

Abstract
High-resolution Cas9 structures have yet to reveal catalytic conformations due to HNH nuclease domain positioning away from the cleavage site. Nme1Cas9 and Nme2Cas9 are compact nucleases for in vivo genome editing. Here we report structures of meningococcal Cas9 homologs in complex with sgRNA, dsDNA, or the AcrIIC3 anti-CRISPR protein. DNA-bound structures represent an early step of target recognition, a later HNH pre-catalytic state, the HNH catalytic state, and a cleaved-target-DNA-bound state. In the HNH catalytic state of Nme1Cas9, the active site is seen poised at the scissile phosphodiester linkage of the target strand, providing a high-resolution view of the active conformation. The HNH active conformation activates the RuvC domain. Our structures explain how Nme1Cas9 and Nme2Cas9 read distinct PAM sequences and how AcrIIC3 inhibits Nme1Cas9 activity. These structures provide insights into Cas9 domain rearrangements, guide-target engagement, cleavage mechanism, and anti-CRISPR inhibition, facilitating the optimization of these genome editing platforms.
Graphical Abstract
eTOC blurb
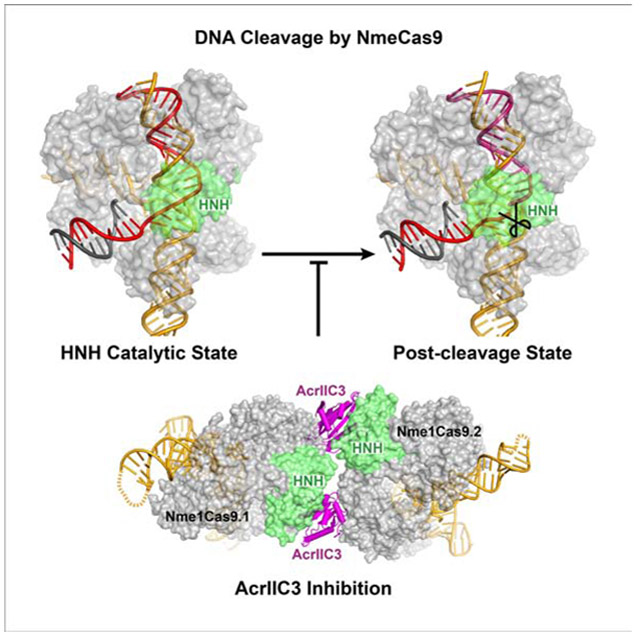
Sun et al. determined the crystal structures of two Neisseria meningitidis Cas9 homologs in sgRNA-loaded, DNA-bound, and AcrIIC3-inhibited states. The structures reveal the catalytically poised conformation as well as pre-catalytic and post-cleavage states. In the AcrIIC3-inhibited conformation, two AcrIIC3 monomers tether two Nme1Cas9 RNPs together.
Introduction
Cas9, the signature protein of Type II CRISPR-Cas systems, is an RNA-programmable DNA endonuclease that cleaves target dsDNA via RuvC and HNH domains (Gasiunas et al., 2012; Jinek et al., 2012). CRISPR-Cas9 protects bacteria against invasive nucleic acids and also provides a revolutionary RNA-guided genome editing platform (Komor et al., 2017). Cas9 activity requires a crRNA guide, a tracrRNA cofactor, and a protospacer adjacent motif (PAM) sequence in the DNA target (Jinek et al., 2012). The crRNA and tracrRNA can be fused into a single-guide RNA (sgRNA) (Jinek et al., 2012). Multiple Cas9 structures in different binding forms have been reported (Anders et al., 2014; Jinek et al., 2014; Nishimasu et al., 2014; Jiang et al., 2015; Jiang et al., 2016; Hirano et al., 2016; Yamada et al., 2017; Hirano et al., 2019; Zhu et al., 2019a). However, in currently available DNA-bound, pre-cleavage Cas9 structures, the HNH active site is distant from the scissile phosphodiester of the crRNA-complementary target strand (TS), suggesting that the HNH domain undergoes significant conformational changes from the determined structures before cleaving the TS. To understand the mechanism of TS cleavage, including the enzyme’s detection of guide-TS complementarity that is sufficient for nuclease activation, it is critical to elucidate the structures of DNA-bound Cas9 complexes in their catalytically competent states.
Two Cas9 homologs from Neisseria meningitidis [Nme1Cas9 and Nme2Cas9, Type II-C (Mir et al., 2018)] have been developed as genome-editing tools (Amrani et al., 2018; Bolukbasi et al., 2018; Edraki et al., 2019; Esvelt et al., 2013; Hoffmann et al., 2019; Hou et al., 2013; Ibraheim et al., 2018; Lee et al., 2016, 2019). At 1,082 amino acids, they are both smaller than the commonly used Type II-A ortholog SpyCas9 (1,368 amino acids), enabling delivery of Nme1Cas9 or Nme2Cas9, along with sgRNA, with single adeno-associated virus (AAV) vectors (Edraki et al., 2019; Hoffmann et al., 2019; Ibraheim et al., 2018). The PAM-interacting (PI) domains of Nme1Cas9 and Nme2Cas9 are highly diverged (52% identity) even though the other portions of the proteins are >98% identical. This divergence leads to distinct PAM specificities: N4GAYW/N4GYTT/N4GTCT for Nme1Cas9 (Amrani et al., 2018; Esvelt et al., 2013; Hou et al., 2013; Lee et al., 2016; Zhang et al., 2015), and N4CC for Nme2Cas9 (Edraki et al., 2019) [PAM sequences are 5’ to 3’ on the non-target strand (NTS)]. Both can also be inhibited by anti-CRISPR (Acr) proteins encoded by mobile genetic elements (Edraki et al., 2019; Hoffmann et al., 2019; Lee et al., 2018, 2019; Pawluk et al., 2016), representing potentially powerful off-switch strategies to control Cas9 (Pawluk et al., 2018), including in the tissues of adult mammals (Lee et al., 2019). Five anti-CRISPRs (AcrIIC1-AcrIIC5) have been identified that inhibit Nme1Cas9 (Lee et al., 2018; Pawluk et al., 2016), and all but AcrIIC5Smu also inhibit Nme2Cas9 (Edraki et al., 2019). Nme1Cas9 and Nme2Cas9 both cleave mammalian genomes with little or no off-targeting (Amrani et al., 2018; Edraki et al., 2019; Ibraheim et al., 2018; Lee et al., 2016). However, the complete structures of Nme1Cas9 and Nme2Cas9 have not been reported.
Here we report the structures of Nme1Cas9 and Nme2Cas9 in five distinct functional conformations: (i) Nme1Cas9-sgRNA, (ii) Nme1Cas9 bound to a seed-matched DNA with 8 bp, (iii) Nme2Cas9-sgRNA in a fully complementary DNA-bound but pre-catalytic state, (iv) Nme1Cas9-sgRNA in a fully complementary DNA-bound and catalytically activated state, and (v) Nme1Cas9-sgRNA bound to a fully complementary DNA after TS cleavage. In addition, we present structures of a potent anti-CRISPR (AcrIIC3Nme, Pawluk et al., 2016) in complex with Nme1Cas9-sgRNA, in both DNA-unbound and DNA-bound states. Our structure of Nme1Cas9-sgRNA-dsDNA represents the first high-resolution view of a Cas9 ortholog in the catalytically poised state, with extensive readout of the guide-TS heteroduplex backbone and the HNH domain active site positioned within angstroms of the TS scissile phosphate. These studies will provide a wealth of new structural and mechanistic insight to apply to the Cas9 protein family.
Results
Structure of Nme1Cas9 in Complex with sgRNA
To understand how Nme1Cas9 assembles with the sgRNA (Figure 1A), we determined the crystal structure of Nme1Cas9, in complex with a 135-nucleotide (nt) truncated sgRNA, at 2.95 Å resolution (Figure 1B and Table S1). Nme1Cas9 adopts a bi-lobed architecture consisting of a REC lobe and a NUC lobe, similar to that reported for other Cas9 homologs (Figure 1C). The REC lobe comprises REC1 and REC2 domains. The REC2 domain of Type II-C Cas9s is similar to the REC3 domain of SpyCas9, with the REC2 subdomain of SpyCas9 being absent from Type II-C and other small Cas9s. The NUC lobe consists of the RuvC, HNH, WED and PI domains. The REC and NUC lobes are connected by an arginine-rich bridge helix (BH).
Figure 1. The overall structure of Nme1Cas9 in complex with truncated sgRNA.

A. Domain organization of Nme1Cas9.
B. Schematic representation of the 135-nt sgRNA used for crystallization. The repeat:anti-repeat region is highlighted in green background. The stem-loops 1 and 2 are highlighted in pink and grey background, respectively. The black and blue dashed-line boxes indicate the truncated nts of the 122-nt and 102-nt sgRNAs, respectively. The cartoon diagram represents the state of Nme1Cas9 in this structure.
C. Crystal structure of the Nme1Cas9-sgRNA binary complex. Individual Nme1Cas9 domains are colored according to the scheme in Figure 1A.
D. DNA cleavage by wild-type and mutant Nme1Cas9 using linearized plasmid DNA containing a target sequence fully complementary to the sgRNA, and various sgRNAs containing different truncations.
E. Top: schematic representation of the partially double-stranded DNA target with its TS complementary to guide nts 17-24 (seed only). Bottom: crystal structure of the seed-only Nme1Cas9-sgRNA-dsDNA complex.
F. Conformational changes of Nme1Cas9 before (C) and after (E) PAM recognition and seed pairing.
See also Figures S1, S2, and Table S1.
The sgRNA, which consists of a guide region (nts 1-24), a repeat region (25-42) and the tracrRNA (47-135), is positioned in the positively-charged cleft formed by the REC and NUC lobes. The tracrRNA 3' tail contains two stem-loops (stem-loops 1 and 2), like that of SauCas9 (Nishimasu et al., 2015; Ran et al., 2015) but differing from the SpyCas9 tracrRNA that contains three stem-loops (Deltcheva et al., 2011). Nme1Cas9 makes extensive hydrogen-bond contacts and stacking interactions with the sgRNA (Figure S1). We observed unambiguous electron density only for guide nts 15-24, all of which are located in the seed region, whereas nts 1-14 are disordered. This observation suggests that in the absence of target DNA, the Nme1Cas9 sgRNA seed is pre-ordered in a nearly A-form conformation, similar to that of SpyCas9’s sgRNA (Jiang et al., 2015). The sugar-phosphate backbone of the ordered seed region forms extensive interactions with the arginine-rich BH.
Only 13 bp of the repeat:anti-repeat duplex contact the REC1 and WED domains, whereas the other end of that duplex lacks interactions with Cas9, suggesting that they are not critical for Cas9 activity. We tested this by making different deletions in the sgRNA and found that truncation from full-length 147 nt to 135 nt is compatible with Nme1Cas9 activity in vitro (Figure 1D), in agreement with previous results in bacteria (Zhang et al., 2015). Stem-loop 2, which has 46 nts, is located in the cleft between the RuvC and PI domains. U127 (U139 in the full-length sgRNA) is flipped out and stabilized by stacking on the side-chain of Met838 and by hydrogen bonding with Arg949 and Pro1080 (Figure S2A). Truncation of stem-loop 2 reduced Nme1Cas9 activity, whereas the sgRNA lacking stem-loop 2 exhibited no activity (Figure 1D).
The repeat:anti-repeat duplex, as well as stem-loops 1 and 2, are sequence-specifically recognized by the WED, REC1, BH and RuvC domains of Nme1Cas9. Residues Lys909 and Arg880 form hydrogen bonds with nts C34 and C36 in the repeat region, respectively. Arg880 and Asn108 form base contacts with C53 and U59 in the anti-repeat region, respectively (Figures S2B and S2C). U132 in stem-loop 2 is sequence-specifically recognized by Arg62 in the BH via hydrogen bonding (Figure S2D). A65 in stem-loop 1 forms base contacts with the main-chains of Gly840 and Gly842 (Figure S2E).
Crystal Structure of a Seed-paired Nme1Cas9-sgRNA-dsDNA Ternary Complex
To illuminate the basis for TS recognition by the crRNA seed, we solved the crystal structure at 3.3 Å resolution of Nme1Cas9 in complex with the sgRNA and a partially duplexed DNA that contains 8 nts of the PAM-proximal TS, as well as an optimal N4GATT PAM (Figures 1E and S2F). The 8 nts of the TS base pair with seed nts 17-24 of the sgRNA guide region to form an RNA-DNA heteroduplex. The 8-bp heteroduplex occupies the cleft between the REC and NUC lobes. Guide nts 1-16, as well as parts of the REC2 domain, are disordered. The HNH domain is located similarly to its position in the Nme1Cas9-sgRNA binary complex, including direct interactions with the RuvC and REC2 domains. In contrast, the resolvable portion of the REC2 domain is rotated ~24° away from the REC1 domain after seed pairing is established (Figure 1F). The outward rotation of the REC2 domain generates a wider groove between the REC and NUC lobes, even though the 8-bp RNA-DNA duplex does not reach the REC2 domain. This structure represents an early step of guide-TS recognition and suggests that the REC2 domain rearranges during the initial phases of heteroduplex formation, in a manner that likely facilitates PAM-distal propagation of base pairing.
Structure of a Fully Paired Nme1Cas9-sgRNA-dsDNA Ternary Complex
To investigate the molecular mechanisms responsible for the ability of Nme1Cas9 to cleave target dsDNA, we determined the crystal structure at 3.1 Å resolution of Nme1Cas9 His588Ala bound to full-length 147-nt sgRNA and a partial-duplex DNA containing all 24 guide-complementary nts of the TS as well as an N4GATT PAM (Figures 2A, 2B, S3A and S3B). The mutation of the HNH domain catalytic residue His588 to alanine (Esvelt et al., 2013; Hou et al., 2013; Zhang et al., 2013, 2015) was included to prevent DNA cleavage in the TS. The overall domain organization of fully paired, DNA-bound Nme1Cas9 is similar to that of the CjeCas9-sgRNA-dsDNA and CdiCas9-sgRNA-dsDNA Type II-C structures (Yamada et al., 2017; Hirano et al., 2019), except for the HNH domain, which was deleted in both. As observed in SpyCas9 and SauCas9 (Anders et al., 2014; Nishimasu et al., 2014, 2015; Jiang et al., 2016), the guide RNA and the TS form a heteroduplex that resides in the central channel formed by the REC and NUC lobes and that is stabilized by the REC2, BH, HNH and RuvC domains (Figure 2C). In contrast to the Nme1Cas9-sgRNA binary and seed-paired Nme1Cas9-sgRNA-DNA ternary structures, in which the 5' end of the guide RNA was disordered, density was visible for the entire 24-nt spacer segment. The sugar-phosphate backbone of the PAM-distal heteroduplex region (involving guide nts 1-12) interacts with the REC2 and RuvC domains as well as the L1 linker (Figure 2C). The PAM-proximal, seed-containing half of the guide-TS heteroduplex is extensively stabilized by the RuvC, REC1, REC2 and HNH domains as well as the BH. The phosphate lock loop (Glu845-Thr846) in the WED domain contacts the phosphodiester linkage that connects the variable, 24-nt guide sequence to the invariant repeat region of the sgRNA.
Figure 2. Overall structure of the HNH-inactivated, fully complementary Nme1Cas9-sgRNA-dsDNA complex.

A. Schematic diagram of the sgRNA and DNA target used for co-crystallization. Guide RNA, TS DNA and NTS DNA are in orange, red and black, respectively.
B. Cartoon and surface representation of Nme1Cas9 His588Ala in complex with sgRNA and a fully complementary DNA duplex bearing a 5'-N4GATT-3' PAM sequence.
C. Contacts between Nme1Cas9 residues and the guide-TS heteroduplex in the catalytically poised structure. Residues are color-coded by domain as in Figures 1A and 2B.
D. Zoomed-in view of the PAM-binding region in Nme1Cas9.
E. DNA endonuclease activity of wild-type (WT) and mutant Nme1Cas9 using a linearized plasmid DNA containing a target sequence fully complementary to the sgRNA shown in (A).
Recognition of the N4GATT PAM
The N4GATT double-stranded PAM region of the DNA target inserts between the PI and WED domains. Unlike SpyCas9 and SauCas9, which recognize the PAM via the NTS (Anders et al., 2014; Nishimasu et al., 2015), Nme1Cas9 reads the sequence of both the target and non-target DNA strands (Figure 2D), explaining previous results from PAM mutagenesis studies (Zhang et al., 2015). The side-chain of His1024 forms two hydrogen bonds with the base of NTS nt G(−5)', and Thr1027 forms a hydrogen bond with the base of NTS nt A(−6)'. The side-chains of Asn1029 and Gln981 form base contacts with nts A(−7) and A(−8) in the TS, respectively. Our mutagenesis studies showed that His1024Ala completely abolished Cas9 activity in vitro, and Gln981Ala, Thr1027Ala and Asn1029Ala substitutions substantially reduced activity (Figure 2E).
Considerable variability from Nme1Cas9’s optimal N4GATT PAM is already known to be compatible with genome editing (Amrani et al., 2018). In the DNA-bound Nme1Cas9 complex, the interaction of Asn1029 with A(−7) in the TS involves hydrogen bond donation to the N7 position of the purine ring (Figure 2D). This N7 is shared by both adenine and guanine, explaining how certain PAMs with either purine in this position can function with Nme1Cas9.
The HNH Nuclease Domain of Nme1Cas9 is in the Catalytic State
The HNH domains of Cas9 orthologs use a metal ion cofactor to catalyze TS cleavage between nts 3 and 4 of the protospacer (counting from the PAM-proximal end). In previously reported structures of Cas9-sgRNA complexes containing uncleaved target DNA, the active site of the HNH domain is neither pointed toward nor located near the TS cleavage site (Anders et al., 2014; Nishimasu et al., 2014, 2015; Hirano et al., 2016; Jiang et al., 2016). In the CjeCas9-sgRNA-dsDNA and CdiCas9-sgRNA-dsDNA structures, the HNH domain was deleted altogether to facilitate crystallization (Yamada et al., 2017; Hirano et al., 2019). In contrast to these structures, the intact HNH domain in our dsDNA-bound Nme1Cas9 complex is positioned close to the target DNA. The HNH domain and Linker L1 form extensive hydrogen-bond contacts with the backbone of the guide-TS duplex in and just beyond the seed region. The side-chains of Asn526, Arg530 and Arg557 form hydrogen bonds with the phosphate groups of guide nts 12 and 13, respectively (Figure 2C). The side-chains of Lys549, Lys555, Lys581, Ser593, Asn611 and Gln612, and the main-chains of residues Ile586 and His588, form hydrogen bonds with the sugar-phosphate backbone of TS seed nts 3-8 (Figure 3A). Lys555, Ser593 and Gln612 are modestly conserved in the HNH domain. These interactions between the HNH domain and the guide-TS heteroduplex keep the HNH domain poised at the cleavage site of the TS.
Figure 3. HNH catalytic conformation.

A. The interaction between the HNH domain in the catalytic state and the TS.
B. Close-up view of the catalytic site of the HNH domain and the TS cleavage site (the phosphate between TS 3 and 4). The Mg2+ is shown as a magenta sphere. The distances between the Mg2+ and the coordinated atoms are 2.1-2.3 Å.
C. The interaction of Linker L1 and the RNA-DNA duplex.
D-E Structural comparison of the HNH domain in the seed-matched (D) and fully paired (E) states showing that the formation of the RNA-DNA heteroduplex drives the conformational change of the HNH domain.
F-G. The DNA cleavage assay using linear plasmid DNA.
H. The schematic representation of the Cy3- and Cy5- labeled short DNA (TS and NTS, respectively).
I-J. DNA cleavage of the Cy3- and Cy5-labeled DNA with Nme1Cas9 mutants at 25°C.
See also Figure S3.
Importantly, all catalytic amino acids (Asp587, His588Ala and Asn611) are located close to TS nts 3 and 4 (Figure 3B), explaining why the HNH domain cleaves the TS between these nts. The distance between the catalytic residue Asp587 and the scissile phosphate is ~3.4 Å, sufficiently close to support the catalytic activity of Nme1Cas9. As shown recently with SpyCas9, binding to a divalent cation is a prerequisite for HNH activation (Dagdas et al., 2017; Gong et al., 2018). In our DNA-bound Nme1Cas9 complex, a Mg2+ ion is located in the catalytic pocket of the HNH domain, coordinating with the side-chains of the catalytic residues Asp587 and Asn611, as well as two oxygen atoms of the scissile phosphate of TS nts 3 and 4. This observed binding of Mg2+ is consistent with the requirement for one metal ion for HNH activity (Gong et al., 2018) and with cleavage products having 5'-phosphate and 3'-OH termini (Li et al., 2003). The Cβ atom of His588Ala points towards the scissile phosphate, suggesting that the side-chain of His588 facilitates hydrolysis of the phosphate during target cleavage. We observed that the target DNA is not cleaved in the DNA-bound ternary complex, confirming that His588 is essential for HNH domain activity (Zhang et al., 2015). Together, these results clearly indicate that the HNH domain in our dsDNA-bound Nme1Cas9 structure adopts a catalytically active conformation. These results also suggest that the metal is critical for the HNH domain to adopt an active conformation, whereas the protospacer region of the NTS is not essential.
The structure of the catalytically poised complex also suggests that linkers L1 and L2, which connect the HNH and RuvC domains, are essential for the dynamics of the HNH domain. In this structure, L1 forms a long α-helix and is positioned across ~10 bp the RNA-DNA duplex (Figure 3C). Positively charged residues (Arg516, Lys517 and Gln523) form hydrogen bonds with the TS, stabilizing the conformation of the HNH domain.
Guide-target Duplex Propagation Triggers Conformational Changes of the L1 Linker and the RuvC, REC2 and HNH Domains
Current models of Cas9 target recognition involve the initial establishment of guide-TS seed pairing followed by the PAM-distal propagation of the heteroduplex to the 5' end of the guide RNA. To identify domain rearrangements that accompany guide-TS heteroduplex propagation, we compared the DNA-bound Nme1Cas9 complexes in the partially (Figure 1E) and fully (Figure 2B) base-paired states by aligning the REC1, PI and WED domains. Our structural comparison showed that the HNH domain undergoes significant conformational changes during or after guide RNA-DNA duplex propagation (Figures 3D, 3E and S3C). The HNH domain movement that we observe is consistent with the dynamics inferred from SpyCas9 fluorescence measurements and molecular simulations (Chen et al., 2017; Dagdas et al., 2017; Palermo et al., 2017; Sternberg et al., 2015; Yang et al., 2018; Zuo and Liu, 2017), and with a very recent SpyCas9 post-cleavage cryo-EM structure (Zhu et al., 2019a). In the absence of dsDNA and in the seed-only complex, the HNH domain contacts the RuvC domain, far from the sgRNA seed region (Figures 3D and S3D). Upon forming the complete, 24-bp sgRNA-DNA duplex, the HNH domain rotates towards the guide-TS heteroduplex, resulting in a structure with the domain properly positioned to cleave the target DNA strand (Figure 3E). This heteroduplex-docked conformation also includes new contacts (not observed in the pre-catalytic structures) between the HNH and REC1 domains.
A striking aspect of our catalytically poised structure is the dense collection of contacts between multiple Nme1Cas9 domains and the entire length of the guide-TS heteroduplex backbone (Figure 2C). These contacts extend all the way out to the three most PAM-distal base pairs, which interact with RuvC residues. Guide-TS base pairs 4-9 (counting from the 5’ end of the guide) interact extensively with the REC2 domain as well as with additional RuvC residues, and base pairs 10-12 contact the long L1 helix described above (Figure 3C). The formation of these interactions, presumably made possible by the PAM-distal propagation of guide-target base pairing, likely help to drive the extensive rearrangements of the RuvC and REC2 domains and the L1 linker (Figure S3C), which in turn enable the large-scale rotation and translocation that docks the HNH domain onto the scissile phosphodiester linkage. This may be particularly true for the RuvC domain, since its formation of PAM-distal heteroduplex contacts could help drive the disruption of its direct interactions with the HNH domain, freeing the latter for its conformational transition into the catalytically poised state. Intriguingly, previous Nme1Cas9 sgRNA truncations reveal a loss of genome editing activity when the 5’ end of the guide is shortened by 4-6 residues (Amrani et al. 2018), corresponding to the likely loss of many of the PAM-distal RuvC and REC2 contacts. Consistent with this, in vitro cleavage assays with linear dsDNA show that 4 mismatches at the PAM-distal end reduce cleavage by Nme1Cas9, and 6 or more terminal mismatches abolish cleavage (Figure S3E). The combined structural and functional results strongly suggest that the PAM-distal propagation of guide-TS pairing into a nearly full-length (20-24 base pairs) heteroduplex supports L1, REC2 and RuvC substrate interactions that in turn enable the HNH domain's transition to the catalytic conformation.
Nme1Cas9 Mutants with Increased DNA Cleavage Efficiency
The HNH domain is flexible and exhibits a conformational equilibrium that usually favors positioning distant from the scissile phosphodiester (Jiang and Doudna, 2017). We reasoned that stabilizing the HNH catalytic conformation via strengthened interaction between the HNH domain and the guide-TS heteroduplex may improve the DNA cleavage activity of Cas9. To test this, we replaced some amino acids positioned close to the TS by Arg, Lys or Gln, which are often employed for side-chain interactions with nucleic acid backbone groups. We observed that single or double mutations of amino acids Ser593 and Trp596 dramatically increase the extent of dsDNA cleavage by Nme1Cas9 (Figure 3F). Double mutants Ser593Gln/Trp596Arg and Ser593Gln/Trp596Lys cleaved the substrate to a similar extent as SpyCas9 under these conditions and were more efficient than the similarly compact SauCas9 (Figure 3G). The increase in cleavage efficiency in vitro by the Ser593/Trp596 mutants was much more substantial at 25°C than at 37°C (Figures 3F and S3F). These temperature-dependent effects may be due to decreased mobility of the HNH domain at the lower temperature, limiting the frequency of its transition into the catalytic state. The effects of the additional interactions that stabilize the HNH domain’s catalytic conformation may therefore become unmasked at the lower temperature. It should be noted that increased DNA cleavage activity might lead to a corresponding increase in off-target as well as on-target editing. Nonetheless, these results suggest that stabilizing the HNH domain in the catalytic conformation can improve the DNA cleavage activity of Nme1Cas9, providing new insights into the design of Cas9 variants with improved properties. These results also suggest that distinct strategies will likely be needed to improve Nme1Cas9 activity during mammalian genome editing applications, especially if a different step [e.g. initial DNA unwinding (Gong et al., 2018; Ma et al., 2015)] limits the overall efficiency.
The HNH Catalytic Conformation Promotes Activation of the RuvC Domain
To test whether the strengthened HNH-TS interactions affect the activity of both the HNH and RuvC domains, we performed DNA cleavage assays using the Cy3- and Cy5-labeled short dsDNA to test strand-specific effects on DNase activity (Figure 3H). The single or double mutation of residues Ser593 and Trp596 improved both TS and NTS cleavage to a similar extent (Figure 3I). Conversely, destabilization of the HNH catalytic conformation via alanine substitution of L1 residues Arg516 and Lys517, which contact a TS backbone phosphate, displayed reduced cleavage efficiency of the NTS as well as the TS. In contrast, alanine substitutions of HNH catalytic residues, which do not directly contact the TS by their side-chains, are strand-specific: they have no effect on NTS cleavage efficiency even though they abolish TS cleavage (Figure 3J). Taken together, our results indicate that stabilizing the HNH catalytic conformation increases the DNase activities of both HNH and RuvC domains, whereas destabilizing the HNH catalytic conformation reduces the enzymatic activity of both nuclease domains. Because mutations in HNH catalytic residues (e.g. His588Ala) do not prevent the adoption of the catalytic conformation (Figure 2B), they do not affect NTS cleavage by the RuvC domain. It is noteworthy in this context that kinetic analyses of SpyCas9 show that TS cleavage by the HNH domain usually precedes NTS cleavage by the RuvC domain (Gong et al., 2018), implying that the HNH catalytic state is usually traversed first. These results are consistent with a model in which the dynamics of the HNH domain regulate the activity of the RuvC domain, and that the catalytic conformation of the HNH domain is crucial for activating the RuvC domain, as suggested previously by ensemble and single-molecule studies (Sternberg et al., 2015).
Structure of a Product-bound Nme1Cas9 Complex
To capture the structure of a DNA-product-bound state, we solved the structure of wild-type Nme1Cas9 in complex with a 135-nt sgRNA and partially duplexed DNA, in the presence of MgCl2, at 2.9 Å resolution. In contrast to the His588Ala mutant complex, in which the TS is intact, the phosphodiester bond between nts 3 and 4 of the TS is broken in the DNA-bound, wild-type Nme1Cas9 complex (Figures 4A and 4B), indicating that this structure represents the TS-cleaved, product-bound state.
Figure 4. The crystal structure of Nme1Cas9-sgRNA in complex with the cleaved, partial-duplex DNA.

A. Overall structure of the product-bound wild-type Nme1Cas9.
B. Close-up view of the catalytic site of the HNH domain and the RNA-DNA duplex. The 3'-product of the cleaved DNA is in light magenta (left panel). The Fo-Fc electron density omit map of nts 2-5 in the target strand (contoured at 3.0 σ) is shown.
C. Interactions between Nme1Cas9 and the RNA-DNA duplex. The two fragments of broken DNA are in red and light magenta.
D. Structural comparison of the Linker L1 and HNH domain in the HNH catalytic state (in grey) and the post-cleavage state.
E. Conformational change of Nme1Cas9 upon TS cleavage.
F. RNA-DNA duplex comparison in the HNH catalytic state (in grey) and the product-bound state showing the shift of the cleaved TS. The colors in the product-bound state are identical to those used in (A).
See also Table S1.
The PAM-proximal fragment of the cleaved TS has a 5’-phosphate group at the cleaved site, whereas the PAM-distal fragment has a 3'-OH group. Both fragments maintain their base pairing with the sgRNA. In the product-bound Nme1Cas9 complex, the HNH domain rotates away from the guide-TS heteroduplex and disengages from the direct substrate contacts observed in the HNH catalytic state (Figures 4C-4F). Compared with the intact guide-TS heteroduplex in the His588Ala mutant complex, protospacer nts 2 and 3 (complementary to guide nts 23 and 22) translate away from the sgRNA by ~3.7 Å (Figure 4F). The distance between the scissile phosphate and Cα of His588 is 11.1 Å in the product-bound state, whereas this distance is only 5.2 Å in the HNH catalytic state. In addition, HNH-REC1 interactions that are observed in the HNH catalytic state are lost in the product state. Collectively these results suggest the existence of conformational responses to TS hydrolysis by the HNH domain. These results are consistent with kinetic analyses suggesting that a likely conformational step intervenes between HNH and RuvC cleavage events, accounting for the slower rate of the latter (Gong et al., 2018).
Structure of an Nme2Cas9-sgRNA-dsDNA Complex in a Pre-catalytic State
The high target site density of Nme2Cas9 afforded by its dinucleotide (N4CC) PAM makes it particularly useful as an AAV-deliverable genome editing platform (Edraki et al., 2019; Lee et al., 2019). Although the structures and interactions of the N-terminal 820 amino acids of Nme2Cas9 should be highly similar to those of Nme1Cas9 (due to their >98% identity in these regions), the PI and WED domains of the two orthologs are far more divergent, resulting in their distinct PAM specificities. To investigate the molecular mechanism of Nme2Cas9 PAM recognition, we determined the crystal structure of Nme2Cas9 in complex with a 135-nt truncated sgRNA, together with a partially duplexed target DNA containing a N4CC PAM sequence (Figures S4A and S4B). Nme2Cas9 Asp16Ala/His588Ala mutants were used to prevent potential DNA cleavage. We solved the co-crystal structures with two distinct dsDNAs with a complete protospacer in the TS, but with either no protospacer or a 5-nt protospacer in the NTS (Figure S4C). These structures were solved at 3.15 Å and 2.93 Å resolution, respectively. The two structures of DNA-bound Nme2Cas9 complexes are similar overall, and the HNH domain is completely disordered in both (Figure 5A and Table S2). Only two NTS protospacer nts are observed, while the other three are disordered in the 5-nt protospacer DNA-bound Nme2Cas9 complex. The dsDNA is positioned in the cleft formed by the PI and WED domains, as seen in Nme1Cas9. In the PAM region, the side-chain of Asp1028 accepts a hydrogen bond from the exocyclic amino group of C(−5)' of the NTS (Figures 5B and S4D). T or G residues in this position would present a keto oxygen that cannot act as a hydrogen bond donor. In addition, if adenine were to replace cytosine, the distance between the side-chain of Asp1028 and the exocyclic -NH2 group would be too great to form a favorable hydrogen bond. These observations therefore explain the strong requirement for a C at that position (Edraki et al., 2019). In contrast to NTS recognition of C(−5)', the side-chain of Arg1033 forms a pair of hydrogen bonds with the base of G(−6) on the TS, indicating that Nme2Cas9 (like Nme1Cas9) uses both the TS and the NTS for PAM recognition. The other nts within the duplex dsDNA have no base contacts with Nme2Cas9.
Figure 5. The crystal structure of the Nme2Cas9-sgRNA-dsDNA complex, and requirements for Nme1Cas9/Nme2Cas9/sgRNA residues during genome editing.

A. The overall structure of the Nme2Cas9-sgRNA-dsDNA complex.
B. Detailed view of the interaction between the PI domain of Nme2Cas9 and the 5'-N4CC-3' PAM sequence.
C. Editing efficiency of wild-type and mutant Nme1Cas9 at two genomic sites in HEK293T cells.
D. Nme1Cas9 editing efficiency of wild-type and mutant sgRNA at two genomic sites in HEK293T cells.
E. Editing efficiency of wild-type and mutant Nme2Cas9 at two genomic sites in HEK293T cells.
Editing efficiency values are mean ± s.d. from three biological replicates.
See also Figures S4, S5 and Table S2.
In both structures of Nme2Cas9, the HNH domain and Linker L1 are disordered. In contrast, in the fully paired, HNH-inactivated Nme1Cas9-sgRNA-DNA complex, L1 is well ordered and interacts with the guide-TS heteroduplex. Superposition of DNA-bound Nme1Cas9 and Nme2Cas9 complexes shows that L2 also adopts a different conformation (Figure S4E). Together, these observations indicate that the DNA-bound Nme2Cas9 complex represents a likely pre-catalytic state.
PAM-interacting Residues and sgRNA-protein Contacts are Important for Mammalian Genome Editing Activity
To test the importance of specific Nme1Cas9 amino acids in mammalian genome editing, we mutated Arg880 and Lys909 (implicated in repeat:anti-repeat duplex recognition; Figure S2B) and Gln981, His1024, Thr1027 and Asn1029 (implicated in PAM recognition; Figure 2D) to alanine in HA-tagged Nme1Cas9 expression plasmids. We then co-transfected wild-type and mutant Nme1Cas9 constructs, along with sgRNA-expressing plasmids targeting previously validated genomic editing sites (Figure S5A), into HEK293T cells for genome editing. Consistent with previous results (Amrani et al., 2018), anti-HA western blots indicated that Nme1Cas9 accumulation requires co-expression with the cognate sgRNA (Figure S5B). All mutant proteins were expressed at wild-type levels except Lys909Ala, suggesting that this mutant fails to fold properly, or fails to stably load the sgRNA, or both. Indel efficiencies at the genomic editing sites [measured by PCR amplification, Sanger sequencing, and Inference of CRISPR Edits (ICE) analysis] revealed that, as expected, the Lys909Ala substitution abolished editing at both target sites tested [DTS3 (in ARHGEF9) and NTS55 (in CYBB)] (Figure 5C). In contrast, mutation of residue Arg880 shows only a modest reduction in editing efficiency relative to wild-type. Among the PAM-interacting residues tested, mutation of His1024 reduced editing to background levels (Figure 5C), consistent with in vitro results (Figure 2E). Alanine mutations in Gln981, Thr1027 and Asn1029 exhibited more modest effects on editing efficiency (Figure 5C). These results are consistent with functional roles for the PAM interactions (especially those of His1024) revealed by our crystallographic analyses (Figure 2D).
Our Nme1Cas9-sgRNA structure shows that, unlike most stem-loop 2 sgRNA nts, U139 of the full-length guide RNA [Figure 2A; equivalent to U127 in the truncated version (Figure 1B)] is flipped out and forms direct interactions with Arg949 and Pro1080 of Nme1Cas9 (Figure S2A). To assess the importance of this interaction, we deleted U139 and also mutated it to A, C or G. ICE analysis revealed that U139 deletion partially compromised editing efficiency in mammalian cells (Figure 5D), whereas the substitutions had little or no effect. These results indicate that U139 interactions play non-essential roles in Nme1Cas9 sgRNA loading.
To test the importance of the apparent Nme2Cas9 PAM-interacting residues Asp1028 and Arg1033, as well as a neighboring residue (Asn1031) as a negative control, we mutated each to alanine and examined editing efficiencies in transiently transfected HEK293T cells. The Asp1028Ala and Arg1033Ala mutations completely abolished Nme2Cas9 editing, whereas the Asn1031Ala mutation had no effect (Figures 5E and S5C). These results confirm the importance of the Asp1028 and Arg1033 interactions in PAM recognition and reveal the basis for differential PAM recognition by Nme1Cas9 and Nme2Cas9.
Two AcrIIC3 Proteins Tether Two Nme1Cas9 Proteins Together
AcrIIC3 was recently shown to be active as a genome editing off-switch in the tissues of adult mammals, enabling suppression of off-tissue mutagenesis (Lee et al., 2019). To define the state of Nme1Cas9 that binds to AcrIIC3, we performed size-exclusion chromatography (SEC). Our results showed that AcrIIC3 bound to all three of the Nme1Cas9 apo, sgRNA-loaded, and DNA-bound states (Figure 6A), indicating that AcrIIC3 binding neither depends upon nor prevents sgRNA loading and dsDNA binding. In addition, we observed similar results for Nme2Cas9 (Figure S6A), suggesting that AcrIIC3 inhibits the cleavage activities of Nme2Cas9 and Nme1Cas9 in a similar manner.
Figure 6. Structures of Nme1Cas9-sgRNA in complex with AcrIIC3 or with both AcrIIC3 and dsDNA.

A. AcrIIC3 binds to Nme1Cas9 in the apo, sgRNA-bound, and sgRNA-dsDNA-bound states.
B. Architecture of the Nme1Cas9-sgRNA in complex with AcrIIC3, showing that two AcrIIC3 monomers fasten two Nme1Cas9-sgRNA complexes together.
C. Architecture of the Nme1Cas9-sgRNA in complex with AcrIIC3 and dsDNA, showing that AcrIIC3 does not fully prevent target DNA binding.
We then determined the crystal structure of Nme1Cas9 in complex with sgRNA and AcrIIC3 at 3.46 Å resolution. In the Nme1Cas9-sgRNA-AcrIIC3 complex, two AcrIIC3 proteins are bound to two Nme1Cas9-sgRNA complexes (named Nme1Cas9.1 and NmeCas9.2) (Figure 6B). The two AcrIIC3 subunits do not contact with each other, suggesting that AcrIIC3 functions as a monomer. Similarly, the two sgRNA-bound Nme1Cas9 complexes in the AcrIIC3-bound structure have no direct interactions with each other, indicating that the two Nme1Cas9s are also independently bound monomers.
To investigate how AcrIIC3 affects the conformation of DNA-bound Nme1Cas9, we then determined the crystal structure of a Nme1Cas9-sgRNA-AcrIIC3-dsDNA complex at 3.06 Å resolution (Figure 6C and Table S3). To facilitate crystallization, the dsDNA in this quaternary complex has a forked structure bearing an N4GATT PAM sequence and a 24-nt protospacer region (Figure S6B). In the Nme1Cas9-sgRNA-AcrIIC3-dsDNA complex, the TS base pairs with the sgRNA to form a heteroduplex as in the AcrIIC3-free complex (Figure 2B). Seven nts in the spacer region of the NTS are observed, and the remainder is disordered. Two AcrIIC3 proteins interact with the two Nme1Cas9-sgRNA-dsDNA complexes in a similar manner as that seen in the Nme1Cas9-sgRNA-AcrIIC3 complex. Thus, two AcrIIC3 proteins are able to fasten two Nme1Cas9 proteins together, but do not promote the direct dimerization of Nme1Cas9 that was suggested by electron microscopy (Harrington et al., 2017).
AcrIIC3 Interacts with the HNH and REC2 Domains from Two Nme1Cas9s
In the AcrIIC3-bound Nme1Cas9 complexes, each AcrIIC3 monomer interacts with the HNH domain of one Nme1Cas9 and the REC2 domain of the other, thereby bridging the two Nme1Cas9s (Figure 7A). The HNH domain forms extensive hydrogen bond networks and aromatic stacking interactions with AcrIIC3. Residues Arg4, Tyr39, Glu91, Leu97 and Arg102 of AcrIIC3 interact with Glu560, Tyr559, Arg643, His563 and Lys565 of the HNH domain, respectively (Figure 7B). The L1 linker that connects the RuvC and HNH domains also forms numerous contacts with AcrIIC3 (Figure 7C). Simultaneously, the REC2 domain of the second Nme1Cas9 forms extensive contacts with this AcrIIC3 protein (Figure 7D). Thus, these two AcrIIC3 proteins tether two Nme1Cas9s, locking the positions of the REC2 and HNH domains in each.
Figure 7. Two AcrIIC3 monomers interact with the HNH and REC2 domains from two Nme1Cas9 complexes, inhibiting the HNH domain from rotating to the scissile site.

A. AcrIIC3 interacts with the HNH and REC2 domains from two Nme1Cas9 complexes.
B-D. The detailed interactions between AcrIIC3 and the HNH domain in Nme1Cas9.1 (B), Linker L1 in Nme1Cas9.1 (C), and the REC2 domain in Nme1Cas9.2 (D).
E. The HNH domain in the AcrIIC3-bound Nme1Cas9 complex is located distant from the cleavage site (highlighted by a magenta sphere) on the target strand. The RuvC and PI domains are omitted for clarity.
F. Schematic representations of interactions that enable AcrIIC3 inhibition of DNA cleavage by Nme1Cas9 and Nme2Cas9.
See also Figure S7.
AcrIIC3 Traps the HNH Domain Away from the Cleavage Site
Structural comparison of the two AcrIIC3-bound Nme1Cas9 complexes shows that the HNH domain in each adopts a conformation similar to that within the Nme1Cas9-sgRNA binary complex (Figures S6C and S6D). In the presence of AcrIIC3 the HNH domain remains distant (>50 Å) from the scissile phosphodiester and the catalytic residues of the HNH domain do not face that linkage (Figure 7E). In contrast, in the absence of AcrIIC3, the HNH domain rotates to the scissile phosphodiester upon binding to target dsDNA (Figure 2B). These observations suggest that AcrIIC3 inhibits the HNH domain structural rearrangement that is normally triggered by dsDNA binding.
To test the impact of AcrIIC3 binding on HNH and RuvC domain function, we performed cleavage assays with a short, Cy3- and Cy5-labeled target dsDNA (Figure S6E). The results show that AcrIIC3 inhibits the cleavage of both the TS and NTS, suggesting that activities of both the HNH and RuvC domains are inhibited in the presence of AcrIIC3. One side of AcrIIC3 binds to the HNH domain of Nme1Cas9.1 and the opposite side contacts the REC2 domain from Nme1Cas9.2. As a consequence, interactions between AcrIIC3 and REC2 of Nme1Cas9.2 substantially reduce the mobility of the HNH domain of Nme1Cas9.1, blocking the HNH domain’s movement towards the TS cleavage site to adopt the cleavage-component state. Furthermore, the inactive HNH conformation fails to activate the RuvC domain, resulting in the inhibition of NTS cleavage (Figure 7F). Taken together, we conclude that AcrIIC3 can bind the NmeCas9-sgRNA complex either before or after DNA binding, and that the AcrIIC3-NmeCas9-sgRNA complex is not fully precluded from DNA binding. Nonetheless, in all of these cases, the HNH domain is held in an inactive conformation, preventing DNA cleavage.
Discussion
Nme1Cas9 and Nme2Cas9 are single-AAV-deliverable genome engineering platforms that exhibit remarkably little off-target mutagenesis (Amrani et al., 2018; Edraki et al., 2019; Ibraheim et al., 2018; Lee et al., 2016, 2019). Furthermore, the dinucleotide PAM (N4CC) of Nme2Cas9 (Edraki et al., 2019) provides a target site density comparable to that of the widely used SpyCas9. However, the structures and mechanisms of Nme1Cas9 and Nme2Cas9 have not been well-defined. In addition, to our knowledge, no Cas9 homolog has been structurally solved at high resolution in a catalytically active, pre-cleavage state. Finally, of the five anti-CRISPRs reported to inhibit one or both of these meningococcal Cas9 homologs (Lee et al., 2018; Pawluk et al., 2016; Edraki et al. 2019), structural insights have only been obtained with Nme1Cas9 subfragments (Harrington et al., 2017; Zhu et al., 2019b; Thavalingam et al., 2019). We have solved the structures of the Nme1Cas9-sgRNA binary complex as well as seed-matched, fully paired pre-catalytic, fully paired catalytic, and post-TS-cleavage DNA-bound Nme1/2Cas9 ternary complexes. In the catalytic complex, HNH active site residues are close to the cleavage site of the target DNA, suggesting that the HNH domain is structurally ready for cleavage between nts 3 and 4 of the TS protospacer (Figure S7A). Finally, we have solved two structures of AcrIIC3-bound Nme1Cas9-sgRNA complexes with or without dsDNA. Our results provide a high-resolution view of the engagement of a Cas9 HNH domain (in this case, Nme1Cas9) with the guide-TS heteroduplex, including the hydrogen-bonding and salt bridge interactions with the backbone that enable non-sequence-specific TS binding and cleavage. Furthermore, these structures show important structural rearrangements of the REC2 and HNH domains of Nme1/2Cas9 during target DNA binding and cleavage (Figure S7B and Video S1).
Nme1Cas9 and Nme2Cas9 use identical sgRNA scaffolds to guide DNA target binding and cleavage, and they are >98% identical through their N-terminal 840 amino acids (Edraki et al., 2019). In contrast, their PI domains are only ~52% identical, in keeping with their distinct PAM specificities. Our analyses provide the structural basis for differential PAM recognition by the two orthologs, and mutational analyses in vitro and in mammalian editing applications confirm the importance of key PAM recognition determinants identified crystallographically. Nme1Cas9 was solved as a binary complex with its sgRNA and in three sgRNA-loaded, DNA-bound ternary complexes that reflect distinct functional states (including a catalytically competent conformation). In contrast, the Nme2Cas9-sgRNA-dsDNA structure was obtained only in an apparent pre-catalytic conformation, as observed with other Cas9 orthologs. Abundant evidence from SpyCas9 (Palermo et al., 2018; Zhu et al., 2019a) indicates that catalytic activation requires that the HNH domain swing from the distant position we observe in the seed-only Nme1Cas9-sgRNA-dsDNA structure (Figure 1E), to the cleavage-site-proximal position we observe in the fully complementary, Nme1Cas9-sgRNA-dsDNA structure (Figure 2B). Given the near identity between the two meningococcal homologs (and their guides) outside of the WED and PI domains, the Nme1Cas9 and Nme2Cas9 structures can be readily compared, giving rise to views of five important functional states: before DNA target binding (Nme1Cas9-sgRNA, Figure 1C); after PAM recognition, DNA unwinding and guide-TS seed pairing (Figure 1E); after heteroduplex propagation but before catalytic activation (Nme2Cas9-sgRNA-dsDNA, Figure 5A); after the structural rearrangements that accompany catalytic activation (Nme1Cas9-sgRNA-dsDNA, Figure 2B); and after TS cleavage by the HNH domain (Figure 4A). Our ability to visualize these distinct functional states, but within a common structural framework (outside of the WED and PI domains) and at high resolution, will enable deeper understanding of the multiple steps along the Cas9 reaction pathway.
A crucial aspect of Cas9 function is the assessment of guide-TS pairing along nearly the entire length of the heteroduplex, and the restraint of the nuclease domains from close substrate engagement until and unless sufficient complementarity has been detected. A noteworthy feature of our catalytically poised structure is the exceptionally dense collection of protein contacts with the guide-TS heteroduplex backbone along its entire length. With the exception of the PI domain, every major domain of Nme1Cas9 — the phosphate lock loop of the WED domain, as well as five or more residues within each of the REC1, REC2, BH, L1, HNH, and RuvC domains — is involved in heteroduplex contacts. On both the guide and TS sides of the heteroduplex, no backbone segment longer than two phosphodiester linkages lacks protein contacts. In each of the previous pre-catalytic Cas9-sgRNA-DNA structures (Anders et al., 2014; Nishimasu et al., 2014, 2015; Hirano et al., 2016, 2019; Jiang et al., 2016), some but not all of these interactions are present, and longer stretches of heteroduplex backbone lack protein contacts. Formation of some of these extensive, A-form-complementary, sequence-independent interactions likely enable the domain rearrangements that promote HNH domain disengagement from its RuvC interaction and subsequent docking onto the scissile linkage of the guide-TS heteroduplex (accompanied by new interactions between the HNH and REC1 domains), as we observe in our catalytically activated Nme1Cas9-sgRNA-dsDNA structure. These guide-TS interactions are therefore strong candidates for complementarity readouts that help restrict off-target editing in eukaryotic cells (Amrani et al., 2018; Edraki et al., 2019; Ibraheim et al., 2018; Lee et al., 2016).
Our structures also illuminate mechanisms of Cas9 inhibition by anti-CRISPR proteins. AcrIIC1 proteins have been previously revealed as broad-spectrum Type II-C Cas9 inhibitors that bind a conserved surface on the HNH domain and block access to the nuclease active site; sgRNA-guided DNA binding is not affected by AcrIIC1, but catalytic activation is prevented (Harrington et al., 2017). In contrast, AcrIIC3 was reported to decrease sgRNA-guided DNA binding affinity by ~12-fold, with a narrower phylogenetic spectrum of Cas9 inhibition (Harrington et al., 2017). The reported reduction of DNA-binding activity in vitro is consistent with previous observations that AcrIIC3 co-expression prevents fluorescently tagged Nme1Cas9 from localizing to genomic target sites in human cells (Pawluk et al., 2016). Electron microscopic analyses indicated that AcrIIC3 induces dimerization of Nme1Cas9-sgRNA complexes, with AcrIIC3 protein suggested to bridge between them (Harrington et al., 2017). Our studies demonstrate that AcrIIC3 does not fully prevent target DNA binding, nor does a single AcrIIC3 protein induce dimerization of two Nme1Cas9-sgRNA complexes. Instead, two independent AcrIIC3 proteins bridge the HNH and REC2 domains of two similarly independent Nme1Cas9-sgRNA complexes, with each AcrIIC3 monomer in the opposite orientation relative to the two bridged Nme1Cas9-sgRNA complexes. The overall structure is effectively a four-membered ring of proteins, with the Nme1Cas9-sgRNA constituents retaining some sgRNA-guided DNA-binding affinity. Consistent with our structural observations, AcrIIC3 association with both the HNH domain and an undefined segment of the REC lobe was recently demonstrated, with binding studies also suggesting a 2:2 AcrIIC3:Nme1Cas9 stoichiometry (Zhu et al., 2019b).
Our results show that AcrIIC3 can bind to the apo, sgRNA-loaded, and DNA-bound states, and are consistent with the retention of some DNA binding activity by the AcrIIC3-Nme1Cas9-sgRNA complex (Harrington et al., 2017). The structure of the Nme1Cas9-sgRNA-AcrIIC3-dsDNA complex reveals likely inhibitory mechanisms that prevent DNA cleavage even under conditions in which target binding occurs. AcrIIC3 binding to the HNH domain while the latter is distant from the TS cleavage site appears to restrict the domain’s rearrangement into a conformation that can engage the scissile phosphate. Furthermore, in the AcrIIC3-bound complex, the NTS likely binds in a channel different from that in the absence of AcrIIC3, forcing the NTS into a position too far away to be cleaved by the RuvC domain. These interactions prevent cleavage of the TS and NTS by two distinct but nonetheless coordinated strategies and enable DNA cleavage inhibition even under conditions when DNA binding by the AcrIIC3-Nme1Cas9-sgRNA complex occurs.
Collectively, our structural and functional analyses provide an improved framework for understanding the mechanistic basis for Cas9 function and genome editing, especially by Type II-C systems, which are less well understood than their Type II-A counterparts such as SpyCas9 and SauCas9. A potentially valuable feature of some Type II-C Cas9s (e.g. CjeCas9, Nme1Cas9 and Nme2Cas9) is the resistance of the wild-type proteins to off-target mutagenesis in eukaryotic cells (Amrani et al., 2018; Edraki et al., 2019; Ibraheim et al., 2018; Kim et al., 2017; Lee et al., 2016), perhaps by virtue of lower catalytic efficiencies (Ma et al., 2015) that limit cleavage at nearcognate sites more drastically than at cognate sites (Bisaria et al., 2017). The structural understanding afforded by our results, including the inferred conformational transitions along the reaction pathway, will facilitate exploration of the mechanistic basis of on- vs. off-target discrimination, catalytic activation, anti-CRISPR inhibition, and other features that are critical for the ongoing development of genome editing technology.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for reagents and further information may be directed to, and will be fulfilled by Yanli Wang (ylwang@ibp.ac.cn).
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli Rosetta (DE3) (Novagen) was grown in LB. HEK293T cells were cultured in DMEM.
METHOD DETAILS
Protein expression and purification
Full-length ORFs of Nme1Cas9 and Nme2Cas9 (encoding residues 1-1082), as well as AcrIIC3 (encoding residues 1-116), were purchased from Sangon Biotech. Nme1Cas9 and Nme2Cas9 were cloned into an expression vector pET28a-Sumo with a His6-Sumo tag at the N-terminus, or pET-30b with a His8-tag at the C-terminus. Mutants were constructed using a site-directed mutagenesis kit. All proteins were overexpressed in E. coli Rosetta (DE3) (Novagen) cells and were induced with 0.2 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) at OD600 = 0.6 for 12 h at 18°C.
Cells expressing Nme1Cas9 or Nme2Cas9 were lysed by sonication in buffer containing 20 mM Tris-HCl (pH 7.5) and 0.5 M NaCl at 4°C. After centrifugation, the supernatant was incubated with Ni2+-Sepharose resin (GE Healthcare), and the bound protein was eluted with lysis buffer supplemented with 200 mM imidazole. Eluted Nme1Cas9 and Nme2Cas9 proteins with the His6-Sumo tag were digested with His-tagged ubiquitin-like protein 1 (Ulp1) protease and dialyzed against buffer containing 20 mM Tris-HCl (pH 7.5), 0.3 M NaCl for 2 h at 4°C. Nme1Cas9 and Nme2Cas9 proteins were further purified by a Ni2+-Sepharose column to remove the His6-Sumo tag. The flow-through collections were loaded into an SP column (GE Healthcare) eluting with buffer containing 20 mM Tris-HCl (pH 7.5) and 1 M NaCl. The Nme1Cas9 and Nme2Cas9 proteins with a His8-tag at the C-terminus were purified using an SP column following the first Ni2+-Sepharose column purification, using the same protocol as described above.
Cells expressing His6-Sumo-AcrIIC3 were lysed in 20 mM Tris-HCl (pH 7.5), 0.5 M NaCl buffer at 4°C. First, AcrIIC3 protein was puri fied using His-select Ni2+-Sepharose resin. The protein was then digested by His-tagged Ulp1 protease and dialyzed against 20 mM Tris-HCl (pH 7.5), 0.3 M NaCl for 2 h at 4°C. AcrIIC3 protein was further purified using Ni2+-Sepharose and Heparin columns (GE Healthcare) and then eluted with buffer containing 20 mM Tris-HCl (pH 7.5) and 1 M NaCl.
In vitro transcription and purification of sgRNA
In vitro transcription with T7 RNA polymerase and linearized plasmid DNAs as templates were employed for synthesis of the 147-, 135-, 122-, and 102-nucleotide sgRNAs. Transcription reactions were performed at 37°C for 4 h in buffer containing 100 mM HEPES-KOH (pH 7.9), 25 mM MgCl2, 30 mM DTT, 2 mM each NTP, 2 mM spermidine, 0.1 mg/ml T7 RNA polymerase, and 35 ng/μl linearized plasmid DNA template. The sgRNA was then purified by gel electrophoresis on a 12% denaturing (8 M urea) polyacrylamide gel and an Elutrap system. Finally, the sgRNA was resuspended in DEPC (diethylpyrocarbonate) H2O.
Preparation of DNA
All short DNAs used in this study were purchased from Sangon Biotech. The DNA sequences used for crystallization and cleavage assays are shown in Table S4. Both the TS and NTS DNAs were dissolved in buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2]. Equimolar amounts of TS and NTS were pre-denatured at 95°C for 10 min and then annealed at room temperatu re.
Reconstitution of Nme1Cas9-sgRNA, Nme1Cas9-sgRNA-dsDNA and Nme2Cas9-sgRNA-dsDNA complexes
To prepare the Nme1Cas9-sgRNA binary complex, purified Nme1Cas9-His8 protein was mixed with 135-nucleotide sgRNA at a molar ratio of 1:1.05, incubated on ice for 30 min, and purified by gel filtration chromatography (Superdex 200 increase 10/300 GL, GE Healthcare). To prepare the DNA-bound Nme1Cas9-sgRNA complexes, either Nme1Cas9 His588Ala mutant or wild-type Nme1Cas9, 147- or 135-nucleotide sgRNA, and dsDNA was mixed at a molar ratio of 1:1.05:1.2. Components were added in the order listed and incubated for 30 min on ice before adding the next component. The resulting complex was purified by gel filtration chromatography. The Nme2Cas9-sgRNA-dsDNA complex was reconstituted by incubation of purified Nme2Cas9 (Asp16Ala/His588Ala), 135-nucleotide sgRNA, and dsDNA at a molar ratio of 1:1.05:1.2 on ice. The resulting complex was purified by gel filtration chromatography. Finally, complexes were concentrated to an A280 absorbance to ~15, as measured by Nanodrop 2000, before crystallization.
Preparation of Nme1Cas9-sgRNA-AcrIIC3 ternary and Nme1Cas9-sgRNA-AcrIIC3-DNA quaternary complexes
The complex of Nme1Cas9-His8, 135-nucleotide sgRNA, and AcrIIC3 was reconstituted by incubation of each purified component at a molar ratio of 1:1.05:1.05, respectively, on ice. The Nme1Cas9-sgRNA-AcrIIC3-DNA complex was reconstituted by incubation of purified Nme1Cas9 (Asp16Ala/His588Ala), 135-nucleotide sgRNA, AcrIIC3 and dsDNA-24n at a molar ratio of 1:1.05:10:1.2 on ice. Each component was added sequentially, with an intermittent incubation of 30 min before adding the next component. The resulting complexes were purified by an additional gel filtration chromatography step (Superdex 200 increase 10/300 GL, GE Healthcare). Subsequently, the two complexes (DNA-free and DNA-bound) were concentrated to a respective A280 absorbance of ~15 and ~18, as measured by Nanodrop 2000, before crystallization.
Crystallization, data collection and structure determination
The hanging-drop vapor-diffusion method was used for crystal growth. All crystals were obtained by mixing 1 μL Nme1Cas9/Nme2Cas9 complex with 1 μL reservoir solution and incubated at 16°C.
Crystals of the Nme1Cas9-sgRNA complex were grown from 0.1 M Tris-HCl (pH 8.5), 0.12 M K2SO4, and 18.5% PEG 6000. Crystals of the seed-paired Nme1Cas9-sgRNA-DNA complex were grown from 0.2 M sodium nitrate, 0.1 M Bis-Tris propane (pH 8.0), 20% PEG 3350. Crystals of the Nme1Cas9-sgRNA-DNA complexes in the catalytic state were grown from 0.2 M sodium citrate, 0.1 M HEPES-NaOH (pH 8.0), and 18.5% PEG 3350. Crystals of the Nme1Cas9-sgRNA-DNA in the post-cleavage state were grown from 0.2 M sodium citrate, 0.1 M Bis-Tris propane (pH 7.2), 19% PEG 3350. Crystals of Nme2Cas9-sgRNA-dsDNA with no protospacer in the NTS were grown from 1.2 M LiCl, 0.1 M citric acid (pH 5.6), 4% 1,1,1,3,3,3-Hexafluoro-2-propanol, and 10% PEG 6000. Crystals of Nme2Cas9-sgRNA-dsDNA with a 5-nucleotide protospacer in the NTS in the pre-catalytic state were grown from 0.1 M sodium citrate (pH 5.2), 1 M LiCl, 0.01 M spermidine, and 10% PEG 6000. All crystals above were cryoprotected using the corresponding reservoir solution, supplemented with glycerol, and flash-frozen in liquid nitrogen.
Crystals of Nme1Cas9-sgRNA-AcrIIC3 were grown from 0.21 M NaAc, 0.1 M NaAc (pH 5.5), 0.04 M Tris-HCl (pH 7.5), 32% pentaerythritol propoxylate (5/4 PO/OH). Crystals of Nme1Cas9-sgRNA-AcrIIC3-dsDNA were grown from 1 M NaCl, 0.1 M sodium citrate (pH 5.8), 30% PEG 600, and 9.6% glycerol. All crystals above were flash-frozen in liquid nitrogen without any additional cryoprotectant.
All diffraction datasets were collected at beamline BL17U1 or BL19U1 at the Shanghai Synchrotron Radiation Facility (SSRF), or at beamline BL41XU at Spring-8 in Japan, and processed with XDS or HKL2000 (Otwinowski and Minor, 1997). The data of the Nme1Cas9-sgRNA-AcrIIC3 complex was collected at Spring-8 BL41XU. All other complexes were collected at SSRF BL19U1 and BL17U1. All structures were solved by molecular replacement (MR) using PHENIX PHASER (McCoy et al., 2007). The first search model for the Nme1Cas9-sgRNA-DNA complex in the catalytic state was the CjeCas9-ΔHNH ternary complex structure (PDB ID: 5X2G). The resolved Nme1Cas9 complex structure was used as the starting model for other sgRNA-bound or dsDNA-bound Nme1Cas9 or Nme2Cas9 complexes. The Nme1Cas9-sgRNA complex structure was used as the search model for the Nme1Cas9-sgRNA-AcrIIC3 complex. AcrIIC3 was built manually in COOT (Emsley et al., 2010). The determined Nme1Cas9-sgRNA-AcrIIC3 structure was the starting model for the Nme1Cas9-sgRNA-AcrIIC3-dsDNA. After the initial phases were obtained by MR, the atomic models were manually built and adjusted using the program COOT (Emsley et al., 2010). Iterative cycles of crystallographic refinement were performed using PHENIX (Adams et al., 2002). All structural figures were prepared using PyMOL.
In vitro cleavage assay
The in vitro DNA cleavage assays were performed with either plasmid target or with Cy3- and Cy5-labeled dsDNA (5’-Cy3-labeled TS and 5’-Cy5-labeled NTS). The pUC19 target DNA plasmid (35 bp target DNA cloned into the pUC19 vector) was linearized by ScaI digestion before the cleavage reaction. The Nme1Cas9-sgRNA or Nme1Cas9-sgRNA-AcrIIC3 complexes were incubated with 300 ng pUC19 target DNA in 10 μL reaction buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 10 mM MgCl2, 1 mM DTT and 5% glycerol for 10 min at the temperatures indicated in figure legends. Reactions were quenched by adding 2 μl gel-Loading dye containing 10 mM EDTA (NEB). The reaction products were run on 1% agarose gels stained with ethidium bromide for production detection.
For the reaction with 5'-Cy3- and 5'-Cy5- labeled dsDNA substrate, 200 to 500 nM Nme1Cas9-sgRNA complexes either with or without AcrIIC3 were incubated with the substrate dsDNA in 10 μL reaction buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 10 mM MgCl2, 1 mM DTT and 5% glycerol for 10 min at the temperatures indicated in figure legends. All reactions were stopped by adding 1 μL 0.5 M EDTA and 1 μL 0.1 mg/ml proteinase K for 30 min at room temperature. Products were separated on a 20% urea denaturing polyacrylamide gel. DNA was visualized using a FluorChem system.
Mammalian cell expression and genome editing
For genome editing in mammalian cells, the Nme1Cas9 and Nme2Cas9 orthologs were expressed from the pCS2-Dest Gateway plasmid under the control of the CMV IE94 promoter as described previously (Amrani et al., 2018; Edraki et al., 2019; Pawluk et al., 2016). All Nme1Cas9, Nme2Cas9, and sgRNA mutants were created by site-directed mutagenesis via PCR and ligation, using primers listed in Table S5. All sgRNAs were driven by the U6 promoter in pLKO.1-puro plasmids as described previously (Amrani et al., 2018; Edraki et al., 2019; Pawluk et al., 2016). Plasmids are also listed in Table S5.
HEK293T cells were cultured in DMEM with 10% FBS at 37°C with 5% CO2. Approximately 150 × 103 cells were transfected with 200 ng Nme1Cas9- or Nme2Cas9-expressing plasmids and 200 ng sgRNA-expressing plasmids using Polyfect transfection reagent (Qiagen) in a 24-well plate according to the manufacturer’s protocol. After 72 hours, genomic DNA was extracted using the DNeasy Blood and Tissue kit (QIAGEN) according to the manufacturer’s protocol. For genome editing analyses, 50 ng of genomic DNA was PCR-amplified [High Fidelity 2× PCR Master Mix (New England Biolabs)] with primers flanking the targeted sites (Table S5). The PCR product was then purified using DNA Clean & Concentrator kit (Zymo) following the manufacturer’s instructions. The purified PCR product was Sanger-sequenced (Genewiz) and editing was measured by Inference of CRISPR Edits (ICE) analyses (https://ice.synthego.com/#/).
For western analyses of Nme1Cas9 and Nme2Cas9 accumulation in HEK293T cells, the cells were harvested 48 h post-transfection and lysed with 50 μl of RIPA buffer containing protease inhibitors. ~10 μg total protein was used for SDS-PAGE and blotting. The blots were probed with anti-HA (Sigma, H3663) and anti-GAPDH (Abcam, ab9485) as primary antibodies, and then with horseradish peroxidase-conjugated anti-mouse IgG (ThermoFisher, 62-6520) or anti-rabbit IgG (Biorad, 1706515) secondary antibodies, respectively. Blots were visualized using the Clarity Western ECL substrate (Biorad, 170-5060).
QUANTIFICATION AND STATISTICAL ANALYSIS
Biochemical assays were repeated three times, and representative results are shown. Crystallographic data collection and refinement statistics are listed in Tables S1-S3.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession numbers for the atomic coordinates reported in this paper are PDB: 6JDQ (crystal structure of Nme1Cas9-sgRNA complex), 6KC7 (Nme1Cas9-sgRNA-DNA in seed-only base-paired state), 6JDV [Nme1Cas9 (His588Ala)-sgRNA-DNA in the catalytic state], 6KC8 [Nme1Cas9(WT)-sgRNA-DNA in a post-cleavage state], 6JFU (Nme2Cas9-sgRNA-dsDNA-0nt), 6JE3 (Nme2Cas9-sgRNA-dsDNA-5nt), 6JE9 (Nme1Cas9-sgRNA-AcrIIC3) and 6JE4 (Nme1Cas9-sgRNA-AcrIIC3-dsDNA).
Supplementary Material
Video S1. Conformational changes of Nme1Cas9 during target DNA recognition and cleavage. Related to Figure 7. The blue cross indicates the cleavage site in the target strand.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| HEPES | Sigma-Aldrich | H3375-250G |
| Sodium citrate tribasic dihydrate | Sigma-Aldrich | C8532-1KG |
| Citric acid | Sigma-Aldrich | C0759-500G |
| Sodium acetate | Sigma-Aldrich | S7545-1KG |
| Magnesium chloride hexahydrate | Sigma-Aldrich | M2393-500G |
| Potassium sulfate | Sigma-Aldrich | P9458-250G |
| Lithium chloride | Sigma-Aldrich | L9650-500G |
| glycerol | Sigma-Aldrich | G5516-1L |
| DL-Dithiothreitol (DTT) | Sigma-Aldrich | D9163-25G |
| Pentaerythritol propoxylate (5/4 PO/OH) | Sigma-Aldrich | 41-8749-500ML |
| PEG 600 | Sigma-Aldrich | 202401-500G |
| PEG 3,350 | Sigma-Aldrich | 88276-1KG-F |
| PEG 6,000 | Sigma-Aldrich | 81255-1KG |
| T7 RNA polymerase | Home-made | N/A |
| Ulp1 | Home-made | N/A |
| Toluidine Blue | AMRESCO | 0672-25G |
| Urea | AMRESCO | 0378-5KG |
| Spermidine | Sigma-Aldrich | S2626-1G |
| ATP | Sigma-Aldrich | A2383 |
| UTP | Sigma-Aldrich | U6625 |
| CTP | Sigma-Aldrich | C1506 |
| GTP | Sigma-Aldrich | G8877 |
| AxyPrep Plasmid Maxiprep Kit 25-prep | Axygen | AP-MX-P-25G |
| Critical Commercial Assays | ||
| Chelating Sepharose Fast Flow | GE Healthcare | Cat# 17-0575-02 |
| HiTrap Heparin HP | GE Healthcare | Cat# 17-0407-03 |
| HiTrap SP HP | GE Healthcare | Cat# 17-1152-01 |
| Superdex 200 Increase, 10/300 GL | GE Healthcare | Cat# 28-9909-44 |
| Deposited Data | ||
| The coordinates of Nme1Cas9-sgRNA complex | This paper | PDB: 6JDQ |
| The coordinates of Nme1Cas9 (His588Ala)-sgRNA-DNA in catalytic state | This paper | PDB: 6JDV |
| The coordinates of Nme1Cas9-sgRNA-DNA in seed-base paring state | This paper | PDB: 6KC7 |
| The coordinates of Nme1Cas9(WT)-sgRNA-DNA in post-cleavage state | This paper | PDB: 6KC8 |
| The coordinates of Nme2Cas9-sgRNA-dsDNA-0nt complex | This paper | PDB: 6JFU |
| The coordinates of Nme2Cas9-sgRNA-dsDNA-5nt complex | This paper | PDB: 6JE3 |
| The coordinates of Nme1Cas9-sgRNA-AcrIIC3 complex | This paper | PDB: 6JE9 |
| The coordinates of Nme1Cas9-sgRNA-AcrIIC3-dsDNA complex | This paper | PDB: 6JE4 |
| The coordinates of SpyCas9-sgRNA-DNA complex | Anders et al., 2014. | PDB: 4UN3 |
| The coordinates of SauCas9-sgRNA-DNA complex | Nishimasu et al., 2015. | PDB: 5CZZ |
| The coordinates of SpyCas9-sgRNA-DNA complex | Jiang et al., 2016. | PDB: 5F9R |
| Experimental Models: Organisms/Strains | ||
| Escherichia coli Rosetta (DE3) | Novagen | Cat# 70954-3 |
| Recombinant DNA | ||
| pET28a-Nme1Cas9, various mutants | This paper | N/A |
| pET28a-Nme2Cas9, various mutants | This paper | N/A |
| pET28a-AcrIIC3 | This paper | N/A |
| pET30b-Nme1Cas9 | This paper | N/A |
| Oligonucleotides | ||
| 147-nt SgRNA sequence. GGUCACUCUGCU AUUUAACUUUACGUUGUAGCUCCCUUUC UCAUUUCGGAAACGAAAUGAGAACCGUUG CUACAAUAAGGCCGUCUGAAAAGAUGUGC CGCAACGCUCUGCCCCUUAAAGCUCCUGC UUUAAGGGGCAUCGUUUAUC |
This paper | N/A |
| 135-nt SgRNA sequence. GGUCACUCUGCUAU UUAACUUUACGUUGUAGCUCCCUUUCUCG AAAGAGAACCGUUGCUACAAUAAGGCCGUC UGAAAAGAUGUGCCGCAACGCUCUGCCCC UUAAAGCUCCUGCUUUAAGGGGCAUCGU UUAUC |
This paper | N/A |
| 122-nt SgRNA sequence. GGUCACUCUUCUAU UUACCUAUACGUUGUAGCUCCCUUUCUCAUU UCGGAAACGAAAUGAGAACCGUUGCUACAAUA AGGCCGUCUGAAAAGAUGUGCCGCAACGCUC UUUCUAGAGCGUUC |
This paper | N/A |
| 102-nt SgRNA sequence. GGACUCACGACGCA UUACCUUUACGUUGUAGCUCCCUUUCUCAUU UCGGAAACGAAAUGAGAACCGUUGCUACAAUA AGGCCGUCUGAAAAGAUGUGCCAUC |
This paper | N/A |
| SpyCas9 sgRNA sequence. GGAAAUUAGGUGCGC UUGGCGUUUUAGAGCUAGAAAUAGCAAGUUAAAA UAAGGCUAGUCCGUUAUCAACUUGAAAAAG UGGCACCGAGUCGGUGCUUC |
This paper | N/A |
| SauCas9 sgRNA sequence. GGUCUGCUAUUUCUA UUUACGUUUUAGUACUCUGGAAACAGAAUCUAC UAAAACAAGGCAAAAUGCCGUGUUUAUC UCGUCAACUUGUUGGCGAGAUC |
This paper | N/A |
| DNA sequences | This paper | Table S4 |
| Plasmids and primers | This paper | Table S5 |
| Software and Algorithms | ||
| PHENIX | Adams et al., 2002 | http://www.phenix-online.org/ |
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com/ |
| COOT | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Molecular Graphics System, Version 1.8 Schrödinger, LLC | https://www.pymol.org/2/ |
| PHASER | McCoy et al., 2007 | https://www-structmed.cimr.cam.ac.uk/phaser_obsolete/ |
| Other | ||
| Amicon Ultra Centrifugal Filters Ultracel-50K | Merck Millipore Ltd | UFC903096 |
| Amicon Ultra Centrifugal Filters Ultracel-10K | Merck Millipore Ltd | UFC801096 |
| 3 Well Midi Crystallization Plate (Swissci) | HAMPTON RESEARCH | Cat# HR3-125 |
Highlights:
Five structures of Neisseria meningitidis Cas9s show distinct reaction states
An Nme1Cas9 structure reveals the catalytically activated HNH domain conformation
The HNH catalytic conformation promotes activation of the RuvC domain
AcrIIC3 inhibits Nme1Cas9 by tethering two Nme1Cas9 complexes together
Acknowledgements
We are grateful to the staff of the BL-17U1 and BL-19U1 beamlines at the Shanghai Synchrotron Radiation Facility, the BL41XU beamline at SPring-8 (2018A2581, 2018A2711), and Alan Davidson and Karen Maxwell (U. of Toronto) and Nathan Bamidele and Scot Wolfe (UMass Medical School) for helpful discussions. This work was supported by grants from the Chinese Ministry of Science and Technology (2017YFA0504203), the Natural Science Foundation of China (31725008, 31630015, 31571335 and 31700662), and the Chinese Academy of Sciences (QYZDY-SSW-SMC021) to Y.W., and by U.S. National Institutes of Health grants F31 DK120333 (to R.I.), R01 GM125797 (to E.J.S.), and UG3 TR002668 (to E.J.S.).
Footnotes
Declaration of Interests
N.A., R.I., A.E. and E.J.S. are co-inventors on intellectual property applications related to Nme1Cas9, Nme2Cas9, and AcrIIC3. E.J.S. is a co-founder, scientific advisor, and shareholder of Intellia Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung L-W, loerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Amrani N, Gao XD, Liu P, Edraki A, Mir A, Ibraheim R, Gupta A, Sasaki KE, Wu T, Donohoue PD, et al. (2018). NmeCas9 is an intrinsically high-fidelity genome-editing platform. Genome Biol. 19, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, and Jinek M (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaria N, Jarmoskaite I, and Herschlag D (2017). Lessons from Enzyme Kinetics Reveal Specificity Principles for RNA-Guided Nucleases in RNA Interference and CRISPR-Based Genome Editing. Cell Syst. 4, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolukbasi MF, Liu P, Luk K, Kwok SF, Gupta A, Amrani N, Sontheimer EJ, Zhu LJ, and Wolfe SA (2018). Orthogonal Cas9-Cas9 chimeras provide a versatile platform for genome editing. Nat. Commun. 9, 4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagdas YS, Chen JS, Sternberg SH, Doudna JA, and Yildiz A (2017). A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv 3, eaao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, Song C-Q, Cao Y, Gallant J, Xue W, Rivera-Pérez JA, et al. (2019). A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell 73, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, and Church GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 109, E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Yu HH, Johnson KA, and Taylor DW (2018). DNA Unwinding Is the Primary Determinant of CRISPR-Cas9 Activity. Cell Rep. 22, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu J-J, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, Nakane T, Ishitani R, Hatada I, Zhang F, et al. (2016). Structure and Engineering of Francisella novicida Cas9. Cell 164, 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Abudayyeh OO, Gootenberg JS, Horii T, Ishitani R, Hatada I, Zhang F, Nishimasu H, and Nureki O (2019). Structural basis for the promiscuous PAM recognition by Corynebacterium diphtheriae Cas9. Nat. Commun 10, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann MD, Aschenbrenner S, Grosse S, Rapti K, Domenger C, Fakhiri J, Mastel M, Borner K, Eils R, Grimm D, et al. (2019). Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res. 47, e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu L-F, Sontheimer EJ, and Thomson JA (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 110, 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraheim R, Song C-Q, Mir A, Amrani N, Xue W, and Sontheimer EJ (2018). All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 19, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, and Doudna JA (2017). CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys 46, 505–529. [DOI] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, and Doudna JA (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Zhou K, Ma L, Gressel S, and Doudna JA (2015). A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang FG, Taylor DW, Sternberg SH, Kaya E, Ma EB, Anders C, Hauer M, Zhou KH, Lin S, et al. (2014). Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 343, 1247997–1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho H-Y, Song DW, Lee KJ, Jung MH, Kim S, et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2017). CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Cradick TJ, and Bao G (2016). The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther 24, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mir A, Edraki A, Garcia B, Amrani N, Lou HE, Gainetdinov I, Pawluk A, Ibraheim R, Gao XD, et al. (2018). Potent Cas9 Inhibition in Bacterial and Human Cells by AcrIIC4 and AcrIIC5 Anti-CRISPR Proteins. MBio 9, e02321–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mou H, Ibraheim R, Liang S-Q, Liu P, Xue W and Sontheimer EJ (2019). Tissue-restricted genome editing in vivo specified by microRNA-repressible anti-CRISPR proteins. RNA, in press (ePub ahead of print: DOI: 10.1261/rna.071704.119). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CL, Hor LI, Chang ZF, Tsai LC, Yang WZ, and Yuan HS (2003). DNA binding and cleavage by the periplasmic nuclease Vvn: a novel structure with a known active site. EMBO J. 22, 4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Harrington LB, O’Connell MR, Zhou K, and Doudna JA (2015). Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 60, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A, Edraki A, Lee J, and Sontheimer EJ (2018). Type II-C CRISPR-Cas9 Biology, Mechanism, and Application. ACS Chem. Biol 13, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, Kurabayashi A, Ishitani R, Zhang F, and Nureki O (2015). Crystal Structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Palermo G, Chen JS, Ricci CG, Rivalta I, Jinek M, Batista VS, Doudna JA, and McCammon JA (2018). Key role of the REC lobe during CRISPR-Cas9 activation by “sensing”, “regulating”, and “locking” the catalytic HNH domain. Q. Rev. Biophys 51, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. (2016). Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167, 1829–1838.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Davidson AR, and Maxwell KL (2018). Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol 16, 12–17. [DOI] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, and Doudna JA (2015). Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavalingam A, Cheng Z, Garcia B, Huang X, Shah M, Sun W, Wang M, Harrington L, Hwang S, Hidalgo-Reyes Y, et al. (2019). Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun 10, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Watanabe Y, Gootenberg JS, Hirano H, Ran FA, Nakane T, Ishitani R, Zhang F, Nishimasu H, and Nureki O (2017). Crystal Structure of the Minimal Cas9 from Campylobacter jejuni Reveals the Molecular Diversity in the CRISPR-Cas9 Systems. Mol. Cell 65, 1109–1121.e3. [DOI] [PubMed] [Google Scholar]
- Yang M, Peng S, Sun R, Lin J, Wang N, and Chen C (2018). The Conformational Dynamics of Cas9 Governing DNA Cleavage Are Revealed by Single-Molecule FRET. Cell Rep. 22, 372–382. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, and Sontheimer EJ (2013). Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 50, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rajan R, Seifert HS, Mondragón A, and Sontheimer EJ (2015). DNase H Activity of Neisseria meningitidis Cas9. Mol. Cell 60, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Clarke R, Puppala AK, Chittori S, Merk A, Merrill BJ, Simonovíc M and Subramaniam S (2019a). Cryo-EM structures reveal coordinated domain motions that govern DNA cleavage by Cas9. Nat. Struct. Mol. Biol 26, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, Liu S, Gao G, Serganov A, and Gao P (2019b). Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol. Cell 74, 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z, and Liu J (2017). Structure and Dynamics of Cas9 HNH Domain Catalytic State. Sci Rep. 7, 17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Conformational changes of Nme1Cas9 during target DNA recognition and cleavage. Related to Figure 7. The blue cross indicates the cleavage site in the target strand.
Data Availability Statement
Data Resources
The accession numbers for the atomic coordinates reported in this paper are PDB: 6JDQ (crystal structure of Nme1Cas9-sgRNA complex), 6KC7 (Nme1Cas9-sgRNA-DNA in seed-only base-paired state), 6JDV [Nme1Cas9 (His588Ala)-sgRNA-DNA in the catalytic state], 6KC8 [Nme1Cas9(WT)-sgRNA-DNA in a post-cleavage state], 6JFU (Nme2Cas9-sgRNA-dsDNA-0nt), 6JE3 (Nme2Cas9-sgRNA-dsDNA-5nt), 6JE9 (Nme1Cas9-sgRNA-AcrIIC3) and 6JE4 (Nme1Cas9-sgRNA-AcrIIC3-dsDNA).