

SUMMARY
Flaviviruses cause systemic or neurotropic-encephalitic pathology in humans. The flavivirus nonstructural protein 1 (NS1) is a secreted glycoprotein involved in viral replication, immune evasion, and vascular leakage during dengue virus infection. However, the contribution of secreted NS1 from related flaviviruses to viral pathogenesis remains unknown. Here, we demonstrate that NS1 from dengue, Zika, West Nile, Japanese encephalitis, and yellow fever viruses selectively binds to and alters permeability of human endothelial cells from lung, dermis, umbilical vein, brain, and liver in vitro and causes tissue-specific vascular leakage in mice, reflecting the pathophysiology of each flavivirus. Mechanistically, each flavivirus NS1 leads to differential disruption of endothelial glycocalyx components, resulting in endothelial hyperpermeability. Our findings reveal the capacity of a secreted viral protein to modulate endothelial barrier function in a tissue-specific manner both in vitro and in vivo, potentially influencing virus dissemination and pathogenesis and providing targets for antiviral therapies and vaccine development.
In Brief
Puerta-Guardo et al. discover that five flavivirus NS1 proteins trigger hyperpermeability and vascular dysfunction in human endothelial cells and mice in a manner reflecting disease tropism. This tissue-specific tropism is partially determined by the capacity of NS1 to bind endothelial cells and is characterized by disruption of endothelial glycocalyx components.
Graphical Abstract
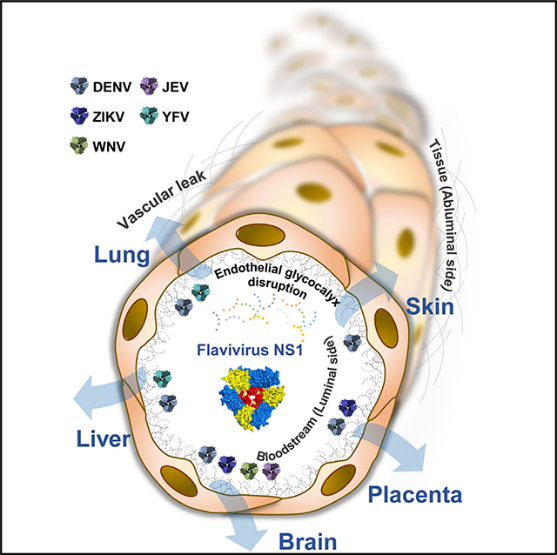
INTRODUCTION
The Flavivirus genus consists of enveloped, positive-sense RNA viruses (~11-kb genome) that are transmitted by arthropod vectors, collectively causing hundreds of millions of infections each year. Flaviviruses include medically important mosquito-borne pathogens such as the four dengue virus serotypes (DENV1-DENV4), Zika virus (ZIKV), West Nile virus (WNV), Japanese encephalitis virus (JEV), and yellow fever virus (YFV) (Calisher and Gould, 2003). In humans, flavivirus infections result in a range of outcomes, from clinically inapparent infections to severe, sometimes fatal disease, characterized by hemorrhagic manifestations and vascular leakage with organ failure (DENV and YFV), meningomyeloencephalitis (JEVand WNV), and congenital Zika syndrome in pregnant women and Guillain-Barré syndrome in adults associated with ZIKV infection (Gould and Solomon, 2008; Krauer et al., 2017).
The flavivirus genome encodes three structural proteins (C, prM/M, and E) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). NS1 is a multifunctional viral protein that is secreted as a hexamer by infected cells and circulates in the host bloodstream (Muller and Young, 2013). Inside infected cells, NS1 acts as a cofactor for viral replication and assembly, while the secreted form plays a role in immune evasion by binding and triggering degradation of the complement protein C4 (Avirutnan et al., 2010; Mackenzie et al., 1996; Scaturro et al., 2015) and in dengue pathogenesis via activation of complement (Avirutnan et al., 2006). Additionally, NS1 has been shown to contribute to dengue pathogenesis by directly triggering endothelial hyperpermeability as well as by inducing the release of vasoactive cytokines from peripheral blood mononuclear cells (PBMCs) via activation of TLR4, both leading to vascular leak (Beatty et al., 2015; Modhiran et al., 2015). Further, DENV NS1 has been shown to directly alter the barrier function of pulmonary endothelial cell monolayers through disruption of the endothelial glycocalyx-like layer (EGL) by triggering the activation of endothelial sialidases, cathepsin L, and heparanase, enzymes responsible for degrading sialic acid and heparan sulfate proteoglycans (Puerta-Guardo et al., 2016). Importantly, disruption of endothelial glycocalyx components has been shown to correlate with plasma leakage during severe DENV infection (Suwarto et al., 2017; Tang et al., 2017). More recently, the contribution of these DENV NS1-induced endothelial cell-intrinsic pathways to NS1-mediated vascular leakage was demonstrated to be independent of inflammatory cytokines but dependent on the integrity of endothelial glycocalyx components both in vitro and in vivo (Glasner et al., 2017).
NS1 is well conserved among flaviviruses, exhibiting 20%–40% identity and 60%–80% similarity (Song et al., 2016; Xu et al., 2016). During the acute phase of illness in patients with severe dengue disease, DENV NS1 has been shown to circulate at high levels (1–2 μg/mL), correlating with DENV viremia (Libraty et al., 2002). Human studies describing the kinetics of circulating NS1 levels in other flavivirus infections are lacking, although NS1 has been detected in the serum of mice experimentally infected with ZIKV, WNV, JEV, and YFV (Chung and Diamond, 2008; Liu et al., 2016, 2017; Macdonald et al., 2005). Notably, ZIKV NS1 has been found in placental tissues infected with ZIKV (Jurado et al., 2016). This suggests that, in addition to the effect of NS1 circulating in serum on the luminal side of endothelial cells lining blood vessels, the action of NS1 in relation to vascular leak may also occur in capillary beds of tissues, where the cumulative effect of NS1 is likely amplified and/or concentrated.
Here, we demonstrate that NS1 proteins from dengue, Zika, West Nile, Japanese encephalitis, and yellow fever viruses selectively alter the permeability of monolayers of endothelial cell lines derived from distinct human tissues. Additionally, we find that flavivirus NS1 proteins differentially induce disruption of EGL components, critical determinants of endothelial barrier function (Reitsma et al., 2007). Further, NS1 proteins enhance the extravasation of fluids in vivo in a tissue-dependent manner, which correlates with flavivirus pathophysiology. Together, our findings reveal a previously unidentified ability of flavivirus NS1 proteins to modulate endothelial permeability in a tissue-specific manner both in vitro and in vivo, potentially influencing flavivirus dissemination, pathogenesis, and disease.
RESULTS
Flavivirus NS1 Proteins Trigger Endothelial Barrier Dysfunction in a Tissue-Specific Manner
Previously, we demonstrated that NS1 proteins from all four DENV serotypes, but not the related WNV, cause a dose-dependent increase in endothelial permeability of human pulmonary microvascular endothelial cells (HPMECs) (Beatty et al., 2015; Puerta-Guardo et al., 2016). Further, we showed that DENV2 NS1 alone is able to induce vascular leakage in vivo (Beatty et al., 2015; Glasner et al., 2017). Here, we used a trans-endothelial electrical resistance (TEER) assay to evaluate the ability of DENV, ZIKV, WNV, JEV, and YFV NS1 proteins to trigger endothelial barrier dysfunction in distinct human endothelial cell monolayers, including pulmonary (HPMECs), dermal (human microvascular dermal endothelial cells [HMEC-1]), umbilical vein (human umbilical vein endothelial cells [HUVECs]), brain (human brain microvascular endothelial cells [HBMECs]), and liver (human liver sinusoidal microvascular endothelial cells [HLSECs]). Recombinant flavivirus NS1 proteins used in all experiments were produced in mammalian cells (HEK293) and were shown to be >95% pure and oligomeric, as demonstrated by native PAGE followed by silver staining or western blot analyses (Figure S1A). NS1 protein concentrations used in these experiments were based on levels of NS1 circulating in patients with severe dengue disease and on previous studies demonstrating the effect of DENV NS1 on human endothelial cells (Beatty et al., 2015; Glasner et al., 2017; Puerta-Guardo et al., 2016). Interestingly, our results show that the different NS1 proteins trigger hyperpermeability in endothelial cells derived from tissues associated with the viral tropism of each flaviviral disease (Figures 1 and S1B-S1F). Specifically, NS1 from DENV, which causes systemic disease, induces endothelial hyperpermeability in all endothelial cells tested, while NS1 from ZIKV, which targets the placenta and developing brain, induces hyperpermeability only in umbilical vein and brain endothelial cells. NS1 from both Uganda and Suriname strains behaved similarly (Figures S2A and S2B). NS1 from WNV and JEV, which cause encephalitis, induce hyperpermeability only in brain endothelial cells, whereas NS1 from YFV, which is systemic but primarily causes hepatic damage, exerted the strongest effect in liver endothelial cells, with a slight increase of permeability in lung endothelial cells. All endothelial cells demonstrated increased permeability when exposed to the pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α) as a positive control. However, no increase in permeability was observed with recombinant DENV2 E protein, demonstrating that the hyperpermeability induced is an NS1-specific effect (Figure 1). The TEER results were confirmed in a solute flux assay using 70-kDa dextran conjugated to fluorescein isothiocyanate (FITC) as a tracer in HUVEC monolayers (Figure S2C).
Figure 1. Flavivirus NS1 Proteins Trigger Endothelial Barrier Dysfunction In Vitro in a Tissue-Specific Manner.

(A-E) Human endothelial cells from different tissues (A, lung; B, skin; C, umbilical vein; D, brain; and E, liver) were grown on Transwell semi-permeable membranes (0.4 μm pore size), and distinct flavivirus NS1 proteins (5 μg/mL, 1.5 μg total protein) were added to the apical chamber. TNF-α (10 ng/mL) was used as a positive control, and DENV2 recombinant envelope (E) protein (5 μg/mL) was used as a negative control. A TEER assay was used to evaluate the effect of these NS1 proteins on endothelial permeability at indicated time-points over 48 h.^ represents media change. Relative TEER values from two independent experiments performed in duplicate are plotted. Error bars indicate SEM. Statistically significant differences between distinct groups (all time points combined for each group) compared to the untreated groups were determined by a two-way ANOVA analysis using Dunnett’s test for multiple comparisons, with ****p < 0.0001 and ns (not significant), p > 0.05.
In addition to our endothelial permeability assays, we assessed the ability of each flavivirus NS1 protein to interact with the surface of the different human endothelial cells in vitro by performing NS1 binding assays. Briefly, NS1 proteins were added to confluent monolayers of human endothelial cells, and the amount of NS1 bound to the surface of the endothelial cell monolayers was determined by immunofluorescence microscopy. At 1 h post-treatment (hpt), all NS1 proteins showed a binding pattern that reflected their capacity to selectively induce endothelial hyperpermeability as measured by TEER (Figures 2A–2E and S3A). DENV NS1 bound in a dose-dependent manner to the surface of all endothelial cells, showing maximum binding to pulmonary endothelial cells. ZIKV NS1 showed highest binding to umbilical vein endothelial cells and brain endothelial cells. WNV NS1 and JEV NS1 only exhibited increased binding capacity to brain endothelial cells. Surprisingly, YFV NS1 bound to all endothelial cells; however, significantly higher amounts were found on liver endothelial cells (Figures 2 and S3). These binding patterns were confirmed by western blot analysis of cell lysates from confluent monolayers of human endothelial cells exposed to individual flavivirus NS1 proteins (Figures 2F–2J and S3B-S3F).
Figure 2. Flavivirus NS1 Proteins Bind Differentially to the Surface of Distinct Human Endothelial Cells.

Binding of flavivirus NS1 proteins to the surface of different human endothelial cell monolayers as measured by (A-E) confocal microscopy or (F-J) western blot. (A-E) (A) HPMECs, (B) HMEC-1, (C) HUVECs, (D) HBMECs, and (E) HLSECs were grown on coverslips and treated with 5 μg/mL NS1 or10 μg/mL NS1 (1.5 μg or 3 μg of total protein, respectively) as indicated (also see Figure S3); binding was evaluated 1 h post-treatment (hpt). The amount of bound NS1 was quantified and expressed as MFI.
(F-J) Cell lysates from (F) HPMECs, (G) HMEC-1, (H) HUVECs, (I) HBMECs, and (J) HLSECs treated with 10 μg/mL (3 μg total protein) of flavivirus NS1 protein and collected 1 hpt and analyzed by western blot (Figure S3). All graphs show the average of quantification from two independent experiments run in duplicate. Error bars indicate SEM.
In (A)-(E), statistically significant differences between distinct treatment groups compared to the untreated group [NS1(−)] were determined by a two-way ANOVA analysis using Dunnett’s test for multiple comparisons, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Further, the specificity of the interactions of NS1 with the surface of human endothelial cells was corroborated by testing the effect of an anti-6x-His-tag monoclonal antibody (mAb) targeting the tag on the NS1 proteins and an NS1-specific mAb on DENV-NS1-induced barrier dysfunction of HPMECs, as measured by TEER (Figure S3G), and on NS1’s interaction with HPMEC and 293F cells, as measured by immunofluorescence microscopy and flow cytometry, respectively (Figures S3H and S3I). As expected, the anti-His-tag mAb did not prevent either the DENV-NS1-induced hyperpermeability of HPMECs or NS1 binding to HPMECs and 293F cells. However, the anti-NS1 mAb blocked the barrier dysfunction induced by NS1 in HPMECs as well as NS1 binding to HPMECs and 293F cells. These results indicate that the different flavivirus NS1 proteins have the ability to bind to distinct tissue-specific endothelial cells, potentially leading to the endothelial dysfunction and hyperpermeability identified in the TEER assay (Figure 1).
To identify a specific region of the flaviviral NS1 protein necessary for the distinct endothelial cell binding patterns observed (Figure 2), we constructed a chimeric mutant comprised of a DENV NS1 backbone containing amino acids 101–135 from WNV NS1 (DENVWNV101–135). We had previously identified this region of DENV NS1, known as the wing domain, as immunodominant in mice and humans (Hertz et al., 2017), and another study implicated this region as critical for antibody-mediated inhibition of NS1-induced pathogenesis in humans (Lai et al., 2017). We hypothesized that if this region of the wing domain contributed to the differential ability of DENV and WNV NS1 to bind to HPMECs, then this chimeric mutant would behave like WNV and display diminished binding to HPMECs. Using our 293-protein expression system, we produced and isolated this chimeric mutant. As expected, when we compared the ability of DENV, WNV, and DENVWNV101–135 NS1 proteins to bind to HPMECs, we observed that DENVWNV101–135 NS1 bound significantly less than DENV NS1, binding comparably to WNV NS1 (Figures S4A and S4B). These data suggest that the wing domain of NS1, particularly the motif between amino acids 101 and 135, is critical for the differential NS1 binding patterns observed on HPMECs.
Intriguingly, the differential binding patterns of the NS1 proteins (Figure 2) correlated with the capacity to trigger endothelial hyperpermeability (Figure 1) for all NS1 proteins except YFV NS1. This suggests that binding to endothelial cells is not sufficient for NS1 to trigger endothelial hyperpermeability. Because DENV NS1 was previously shown to be internalized by hepatocytes (Alcon-LePoder et al., 2005), we hypothesized that YFV NS1 bound to but was not internalized by all endothelial cell lines. To test this hypothesis, we compared internalization of DENV and YFV NS1 into HBMECs (where DENV, but not YFV NS1, triggered hyperpermeability) and HLSECs (where both NS1 proteins induced hyperpermeability), as determined by colocalization of NS1 with the early endosome marker Rab5. We found that while DENV NS1 was efficiently internalized into both cell types, YFV NS1 was only significantly internalized into HLSECs (Figures S4C and S4D). This selective internalization of YFV NS1 suggests that both binding and internalization are required for NS1 to trigger endothelial hyperpermeability.
Flavivirus NS1 Proteins Induce Degradation of EGL Components after Triggering Sialidase and Cathepsin L-Heparanase Activation in Human Endothelial Cells In Vitro
The EGL is a network of glycosaminoglycans (GAGs) and membrane-bound proteoglycans and glycoproteins expressed on the endothelium that lines the luminal surface of blood vessels and contributes to barrier function critical for vascular homeostasis (Weinbaum et al., 2007). Previously, we demonstrated that DENV NS1, but not WNV NS1, activates endothelial cell-intrinsic pathways that lead to barrier dysfunction of HPMECs in vitro. NS1 triggered loss of sialic acid from the cell surface upon activation of endothelial sialidases as well as the shedding of heparan sulfate proteoglycans by activating endothelial cathepsin L and heparanase (Puerta-Guardo et al., 2016). To establish whether different flavivirus NS1 proteins induce endothelial cell-specific barrier dysfunction via activation of similar mechanisms, polarized monolayers of HPMECs, HMEC-1, HUVECs, HBMECs, and HLSECs were exposed to the different flavivirus NS1 proteins. The integrity of EGL components was evaluated 6 hpt using immunofluorescence staining for sialic acid (Figures 3 and S5A), heparan sulfate (Figures 4 and S5B), and syndecan-1, a major heparan-sulfate-bearing proteoglycan that supports the integrity of the EGL (Choi et al., 2011) (Figures S6A and S6B). DENV NS1 induced disruption of sialic acid on all endothelial cells, with significant reductions in sialic acid expression that ranged from 20% to 80% depending on the endothelial cell type. Reflecting the pattern observed in endothelial permeability (Figure 1), ZIKV NS1 disrupted sialic acid in both HUVECs and HBMECs, whereas WNV NS1 and JEV NS1 only disrupted sialic acid on HBMECs. A significant reduction in expression of sialic acid on HLSECs was only observed in response to YFV NS1 (Figure 3). Regarding expression of heparan sulfate and syndecan-1, degradation of heparan sulfate after treatment with the distinct flavivirus NS1 proteins was detected on the surface of the different human endothelial cells after 6 hpt following a pattern that reflected our observations for sialic acid expression (Figure 4). Of note, treatment of endothelial cells with recombinant envelope protein of DENV did not change the surface levels of these EGL components (Figures S5A and S5B). In parallel, accumulation of syndecan-1, whose ectodomain binds back to the surface of endothelial cells after being shed (Choi et al., 2011), was detected on the surface of the different endothelial cells 6 hpt in a similar pattern (Figures S6A and S6B). Thus, disruption of EGL components was also determined by the flavivirus NS1 protein and the tissue-specific endothelial cell.
Figure 3. Flavivirus NS1 Proteins Alter the Expression of Sialic Acid on the Surface of Endothelial Cells in a Cell-Type-Dependent Manner.

(A) Sialic acid expression on human endothelial cell monolayers grown on coverslips 6 hpt with different flavivirus NS1 proteins (5 μg/mL, 1–5 μg total protein), examined by confocal microscopy. Sialic acid was stained with wheat germ agglutinin conjugated to Alexa Fluor 647 (red). Nuclei were stained with Hoechst (blue). Images (20×) are representative of three independent experiments. Scale bar, 10 μm.
(B) Quantification of MFI in (A) from three independent experiments. Error bars indicate SEM. Reduction of sialic acid expression in NS1-treated monolayers was normalized to untreated controls (dotted line). Statistically significant differences between distinct treatment groups compared to the untreated group were determined by a two-way ANOVA analysis using Dunnett’s test for multiple comparisons, with *p < 0.05, ***p < 0.001, and ****p < 0.0001.
Figure 4. Flavivirus NS1 Proteins Alter the Expression of Heparan Sulfate on the Surface of Endothelial Cells in a Cell-Type-Dependent Manner.

(A) Heparan sulfate expression on human endothelial cell monolayers grown on coverslips 6 hpt with different flavivirus NS1 proteins (5 μg/mL, 1–5 μg total protein), examined by confocal microscopy. Heparan sulfate was stained with anti-heparan sulfate mAb (green). Nuclei were stained with Hoechst (blue). Images (20×) are representative of three independent experiments. Scale bar, 10 μm.
(B) Quantification of MFI in (A) from three independent experiments. Error bars indicate SEM. Reduction of heparan sulfate expression in NS1-treated monolayers was normalized against untreated controls (dotted line). Statistically significant differences between distinct treatment groups compared to the untreated group were determined by a two-way ANOVA analysis using Dunnett’s test for multiple comparisons, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
We also evaluated the effect of flavivirus NS1 proteins on activity and expression of endothelial cathepsin L and expression of the endoglycosidase heparanase, enzymes involved in the degradation of EGL components (Glasner et al., 2017; Puerta-Guardo et al., 2016). Compared to untreated monolayers, DENV NS1 increased the expression and activity of cathepsin L in all endothelial cells (Figures 5A, 5B, S5C, S6C, and S6D). ZIKV NS1 increased levels of cathepsin L expression and proteolytic activity in cultures of umbilical vein endothelial cells and brain endothelial cells. NS1 proteins from DENV, ZIKV, WNV, and JEV, but not YFV, modulated expression levels of cathepsin L and increased its activity in brain endothelial cells. Finally, YFV NS1 stimulated the proteolytic activity of cathepsin L in liver endothelial cells to levels 25-fold higher than untreated controls, whereas ZIKV NS1, WNV NS1, and JEV NS1 triggered no response (Figures 5A, 5B, S5C, S6C, and S6D). We also examined the maturation state of cathepsin L on different endothelial cells exposed to distinct NS1 proteins at 6 hpt. Densitometry analyses of western blots revealed expression of the pro-cathepsin L form in all endothelial cells exposed to different NS1 treatments but increased appearance of the active form of cathepsin L (Figures 5C–5E) that reflected the results obtained using the fluorescence proteolytic activity assay (Figures 5A, 5B, and S6D) and correlated with the patterns of hyperpermeability observed in Figure 1. As before, the activation of cathepsin L was specific to NS1, as the DENV E protein failed to significantly trigger maturation of cathepsin L (Figure S5C). Because proteolytic activity of cathepsin L is required for the activation of heparanase and subsequent trimming of heparan sulfate from the EGL (Abboud-Jarrous et al., 2008), we also examined the expression of heparanase in endothelial cells after NS1 stimulation using immunofluorescence microscopy. As before, we observed a similar cell-type- and NS1-dependent pattern of heparanase expression compared to the cathepsin L results (Figures 5F and S6C).
Figure 5. Flavivirus NS1 Proteins Modulate the Expression and Activity of Endothelial Cathepsin L and Heparanase in a Cell-Type-Dependent Manner.

(A and B) Fold change over untreated controls (dotted lines) of the quantification of MFI from three independent confocal microscopy experiments examining (A) expression and (B) activity of cathepsin L (Magic Red reagent) in human endothelial cell monolayers 6 hpt with different flavivirus NS1 proteins (5 μg/mL, 1–5 μg total protein) (Figure S6).
(C) Western blot data of pro- and mature- cathepsin L expression in human endothelial cell monolayers 6 hpt with different flavivirus NS1 proteins (5 μg/mL, 1.5 μg total protein).
(D and E) Densitometry of bands from (C) for (D) pro-cathepsin L and (E) mature cathepsin L with values normalized to GAPDH and expressed as fold change from untreated monolayers.
(F) Fold change over untreated controls (dotted line) of the quantification of MFI from three independent confocal microscopy experiments examining the expression of heparanase in human endothelial cell monolayers 6 hpt with different flavivirus NS1 proteins (5 μg/mL, 1.5 μg total protein) (Figure S6).
For all graphs, error bars indicate SEM. Statistically significant differences between distinct treatment groups compared to the untreated group were determined by a two-way ANOVA analysis using Dunnett’s test for multiple comparisons, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Additionally, expression of endothelial sialidases (Neu1, Neu2, and Neu3), glycosidases that catalyze the removal of terminal sialic acids of glycoproteins and glycolipids, was evaluated on endothelial cells treated with the different flavivirus NS1 proteins (Figures S7A-S7D). Compared to untreated cells, flavivirus NS1 proteins led to increased expression of Neu1, Neu2, and Neu3 in a cell-type-dependent manner at 6 hpt. In the case of DENV NS1, which increased the permeability of all the evaluated endothelial cells, Neu2 was the most upregulated sialidase in all endothelial cells, followed by Neu3 and Neu1. ZIKV NS1 upregulated the expression of Neu2 and Neu3 in HUVECs. Regarding the encephalitic flaviviruses, only HBMECs showed significant changes in the expression of the three sialidases in response to WNV and JEV NS1. Finally, for YFV NS1, which significantly increased the permeability of liver endothelial cells, the expression of Neu2 and Neu3 was significantly upregulated only in HLSECs, with some increase in Neu2 in HPMECs, also consistent with the patterns observed above.
Flavivirus NS1 Proteins Cause Increased Vascular Leakage in a Tissue-Dependent Manner and Induce Disruption of Endothelial Glycocalyx Components In Vivo
We next examined the ability of NS1 proteins from different flaviviruses to cause localized dermal or systemic vascular leakage in vivo using murine models. To evaluate dermal leak, we performed a fluorescent dextran-adapted Miles assay as previously described (Glasneretal., 2017). Briefly, following intradermal injection of recombinant NS1 proteins, fluorescently labeled (Alexa Fluor 680) dextran was delivered intravenously, and 2 h later, leakage in the dorsal dermis was visualized using a fluorescence detection system. We found that DENV NS1, but not ZIKV, WNV, JEV, or YFV NS1, induced a significant increase in vascular leakage in the murine dermis compared to injection of PBS (Figures 6A and 6B). To characterize the systemic effects of flavivirus NS1, we used a traditional Miles assay to evaluate induction of vascular leak in organs, including the brain, lungs, and liver. Mice were inoculated with 10 mg/kg flavivirus NS1, an amount that leads to circulating levels of NS1 equivalent to human dengue hemorrhagic fever (DHF)-dengue shock syndrome (DSS) cases (Beatty et al., 2015; Libraty et al., 2002). Three days post-inoculation (dpi), animals were administered Evans blue dye or Alexa-Fluor-680-labeled dextran for subsequent tissue extraction or fluorescent imaging, respectively. Following inoculation of DENV NS1, increased amounts of Evans blue dye were detected in the lungs and liver (Figures 6C and 6D). However, consistent with previous studies (Beatty et al., 2015), no significant extravasation of Evans blue dye was observed after inoculation of 10 mg/kg ovalbumin (OVA). YFV NS1 was the only other NS1 protein to induce significantly higher levels of Evans blue dye leakage in the liver (Figure 6D).
Figure 6. Flavivirus NS1 Proteins Induce Differential Local and Systemic Vascular Leakage In Vivo.

(A-H) Wild-type C57BL/6 mice were used to evaluate (A and B) local or (C-H) systemic vascular leakage induced by flavivirus NS1 proteins injected either (A and B) intradermally (dorsal dermis) or (C-H) intravenously with 15 μg and 200 μg (10 mg/kg) of total protein, respectively. The magnitude of the vascular leakage induced by these proteins was evaluated by using (A, B, and E-H) Alexa-Fluor-680-conjugated dextran (10 mg/mL) or (C and D) Evans blue dye (0.5%) and quantified by (A, B, and E-H) fluorescent imaging or (C and D) spectrophotometric analyses. (A, E, and F) Images (LI-COR Odyssey) and (B-D, G, and H) MFI values are representative of two individual experiments (n = 3–6 per group). Ovalbumin (OVA; 10 mg/kg) and PBS were used as controls for systemic and local vascular leakage experiments, respectively.
(I) Levels of heparan sulfate detected in serum of mice (n = 3 per group) treated with ovalbumin (OVA), DENV2 NS1, WNV NS1, or YFV NS1 (all 10 mg/kg) as measured by ELISA. Untreated mice were used as a steady-state control.
For dextran-adapted dermal Miles assay, data were derived from three independent experiments and expressed as the fold-change ratio of the MFI obtained in each individual treatment group compared to PBS injection (dotted line in B). MFI was measured inside the leakage area by drawing a standard circular area in each treatment condition using ImageJ tools. Statistically significant differences between distinct treatment groups compared to the control groups were determined by a one-way ANOVA analysis using Dunnett’s test for multiple comparisons, with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns (not significant), p > 0.05 . Error bars indicate SEM. Scale bars represent (A) 2.5 cm and (E and F) 0.5 cm.
We also examined vascular leak in the brain and lung by measuring the accumulation of fluorescently labeled dextran. We used fluorescently labeled dextran instead of Evans blue dye to measure vascular leak in the brain, because this reagent showed greater sensitivity in our assay compared to Evans blue dye. Further, because both WNV and JEV are neurotropic flaviviruses, we only included WNV in subsequent mouse experiments to minimize the number of mice per experiment. Intravenous administration of DENV, ZIKV, or WNV NS1 proteins resulted in significant accumulation and extensive leakage of dextran in brain tissue compared to OVA and YFV NS1 treatments (Figure 6E–6G). In the lungs, accumulation of dextran following inoculation with DENV NS1 resembled the extravasation pattern observed with Evans blue dye in the Miles assay, with large areas of increased mean fluorescence intensity (MFI) that result from the accumulation of dextran in tissue (Figures 6F–6H). In contrast, administration of WNV NS1 and OVA (used as a protein control) resulted in no dextran accumulation (Figures 6F–6H). Using this assay, mice inoculated with YFV and ZIKV NS1 proteins showed areas of leakage in the lung with increased MFI for dextran compared to OVA but substantially less than DENV NS1, suggesting a minimal level of vascular leak from the inoculation (Figures 6F–6H). Depending on their molecular size, dextran molecules can be used to test solute and ion permeability (low-molecular-weight dextran), indicating paracellular pathways. Therefore, leakage in the lung induced by YFV NS1 may be preferentially measured by smaller solute tracers, suggesting a potential increased paracellular transport of solutes between endothelial cells. Finally, to evaluate the impact of flavivirus NS1 proteins on vascular leakage in vivo, we investigated the effect of intravenous administration of NS1 proteins on the circulation of heparan sulfate, a major GAG used as an indirect marker of endothelial glycocalyx integrity in vivo. For this assay, to minimize numbers of mice per experiment, we included one systemic flavivirus (DENV), one encephalitic virus (WNV), and one hepatotropic virus (YFV). Compared to untreated mice and mice treated with OVA (used as a protein control), administration of DENV, YFV, and, to a lesser extent, WNV NS1 proteins led to increased serum levels of heparan sulfate (DENV, p = 0.0002; YFV, p = 0.0233; WNV, p = 0.0231) (Figure 6I), suggesting that shedding of heparan sulfate may indeed be a consequence of activation of the cathepsin L-heparanase pathway by NS1.
DISCUSSION
In this study, we describe the capacity of NS1 proteins from DENV, ZIKV, WNV, JEV, and YFV to induce hyperpermeability of cultured polarized human endothelial cells derived from different tissues, including lung, skin, umbilical vein, brain, and liver, as measured by TEER. These findings are supported by in vivo data collected using both Evans blue dye and fluorescently labeled dextran, showing that flavivirus NS1 proteins can differentially and selectively induce vascular leakage in distinct organs, including lung, skin, brain, and liver, reflecting respective flaviviral pathophysiology. We found that DENV NS1 induced hyperpermeability in all tested human endothelial cells, with maximal effects observed on pulmonary endothelial cells. DENV NS1 also stimulated vascular leak in all evaluated organs upon inoculation into mice. ZIKV NS1 increased the permeability of umbilical vein and brain endothelial cells in vitro and caused vascular leakage in the brain in vivo, whereas WNV NS1 and JEV NS1 only induced hyperpermeability of brain endothelial cells and substantial vascular leakage in the mouse brain. NS1 from YFV significantly increased the permeability of endothelial cells derived from liver and, to a lesser extent, lung, and this same pattern was seen in murine organs in vivo. Mechanistically, the observed hyperpermeability in vitro is related to disruption of the EGL components sialic acid, heparan sulfate, and syndecan-1 following upregulation of sialidases as well as cathepsin L expression and activity and subsequent expression of heparanase, enzymes known to contribute to the degradation of cellular GAGs and proteoglycans in the EGL. Taken together, our results indicate that flavivirus NS1 proteins modulate endothelial permeability in a tissue-specific manner both in vitro and in vivo, potentially influencing flavivirus dissemination, pathogenesis, and disease (Figure 7).
Figure 7. Model of Flavivirus NS1-Induced Endothelial Cell-Type-Dependent Hyperpermeability and Tissue-Specific Vascular Leakage and Its Potential Contribution to Flavivirus Pathogenesis and Disease.

Flavivirus infections can affect specific tissues, including brain, lung, skin, liver, and the placenta during pregnancy, to cause neurotropic-encephalitic or systemic diseases in humans. Infection with any of the flaviviruses leads to secretion of NS1, a soluble viral protein that directly triggers endothelial hyperpermeability and vascular leakage associated with the disruption of key components of the endothelial glycocalyx layer, which lines the luminal surface of endothelial cells in the vasculature. The EGL is composed of monosaccharides such as sialic acid (Sia), glycosaminoglycans (GAGs) such as heparan sulfate, and heparan sulfate proteoglycans (HSPGs) such as syndecan-1. Here, we describe an endothelial cell-type-dependent increase in permeability and vascular leakage induced by different flavivirus NS1 proteins that reflect the disease pathogenesis of distinct flavivirus infections. DENV NS1 increases the permeability of endothelial cells from multiple tissues, such as lung, skin, umbilical vein, brain, and liver, consistent with the systemic disease caused by DENV. ZIKV NS1 induces endothelial dysfunction of umbilical vein and brain endothelial cells, reflecting severe congenital and neurological defects associated with Zika, while WNV and JEV NS1 only affect the barrier function of brain endothelial cells, in line with the neurotropic and encephalitic nature of WNV and JEV disease. Finally, YFV NS1 increases the permeability of lung and especially liver endothelial cells, consistent with hepatic and systemic pathology of yellow fever virus. This selective permeability may contribute to the pathogenesis of the different flaviviruses by either inducing extravasation of fluids that result in inflammation of tissues or facilitating virus dissemination into target organs that may lead to enhanced viral infection and disease.
Clinically, severe WNV and JEV infections, as well as fetal ZIKV infections during pregnancy, primarily result in cerebrovascular disease with encephalitis or congenital brain abnormalities with neurological complications (Gould and Solomon, 2008; Krauer et al., 2017). In contrast, severe DENV and YFV infections are generally associated with systemic disease, characterized primarily by increased vasculopathy and plasma leakage from blood vessels into tissues, as well as hepatic damage/dysfunction (World Health Organization, 2009). However, there are multiple reports describing DENV-associated neurological complications such as encephalitis and DENV infection in the human brain in autopsies, as well as detection of viral antigens in placental tissues from DENV-infected mothers (Balsitis et al., 2009; Kumar et al., 2008; Ribeiro et al., 2017; Soni et al., 2017). These findings indicate that DENV may also breach the blood-brain barrier as well as reach the placenta during pregnancy. Overall, our data here demonstrate a tissue-specific pattern of interaction between flavivirus NS1 and endothelial cells, where NS1-mediated effects mirror the clinical pathophysiology of the corresponding flaviviral disease in humans. Data are lacking regarding levels of flavivirus NS1 in humans (other than DENV), especially in tissues, where vascular leak occurs. Regardless of levels of circulating NS1 antigenemia, the cumulative effect of NS1 is likely amplified and/or concentrated in the capillary beds of tissues, the primary sites of leakage.
Vascular leak is apparently mediated by the specific interaction and subsequent internalization of NS1 proteins in different endothelial cells. Our data showed that DENV NS1 binds to the surface of all human endothelial cells we studied but displayed a greater interaction with lung and liver endothelial cells, correlating with the pathophysiology of severe DENV disease (i.e., pleural effusion and liver dysfunction) (World Health Organization, 2009). ZIKV NS1 was found only on the surface of umbilical vein and brain endothelial cells. Interestingly, ZIKV infects fetal macrophages (Hofbauer cells) and placental villous fibroblasts, resulting in increased expression and secretion of ZIKV NS1 in placenta villus cores where fetal endothelial cells are present in blood vessels (Jurado et al., 2016; Tabata et al., 2016). Additionally, ZIKV infects human PBMCs in vivo and neural progenitor cells in vitro, suggesting that ZIKV NS1 may circulate in the blood of infected patients and be secreted in brain tissues (Michlmayr et al., 2017; Mlakar et al., 2016; Tang et al., 2016). WNV NS1 and JEV NS1 only interacted with brain endothelial cells, which correlated with their ability to induce hyperpermeability of these human endothelial cells as well as with their well-described neurotropism, resulting in neurological complications and encephalitis (Diamond and Klein, 2004). NS1 from YFV, most often associated with severe liver disease and systemic organ failure (Monath and Barrett, 2003), was unexpectedly found to bind to some degree to all human endothelial cells. However, it bound to liver endothelial cells to the highest level, which was the only cell type that displayed a high degree of YFV-NS1-induced hyperpermeability. This suggests that binding of NS1 to endothelial cells is not sufficient to explain NS1-triggered vascular leak and that further downstream steps, such as internalization, may further dictate this tissue-specific tropism. This concept is supported by our data showing that YFV NS1 can bind to but is not significantly internalized by HBMECs, whereas it binds to and is internalized by HLSECs. This indicates that internalization of NS1 may well be important for NS1-triggered endothelial hyperpermeability. These results further suggest that NS1 binding specificity may rely on the tissue-specific expression of surface molecules on different endothelial cells that may facilitate the early stages of NS1 binding via protein-protein or protein-glycan interactions. Preliminary studies to investigate the specificity of NS1 binding and its role in endothelial cell- type- and tissue-specific hyperpermeability suggest a very complicated interplay between certain EGL-associated cellular attachment factors, such as GAGs and proteoglycans, and the different flavivirus NS1 proteins, modulated by the level of sulfation and complexity of glycan moieties in the EGL as well as particular motifs on the NS1 proteins. With regard to the molecular determinants of DENV NS1 required to bind to endothelial cells, results obtained using the chimeric mutant DENVWNV101–135 NS1 implicates a particular region of the wing domain of NS1 as crucial for endothelial cell binding and ultimately endothelial hyperpermeability. Further characterization of this and other regions of NS1 is critical to decipher the biochemical mechanism behind NS1 binding from the viral side and is the focus of additional studies. From the host side, the cognate NS1 attachment factor(s) and receptors) remain elusive, and their identification requires numerous additional experiments in the fields of glycobiology, proteomics, and molecular genetics.
The vascular endothelium is composed of a single layer of endothelial cells that form blood vessels and modulate vascular homeostasis. The negative charge provided by EGL components, including glycoproteins containing sialic acid residues and GAGs, such as heparan sulfate (more than 50% of the total GAG pool), chondroitin sulfate, and hyaluronic acid, contribute to the maintenance of endothelial barrier function (Weinbaum et al., 2007). Degradation of the EGL has been linked to disease severity in several viral hemorrhagic fever diseases (Connolly-Andersen et al., 2014), including dengue, where increased levels of heparan sulfate, hyaluronic acid, and syndecan-1 have been found in the blood of patients with severe dengue disease as compared to controls with milder forms of dengue (Suwarto et al., 2017; Tang et al., 2017; Trung and Wills, 2010). However, the importance of vascular permeability and the role of EGL components in the pathogenesis of other flavivirus infections is still unknown. Here, we found that NS1-induced endothelial hyperpermeability correlates with increased disruption of key EGL components, regardless of the tissue origin of the human endothelial cells. The interaction of distinct flavivirus NS1 proteins with the surface of different human endothelial cells led to significantly decreased expression of sialic acid and heparan sulfate, suggesting degradation of these components. This interaction also resulted in increased deposition and accumulation of syndecan-1, which, after being shed, can bind back to the endothelial surface, inducing endothelial dysfunction (Choi et al., 2011; Puerta-Guardo et al., 2016). Importantly, inoculation of these recombinant NS1 proteins in a mouse model, particularly DENV, YFV, and WNV, led to a systemic increase in the levels of heparan sulfate, confirming the role of NS1 in remodeling the integrity of EGL components in endothelial beds in vivo and is consistent with increased heparan sulfate circulation in severe human dengue cases with vascular leak (Tang et al., 2017). Notably, NS1-induced EGL alterations in vitro were associated with increased expression and activation of endothelial sialidases (Neu1, Neu2, and Neu3), the cysteine protease cathepsin L, and the endoglycosidase heparanase, which have been implicated in the degradation of the extracellular matrix as well as cleavage of proteoglycans mainly expressed on the luminal surface of endothelial cells (Abboud-Jarrous et al., 2008; Choi et al., 2011; Weinbaum et al., 2007). The pattern revealed is consistent with the flavivirus NS1- and tissue-specific profiles reported above. However, the molecular mechanisms leading to upregulation and activation of the mammalian sialidases are still unknown. In turn, cathepsin L is known to activate heparanase after proteolytic processing of the non-active precursor into an active protein. We previously showed that inhibitors of sialidases, heparanase, and cathepsin L prevent NS1-induced disruption of the EGL in vitro and vascular leak in vivo (Glasner et al., 2017; Puerta-Guardo et al., 2016). Thus, we found that the cell-type-dependent pattern of endothelial hyperpermeability observed following flavivirus NS1 treatment of human cell monolayers is also reflected in endothelial cell-intrinsic mechanisms implicated in NS1 pathogenesis in vitro and putative disruption of the endothelial glycocalyx in a mouse model.
The assessment of vascular permeability is complicated, because it is affected by multiple factors, including the type of microvessels, endothelial cells, the size and charge of extravasating molecules, and the pathway that molecules take to traverse the endothelium. In addition to the glycocalyx layer, the hemostasis of the endothelium is maintained by the presence of intercellular junctional complex composed of protein-protein interactions that help to preserve the cell-to-cell contacts (Dejana, 2004). In these studies, we utilized five distinct measures of endothelial permeability and vascular leak: TEER and a dextran flux assay in vitro, Evans blue dye and fluorescently labeled dextran in a systemic Miles assay in vivo, and fluorescently labeled dextran in a modified local Miles assay in vivo. These assays all assess endothelial permeability, but because of the nature of the assays, they potentially measure different pathways. In vitro, TEER measures the ohmic resistance of a barrier, whereas flux assays measure the direct passage of a macromolecule from the apical to the basolateral chamber. Both of these methods represent a composite of both paracellular and transcellular pathways. In vivo, Evans blue binds to albumin, which does not cross the endothelium in the absence of inflammation or stimulus that disrupts normal barrier function. On the other hand, 10-kDa dextran should extravasate from the blood into tissue even during basal conditions (Egawa et al., 2013). As with the in vitro models, these assays most likely measure a composite of paracellular and transcellular pathways. The results from each of these models support our overarching hypothesis that NS1 exerts a virus- and tissue-specific effect on the human endothelium, leading to barrier dysfunction and hyperpermeability, but the amount of leakage due to transcellular versus paracellular pathways remains unknown, and further studies are needed to elucidate the molecular mechanisms driving this phenomenon. As the primary determinant of paracellular permeability, intercellular junctions are important regulators of endothelial barrier function, though it is likely that both the glycocalyx and intercellular junctions are involved in NS1-induced barrier dysfunction and that multiple factors work in synergy, resulting in endothelial hyperpermeability (Chen et al., 2016; Glasner et al., 2018). Discerning the relative contributions of the glycocalyx and intercellular junctions will undoubtedly prove to be challenging, but a more complete understanding of the dynamics and kinetics involved in disruption of the endothelial barrier by NS1 may inform clinical management and treatment protocols for flavivirus disease.
Flavivirus infection has been shown to compromise the integrity of many biological barriers, including the pulmonary microvascular endothelium and the blood-brain barrier, which are usually able to protect against virus infection (Li et al., 2015; Puerta-Guardo et al., 2013; Spindler and Hsu, 2012; Wang et al., 2004). So far, this process has been primarily attributed to exacerbated host immune responses that lead to increased permeability of endothelial cells in the different microvascular beds. However, the role of secreted flaviviral factors such as NS1 proteins in the pathophysiology of endothelial cells has not been reported, other than DENV NS1. Despite the mounting evidence showing that high levels of NS1 antigenemia during the acute phase of DENV infection correlate with increased risk of developing severe dengue disease, including vascular leakage (Song et al., 2016), little is known about circulating levels of NS1 from other flavivirus infections, including ZIKV, WNV, JEV, and YFV. Here, our data suggest a potential role for flavivirus NS1 proteins in causing tissue-specific pathology associated with endothelial dysfunction. We hypothesize that NS1 may induce selective hyperpermeability of different microvascular beds as a means of facilitating virus dissemination into target organs where permissive cells critical for local replication are found, resulting in virus amplification. Numerous studies of flavivirus infection in different animal models as well as human autopsies have shown a selective tropism of distinct groups of flaviviruses that target different tissues, leading to systemic versus neurotropic-encephalitic pathology (Gould and Solomon, 2008; Muller and Young, 2013). Despite studies describing the potential infection of endothelial cells by flaviviruses in vitro, flavivirus infection of endothelial cells in vivo is still controversial. In this study, infection of endothelial cells by DENV and ZIKV does not occur during the time frame of the experiment (12–24 h) (data not shown). Thus, we posit that NS1 is a virulence factor that, in addition to facilitating virus replication and immune evasion, may also facilitate virus dissemination from the bloodstream into tissues, influencing virus tissue tropism and disease manifestations.
Taken together, we report a previously undescribed function of different flavivirus NS1 proteins in modulating endothelial barrier function in vitro and in vivo in a tissue-dependent manner. Mechanistically, we found that all NS1 proteins were able to alter the integrity of EGL components on the surface of endothelial cells, leading to endothelial hyperpermeability. This evidence strongly suggests that distinct flavivirus NS1 proteins may modulate virus dissemination and thus influence viral pathogenesis and disease. These findings provide insight into the biology of NS1 and contribute to a better understanding of flavivirus pathogenesis, supporting the inclusion of NS1 protein in flavivirus vaccine development and generating new targets for future therapies against flavivirus infections.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eva Harris (eharris@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cultured human endothelial cells
Human pulmonary microvascular ECs (HPMEC) (line HPMEC-ST1.6R) were kindly donated by Dr. J.C. Kirkpatrick at Johannes Gutenberg University, Germany. HPMEC were isolated from adult male human donors, and the immortalized-selected clone (HPMEC-ST1.6R) displayed all major constitutively expressed and inducible phenotypic markers for endothelial cells. Human umbilical vein ECs (HUVEC) were a kind gift from Melissa Lodoen at the University of California, Irvine. HUVEC are primary endothelial cells obtained from a single female donor (Lonza). Both cell lines, HPMEC and HUVEC, were propagated (passages 5–10) and maintained in endothelial cell basal medium-2 supplemented with growth factors: FBS (5%), hydrocortisone (0.2 ml), R3-IGF-1 (0.5 ml), Ascorbic Acid (0.5 ml), hEGF (2 ml), and gentamicin-1000 (0.5 ml) as per the manufacturer’s specifications (Lonza). The human dermal microvascular endothelial cell line HMEC-1 (isolated from a newborn male), kindly donated by Dr. Matthew Welch, University of California, Berkeley, was propagated (passages 18–25) and maintained using MCDB 131 medium (Sigma) supplemented with 1% penicillin/streptomycin (Life Technologies), 0.2% Epidermal Growth Factor (Life Technologies), 0.4% hydrocortisone (Sigma), and 5% Fetal Bovine Serum (Corning). The human brain microvascular endothelial cells (HBMEC) were donated by Ana Rodriguez at New York University. HBMEC were obtained from ScienCell Research Laboratories (gender not available) and maintained at low passages (passages 8–10) using endothelial cell medium supplemented with FBS (5%), 5 mL of endothelial cell growth supplement (ECGS, Cat. No. 1052), and 5 mL of penicillin/streptomycin (ScienCell Research Labs). The primary human liver sinusoidal microvascular endothelial cells (HLSEC) were acquired from Cell Biologics (adult male donor) and maintained using complete human EC medium and EC medium supplement kit according to the manufacturer’s recommendations. All human endothelial cells were tested to be mycoplasma free, kept at low passage at 37° C in humidified air with 5% CO2, and routinely passaged prior to reaching 100% confluence. FreeStyle 293F suspension cells (Thermo Fisher Scientific) used to produce in house recombinant NS1 protein and to examine the binding of NS1 by flow cytometry were both cultured in FreeStyle 293 Expression medium (Thermo Fisher Scientific) supplemented with 1% penicillin/streptomycin and maintained inside a humid chamber at 37°C and 8% CO2 at a density of 0.15–1.2×106 cells/ml on a cell shaker at 135 rpm. 293 cells are human-female embryonic kidney epithelial cells.
Mice
Five- to eight-week old wild-type C57BL/6 male mice were purchased from the Jackson Laboratory (Sacramento, CA) and maintained under specific pathogen-free conditions at the University of California, Berkeley, Animal Facility. Mice were housed in a controlled temperature environment on a 12-hour light/dark cycle, with food and water provided ad libitum. All experimental procedures involving animals were pre-approved by the Animal Care and Use Committee (ACUC) of the University of California, Berkeley.
METHOD DETAILS
Production and purification of recombinant proteins
Recombinant NS1 proteins from DENV2 (Thailand/16681/84), ZIKV (Uganda MR766 and Suriname Z1106033), WNV (NY99), JEV (SA-14), and YFV (17D) were commercially acquired from The Native Antigen Company (Oxfordshire, United Kingdom). These proteins were produced in HEK293 cells with a purity greater than 95%, composed mostly of oligomeric forms as confirmed by silver staining and western blot assays (Figure S1) and certified by the manufacturer to be free of endotoxin contaminants. The recombinant DENV2/WNV chimera (aa101–135WNV), was generated in house using FreeStyle 293F suspension cells (Thermo Fisher Scientific) and the primers: 1r: CTC TTT CCC CAG GCC TTC CAG CCA ATT TCC AAT TTT TCC GTG GTG GCG GTG AGG CGT TTA GGT CCT GCC TGC ATG ATT CCT TTG ATG and 2f: TGG AAG GCC TGG GGA AAG AGT ATT TTA TTT GCA CCA GAA CTC GCC AAC AAC ACC TTT GTG GTT GAT GGC CCC GAA ACA GCA GAA. Transfections of 293F cells were performed using FreeStyle MAX transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol, and supernatant containing NS1 was collected 48 hours post-transfection. The 6x-His-tagged recombinant DENV NS1 proteins were purified by batch method using nickel nitrilotriacetic acid resin agarose beads (Thermo Fisher Scientific). Recombinant E protein from DENV2 [recE80% (aa 1–3970)] was kindly donated by Dr. Aravinda M. de Silva at the University of North Carolina, Chapel Hill. RecE was expressed and purified from supernatants of Sf21 cells infected with a baculovirus system. All recombinant protein preparations were confirmed to be endotoxin-free (< 0.001) using the Endpoint Chromogenic Limulus Amebocyte Lysate QCL-1000TM kit (Lonza).
Trans-endothelial electrical resistance (TEER) to measure endothelial permeability
The effect of recombinant flavivirus NS1 proteins on endothelial permeability was evaluated by measuring TEER of endothelial cell (EC) monolayers grown on a 24-well Transwell polycarbonate membrane system (Transwell® permeable support, 0.4 μm, 6.5 mm insert; Corning Inc.). Briefly, human endothelial cells (80%–90% confluency) cultured in vented-75 cm2 flasks (Corning®) were detached using a combination of three washing steps using sterile 1X PBS supplemented with EDTA (2 mM) (lifting buffer), followed by two additional washing steps (~30 s total) using a solution of trypsin-EDTA (0.25%) (GIBCO®, Thermo Scientific). Detached cells were resuspended using fresh culture media and then counted using a tissue-culture hemocytometer. A total of 60,000 cells for HPMEC, HMEC-1, HUVEC, and HBMEC or 80,000 cells (HLSEC) were seeded in the apical side of Transwell inserts (top chamber), using a final volume of 300 μL per well (top chamber). Each Transwell was transferred inside a 24-well format plate containing 1.5 mL of endothelial cell culture media, which represents the basolateral side of each Transwell. Transwells containing endothelial cells were incubated at 37°C and 5% CO2 for 3 days, and 50% of culture medium was changed in each well 48 hours post-seeding. Cells were grown until TEER values between 150 and 180 Ohms (Ω) were reached, depending on cell type, indicating 100% cell confluency. After this, a single dose of recombinant protein including different flavivirus NS1 proteins (5 and 10 μg/ml, 1.5 and 3 μg total protein, respectively), recE (5 μg/ml, 1.5 μg total protein), and TNF-α (10 ng/ml, 3 ng total protein), were added to the apical side of the Transwell insert (top chamber, 300 μl) containing the cell monolayer. TEER values, expressed in Ohms (Ω), were collected at sequential 2-hour time-points following the addition of test proteins using an Epithelial Volt Ohm Meter (EVOM) with “chopstick” electrodes (World Precision Instruments). Endothelial permeability was expressed as relative TEER which represents a ratio of resistance values (Ω) as follows: (Ω experimental condition - Ω medium alone)/(Ω non-treated endothelial cells - Ω medium alone). After 24 hours of treatment, 50% of upper and lower chamber media was replaced by fresh endothelial cell medium. Then, TEER was measured 3 hours (27 hpt) and 24 hours later (48 hpt).
Solute flux assay
To evaluate the effect of different flavivirus NS1 proteins on the passage of macromolecules through a polarized monolayer of human ECs, HUVEC (60,000 cells/Transwell insert) were grown on collagen-coated Transwell inserts (Corning Transwell-COL collagen-coated membrane inserts, 24-well, 0.4 μm, 6.5 mm insert; Corning Inc.). After three days of culture using complete endothelial cell media (Lonza), 50% of upper and lower chamber media was replaced with fresh EC Growth Media-2 (PromoCell) for 24 hours before beginning the experiment. Test proteins (10 μg/ml, 3 μg total protein) were added to the apical chamber of the Transwell inserts and incubated for 6 hours at 37°C. At 6 hpt, 70-kDa dextran conjugated to FITC (Sigma) was added to the apical chamber of the Transwell inserts at a final concentration of 1 mg/ml and allowed to circulate for one hour at 37°C. Transwell inserts were removed, and 100 μL from each well was collected from duplicate wells and transferred to a 96-well flat-bottom plate. Fluorescence was measured on a plate reader, and the concentration of dextran-FITC that passed from the apical to the basolateral chamber was determined using a standard curve (0.488 – 1000 μg/ml). TNF-α (100 ng/ml, 30 ng total protein) was used as a positive control, and untreated cell monolayers were used as a baseline control.
Flavivirus NS1 protein binding assay
To assess the ability of each flavivirus NS1 protein to interact with the surface of human endothelial cells, we used two different approaches: an immunofluorescence assay (IFA) to visualize the extracellular NS1 protein (5 and 10 μg/ml; 1.5 and 3 μg total protein, respectively) on non-permeabilized monolayers of endothelial cells grown on 0.2% gelatin-coated coverslips (Sigma); and a western blot assay using total protein extracts collected from confluent endothelial cell monolayers exposed to 10 μg/ml (3 μg) of each NS1 protein. For both assays, individual flavivirus NS1 proteins were incubated for one hour at 37°C and 5% CO2 in a total volume of 300 μl. All flavivirus NS1 proteins used in this study contain a C-terminal 6x-His-tag. NS1 protein bound to the cell surface was detected using an anti-6x-His-tag mAb (HIS.H8) conjugated to Alexa Fluor 647 (Thermo Scientific) followed by confocal microscopy analyses. Briefly, after one-hour post-treatment with NS1 proteins, the anti-6x-His-tag mAb conjugated to Alexa Fluor 647 was added into the same culture media at a dilution of 1:200 and incubated for 30 minutes protected from light. Both unbound NS1 and anti-His antibody were removed, and cell monolayers were washed twice with washing buffer (1X PBS + 2% FBS) before being fixed with 4% formaldehyde (Thermo Scientific) diluted in washing buffer. Untreated cells were used as a negative background control for non-specific anti-His antibody binding to the surface of endothelial cells. Hoechst 33342 (0.125 μl/slide) was used to stain the nuclei. ImageJ was used to estimate the amount of NS1 bound to the cell surface after image processing and analysis, expressed as mean fluorescence intensity values (MFI). Additionally, the binding ability of DENV NS1 (5 μg/ml) was tested on 293F cells in the presence and absence of the anti-6x-His-tag antibody (10 μg/ml) measured by flow cytometry using an anti-NS1 mAb in house-conjugated to Alexa Fluor 568. DENV NS1 binding was estimated from the positive signal obtained from non-NS1 treated cells, used as a negative control for NS1-treated cells equally stained with the Alex Fluor 568-conjugated anti-NS1 mAb. Flow cytometry data analyses of NS1 positive cells was performed by FlowJo V10.
Alternatively, a combination of anti-6x-His-tag mAb with an anti-mouse IgG (H+L) conjugated to Alexa Fluor 680 was used to detect the presence of NS1 protein in total protein extracts by western blot using an infrared detection system (LI-COR). Briefly, pre-chilled culture plates (15 minutes at 4°C) containing confluent monolayers of human endothelial cells (4–5 days in culture) were treated with a single dose of distinct flavivirus NS1 proteins (10 μg/ml). Culture plates were then immediately transferred to 4°C for additional 45 minutes of incubation. Later, plates were transferred to 37°C for 1.5 hours. Monolayers were then washed twice with cold 1X PBS followed by two subsequent treatments with a solution of pre-warmed trypsin-EDTA(0.25%) (GIBCO®, Thermo Scientific) until cells rounded. At this point, the trypsin solution was aspirated away gently, and monolayers were detached using cold 1X PBS + 2% FBS. Cells were transferred into low protein binding microcentrifuge tubes (0.6 ml, Fisher Scientific) and pelleted by centrifugation at 4°C at 1,500 rpm for 5 minutes. The cell pellet was rinsed twice with cold sterile 1X PBS. The supernatant was carefully discarded after every wash step. Cell pellets were resuspended in 50 mL of cold 1X PBS, transferred into a new tube containing ~500 μl of 1X PBS, and centrifuged at 1,500 rpm at 4°C for 5 minutes. Supernatant was discarded, and cell lysis was performed by directly adding 60 μLof Laemmli sample buffer (60 mMTris-Cl pH 6.8,2% SDS, 10% glycerol, 100 mM DTT, and 0.01% bromophenol blue) to the cell pellet. Equal volumes of cell lysates (30 μl/well) were loaded and separated under reducing conditions using SDS-PAGE polyacrylamide gradient gel (4%–20% Criterion TGX Precast Midi Protein Gel, BioRad), and running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS) at a constant voltage of 100 V for 1.5 hours. Blotting was performed using Trans-Blot Turbo Mini PVDF Transfer Packs (Cat# 1704156, BioRad) and Trans-Blot Turbo Transfer System (BioRad). PVDF membranes were blocked using a mix of skimmed milk (5%) and bovine serum albumin (BSA, 1%) diluted in TBS-T (Tris-buffered saline 20mM pH 7.4; NaCl 150 mM, and 0.1% Tween-20) for 45 minutes at room temperature and gentle oscillation. The amount of NS1 bound to the cell surface was measured using an anti-6x-His-tag primary antibody (1:500 diluted in TBS-T overnight at 4°C), followed by an anti-mouse IgG conjugated to Alexa Fluor 750 used as the secondary antibody (1:5000 diluted in TBS). NS1 was detected after infrared detection analyses using LICOR imaging software. GAPDH was used as a protein loading control to normalize the relative amount of NS1 protein bound to the surface of each endothelial cell. An anti-rabbit IgG conjugated to Alexa Fluor 680 was used as a secondary antibody for GAPDH. Untreated monolayers were used as a negative control for background subtraction. Densitometry analyses was performed using Image Studio Lite software v 5.2 (LI-COR Biosciences).
Flavivirus NS1 internalization assay
To examine the internalization of recombinant NS1 proteins from DENV and YFV by human endothelial cells, pre-chilled monolayers of HBMEC and HLSEC grown in 300 μL of cell culture media on coverslips pre-coated with 0.2% gelatin (Sigma) were exposed to 10 μg/ml of individual NS1 proteins (3 mg total protein) and incubated for 45 minutes at 4°C. Plates were then transferred to 37°C and incubated for 1.5 hours to facilitate NS1 protein internalization into endothelial cells. Inoculum was removed and monolayers were rinsed twice with 1X PBS + 2% FBS. Cells were then fixed for 20 minutes in 4% formaldehyde diluted in 1X PBS + 2% FBS and then for 20 minutes in 1 mL of cold 100% methanol added on top of the formaldehyde, followed by a 20-minute permeabilization/blocking step in 1X PBS + 0.2% saponin + 2% FBS + 1% BSA. Internalization of NS1 was demonstrated by co-staining NS1 (6x-His-tag mAb; 1:50 in 1X PBS) and an early endosome marker, Rab5 (1:50 in 1X PBS), using immunofluorescence microscopy. Both primary antibodies were incubated overnight at 4°C. Anti-mouse IgG Alexa Fluor 568 and anti-rabbit IgG Alexa Fluor 488 (1:100 dilution in 1X PBS) were used as secondary antibodies, incubated for 2 hours at room temperature with light protection. Hoechst 33342 (0.125 μl/slide) was used to stain the nuclei. For staining, coverslips were incubated on parafilm containing ~15 μl/slide of the primary or secondary antibody diluted in 1X PBS with the cell side of the coverslip facing down onto the antibody containing drop inside a humid chamber. Coverslips were rinsed 4 times with 1X PBS between antibody probing steps. Coverslips were mounted on glass slides onto a drop of ProLong® Gold Antifade Mountant (Thermos Scientific, Cat# P36934). ImageJ was used for image processing and analyses. Quantification of colocalization between NS1 and Rab5 was determined by manually counting NS1-positive cells and evaluating them for colocalization with Rab5 puncta. At least 50 NS1-positive events were quantified for two individual experiments.
Fluorescence microscopy and analysis of EGL components on endothelial cells
For imaging experiments, all human endothelial cells were grown on sterile coverslips coated with 0.2% gelatin (Sigma) and imaged on a Zeiss LSM 710 Axio Observer inverted fluorescence microscope equipped with a 34-channel spectral detector (CRL Molecular Imaging Center, UC Berkeley). To visualize the effect of all flavivirus NS1 proteins on the expression of EGL components such as sialic acid, heparan sulfate, and syndecan-1, as well as expression of endothelial sialidases (Neu1, Neu2, and Neu3), cathepsin L expression and activity, and heparanase expression, confluent endothelial cell monolayers were treated with 5 μg/ml of each flavivirus NS1 protein. After 6 hpt, cell monolayers were rinsed twice with 1X PBS + 2% FBS before fixing with cold 4% formaldehyde (FA) diluted in the same buffer. Untreated monolayers were used as a control for normal expression/distribution of all EGL components and endothelial enzymes mentioned above. For staining sialic acid, wheat germ agglutinin (WGA) conjugated to Alexa Fluor 647 (5 μg/ml) was incubated with live cells (300 μl) for 30 minutes at 37°C and 5% CO2. Next, coverslips were rinsed twice with 1X PBS + 2% FBS and fixed with 4% formaldehyde diluted in the same buffer for 20–30 minutes at 4°C, light protected. To stain heparan sulfate, after fixing the monolayers with 4% formaldehyde, coverslips were incubated overnight at 4°C with an anti-heparan sulfate antibody (1:50 in 1X PBS). Then, an anti-mouse IgM Alexa Fluor 488 (1:100 in 1X PBS) was used as secondary. For other EGL markers, cells were fixed for 20 minutes in 4% formaldehyde diluted in 1X PBS + 2% FBS and then for 20 minutes in 1 mL of cold 100% methanol added on top of the formaldehyde, followed by a 20-minute permeabilization/blocking step in 1X PBS + 0.2% saponin + 2% FBS + 1% BSA. After this, primary antibodies against Neu1, Neu2, and Neu3 (1:50); syndecan-1 (1:50), human heparanase (1:25), and cathepsin L (1:50) were incubated overnight at 4°C in 1XPBS. Detection of primary mAbs was performed with secondary antibody species-specific IgG antibodies conjugated to Alexa fluorophores (568 and 647). Coverslips were processed as described above, incubating primary and secondary antibodies on top of parafilm sheets inside humid chambers. Hoechst 33342 (0.125 μl/slide) was used to stain the nuclei. ProLong® Gold Antifade was used as mounting medium. Images were acquired at 20X magnification using Zeiss Zen 2010 software and were processed and analyzed with ImageJ software. All RGB images were converted to grayscale. Then, mean grayscale values and integrated density from selected areas were taken, along with adjacent background readings, and plotted as mean fluorescence intensity (MFI). For MFI quantification, individual images (708.48 × 708.48 μm) were obtained as RGB composites and split into 4-channels to adjust the threshold for each individual channel. Then, each channel was converted into a grayscale format and equally divided into four framed non-overlapping areas (341.9 × 353.8 mm, ~45–50 nuclei/frame). MFI was measured using an ImageJ plugin. All different experimental conditions were run in duplicate in two individual experiments. Representative images (125.73 × 138.96 μm).
Western blots for Cathepsin L expression/activity and Magic Red assay
Confluent cell monolayers (~300,000 cells/24-well plate, 4–5 days) of human endothelial cells from lung (HPMEC), skin (HMEC-1), umbilical vein (HUVEC), brain (HBMEC), and liver (HLSEC) were treated with 5 μg/ml of individual flavivirus NS1 proteins (DENV, ZIKV, WNV, JEV, and YFV). After 6 hpt, cells were rinsed with cold 1X PBS (three times), then monolayers were lysed using Laemmli sample buffer (60 μl/well; 60 mM Tris-Cl pH 6.8, 2% SDS, 10% glycerol, 100 mM DTT, and 0.01% bromophenol blue). Cell lysates (30 μl/well) was separated by 4%−20% gradient SDS-PAGE (4%−20% Criterion TGX Precast Midi Protein Gel, BioRad) using running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS) at a constant voltage of 100 V for 1.5 hours. For subsequent analyses, proteins were transferred onto PDVF membranes using a Trans-Blot® Turbo Transfer System (BioRad). Then, membranes were blocked in TBS-T with skimmed milk (5%) and bovine serum albumin (BSA, 1%), 45 minutes at room temperature. Primary anti-human cathepsin L antibody (1:50) and anti-GAPDH (1:1000) (used as a housekeeping protein control) were incubated overnight at 4°C using fresh blocking buffer in a humid chamber. Secondary anti-mouse IgG Alexa Fluor 750 and anti-rabbit Alexa Fluor 680 (1:5000 diluted in TBS) were used to detect the expression of these two proteins by Odyssey CLx Infrared Imaging System (LI-COR). Densitometry analyses was performed using Image Studio Lite software v 5.2 (LI-COR Biosciences). Potential activation of cathepsin L determined by western blot was initially identified by the presence of pro-mature Cathepsin L (higher band) versus mature-cathepsin L (lower band). The ratio of the densitometry values obtained from each experimental condition after normalization with its own loading control (GAPDH) and the subtracted background was used to estimate the levels of cathepsin L activation between the different NS1 treatments on the distinct endothelial cells compare to those values obtained from untreated cells used as basal levels for cathepsin L activation, expressed as fold changes.
In addition to this, the activity of cathepsin L in living cells was monitored using the Magic Red Cathepsin L detection kit (Immunochemistry Technologies, Inc.). Briefly, confluent monolayers of different human endothelial cells (4–5 days of culture) grown on coverslips pre-treated with 0.2% gelatin were exposed to flavivirus NS1 proteins (5 μg/ml), and at 6 hpt, a cell membrane-permeant fluorogenic substrate MR-(Phe-Arg)2 (1:250 dilution per well in 300 μl), which contains the cresyl violet fluorophore branded as Magic Red™, was added to each well. After 1 hour of incubation at 37°C and 5% CO2 while protected from light, cell monolayers were rinsed twice with cold 1X PBS + 2% FBS and then fixed with 4% formaldehyde diluted in the same buffer. After 30 minutes of incubation at 4°C under light protection, all coverslips were rinsed with 1X PBS and then mounted onto glass microscope slides containing ProLong® Gold Antifade used as mounting medium. Hoechst 33342 (0.125 μl/slide) was used to stain the nuclei. Images were acquired at 20X magnification using Zeiss LSM 710 AxioObserver and Zeiss Zen 2010 software, then processed and analyzed with ImageJ software. Cultured cell monolayers expressing active cathepsin L catalyze the hydrolysis of the two Phe-Arg (FRFR) target sequences, generating a red fluorescent species that can be detected by immunofluorescence microscopy. Magic Red excites at 540–590 nm (590 nm optimal) and emits at > 610nm (630 nm optimal). Levels of cathepsin L activation obtained by detection of mean fluorescence intensity (MFI) of the whole cell population exposed to different NS1 proteins were expressed as fold changes over untreated cells used as control for basal levels of cathepsin L activity.
In vivo vascular leakage assays
To evaluate the in vivo effect of flavivirus NS1 proteins on vascular hemostasis, we used a conventional Miles assay using Evans blue dye (EBD), as well as a modified version using fluorescently-labeled dextran. Briefly, wild-type C57BL/6 mice were initially intravenously injected with either 10 mg/kg of ovalbumin or each individual flavivirus NS1 protein (10 mg/kg). Three days post-injection, mice were intravenously administrated with 100 μl of 0.5% EBD in PBS and 200 μl of 10-kDa dextran conjugated to Alexa Fluor 680 (dextran-A680; 1 mg/ml). Both compounds were allowed to circulate for 3 or 2 hours, respectively, before euthanizing animals and perfusing tissues with 20 mL of PBS and 10 mL of formalin solution (Sigma) for tissue fixation. To quantify the levels of vascular leakage into the extravascular space of tissue using dextran-A680, tissues were harvested and transferred into a 6-well format tissue culture plate, and the levels of infrared signal present in different tissues including brain and lungs were measured using the LI-COR Odyssey CLx Imaging System. Brain tissues were manually dissected using a disposable scalpel to separate the right and left hemispheres through the longitudinal fissure, and each hemisphere was transferred into a 24-well plate and read on a LI-COR Odyssey CLx Imaging System. Images were acquired using Image Studio Lite software, and the leakage area was quantified from a standard-scanned area selected for each tissue to obtain a Mean Fluorescence Intensity value using ImageJ analyses. Representative images were selected after ImageJ processing and analyses showing the leakage area in brain and lungs in red compare to the autofluorescence signal (in blue) emitted by biological tissues. All RGB images were converted to grayscale. Then, mean grayscale values and integrated density from selected areas were taken, along with adjacent background readings, and plotted as leakage area, mean fluorescence intensity (MFI).
For EBD extraction, in order to remove the excess water/fixative buffer accumulated during perfusion and tissue harvesting, each tissue was initially chopped into small pieces, transferred into an Eppendorf tube (1.5 ml), then vacuum dried, snap-frozen in liquid nitrogen, and pre-weighed before incubation in formamide (1 ml/g of tissue; Sigma) at 65°C for 72 hours. Formamide is a chemical commonly used to extract extravasated, fixed, accumulated-EBD in tissues. Then, supernatant containing extracted EBD from tissue was collected after centrifugation at 10,000–12,000 rpm and transferred to a clean tube. The absorbance of the extravasated dye was measured at 620 nm using a spectrophotometer. EBD concentration was calculated using a standard curve (3.9 to 250 ng/ml in formamide) and linear regression analysis (R2 = 0.998). To determine the effect of flavivirus NS1 proteins at a local microvascular level, we used the Dextran-adapted dermal Miles assay. Briefly, flavivirus NS1 proteins were intradermally injected (15 μg in 50 μl PBS) into the Nair-depilated dorsal dermis of mice (hair removed 3–4 days prior to injection). PBS (50 ml) was used as an injection control, and VEGF (200 ng in 50 μl PBS) was used as positive control for inducing vascular leakage. Following intradermal injections, 200 μl of Alexa Fluor 680-conjugated 10-kDa dextran (5 mg/ml) was delivered by retro-orbital injection. Two hours post-injection, mice were euthanized, and the dorsal dermis was carefully removed and placed in a Petri dish. Tissues were then scanned using the LI-COR Odyssey CLx Imaging System, and images were analyzed using Image Studio Lite software to quantify the leakage area from a standard-scanned area selected for each site of injection to obtain Mean Fluorescence Intensity values.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analysis
All statistical analyses and graphs were performed and generated using GraphPad Prism 6 software. For microscopy analyses, a comparison between the mean fluorescence intensities obtained from three individual experiments, run in duplicate and using untreated cells as baseline controls, was conducted using an ordinary two-way analysis of variance (ANOVA). Comparison between two groups were carried out using t tests (non-parametric). Differences were considered significant for p values < 0.05. For vascular leakage experiments, a comparison between all groups was conducted using an unpaired, nonparametric Mann-Whitney test, and differences in treatment were considered significant for p values < 0.05. For in vivo murine dermis experiments, an ordinary one-way ANOVA with multiple comparisons to the PBS group using Dunnett’s multiple comparison tests was used to determine significance of flavivirus NS1 protein treatments. For TEER experiments, statistical significance was determined using a two-way ANOVA, and differences in treatment were considered significant for p values < 0.05. For mean fluorescence intensities obtained in immunofluorescence assays, ordinary one-way ANOVA and unpaired non-parametric tests were used to evaluate the statistically significant differences between all groups and individual groups, respectively. Statistically significant differences among means were considered as p values < 0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-NS1 (7E11) in house conjugated to Alexa 568 | Gift from R. Putnak (WRA) in-house conjugated | NA |
| Mouse monoclonal IgG anti-His Tag (AD1.1.10’)(AlexaFluor®647) | NOVUS Biologicals | Cat# NB100–64768AF647 |
| Mouse monoclonal IgG anti-human CD138 (Syndecan-1) purified. Clone: DL-101 | eBioscience | Cat#14–1389–82; RRID:AB_467489 |
| Rabbit polyclonal IgG anti-heparanase 1 (HPA1)(H-80) | Santa Cruz Biotechnology | Cat# sc-25825; RRID:AB_2120854 |
| Mouse monoclonal IgG anti-human Cathepsin L Purified. | Invitrogen | Cat# BMS166; RRID:AB_10597594 |
| Rabbit polyclonal IgG anti-neuraminidase 1 (Neu1)(H-300) | Santa Cruz Biotechnology | Cat# sc-32936; RRID:AB_2298197 |
| Mouse monoclonal IgG anti-neuraminidase 1 (Neu1)(F-8) | Santa Cruz Biotechnology | Cat# sc-166824; RRID:AB_2149061 |
| Goat polyclonal IgG anti-human ganglioside sialidase (Neu3)(N-18) | Santa Cruz Biotechnology | Cat# sc-55826; RRID:AB_2149075 |
| Rabbit polyclonal IgG anti-human neuraminidase 2 (Neu2) | Thermo Scientific (Invitrogen™) | Cat# PA5–35114; RRID:AB_2552424 |
| Rabbit polyclonal IgG anti-human neuraminidase 3 (Neu3) | Thermo Scientific (Invitrogen™) | Cat# PA5–45518; RRID:AB_2576484 |
| Rabbit polyclonal IgG anti-human GAPDH (FL-335) | Santa Cruz Biotechnology | Cat# sc-25778; RRID:AB_10167668 |
| Mouse IgM anti-heparan sulfate (F58–10E4) | Amsbio | Cat# 370255–1; RRID:AB_10891554 |
| Mouse monoclonal IgG anti-6x His epitope Tag (HIS.H8) | Thermo Scientific (Invitrogen™) | Cat# MA1–21315; RRID:AB_557403 |
| Rabbit polyclonal anti-Alpha tubulin (loading control) | Abcam | Cat# Ab4074; RRID:AB_2288001 |
| Rabbit polyclonal anti-Rab5 (early endosome marker) | Abcam | Cat# Ab18211; RRID:AB_470264 |
| Goat anti-mouse IgG (AlexaFluor®647) | Abcam | Cat# ab150115; RRID:AB_2687948 |
| Donkey anti-mouse IgG (AlexaFluor®647) | Thermo Scientific (Invitrogen™) | Cat# A31571; RRID:AB_162542 |
| Goat anti-rabbit IgG (AlexaFluor®568) | Abcam | Cat# ab175471; RRID: AB_2576207 |
| Donkey anti-rabbit IgG (AlexaFluor®568) | Abcam | Cat# ab175470 |
| Donkey anti-rabbit IgG (AlexaFluor®647) | Abcam | Cat# ab150075; RRID: AB_2752244 |
| Donkey anti-goat IgG (AlexaFluor®568) | Abcam | Cat# ab175474; RRID:AB_2636995 |
| Donkey anti-goat IgG (AlexaFluor®647) | Abcam | Cat# ab150131; RRID:AB_2732857 |
| Donkey anti-mouse IgM (AlexaFluor®488) | Jackson Immuno Research | Cat# 715–545–140; RRID:AB_2340845 |
| Goat anti-mouse IgG (AlexaFluor®680) | Thermo Scientific (Invitrogen™) | Cat# A21057; RRID:AB_141436 |
| Goat anti-mouse IgG (AlexaFluor®750) | Thermo Scientific (Invitrogen™) | Cat# A21037; RRID:AB_1500644 |
| Donkey anti-mouse IgG (AlexaFluor®680) | Abcam | Cat# ab175774 |
| Donkey anti-mouse IgG (AlexaFluor®750) | Abcam | Cat# ab175738 |
| Goat anti-rabbit IgG (AlexaFluor®680) | Thermo Scientific (Invitrogen™) | Cat# A21076; RRID:AB_141386 |
| Goat anti-rabbit IgG (AlexaFluor®750) | Thermo Scientific (Invitrogen™) | Cat# A21039; RRID: AB_1500687 |
| Donkey anti-rabbit IgG (AlexaFluor®680) | Abcam | Cat# ab186692; RRID:AB_2725787 |
| Donkey anti-rabbit IgG (AlexaFluor®750) | Abcam | Cat# ab175728 |
| Donkey anti-human IgG (AlexaFluor®680) | Jackson Immuno Research | Cat# 709–625–149; RRID:AB_2340582 |
| Goat anti-mouse IgG (AlexaFluor®488) | Abcam | Cat# Ab150117; RRID:AB_2688012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gelatin solution, Type B, 2% in H2O, tissue culture grade | Sigma | Cat# G1393–20ML |
| Wheat germ agglutinin (AlexaFluor®647) | Thermo Scientific (Invitrogen™) | Cat# W32466 |
| Dextran, Alexa Fluor® 680; 10,000 MW, Anionic, Fixable, 5 m | Life Technologies | Cat# D34680 |
| Formamide | VWR Life science | Cat# 0314 |
| Fluorescein isothiocyanate (FITC)–dextran, average mol wt 70,000 | Sigma | Ca# FD70S-100MG |
| Recombinant dengue virus 2 NS1 (strain 16681) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant Zika virus NS1 (Uganda MR766) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant Zika virus NS1 (Suriname Z1106033) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant West Nile virus NS1 (NY99) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant Japanese encephalitis virus NS1 (SA-14) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant Yellow fever virus NS1 (17D) protein produced in 293 human cells. C-terminal 6x His-Tag | The Native Antigen Company | NA |
| Recombinant Dengue 2 Envelope (E) protein (DENV 2 E80%) produced in Sf21 cells. | In-house produced (gift from Aravinda M. de Silva) | NA |
| Recombinant DENV2/WNV Chimera (aa101–135WNV) produced in 293 human cells. C-terminal 6x His-Tag | In-house produced (this paper) | NA |
| Human TNF-alpha Recombinant Protein | Affymetrix/eBioscience | Cat# 14–8329–63 |
| Vascular endothelial growth factor (VEGF) | Sigma | Cat# V7259–10UG |
| Critical Commercial Assays and kits | ||
| MR-(FR)2 Magic Red® Cathepsin-L substrate | Inmunochemistry | Cat# 6138 |
| Corning Transwell Multiple Well Plate with Permeable Polycarbonate Membrane Inserts | Fisher Scientific (Corning) | Cat# 07–200–147 |
| Corning® Transwell®-COL collagen-coated membrane inserts | Sigma (Corning) | Cat# CLS3495–24EA |
| Endpoint Chromogenic LAL QCL-1000 120 Test Kit | Lonza | Cat# 50–647U |
| EBMTM-2 Basal Medium | Lonza | Cat#: CC-3156 |
| EGMTM-2 MV Microvascular Endothelial SingleQuotsTM Kit | Lonza | Cat#: CC-4147 |
| Endothelial Cell Medium | ScienCell | Cat#: 1001 |
| Endothelial Cell Medium | Cell Biologics | Cat# 1166 |
| Endothelial Cell Growth Medium 2 (Ready-to-use) | PromoCell | Cat# C-22011 |
| FreeStyle serum free medium | Thermo Scientific | Cat# 12338018 |
| Alexa Fluor 568 Antibody Labeling Kit | Thermo Scientific (Invitrogen) | Cat# A20184 |
| Experimental Models: Cell Lines | ||
| Human pulmonary microvascular endothelial cell line (HPMEC). Clone HPMEC.ST1–6R | Gift from J.C. Kirkpatrick, Johannes-Gutenberg University | NA |
| Human umbilical vein cord endothelial cells (HUVEC) | Gift from Melisa Lodoen, UC Irvine (Manufacturer: Lonza) | Cat#:C2517A |
| Human dermal microvascular endothelial cell line (HMEC-1) | Gift from Matthew Welch, UC Berkeley | NA |
| Human brain microvascular endothelial cells (HBMEC) | Gift from Ana Rodriguez, NYU (Manufacturer: ScienCell) | Cat#: 1000 |
| Human liver sinusoidal microvascular endothelial cells (HLSEC) | Cell Biologics | Cat# H-6017 |
| Human 293F cells | UC Berkeley Cell Culture facility | Custom order |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Oligonucleotides | ||
| Primer oligonucleotides for Mycoplasma screening sense GPO-3 5′GGGAGCAAACAGGATTAGATACCCT3′ | Eurofins scientific | Custom order |
| Primer oligonucleotides for Mycoplasma screening antisense MGSO 5′TGCACCATCTGTCACTCTGTTAACCTC3′ | Eurofins scientific | Custom order |
| Primer oligonucleotides for generating DENV/WNV Chimera 1r: CTC TTT CCC CAG GCC TTC CAG CCA ATT TCC AAT TTT TCC GTG GTG GCG GTG AGG CGT TTA GGT CCT GCC TGC ATG ATT CCT TTG ATG | Eurofins scientific | Custom order |
| Primer oligonucleotides for generating DENV/WNV Chimera 2f: TGG AAG GCC TGG GGA AAG AGT ATT TTA TTT GCA CCA GAA CTC GCC AAC AAC ACC TTT GTG GTT GAT GGC CCC GAA ACA GCA GAA | Eurofins scientific | Custom order |
| Recombinant DNA | ||
| pmab vector | Gift from Michael Diamond (Washington University in St. Louis) | NA |
| Software and Algorithms | ||
| ImageJ (Image processing and analyses) | NA | https://www.nature.com/articles/nmeth.2089 |
| Image Studio Lite Ver 5.2 | NA | https://www.licor.com/bio/products/software/image_studio_lite/ |
| Graph Pad Prism 6 2D scientific graphing and biostatistics) | NA | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator CC 2018 | Adobe Inc. | http://www.adobe.com |
| Microsoft PowerPoint | Microsoft Corporation | https://www.microsoft.com/en-us/ |
| FlowJo V10 | FlowJo LLC | https://www.flowjo.com/ |
| Other | ||
| Zeiss LSM 710 Axio Observer Zeiss Axio Observer | ZEISS | CRL MIC, UC Berkeley |
| Chopstick Electrode Set for EVOM2 | World Precision Instruments | STX3 |
| EPITHELIAL VOLTOHMMETER 2 | World Precision Instruments | EVOM2 |
Highlights.
Flavivirus NS1 proteins induce endothelial dysfunction in a tissue-specific manner
Tissue tropism of 5 NS1 proteins is partly determined by endothelial cell binding
NS1-induced endothelial dysfunction is mediated by endothelial glycocalyx disruption
Flavivirus NS1 proteins induce tissue-specific leak in mice mirroring viral tropism
ACKNOWLEDGMENTS
We thank Carmel Malvar for her help with expression and purification of the recombinant NS1 protein DENVWNV101−135 and Jeffrey Shu for his assistance with western blots. We thank Aravinda M. de Silva at the University of North Carolina, Chapel Hill for his kind donation of the baculovirus system used to produce the recombinant E protein from DENV2. We also thank B. Lamason, A. Azliyati, and Ana Rodriguez for facilitating transfer of HMEC-1, HPMEC-ST1.6R, and HBMECs, respectively. This work was supported by NIH grant R01 AI24493 (E.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.036.
REFERENCES
- Abboud-Jarrous G, Atzmon R, Peretz T, Palermo C, Gadea BB, Joyce JA, and Vlodavsky I (2008). Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem 283, 18167–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcon-LePoder S, Drouet MT, Roux P, Frenkiel MP, Arborio M, Durand-Schneider AM, Maurice M, Le Blanc I, Gruenberg J, and Flamand M (2005). The secreted form of dengue virus nonstructural protein NS1 is endocytosed by hepatocytes and accumulates in late endosomes: implications for viral infectivity. J. Virol 73, 11403–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C, et al. (2006). Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis 793, 1078–1088. [DOI] [PubMed] [Google Scholar]
- Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, and Atkinson JP (2010). Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med 207, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis SJ, Coloma J, Castro G, Alava A, Flores D, McKerrow JH, Beatty PR, and Harris E (2009). Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hyg 80, 416–424. [PubMed] [Google Scholar]
- Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, and Harris E (2015). Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med 7, 304ra141. [DOI] [PubMed] [Google Scholar]
- Calisher CH, and Gould EA (2003). Taxonomy of the virus family Flaviviridae. Adv. Virus Res 59, 1–19. [DOI] [PubMed] [Google Scholar]
- Chen HR, Chuang YC, Lin YS, Liu HS, Liu CC, Perng GC, and Yeh TM (2016). Dengue virus nonstructural protein 1 induces vascular leakage through macrophage migration inhibitory factor and autophagy. PLoS Negl. Trop. Dis 70, e0004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chung H, Jung H, Couchman JR, and Oh ES (2011). Syndecans as cell surface receptors: Unique structure equateswith functional diversity. Matrix Biol 30, 93–99. [DOI] [PubMed] [Google Scholar]
- Chung KM, and Diamond MS (2008). Defining the levels of secreted nonstructural protein NS1 after West Nile virus infection in cell culture and mice. J. Med. Virol. 80, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly-Andersen AM, Thunberg T, and Ahlm C (2014). Endothelial activation and repair during hantavirus infection: association with disease outcome. Open Forum Infect. Dis 7, ofu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E (2004). Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol 5, 261–270. [DOI] [PubMed] [Google Scholar]
- Diamond MS, and Klein RS (2004). West Nile virus: crossing the blood-brain barrier. Nat. Med 70, 1294–1295. [DOI] [PubMed] [Google Scholar]
- Egawa G, Nakamizo S, Natsuaki Y, Doi H, Miyachi Y, and Kabashima K (2013). Intravital analysis of vascular permeability in mice using two-photon microscopy. Sci. Rep 3, 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasner DR, Ratnasiri K, Puerta-Guardo H, Espinosa DA, Beatty PR, and Harris E (2017). Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLoS Pathog. 73, e1006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasner DR, Puerta-Guardo H, Beatty PR, and Harris E (2018). The good, the bad, and the shocking: the multiple roles of dengue virus nonstructural protein 1 in protection and pathogenesis. Annu. Rev. Virol 5, 227–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould EA, and Solomon T (2008). Pathogenic flaviviruses. Lancet 377, 500–509. [DOI] [PubMed] [Google Scholar]
- Hertz T, Beatty PR, MacMillen Z, Killingbeck SS, Wang C, and Harris E (2017). Antibody epitopes identified in critical regions of dengue virus nonstructural 1 protein in mouse vaccination and natural human infections. J. Immunol 798, 4025–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, Wu M, Lindenbach BD, Abrahams VM, Guller S, and Fikrig E (2016). Zika virus productively infects primary human placenta-specific macrophages. JCI Insight 7, e88461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauer F, Riesen M, Reveiz L, Oladapo OT, Martinez-Vega R, Porgo TV, Haefliger A, Broutet NJ, and Low N; WHO Zika Causality Working Group (2017). Zika virus infection as a cause of congenital brain abnormalities and Guillain-Barre syndrome: systematic review. PLoS Med. 74, e1002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Tripathi S, Tambe JJ, Arora V, Srivastava A, and Nag VL (2008). Dengue encephalopathy in children in Northern India: clinical features and comparison with non dengue. J. Neurol. Sci 269, 41–48. [DOI] [PubMed] [Google Scholar]
- Lai YC, Chuang YC, Liu CC, Ho TS, Lin YS, Anderson R, and Yeh TM (2017). Antibodies against modified NS1 wing domain peptide protect against dengue virus infection. Sci. Rep 7, 6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, Wang C, Wang K, Cui M, and Fu ZF (2015). Viral infection ofthe central nervous system and neuro-inflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J. Virol 89, 5602–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, Vaughn DW, Nisalak A, Ennis FA, and Rothman AL (2002). High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J. Infect. Dis 786, 1165–1168. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Nie K, Du S, Qiu J, Pang X, Wang P, and Cheng G (2016). Flavivirus NS1 protein in infected host sera enhances viral acquisition by mosquitoes. Nat. Microbiol 7, 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu J, Du S, Shan C, Nie K, Zhang R, Li XF, Zhang R, Wang T, Qin CF, et al. (2017). Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 545, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald J,Tonry J, Hall RA, Williams B, Palacios G, Ashok MS, Jabado O, Clark D, Tesh RB, Briese T, and Lipkin WI (2005). NS1 protein secretion during the acute phase of West Nile virus infection. J. Virol 79, 13924–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Jones MK, and Young PR (1996). Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220, 232–240. [DOI] [PubMed] [Google Scholar]
- Michlmayr D,Andrade P, Gonzalez K, Balmaseda A, and Harris E (2017). CD14+CD16+ monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol. 2, 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodušek V, et al. (2016). Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958. [DOI] [PubMed] [Google Scholar]
- Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, Hume DA, Stacey KJ, and Young PR (2015). Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med 7, 304ra142. [DOI] [PubMed] [Google Scholar]
- Monath TP, and Barrett AD (2003). Pathogenesis and pathophysiology of yellow fever. Adv. Virus Res 60, 343–395. [DOI] [PubMed] [Google Scholar]
- Muller DA, and Young PR (2013). The flavivirus NS1 protein: molecularand structural biology, immunology, role in pathogenesisand application asadiagnostic biomarker. Antiviral Res. 98, 192–208. [DOI] [PubMed] [Google Scholar]
- Puerta-Guardo H, Raya-Sandino A, González-Mariscal L, Rosales VH, Ayala-Dávila J, Chávez-Mungía B, Martínez-Fong D, Medina F, Ludert JE, and del Angel RM (2013). The cytokine response of U937-derived macrophages infected through antibody-dependent enhancement ofdengue virus disrupts cell apical-junction complexes and increases vascular permeability. J. Virol 87, 7486–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puerta-Guardo H, Glasner DR, and Harris E (2016). Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. 12, e1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, and oude Egbrink MG (2007). The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro CF, Lopes VGS, Brasil P, Pires ARC, Rohloff R, and Nogueira RMR (2017). Dengue infection in pregnancy and its impact on the placenta. Int. J. Infect. Dis 55, 109–112. [DOI] [PubMed] [Google Scholar]
- Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, and Bartenschlager R (2015). Dengue virus non-structural protein 1 modulates infectious particle production via interaction with the structural proteins. PLoS Pathog. 11, e1005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Qi J, Haywood J, Shi Y, and Gao GF (2016). Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat. Struct. Mol. Biol 23, 456–458. [DOI] [PubMed] [Google Scholar]
- Soni BK, Das DSR, George RA, Aggarwal R, and Sivasankar R (2017). MRI features in dengue encephalitis: a case series in South Indian tertiary care hospital. Indian J. Radiol. Imaging 27, 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler KR, and Hsu TH (2012). Viral disruption ofthe blood-brain barrier. Trends Microbiol. 20, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwarto S, Sasmono RT, Sinto R, Ibrahim E, and Suryamin M (2017). Association of endothelial glycocalyx and tight and adherens junctions with severity of plasma leakage in dengue infection. J. Infect. Dis 215, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, and Pereira L (2016). Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 20, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. (2016). Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TH, Alonso S, Ng LF, Thein TL, Pang VJ, Leo YS, Lye DC, and Yeo TW (2017). Increased serum hyaluronic acid and heparan sulfate in dengue fever: association with plasma leakage and disease severity. Sci. Rep 7, 46191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trung DT, and Wills B (2010). Systemic vascular leakage associated with dengue infections - the clinical perspective. Curr. Top. Microbiol. Immunol 338, 57–66. [DOI] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, and Flavell RA (2004). Toll-like receptor3mediatesWest Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Weinbaum S, Tarbell JM, and Damiano ER (2007). The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng 9, 121–167. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2009). Dengue: Guidelines for Diagnosis, Treatment, Prevention, and Control (World Health Organization; ). [PubMed] [Google Scholar]
- Xu X, Vaughan K, Weiskopf D, Grifoni A, Diamond MS, Sette A, and Peters B (2016). Identifying candidate targets of immune responses in zika virus based on homology to epitopes in other flavivirus species. PLoS Curr 8, ecurrents.outbreaks.9aa2e1fb61b0f632f58a098773008c4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.