FAT | GO:0051716 | Cellular response to stimulus | 46 | 3.22E−02 |
| BP_FAT | GO:0007568 | Aging | 7 | 3.23E−02 |
| BP_FAT | GO:0031577 | Spindle checkpoint | 3 | 3.23E−02 |
| BP_FAT | GO:0051302 | Regulation of cell division | 6 | 3.23E−02 |
| BP_FAT | GO:2000816 | Negative regulation of mitotic sister chromatid separation | 3 | 3.23E−02 |
| BP_FAT | GO:0007010 | Cytoskeleton organization | 13 | 3.36E−02 |
| BP_FAT | GO:0007051 | Spindle organization | 5 | 3.36E−02 |
| BP_FAT | GO:0007052 | Mitotic spindle organization | 4 | 3.36E−02 |
| BP_FAT | GO:0007346 | Regulation of mitotic cell cycle | 10 | 3.36E−02 |
| BP_FAT | GO:0009888 | Tissue development | 18 | 3.36E−02 |
Table 2.
Pathway enrichment analysis of MeDEGs
| Term ID | Term description | Count | False discovery rate |
|---|---|---|---|
| Hypermethylated downregulated genes | |||
| KEGG:hsa04610 | Complement and coagulation cascades | 7 | 2.39E−05 |
| KEGG:hsa00982 | Drug metabolism—cytochrome P450 | 6 | 8.49E−05 |
| KEGG:hsa04976 | Bile secretion | 6 | 8.49E−05 |
| KEGG:hsa04979 | Cholesterol metabolism | 4 | 3.50E−03 |
| KEGG:hsa00830 | Retinol metabolism | 4 | 7.10E−03 |
| KEGG:hsa00980 | Metabolism of xenobiotics by cytochrome P450 | 4 | 9.00E−03 |
| KEGG:hsa01100 | Metabolic pathways | 16 | 9.00E−03 |
| KEGG:hsa00983 | Drug metabolism—other enzymes | 4 | 9.30E−03 |
| KEGG:hsa05020 | Prion diseases | 3 | 9.30E−03 |
| KEGG:hsa05204 | Chemical carcinogenesis | 4 | 9.30E−03 |
| Reactome:HSA-1430728 | Metabolism | 28 | 1.10E−04 |
| Reactome:HSA-211999 | CYP2E1 reactions | 4 | 1.10E−04 |
| Reactome:HSA-9027307 | Biosynthesis of maresin-like SPMs | 3 | 6.50E−04 |
| Reactome:HSA-114608 | Platelet degranulation | 6 | 1.70E−03 |
| Reactome:HSA-76002 | Platelet activation, signaling and aggregation | 8 | 1.70E−03 |
| Reactome:HSA-425366 | Transport of bile salts and organic acids, metal ions and amine compounds | 5 | 2.00E−03 |
| Reactome:HSA-211859 | Biological oxidations | 7 | 2.30E−03 |
| Reactome:HSA-977606 | Regulation of complement cascade | 4 | 2.30E−03 |
| Reactome:HSA-373076 | Class A/1 (Rhodopsin-like receptors) | 8 | 2.90E−03 |
| Reactome:HSA-211945 | Phase I—functionalization of compounds | 5 | 3.10E−03 |
| Hypomethylated upregulated genes | |||
| KEGG:hsa04110 | Cell cycle | 5 | 4.16E−02 |
| Reactome:HSA-8852405 | Signaling by MST1 | 3 | 1.50E−03 |
| Reactome:HSA-1640170 | Cell cycle | 11 | 1.08E−02 |
| Reactome:HSA-453279 | Mitotic G1-G1/S phases | 6 | 1.08E−02 |
| Reactome:HSA-69278 | Cell cycle, mitotic | 10 | 1.08E−02 |
| Reactome:HSA-1538133 | G0 and Early G1 | 3 | 2.07E−02 |
| Reactome:HSA-174143 | APC/C-mediated degradation of cell cycle proteins | 4 | 2.54E−02 |
| Reactome:HSA-425366 | Transport of bile salts and organic acids, metal ions and amine compounds | 4 | 2.54E−02 |
| Reactome:HSA-6806834 | Signaling by MET | 4 | 2.54E−02 |
| Reactome:HSA-6806942 | MET receptor activation | 2 | 2.54E−02 |
| Reactome:HSA-425407 | SLC-mediated transmembrane transport | 6 | 2.74E−02 |
PPI network construction, module analysis, and identification of hub genes
The PPI network of hypomethylation-high expression genes and hypermethylation-low expression genes was constructed using the STRING online database. An interaction score of > 0.4 and P < 0.05 indicated statistical significance. The results are presented in Fig. 3. Using MCODE in Cytoscape, we identified two modules in the PPI network with a standard of combined scores > 3 and nodes ≥ 5 (Fig. 4). Nine hub genes were identified using CytoHubba with a degree score greater than 16, including F2, AHSG, RRM2, AURKB, CCNA2, TOP2A, BIRC5, PLK1, and ASPM.
Fig. 3.

PPI network of methylation-regulated differentially expressed genes. a Hypomethylated upregulated genes. b Hypermethylated downregulated genes
Fig. 4.
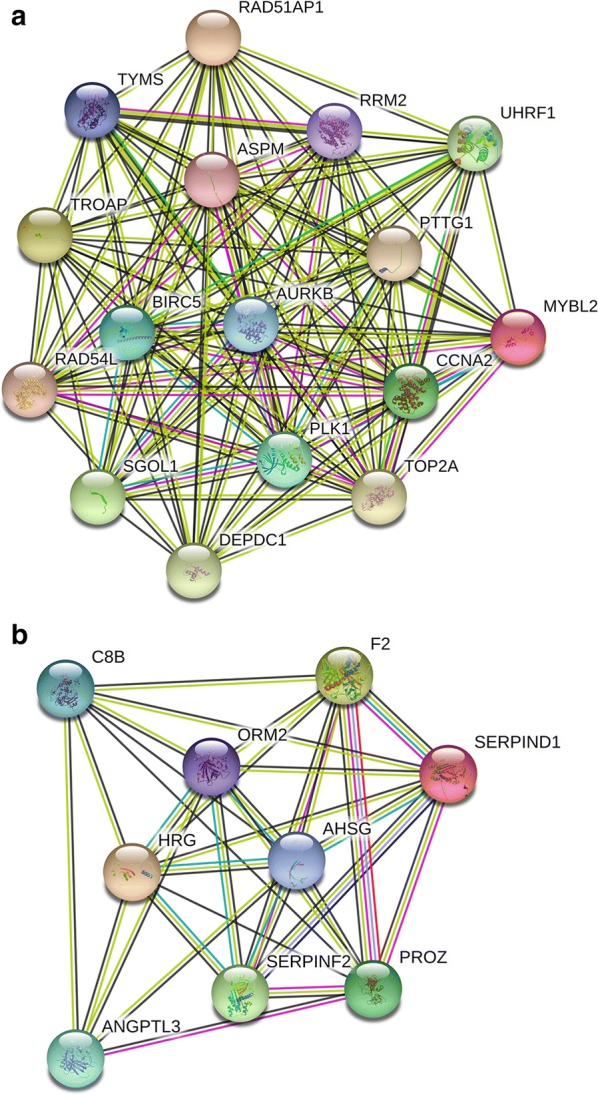
Module analysis of methylation-regulated differentially expressed genes. a Hypomethylated upregulated genes. b Hypermethylated downregulated genes
Verification of hub genes
To further investigate the identified hub genes, the TCGA database was used to validate our results. The expression and DNA methylation data of nine hub genes were obtained using MEXPRESS. The expression levels of seven hypomethylated upregulated genes and two hypermethylated downregulated genes were significantly different in the TCGA database, which were consistent with our results (Table 3). Except for ASPM, the methylation levels of these hub genes were consistent with our findings (Fig. 5a–i: AURKB, PLK1, CCNA2, ASPM, RRM2, TOP2A, BIRC5, F2, and AHSG). In addition, the presence of genetic alterations in the hub genes was examined using the cBioPortal tool. More than 40% of the patient tumors had at least one hub gene alteration and ASPM (29%) was the most frequently altered gene of the nine queried genes (Fig. 6).
Table 3.
Verification of the expression levels of 7 hypomethylated upregulated genes and 2 hypermethylated downregulated genes based on TCGA databases
| Category | Gene ID | logFC | P value |
|---|---|---|---|
| Upregulated | TOP2A | 4.343206 | 2.81E−27 |
| Upregulated | PLK1 | 4.137076 | 2.59E−24 |
| Upregulated | BIRC5 | 3.999543 | 5.92E−23 |
| Upregulated | RRM2 | 3.512025 | 2.17E−20 |
| Upregulated | AURKB | 4.000693 | 2.92E−20 |
| Upregulated | CCNA2 | 3.299934 | 3.11E−19 |
| Upregulated | ASPM | 2.892425 | 1.85E−12 |
| Downregulated | AHSG | − 6.23907 | 1.1E−09 |
| Downregulated | F2 | − 5.61955 | 1.31E−09 |
Fig. 5.

Verification of the methylation levels of 7 hypomethylated upregulated genes and 2 hypermethylated downregulated genes based on TCGA databases. a AURKB. b PLK1. c CCNA2. d ASPM. e RRM2. f TOP2A. g BIRC5. h F2. i AHSG
Fig. 6.

Genetic alterations of hub genes in the TCGA database. a Genetic alteration frequency of nine hub genes in 35 samples. Different colors represent different kinds of genetic alterations. b Summary of alterations per sample. Each sample is presented in a column with each gene in a row. Different kinds of genetic alterations are highlighted in different colors
Discussion
Cholangiocarcinoma (CCA) is a fatal malignancy that arises from cholangiocytes. In recent years, the incidence of CCA has risen, and CCA has become a major health burden worldwide [25]. The majority of patients with CCA present with unresectable or metastatic disease at the time of diagnosis because of the lack of specific symptoms or sensitive indicators [26]. Conventional radiotherapy and current chemotherapy treatments are not very effective [2]. Given the diagnostic difficulty and limited treatment options, the prognosis for patients with CCA is extremely poor [27]. Therefore, identifying novel molecular biomarkers and understanding the underlying mechanisms of carcinogenesis and progression in CCA are critically needed.
DNA methylation is a central epigenetic modification that plays a key role in cellular processes and generally occurs on cytosines that precede a guanine nucleotide [28, 29]. Typically, these cytosine–phosphate–guanine (CpG) sites are concentrated in large clusters (i.e., CpG islands) that are enriched mostly in the promoter region of genes [30]. Because the binding sites for many transcription factors are GC-rich, CpG islands are likely to enhance binding to transcriptional start sites [31]. CpG islands may also enhance the accessibility of DNA by regulating the chromatin structure. Methylation of CpG islands can impair transcription factor binding, recruit repressive methyl-binding proteins, and stably silence gene expression [31]. Thus, CpG islands are normally unmethylated in transcriptionally active genes and methylated in the promoters of silenced genes. To date, numerous studies have demonstrated a crucial role for both hypermethylation of tumor suppressor genes and hypomethylation of oncogenes in cancer development and progression [30]. For example, Wu et al. [32] found that JMJD2C could enhance the metastasis of colorectal cancer by decreasing the histone methylation of the MALAT1 promoter, thereby upregulating MALAT1 expression and enhancing the activity of the β-catenin signaling pathway. Furthermore, Liang et al. [33] identified many novel methylation-regulated differentially expressed genes involved in colon cancer tumorigenesis and progression by identifying hypermethylated downregulated genes and hypomethylated upregulated genes.
The initiation and progression of CCA is a complex and multistage process regulated by both genetic and epigenetic alterations [34]. An increasing number of studies have demonstrated the crucial biological functions of epigenetic modifications, especially DNA methylation, in CCA. For example, significant differences in the methylation levels of OPCML and HOXD9 were observed in serum cell-free DNA from CCA patients, and these two genes can be used for the differential diagnosis between cholangiocarcinoma and other biliary diseases [35]. Moreover, Wang et al. found that DANCR could bind EZH2 and modulate the histone methylation of the FBP1 promoter, thereby regulating the proliferation and migration of CCA cells [25]. However, previous studies mainly focused on specific genes with aberrant DNA methylation or individual gene methylation profiling microarrays. However, a systematic conjoint analysis involving both gene expression and methylation profiling in CCA may yield more accurate and reliable results. Thus, we performed an integrated bioinformatics analysis based on both gene expression and gene methylation profiling (GSE119336 and GSE38860, respectively) to screen for novel biomarkers and therapeutic targets in CCA for future research.
In the present study, we identified a total of 93 hypomethylated, upregulated genes and 98 hypermethylated, downregulated genes by integrating the DEGs and DMGs. Functional and pathway enrichment analysis indicated that the hypomethylated, upregulated genes were mainly enriched in the biological processes of the cell cycle, mitotic cell cycle checkpoint, mitotic sister chromatid separation, spindle assembly, nuclear division, and cell division. These findings are reasonable because it is well known that dysregulation of cell cycle progression, including nuclear division, sister chromatid separation, and spindle assembly, is closely related to cancer 1 [36–39]. Specifically, we identified many upregulated MeDEGs that participate in cell cycle progression, including BIRC5, PLK1, SGOL1, and ASPM. Some of these MeDEGs are involved in tumorigenesis. For instance, SGOL1, a member of the shugoshin family of proteins, is thought to protect centromeric cohesion during mitosis, and its dysregulation at centromeres can lead to chromosome missegregation and miotic arrest [40, 41]. Mu et al. found that SGOL1 expression levels are higher in prostate cancer tissues, and SGLO1 knockdown results in the inhibition of tumor cell proliferation, migration, and invasion [42]. In addition, SGOL1 has been found in other cancers (e.g., breast cancer and glioblastoma) [43–45]. However, few studies have described the methylation state of SGOL1, and research on SGOL1 in CCA has been rarely reported. Therefore, further research on the relationship between these MeDEGs and the cell cycle is warranted.
Pathway analysis also revealed that the hypomethylated, upregulated genes were enriched in MST1 signaling, which has demonstrated significant effects in multiple types of human cancer. MST1 can bind to its specific receptor MST1R, and MST1/MST1R signaling plays an important role in regulating inflammation and stimulating chemotaxis and phagocytosis [46]. The upregulation of MST1R promotes the progression of many epithelial cancers, including pancreatic, lung, and breast cancer [46–48]. We previously demonstrated that MST1R is upregulated and associated with overall survival in CCA [49]. However, the underlying mechanisms regulating MST1R remain largely unknown. In the current study, we found that MST1R was hypomethylated in CCA, which provides a foundation for future research.
The 98 hypermethylated, downregulated genes were mainly enriched in the biological processes of carboxylic acid metabolism, negative regulation of proteolysis, cytolysis, and xenobiotic and monocarboxylic acid metabolism. We also identified the involvement of complement and coagulation cascades, bile secretion, drug metabolism, cholesterol metabolism, and CYP2E1 reactions with these genetic changes. Both the biological processes and pathways analysis showed an abundant enrichment in metabolism, particularly bile metabolism. Among these genes, we found ABCB11, which encodes the bile salt export pump (BSEP), was involved in bile secretion, cholesterol metabolism, bile acid and bile salt metabolism, and recycling of bile acids and salts based on the pathway analysis. BSEP mediates the secretion of bile acids across the canicular membrane of hepatocytes into bile to provide the osmotic pressure for bile flow [50, 51]. Strautnieks et al. found that BSEP is typically absent or greatly reduced due to ABCB11 mutations, and 15% of patients with BSEP deficiency developed hepatocellular carcinoma or CCA [52]. BSEP deficiency may cause CCA through bile-composition shifts or bile-acid damage within cells capable of hepatocytic or cholangiocytic differentiation [53]. Moreover, previous studies also demonstrated a critical role for ABCB11 in lung and ovarian cancer [54, 55]. However, both Srimunta et al. and Fujikura et al. did not find differentially expressed BSEP levels in CCA tissues using immunohistochemistry or real-time RT-PCR, which is inconsistent with our findings [56, 57]. Therefore, further investigation is needed. The cytochrome P450 (CYP) enzymes are membrane-bound hemoproteins that play a pivotal role in the natural product biosynthesis, drug metabolism, cellular metabolism and homeostasis [58, 59]. Anti-cancer drugs and herbs undergo metabolism mediated by CYP enzymes in the body to form numerous stable metabolites and this metabolism may significantly alter their therapeutic potential [60]. Besides, several studies have demonstrated the important role CYP enzymes played in CCA progression. For example, Zhang et al. found that 1,2-dichloropropane, a carcinogenic paint remover, could influence the proliferation and apoptosis of cholangiocytes and this effect is mediated through CYP450 [61]. Moreover, Khenjanta et al. found that CYP39A1 was down regulated in 70% of CCA patients and low expression of CYP39A1 demonstrated a significant correlation with metastasis [62]. However, the mechanisms of methylation regulation mechanism for these CYP enzymes are still unknown.
In the PPI networks generated for hypomethylated, upregulated and hypermethylated, downregulated genes, significantly more interactions than expected were observed with a PPI enrichment P value < 1.0e−16, which indicates that these genes are biologically connected. Additional analysis identified nine hub genes (F2, AHSG, RRM2, AURKB, CCNA2, TOP2A, BIRC5, PLK1, and ASPM), which were validated using the TCGA database. Many of these genes are associated with CCA. For example, Shen et al. found that AURKB is overexpressed in CCA and correlates with overall survival and tumor grade [63]. AURKB is also upregulated and hypomethylated in hepatocellular carcinoma [64, 65]. However, aberrant methylation of AURKB in CCA has not been previously reported.
We explored potential genetic alterations of the identified hub genes using cBioPortal. We observed that more than 40% of the patient tumors analyzed had at least one hub gene alteration. Of the nine evaluated genes, ASPM (Abnormal Spindle Microtubule Assembly) was the most frequently altered (29%). The ASPM protein is involved in mitotic spindle regulation and coordination of mitotic processes [66]. Previous studies have shown that ASPM plays an essential role in tumorigenesis and the development of numerous types of cancers, including pancreatic and breast cancer and clear cell renal cell carcinoma [67, 68]. However, the mechanisms of methylation regulation for ASPM and the role of ASPM in CCA are not known.
Several limitations to the present study should be mentioned. First, our research only focused on upregulated, hypomethylated genes and downregulated, hypermethylated genes. However, contra-regulated genes were not included and need to be studied further. Second, our study was limited to the data of bioinformatics arrays and tools. We did not investigate clinical parameters and prognosis, which may reduce the reliability of our findings. Third, although we validated the identified hub genes using the TCGA database, biological experiments were not performed. Our future research will focus on experimental verification of these results. Finally, we obtained the MeDEGs by overlapping the DEGs and DMGs from two different datasets (expression dataset GSE119336 and DNA methylation dataset GSE38860) but did not include a dataset that included both expression and methylation data for CCA. In addition, the numbers of samples and datasets for the DEG and DMGs were small, which could introduce false positives and reduce the reliability of our findings. Therefore, an independent expression and methylation study for CCA that includes large-scale multicenter clinical samples should be carried out.
Conclusions
In conclusion, using an integrated bioinformatics analysis, our study identified methylation-regulated differentially expressed genes and explored their related pathways and functions in CCA. In addition, we constructed a PPI network that identified nine hub genes. Our findings may deepen the understanding of the methylation-mediated regulatory mechanisms underlying CCA and provide some novel therapeutic targets for further research.
Acknowledgements
We thank BioMed Proofreading (http://www.biomedproofreading.com/) for its linguistic assistance during the preparation of this manuscript.
Abbreviations
- CCA
cholangiocarcinoma
- MeDEGs
methylation-regulated differentially expressed genes
- GEO
Gene Expression Omnibus
- DEGs
differentially expressed genes
- DMGs
differentially methylated genes
- PPI
protein–protein interaction
- TCGA
The Cancer Genome Atlas
- MCODE
Molecular Complex Detection
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- STRING
The Search Tool for the Retrieval of Interacting Genes
- CpG
cytosine–phosphate–guanine
- MGMT
O6-methylguanine-DNA methyltransferase
- ASPM
Abnormal Spindle Microtubule Assembly
Authors’ contributions
The authors contributed to this study and manuscript in the following manner: data collection, CZ and BZ; statistical analysis, CZ and DM; writing and editing, CZ and DM; supervision, CG; funding acquisition, CG. All authors read and approved the final manuscript.
Funding
The study was supported by the Educational Commission of Liaoning Province, People’s Republic of China (Grant Number L2014294).
Availability of data and materials
The data used to support the findings of this study are included in the article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Guo SS, Wang Y, Fan QX. Raddeanin A promotes apoptosis and ameliorates 5-fluorouracil resistance in cholangiocarcinoma cells. World J Gastroenterol. 2019;25(26):3380–3391. doi: 10.3748/wjg.v25.i26.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagante F, Spolverato G, Merath K, et al. Intrahepatic cholangiocarcinoma tumor burden: a classification and regression tree model to define prognostic groups after resection. Surgery. 2019;166:983–990. doi: 10.1016/j.surg.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Bergquist A, von Seth E. Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015;29(2):221–232. doi: 10.1016/j.bpg.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Belkouz A, Labeur TA, Dierks J, et al. Prognostic immunohistochemical biomarkers of chemotherapy efficacy in biliary tract cancer: a systematic review and meta-analysis. Crit Rev Oncol Hematol. 2019;141:82–94. doi: 10.1016/j.critrevonc.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Jung C, Lavole J, Barret M, et al. Local therapy in advanced cholangiocarcinoma: a review of current endoscopic, medical, and oncologic treatment options. Oncology. 2019;97:191–201. doi: 10.1159/000500832. [DOI] [PubMed] [Google Scholar]
- 7.Malpeli G, Innamorati G, Decimo I, et al. Methylation dynamics of RASSF1A and its impact on cancer. Cancers (Basel). 2019;11(7):E959. doi: 10.3390/cancers11070959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasek K, Kubatka P, Samec M, et al. DNA methylation status in cancer disease: modulations by plant-derived natural compounds and dietary interventions. Biomolecules. 2019;9(7):289. doi: 10.3390/biom9070289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magzoub MM, Prunello M, Brennan K, Gevaert O. The impact of DNA methylation on the cancer proteome. PLoS Comput Biol. 2019;15(7):e1007245. doi: 10.1371/journal.pcbi.1007245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang B. Inference of crosstalk effects between DNA methylation and lncRNA regulation in NSCLC. Biomed Res Int. 2018;2018:7602794. doi: 10.1155/2018/7602794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Li Z, Chen J, et al. Downregulation of MGMT promotes proliferation of intrahepatic cholangiocarcinoma by regulating p21. Clin Transl Oncol. 2019 doi: 10.1007/s12094-019-02140-9. [DOI] [PubMed] [Google Scholar]
- 12.Liu P, Zhou TF, Qiu BA, et al. Methylation-mediated silencing of GATA5 gene suppresses cholangiocarcinoma cell proliferation and metastasis. Transl Oncol. 2018;11(3):585–592. doi: 10.1016/j.tranon.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu Y, Zhang M, Liu J, et al. Long non-coding RNA PVT1 promotes cell proliferation and migration by silencing ANGPTL4 expression in cholangiocarcinoma. Mol Ther Nucleic Acids. 2018;13:503–513. doi: 10.1016/j.omtn.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y, Lee K, Jeong S, Wen X, Cho NY, Kang GH. DLEC1 methylation is associated with a better clinical outcome in patients with intrahepatic cholangiocarcinoma of the small duct subtype. Virchows Arch. 2019;475(1):49–58. doi: 10.1007/s00428-018-02511-7. [DOI] [PubMed] [Google Scholar]
- 15.Huang CC, Du M, Wang L. Bioinformatics analysis for circulating cell-free DNA in cancer. Cancers (Basel). 2019;11(6):E805. doi: 10.3390/cancers11060805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20:631–656. doi: 10.1038/s41576-019-0150-2. [DOI] [PubMed] [Google Scholar]
- 17.Kong L, Wu Q, Zhao L, Ye J, Li N, Yang H. Upregulated lncRNA-UCA1 contributes to metastasis of bile duct carcinoma through regulation of miR-122/CLIC1 and activation of the ERK/MAPK signaling pathway. Cell Cycle. 2019;18(11):1212–1228. doi: 10.1080/15384101.2019.1593647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farshidfar F, Zheng S, Gingras MC, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;18(11):2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017;7(10):1116–1135. doi: 10.1158/2159-8290.CD-17-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong Y, Song Y, Deng S. Combined analysis and validation for DNA methylation and gene expression profiles associated with prostate cancer. Cancer Cell Int. 2019;19:50. doi: 10.1186/s12935-019-0753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch A, Jeschke J, Van Criekinge W, van Engeland M, De Meyer T. MEXPRESS update 2019. Nucleic Acids Res. 2019;47(W1):W561–W565. doi: 10.1093/nar/gkz445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koch A, De Meyer T, Jeschke J, Van Criekinge W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genomics. 2015;16:636. doi: 10.1186/s12864-015-1847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang N, Zhang C, Wang W, et al. Long noncoding RNA DANCR regulates proliferation and migration by epigenetically silencing FBP1 in tumorigenesis of cholangiocarcinoma. Cell Death Dis. 2019;10(8):585. doi: 10.1038/s41419-019-1810-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamaoka M, Kozaka K, Matsui O, et al. Early detection of intrahepatic cholangiocarcinoma. Jpn J Radiol. 2019;10:1131–1142. doi: 10.1007/s11604-019-00860-0. [DOI] [PubMed] [Google Scholar]
- 27.Pang Q, Zhou L, Hu XS, et al. Biliary stenting alone versus biliary stenting combined with 125I particles intracavitary irradiation for the treatment of advanced cholangiocarcinoma. Sci Rep. 2019;9(1):11348. doi: 10.1038/s41598-019-47791-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Győrffy B, Bottai G, Fleischer T, et al. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int J Cancer. 2016;138(1):87–97. doi: 10.1002/ijc.29684. [DOI] [PubMed] [Google Scholar]
- 29.Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10:23. doi: 10.1186/s13072-017-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 31.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, Li R, Song Q, et al. JMJD2C promotes colorectal cancer metastasis via regulating histone methylation of MALAT1 promoter and enhancing β-catenin signaling pathway. J Exp Clin Cancer Res. 2019;38(1):435. doi: 10.1186/s13046-019-1439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang Y, Zhang C, Dai DQ. Identification of differentially expressed genes regulated by methylation in colon cancer based on bioinformatics analysis. World J Gastroenterol. 2019;25(26):3392–3407. doi: 10.3748/wjg.v25.i26.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu K, Yin X, Jin Y, Liu F, Gao J. Identification of aberrantly methylated differentially expressed genes in prostate carcinoma using integrated bioinformatics. Cancer Cell Int. 2019;19:51. doi: 10.1186/s12935-019-0763-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasenang W, Chaiyarit P, Proungvitaya S, Limpaiboon T. Serum cell-free DNA methylation of OPCML and HOXD9 as a biomarker that may aid in differential diagnosis between cholangiocarcinoma and other biliary diseases. Clin Epigenetics. 2019;11(1):39. doi: 10.1186/s13148-019-0634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J, Shi R, Zhao S, et al. Cell division cycle 45 promotes papillary thyroid cancer progression via regulating cell cycle. Tumour Biol. 2017;39(5):1010428317705342. doi: 10.1177/1010428317705342. [DOI] [PubMed] [Google Scholar]
- 37.Li W, Wang HY, Zhao X, et al. A methylation-phosphorylation switch determines Plk1 kinase activity and function in DNA damage repair. Sci Adv. 2019;5(3):7566. doi: 10.1126/sciadv.aau7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo L, Mohd KS, Ren H, et al. Phosphorylation of importin-α1 by CDK1-cyclin B controls mitotic spindle assembly. J Cell Sci. 2019 doi: 10.1242/jcs.232314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch LB, Opoku KN, Deng Y, et al. Autophosphorylation is sufficient to release Mps1 kinase from native kinetochores. Proc Natl Acad Sci USA. 2019;116:17355–17360. doi: 10.1073/pnas.1901653116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HS, Lin Z, Chae S, et al. The chromatin remodeler RSF1 controls centromeric histone modifications to coordinate chromosome segregation. Nat Commun. 2018;9(1):3848. doi: 10.1038/s41467-018-06377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang F, Sun Z, Pei Y, Singh RK, Jha HC, Robertson ES. Correction: shugoshin 1 is dislocated by KSHV-encoded LANA inducing aneuploidy. PLoS Pathog. 2019;15(4):e1007732. doi: 10.1371/journal.ppat.1007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mu J, Fan L, Liu D, Zhu D. Overexpression of shugoshin1 predicts a poor prognosis for prostate cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Oncol Targets Ther. 2019;12:1111–1118. doi: 10.2147/OTT.S191157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nasim N, Ghafouri-Fard S, Soleimani S, et al. Assessment of SGO1 and SGO1-AS1 contribution in breast cancer. Hum Antibodies. 2019;27:279–284. doi: 10.3233/HAB-190384. [DOI] [PubMed] [Google Scholar]
- 44.Rahane CS, Kutzner A, Heese K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J Neurooncol. 2019;141(1):57–70. doi: 10.1007/s11060-018-03029-3. [DOI] [PubMed] [Google Scholar]
- 45.Lang F, Sun Z, Pei Y, Singh RK, Jha HC, Robertson ES. Shugoshin 1 is dislocated by KSHV-encoded LANA inducing aneuploidy. PLoS Pathog. 2018;14(9):e1007253. doi: 10.1371/journal.ppat.1007253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babicky ML, Harper MM, Chakedis J, et al. MST1R kinase accelerates pancreatic cancer progression via effects on both epithelial cells and macrophages. Oncogene. 2019;38(28):5599–5611. doi: 10.1038/s41388-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnaswamy S, Bukhari I, Mohammed AK, et al. Identification of the splice variants of Recepteur d’Origine nantais (RON) in lung cancer cell lines. Gene. 2018;679:335–340. doi: 10.1016/j.gene.2018.09.027. [DOI] [PubMed] [Google Scholar]
- 48.Torrezan GT, de Almeida F, Figueiredo M, et al. Complex landscape of germline variants in Brazilian patients with hereditary and early onset breast cancer. Front Genet. 2018;9:161. doi: 10.3389/fgene.2018.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C, Ge C. A simple competing endogenous RNA network identifies novel mRNA, miRNA, and lncRNA markers in human cholangiocarcinoma. Biomed Res Int. 2019;2019:3526407. doi: 10.1155/2019/3526407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu T, Wang RX, Han J, et al. Comprehensive bile acid profiling in hereditary intrahepatic cholestasis: genetic and clinical correlations. Liver Int. 2018;38(9):1676–1685. doi: 10.1111/liv.13714. [DOI] [PubMed] [Google Scholar]
- 51.Sohail MI, Schmid D, Wlcek K, et al. Molecular mechanism of taurocholate transport by the bile salt export pump, an ABC transporter associated with intrahepatic cholestasis. Mol Pharmacol. 2017;92(4):401–413. doi: 10.1124/mol.117.108688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 53.Scheimann AO, Strautnieks SS, Knisely AS, Byrne JA, Thompson RJ, Finegold MJ. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr. 2007;150(5):556–559. doi: 10.1016/j.jpeds.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 54.Sissung TM, Rajan A, Blumenthal GM, et al. Reproducibility of pharmacogenetics findings for paclitaxel in a heterogeneous population of patients with lung cancer. PLoS ONE. 2019;14(2):e0212097. doi: 10.1371/journal.pone.0212097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seborova K, Vaclavikova R, Soucek P, et al. Association of ABC gene profiles with time to progression and resistance in ovarian cancer revealed by bioinformatics analyses. Cancer Med. 2019;8(2):606–616. doi: 10.1002/cam4.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srimunta U, Sawanyawisuth K, Kraiklang R, et al. High expression of ABCC1 indicates poor prognosis in intrahepatic cholangiocarcinoma. Asian Pac J Cancer Prev. 2012;13(Suppl):125–130. [PubMed] [Google Scholar]
- 57.Fujikura K, Yamasaki T, Otani K, et al. BSEP and MDR3: useful immunohistochemical markers to discriminate hepatocellular carcinomas from intrahepatic cholangiocarcinomas and hepatoid carcinomas. Am J Surg Pathol. 2016;40(5):689–696. doi: 10.1097/PAS.0000000000000585. [DOI] [PubMed] [Google Scholar]
- 58.Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets. 2018;19(1):38–54. doi: 10.2174/1389450118666170125144557. [DOI] [PubMed] [Google Scholar]
- 59.Li Z, Jiang Y, Guengerich FP, Ma L, Li S, Zhang W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J Biol Chem. 2019 doi: 10.1074/jbc.REV119.008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sumsakul W, Mahavorasirikul W, Na-Bangchang K. Inhibitory activities of thai medicinal plants with promising activities against malaria and cholangiocarcinoma on human cytochrome P450. Phytother Res. 2015;29(12):1926–1933. doi: 10.1002/ptr.5485. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Zong C, Zhang L, et al. Exposure of mice to 1,2-dichloropropane induces CYP450-dependent proliferation and apoptosis of cholangiocytes. Toxicol Sci. 2018;162(2):559–569. doi: 10.1093/toxsci/kfx272. [DOI] [PubMed] [Google Scholar]
- 62.Khenjanta C, Thanan R, Jusakul A, et al. Association of CYP39A1, RUNX2 and oxidized alpha-1 antitrypsin expression in relation to cholangiocarcinoma progression. Asian Pac J Cancer Prev. 2014;15(23):10187–10192. doi: 10.7314/APJCP.2014.15.23.10187. [DOI] [PubMed] [Google Scholar]
- 63.Shen YC, Hu FC, Jeng YM, et al. Nuclear overexpression of mitotic regulatory proteins in biliary tract cancer: correlation with clinicopathologic features and patient survival. Cancer Epidemiol Biomarkers Prev. 2009;18(2):417–423. doi: 10.1158/1055-9965.EPI-08-0691. [DOI] [PubMed] [Google Scholar]
- 64.Cai C, Wang W, Tu Z. Aberrantly DNA methylated-differentially expressed genes and pathways in hepatocellular carcinoma. J Cancer. 2019;10(2):355–366. doi: 10.7150/jca.27832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sang L, Wang XM, Xu DY, Zhao WJ. Bioinformatics analysis of aberrantly methylated-differentially expressed genes and pathways in hepatocellular carcinoma. World J Gastroenterol. 2018;24(24):2605–2616. doi: 10.3748/wjg.v24.i24.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Z, Zhang Q, Luh F, Jin B, Liu X. Overexpression of the ASPM gene is associated with aggressiveness and poor outcome in bladder cancer. Oncol Lett. 2019;17(2):1865–1876. doi: 10.3892/ol.2018.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsu CC, Liao WY, Chan TS, et al. The differential distributions of ASPM isoforms and their roles in Wnt signaling, cell cycle progression, and pancreatic cancer prognosis. J Pathol. 2019 doi: 10.1002/path.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang J, Lu M, Cui Q, et al. Overexpression of ASPM, CDC20, and TTK confer a poorer prognosis in breast cancer identified by gene co-expression network analysis. Front Oncol. 2019;9:310. doi: 10.3389/fonc.2019.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included in the article.