

Abstract
Fibrous dysplasia (FD) is a nonneoplastic hamartomatous developmental fibro-osseous lesion, with anomaly in bone-forming mesenchyme which manifests as a defect in osteoblastic differentiation and maturation leading to fibro-osseous tissue formation characterized by deformities in the bone, fractures, nerve compression, and bone pain. The clinical behavior and progression of FD make the management of this condition difficult. Here is a case report of a young male patient who was diagnosed as having craniomaxillofacial FD. The diagnosis was based on clinicoradiological and histopathological investigations. In this case, management of FD poses significant challenges to the surgeon.
Keywords: Bones, cranium, fibrous dysplasia, polyostotic
INTRODUCTION
Fibrous dysplasia (FD) of bones was first described by Von Recklinghausen in 1891. Lichtenstein coined the term FD of bone in 1938.[1,2] Many syndromes affect the craniofacial region. One of them which drastically affect the face causing gross deformity is craniofacial FD. Craniofacial FD is described as lesions which are confined to bones of craniofacial skeleton. They are not truly polyostotic as bones other than craniofacial are spared. There are very few cases reported in the literature, seen most often in the first three decades of life.[3] It is caused by somatic-activating mutations encoded by the gene GNAS in the α subunit of the stimulating G protein.[3,4,5,6] It has the potential to cause cosmetic and functional disturbance. Ocular effects are of particular concern. Its compression of the optic nerve resulting in visual impairment is alarming.[7] Below is a case of a 10-year-old male patient of craniofacial polyostotic FD and approach toward treatment.
CASE REPORT
A 10-year-old male patient came to the Department of Oral and Maxillofacial Surgery with the complaint of displacement of right eye inferiorly, swelling over the right forehead region as seen in Figures 1 and 2. The swelling has begun 8 months ago and has noticeably increased in size within this period. There was no history of trauma or any infection to this area. No history of headache, pain over the swelling, and fever. Normal vision was noted. On examination, the patient has 6 cm × 7 cm size bony prominence on the right frontal area as seen in Figure 3. Swelling is not tender and immobile and hard in consistency. No overlying skin changes. No inflammatory changes seen over the swelling. No abnormality seen in his ears, nose, and mouth. The left eye was proptosed and displaced inferolaterally. Pupillary responses were normal in both the eyes. No change in the voice quality. Mouth opening was normal. No lymphadenopathy seen. No thyroid changes noted. No past medical history. Complete blood picture, parathyroid hormone, calcium–phosphorus levels, and serum alkaline phosphatase are within normal limits.
Figure 1.
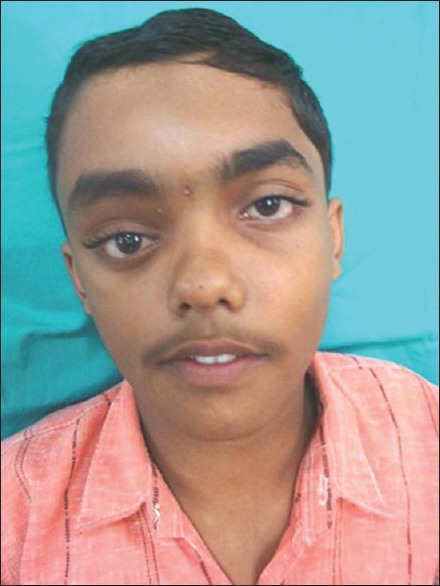
Frontal view: Inferior displacement of the right eye
Figure 2.
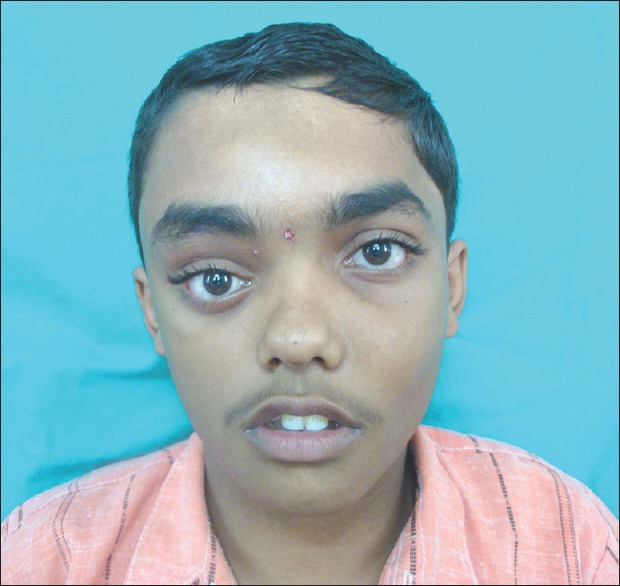
Raise of the left eye superiorly, swelling over the left maxilla
Figure 3.
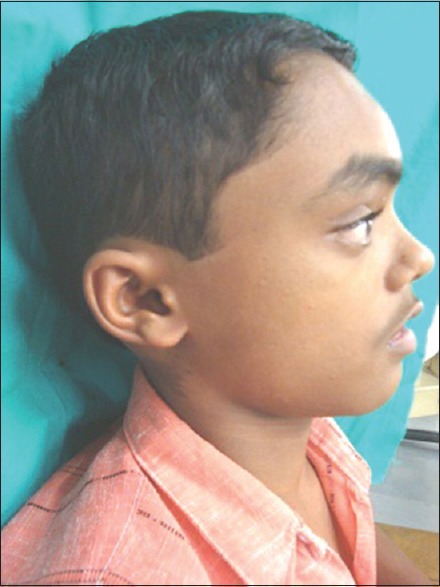
Lateral view: Frontal bossing and proptosis of the eye
Computed tomography (CT) scan reveals asymmetrical increase in bone density with diffuse asymmetrical widening of diploe spaces with thick ground-glass attenuation of matrix is seen involving the right frontal bone, right temporal bone, parietal bone, occipital and sphenoid bones, left maxilla, left zygoma, body and ramus on left and right side of mandible [Figure 4], ethmoid bones, clivus, occipital condyles noted causing asymmetry, and deformation of facial bones. Narrowed sella was seen. Complete sclerosis of mastoid air cells on the right side was seen. Relative narrowing of foramina of skull was noted as seen in Figures 5 and 6.
Figure 4.
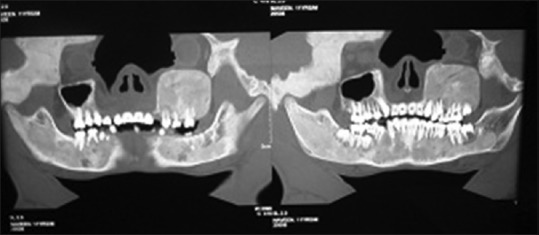
Increase in bone density in the jaw bones
Figure 5.
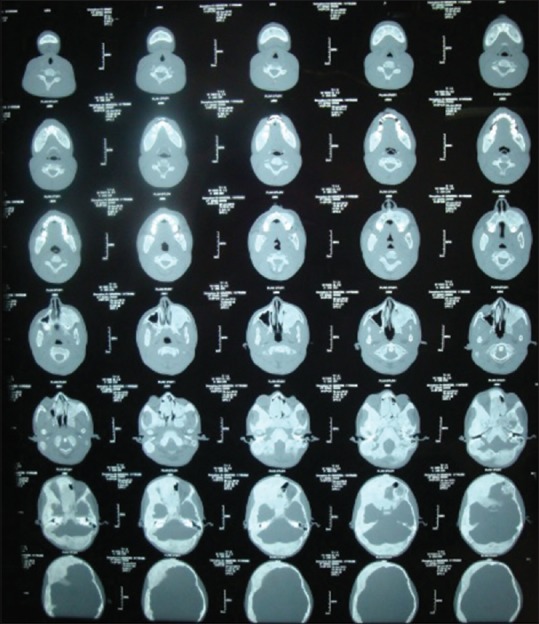
Transverse section of CT scan skuil
Figure 6.
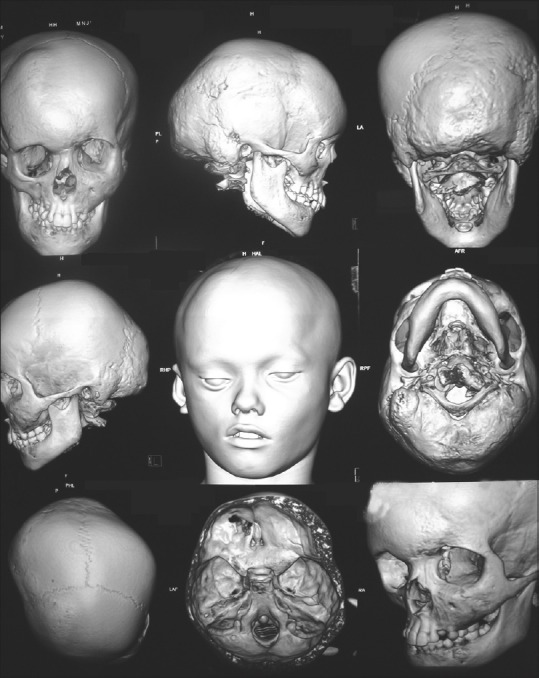
Computed tomography scan shows deformation of the cranial and facial bones
Incisional biopsy was performed in the mandible providing diagnosis of craniofacial polyostotic FD. Histological examination showed connective tissue cell stroma consisting of fibroblasts seen in whorled pattern. Ribbon-like osteoid trabeculae lined by numerous osteoblasts changing into woven bone along with osteoclasts. Multinucleated giant cells noted with areas of hemorrhage in areas of cystic changes.
We have aggressively screened for endocrinopathies, i.e., growth hormone excess. Lesion is not showing rapid growth, no new onset of pain or paresthesia, and visual or hearing changes which warrant immediate surgical referral. In some, cases history, clinical examination, and radiographic diagnosis are adequate for diagnosis of the lesion. Such cases are postponed for surgical treatment until after skeletal maturity and when lesion is quiescent. Rapid change in lesion or any threatening symptoms occur; surgical resection or contouring is warranted. Bisphosphonates are not given as the patient does not complain of pain. This disease requires multidisciplinary execution for optimal care.
Hence, the patient was referred to ophthalmologist for opinion, as the optic nerve compression noted in the CT scan. At present, the patient has no visual impairment. The patient is kept under observation for the symptoms to appear. Each patient presents with variable clinical findings and symptoms. Care of these patients should be customized for the site of lesion and their needs. So in this case, treatment is planned on priority needs. Therapeutic decompression of optic canal was planned when symptoms appear.
DISCUSSION
FD is a nonneoplastic condition where normal bone is replaced by fibrous tissue and haphazardly distributed woven bone. FD is caused by mutations in the α subunit of the stimulatory G protein encoded by the gene GNAS.[4,5] It may involve one bone monostotic or multiple bones polyostotic. McCune–Albright syndrome is a triad of polyostotic FD, café-au-lait skin macules, and endocrinopathies with precocious puberty.[5,8] In polyostotic FD, 90% of cases involve craniofacial region and 95% cases involve the anterior cranial base. FD is a slow-growing mass lesion. The lesions show rapid growth with the cortical bone expansion with displacement of adjacent structures such as the eye and the teeth in young children and prepubertal adolescents. The symptoms include facial asymmetry, visual changes, nasal congestion, pain, paresthesia, hearing impairment, and malocclusion.[3,5,9,10] Malignant change to osteosarcoma has been reported. Functional deficits result due to invasion or compression of vital structures such as the optic nerve, globe and auditory canal, and nasal airway. Neurosurgeons, oral and maxillofacial surgeons, otolaryngologists, and ophthalmologists are consulted based on the symptoms and site of involvement.[9]
The most often found radiological examination shows ground-glass appearance with thin cortical bone and no distinct borders. Its appearance can vary from ground glass to homogenous or mixed radiolucent or radiopaque depending on age of the patient.[3,7,11,12]
The radiographic appearance of FD has three basic patterns:
Type 1: Small unilocular or multilocular radiolucency well-circumscribed border with network of fine trabeculae
Type 2: More opaque and mottled lesion
Type 3: Opaque with delicate trabeculae showing ground-glass appearance or Peau d’orange.[4,13]
CT imaging provides the site of involvement and extensions of the disease in the face and skull bones. Plain films cause overlapping of adjacent structures leading to less diagnostic value in cranial and facial lesions. Lesions around the dentition can be examined and managed by dental radiographs.[11]
Most frequent findings with polyostotic FD around the eye are dystopia hypertelorism due to the involvement of frontal, sphenoid and ethmoid bones, and proptosis. Other findings are difficulty in lid closure, strabismus, optic neuropathy, nasolacrimal duct obstruction, tearing, and trigeminal neuralgia due to skull base involvement.[7,11,14]
In the above case, there was involvement of FD in the cranial and skull base bones and also involving optic nerve, but vision was normal. Our highest concern was for any future vision loss. Regular observation with ophthalmologic examinations in this patient with asymptomatic encasement was treatment option, and optic nerve decompression was not warranted. Studies show that prophylactic decompression may have no improvement in vision or worse postoperative blindness.[5,15,16] Neuroopthalmologic examination for assessment of optic neuropathy includes visual acuity, field examination, color vision, dilated fundus examination, and contrast sensitivity. Others include papillary examination such as afferent pupil, proptosis measurement, exophthalmometry, extraocular movements, lid closure, hypertelorism, tear duct, and puncta examination. Any vision changes the patient has to be referred to neuroopthalmologist.
Various treatment modalities based on site, extent of involvement, and priority of symptoms, there are various categories. Wait and watch technique followed in small asymptomatic lesions which can be kept under observation. Medical therapy includes the use of high-dose glucocorticoids in a new expansile lesions near the optic nerve which could be a aneurysmal bone cyst, which cause immediate decompression and later indicated for resection.[3,9,17]
The use of bisphosphonates such as alendronate, pamidronate, or zoledronic acid is considered as nonsurgical and adjuvant therapy to reduce the pain and rate of growth of the lesion.[3,7,11,17]
Serum alkaline phosphatase and urinary hydroxyproline are useful markers for monitoring the response of nonsurgical therapy and not for diagnosis. Some studies have shown their role in detecting the recurrence of the disease.[3,7,17,18]
Surgical recontouring or remodeling is a conservative method to achieve acceptable esthetics. Stabilization of the lesion occurs when maturation of the bone completes. However, long-term follow-up is important.[17] Surgery is preferred when there is a threat to optic nerve, causing damage to vision. Most surgeons prefer radical surgical therapy with immediate reconstruction to avoid functional disturbances and restore facial form and esthetics.[9] Iliac, rib, and calvarial grafts or revascularized free flaps are used. Some prefer removing thin bone followed by remodeling and reimplant with dysplastic bone grafts for surgical defects. Patients older than 17 years of age where complete resection is not possible, partial resection of the lesion can be considered as regrowth of the lesion is less and importance is given more for aesthesis.
Chen and Noordhoff in 1990 proposed a treatment algorithm for the management of craniomaxillofacial FD. The head and face are divided into four zones based on the unique anatomic considerations for operating in each area and esthetic and functional considerations of the disease at these sites.
Zone 1: Represents the fronto-orbito-malar regions of the face. They recommend radical excision and reconstruction with simple bone grafting techniques, as they are esthetically critical
Zone 2: Represents the hair-bearing scalp. Intervention is optional for the patient
Zone 3: Represents the central skull base; the sphenoid, pterygoid, petrous temporal bone, and mastoid. Recommends observation of lesions, as there is difficulty in obtaining surgical access to these areas
Zone 4: Comprises the maxilla and mandible and tooth-bearing portions of the skull. Recommends conservative management as there is difficulty in reconstructing defects.[3,7,17,19]
CONCLUSION
Most disaster in craniofacial FD is it occurs in the first few decades of life leading to psychological trauma. Growth of the lesion occurs till bone maturation is complete. Early diagnosis and appropriate treatment will increase the period of existence. Long-term follow-up plays a key role. Even with varied treatment options, no procedure is completely successful. Treatment has to be individualized for every patient based on site, extent, and symptoms of the disease. Knowledge regarding the disease is vital for diagnosis, treatment, and follow-up.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Sumita M, Mala K, Karen B. Maxillofacial fibrous dysplasia. Indian J Dent Res. 2005;16:151–2. doi: 10.4103/0970-9290.29905. [DOI] [PubMed] [Google Scholar]
- 2.Amol J, Saurav M, Krutik P, Shaikh A, Shopnil P. Fibrous dysplasia: A case series of five cases. Int J Adv Med. 2016;3:1068–73. [Google Scholar]
- 3.Deepthi A, Jayanth BS, Begum F, Shreyas O, Rao A. Fibrous dysplasia of the craniofacial region – A brief understanding of the disease. J Adv Clin Res Insights. 2016;3:205–8. [Google Scholar]
- 4.Bhavana SM, Nagalaxmi V, Maloth KN, Lakshmi CR, Deshpande PS. Craniofacial fibrous dysplasia – A morbid presentation. J Pak Med Assoc. 2014;64:351–4. [PubMed] [Google Scholar]
- 5.Guru KN, Borle R, Guru RK. An unusual presentation of craniofacial fibrous dysplasia: A case report, review and update on management. Am J Oral Maxillofac Surg. 2015;2:15–22. [Google Scholar]
- 6.Sepúlveda I, Spencer ML, Flores P, Ulloa J. Craniofacial fibrous dysplasia: A case report and literature review. Case Rep Clin Pathol. 2016;4:47. [Google Scholar]
- 7.Chen YR, Chang CN, Tan YC. Craniofacial fibrous dysplasia: An update. Chang Gung Med J. 2006;29:543–9. [PubMed] [Google Scholar]
- 8.Sandhu SV, Sandhu JS, Sabharwal A. Clinicoradiologic perspective of a severe case of polyostotic fibrous dysplasia. J Oral Maxillofac Pathol. 2012;16:301–5. doi: 10.4103/0973-029X.99097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guruprasad Y, Prabhakar C. Craniofacial polyostotic fibrous dysplasia. Contemp Clin Dent. 2010;1:177–9. doi: 10.4103/0976-237X.72787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cdr AA, Capt BC. Craniofacial fibrous dysplasia presenting with visual impairment. Med J Armed Forces India. 2003;59:342–3. [Google Scholar]
- 11.Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, et al. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S2. doi: 10.1186/1750-1172-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bijai LK, Mathew P, Jayaraman V, Austin RD. Fibrous Dysplasia – A case report and review of literature. Int J Dent Sci Res. 2014;2:109–11. [Google Scholar]
- 13.Mahadesh J, Gowda C, Devi L, Kokila G. Fibrous dysplasia of the jaw bones: Clinical, radiographical and histopathological features. Report of two cases. J Dent Sci Res. 2011;2:18–25. [Google Scholar]
- 14.Liakos GM, Walker CB, Carruth JA. Ocular complications in craniofacial fibrous dysplasia. Br J Ophthalmol. 1979;63:611–6. doi: 10.1136/bjo.63.9.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark J, Carson W. A case of craniofacial polyostotic fibrous dysplasia. J Radiol Case Rep. 2010;4:1–6. doi: 10.3941/jrcr.v4i9.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM, Gil Z. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia – A meta-analysis. PLoS One. 2011;6:e25179. doi: 10.1371/journal.pone.0025179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon S, Venkatswamy S, Ramu V, Banu K, Ehtaih S, Kashyap VM. Craniofacial fibrous dysplasia: Surgery and literature review. Ann Maxillofac Surg. 2013;3:66–71. doi: 10.4103/2231-0746.110088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Y, Liu J, Zhang C, Zhang Z, Yang H, Hu J. Maxillofacial fibrous dysplasia: A clinical analysis of 72 cases. Biomed Res. 2017;28:2498–2503. [Google Scholar]
- 19.Rahman AM, Madge SN, Billing K, Anderson PJ, Leibovitch I, Selva D, et al. Craniofacial fibrous dysplasia: Clinical characteristics and long-term outcomes. Eye (Lond) 2009;23:2175–81. doi: 10.1038/eye.2009.6. [DOI] [PubMed] [Google Scholar]