

Abstract
Identifying novel viruses or assessing viral variation by NGS requires high sequencing coverage. More than 90% of total RNA is ribosomal (rRNA), making variant calling, virus discovery or transcriptomic profiling difficult. Current methods to increase informative reads suffer from drawbacks, either they cannot be used for some viruses, are optimized for a single species, or introduce bias. We describe a two-part approach combining reverse-transcription to create RNA/DNA hybrids which are then degraded with RNaseH/DNase sequentially that works for three medically relevant mosquito genera; Aedes, Anopheles, and Culex. We demonstrate depletion of rRNA from different samples, including whole mosquitoes and midgut contents from FTA cards. We describe novel insect-specific virus genomes from field collected mosquitoes. The protocol requires only common laboratory reagents and small oligonucleotides specific to rRNA. This approach can be adapted for other organisms, aiding virus diversity analyses, virus discovery and transcriptomics in both laboratory and field samples.
Keywords: Ribosomal depletion, Next-generation sequencing, Virus discovery, Insect-specific viruses, Mosquito-borne viruses, Arbovirus, Transcriptomics
1. Introduction
The past several decades have witnessed the emergence and expansion of viruses with increasing frequency (Jones et al., 2008). These include H1N1 Influenza in 2009 (Otte et al., 2015), Chikungunya in 2006 (Tsetsarkin et al., 2007), Zika in 2013–14 (Aubry et al., 2017), West Nile in 1999 (Moudy et al., 2007), and MERS (Forni et al., 2015), amongst others. Most, if not all the emerging viruses that pose the greatest threat to human and animal health are RNA viruses. In fact, all 7 of the pathogens identified by the World Health Organization (WHO) in the 2018 annual review of the blueprint list of priority diseases as requiring urgent or serious research were RNA viruses, with the other being unknown pathogens (WHO, 2018). Due to the importance of RNA viruses, it is critical to be able to detect, identify and analyze these pathogens’ genomes using novel high-throughput sequencing methods.
However, total RNA preparations from complex lab and field samples typically contain extremely high levels of ribosomal RNA (rRNA), sometimes 80–90% of the total amount (Eun, 1996). Sequencing data mapping to rRNA is typically removed bioinformatically and therefore represents an economic waste and reduces the number of samples that can be tested in a given sequencing run. To increase the number of reads mapping to sequences of interest, several methods have been employed to either enrich; hybrid-capture (Metsky et al., 2017), amplicon (Metsky et al., 2017; Moratorio et al., 2017), SPIA amplification (Grubaugh et al., 2016) or remove unwanted rRNA sequences; ribosomal RNA depletion most notably (Adiconis et al., 2013; Matranga et al., 2016). Enriching for sequences of interest is highly effective but can only target known sequences, making it difficult to identify novel or divergent viruses. Current ribosomal depletion methods are typically cost-prohibitive or are designed for humans and mice, making depletion in non-model systems highly inefficient.
Here we describe a novel method of ribosomal depletion that utilizes reverse transcription (RT) to specifically target sequences for depletion using the RNA degradation activity of RNase H. During RT, rRNA is converted to cDNA using specific DNA probes which can then be degraded using RNase H. Because this method utilizes small probes to recognize the target sequences for depletion, it is possible to design universal probes that bind to highly divergent species, making depletion of diverse organisms representing several genera possible with the same probes. Additionally, since the probes are effectively reverse primers that are typically used for RT, they are easy to design, cheap and can be quickly designed for any target sequence from any species or genus of interest for which the rRNA sequence is available. Our studies show that using this method we can selectively remove rRNA from mosquitoes from multiple genera which results in increased relevant data recovered from next-generation sequencing. Furthermore, we apply this method to field-caught mosquitoes and show that we can detect multiple novel virus genomes from a highly multiplexed set of samples on a relatively low-output Illumina MiSeq run. Collectively, this work describes an effective method for rRNA depletion that is straight forward, relatively low-cost and highly effective at increasing usable data from high-throughput sequencing experiments.
2. Materials and methods
2.1. Cells, viruses, mosquitoes and sample collection
West Nile virus (strain NY99) was generated from an infectious clone as previously described in BHK-21 cells (Shi et al., 2002). Laboratory colonies of Culex quinquefasciatus, Aedes aegypti and Anopheles gambiae were used for mosquito infections. Mosquitoes were maintained at 26–27 °C and 70–80% relative humidity with a 16:8L:D photoperiod. Water and 10% sucrose were provided ad libitum. For preliminary studies, pools of whole mosquitoes (n = 10) were collected and homogenized in Trizol solution.
For many experiments, we used samples collected using a technique called “xenosurveillance”. This approach uses the natural blood-feeding behavior of female mosquitoes to sample blood from humans and animals (Fauver et al., 2017, 2018; Grubaugh et al., 2015). We have previously used this approach to detect pathogens in the blood collected by mosquitoes in the lab and in the field. In these studies, groups of An. gambiae were exposed to an infectious bloodmeal containing 107 PFU of WNV NY99. The next day, midguts from mosquitoes containing a residual bloodmeal were collected by spreading the midgut contents onto CloneSaver FTA cards (GE Healthcare), and immediately 25 μL of RNAlater (ThermoFisher) was added to facilitate diffusion of blood into the FTA card and stabilize the nucleic acid. The samples placed on the FTA cards were then punched out and nucleic acid was eluted by incubation in RNA rapid extraction solution (ThermoFisher) for 18 h.
2.2. Probe design
In order to design probes that worked for the three most medically relevant mosquito genera, 18S and 28S rRNA sequences from multiple species from each genus were downloaded from the SILVA rRNA database project (Quast et al., 2013). Mitochondrial sequences were also downloaded from NCBI to design against 12S and 16S rRNA. Sequences were aligned using MUSCLE and reverse primer sequences were designed with bases matching at least 95% of the sequences with the Primer3 design tool (all within Geneious v11.0.4). Primers were selected at roughly 200–500 bp intervals. The designed primers were then aligned to both the RefSeq viral database (https://www.ncbi.nlm.nih.gov/genome/viruses/) and the Anopheles transcriptome (RefSeq accession #GCF_000005575.2) to test for off-target hits using Geneious. Primers binding in the reverse orientation were not used further.
2.3. Ribosomal depletion
A detailed protocol is included describing the depletion protocol in Supplemental File 1. Nucleic acids were extracted using either Trizol solution or the Mag-Bind Viral DNA/RNA kit (Omega Bio-Tek, USA) and eluted into 50 μL of water. The samples were then treated with TURBO DNase (ThermoFisher) and purified using RNAClean XP beads (Beckman Coulter). For reverse transcription, the RNA was mixed with oligos specific for rRNA (sequences listed in Supplemental Table 1) and dNTPs and then heat denatured at 95 C for 2 min followed by slow cooling to 50 C at 0.1 C/s. For initial experiments, we tested a panel of reverse transcriptases (RTs), including Tth DNA polymerase (in the presence of Mn2 +, Promega), Superscript III (SSIII, ThermoFisher), Superscript IV (SSIV, ThermoFisher), Avian myeloblastosis virus (AMV, NEB) and Moloney Murine Leukemia Virus (MMLV, NEB). For all RTs, we used the optimal conditions as described by the manufacturer. For all further experiments, AMV RT was then added and incubated at 50 C for 2 h. RNase H (NEB) was then added to destroy the RNA present in the RNA: cDNA hybrid. The samples were then digested with DNase I (NEB) to remove the cDNA and residual oligos. The RNA was then purified using RNAClean XP beads at a 1.8x ratio.
2.4. RNA analysis and qRT-PCR
Input and rRNA depleted RNA were analyzed using a 2100 Bioanalyzer (Agilent) per manufacturer’s protocols with the total RNA Pico kit. The RNA traces were analyzed using Agilent 2100 expert software. Quantitative Reverse-transcriptase PCR (qRT-PCR) was performed using the iTaq universal probes supermix (Biorad) according to the manufacturer. qRT-PCR was performed with the following primers; 18S Forward - AGAGGACTACCATGGTTGCAAC, 18S Reverse - CCTGCTGCCTTCCTTGGATG, 18S Probe - CCGGAGAGGGAGCCTGAGAAATGGC, 28S Forward - AGGTGCGGAGTTTGACTGG, 28S Reverse - TCCTTATGCTCAGCGTGTGG, 28S Probe - AGGTGTCCAAAGGTCAGCTCAGTGTGG, WNV Forward - TCAGCGATCTCTCCACCAAAG, WNV Reverse - GGGTCAGCACGTTTGTCATTG, WNV Probe – TGCCCGACCATGGGAG AAGCTC (Lanciotti et al., 2000). The number of genome copies was generated by fitting the Ct values to a standard curve of RNA specific to each of the primer sets.
2.5. Library preparation and data analysis
Libraries for Illumina sequencing were prepared from input RNA, and samples that were depleted using probes specific to rRNA or in the absence of probes. The libraries were prepared using equal concentrations of RNA as input by using the NEBNext Ultra RNA library prep kit (NEB) and then were sequenced on an Illumina MiSeq using 150 cycles. For data analysis, libraries were first demultiplexed using bcl2fastq (Illumina). Reads were then trimmed for both adapters and quality using BBDuk software (part of the BBMap suite, https://sourceforge.net/projects/bbmap/). For all lab-derived samples, we first normalized the number of clean reads to either contain 1.5 million reads using Reformat (part of the BBMap suite). For transcriptomic analysis of field-derived samples, we first normalized to 200,000 reads for each sample. We did not normalize the number of reads for field-derived samples for calculating the percentage of reads aligning to virus or rRNA, as we are not comparing between the different mosquito species. PCR duplicates were then removed using clumpify (also part of the BBMap suite) and unique reads were mapped to reference genomes using BBSplit (part of the BBMap suite, Langmead and Salzberg, 2012). Removal of PCR duplicates reduces the effects of amplification bias that occurs during the library preparation process and is performed by identifying reads that are identical and removing all, but one read. Read counts for each transcript were generated by using the tag RPKM in BBMap and fold-differences were calculated by comparing against the input RNA. The normalized fastq files were used for these analyses. We then used MultiQC (Ewels et al., 2016) to quantify the number and percentage of reads that mapped to each reference. Data were graphed using GraphPad Prism version 7. To assess intrahost variation, unique reads were mapped to the Bolahun virus reference sequence using BBMap and then variants were called using LoFreq (Wilm et al., 2012). Only variants present at greater than 5% were used for analysis.
For read identification the normalized fastq files were competitively aligned to several reference files simultaneously using BBSplit, which uses the BBMap program for alignment. This method forces the best alignment for each read between the difference references and outputs the percentage of reads that align to each in one file.
2.6. Field mosquito collections
Adult mosquitoes were collected from multiple localities in Chiapas, Mexico over the course of three weeks in August 2016 using CDC gravid traps (John W. Hock Company), CDC Miniature light traps (BioQuip Products) and insectazookas (BioQuip Products). Mosquitoes were euthanized using triethylamine and sorted into pools of up to 25 individuals by species, sex, and collection location (Supplemental Table 2). Mosquitoes were identified to species using morphological keys (Darsie and Ward, 2005). For groups of mosquitoes that could not be identified, multiple individuals of each group were point mounted and preserved for later identification by local experts at the Instituto Nacional de Salud Pública facilities in Mexico. Pools of mosquitoes were preserved in RNAlater (Ambion) and shipped to Colorado State University (CSU).
2.7. Processing of field collected mosquitoes
Prior to homogenization and nucleic acid extraction, mosquito pools were centrifuged, and RNA later was removed. Pools were then processed as described above. All field collected mosquito pools were subjected to rRNA depletion using the same probe mixture as the laboratory experiments. Following rRNA depletion, RNA from pools was prepared for NGS using Nextera XT following manufacturer’s instructions (Illumina). Each library was dual-indexed with a unique barcode to facilitate multiplexing using the Kapa Library Amplification Kit for Illumina (Kapa BioSystems). Libraries were then quantified using the NEBNext Library Quantification Kit for Illumina (New England Biolabs) and pooled together by equal volumes. All libraries were sequenced together on a single Illumina MiSeq run using a 300 cycle (2 × 150) MiSeq v3 kit.
2.8. Identification and characterization of viral sequences
Virus contigs were identified using a previously described pipeline (Cross et al., 2018; Fauver et al., 2018) (found online at https://github.com/stenglein-lab/taxonomy_pipeline). No host filtering was conducted prior to the generation of contigs, as most genera sequenced to do not have a reference genome. Amino acid similarity to other virus or virus-like sequences was determined using NCBI Blastx tool against the nr database (Altschul et al., 1990) (Supplemental Table 3). Virus contigs greater than 500 b.p. were sorted into high-level clades according to Shi et al. (2016). Contigs from the same species of mosquito aligning to similar viral clades were binned together in Geneious v11.0.4 and assessed for open-reading frames (ORFS) using the Find ORFs tool (Kearse et al., 2012). Following translation of complete ORFs, amino acid sequences were queried against the Conserved Domain Database v3.16 using HHpred (Zimmermann et al., 2018). Predicted domains with an e-value > 1e-5 were used for annotation. All putative virus genomes were described entirely using computational methods and virus isolation was not attempted.
Phylogenetic trees were created for coding complete virus genomes. The RNA dependent RNA polymerase (RDRP) gene (Luteo-Sobemo, Levi-Narna) or the whole genome (Negevirus) was used as input for blastp, and all hits with an e-value > 1e-5 were downloaded in fasta format from NCBI. CD-Hit -c 0.90 was used to rid dataset of similar viral RDRPs sequences (Li and Godzik, 2006). Amino acid sequences were aligned using MAFFT v7.308 -auto (Katoh and Standley, 2013). Gaps and poorly aligned sequences in the multiple alignment were removed using trimAl under default settings (Capella-Gutiérrez et al., 2009). The resulting alignments were used as input to generate phylogenetic trees using PHYML with the LG substitution model and 1000 bootstraps (Guindon et al., 2010). Separately, a neighbor-joining approach with 1000 bootstraps using Geneious Tree Builder was conducted and tree topologies were compared. No major differences in topologies were observed between these methods and the results of the PHYML analysis are presented. In addition, genomic sense was inferred based on placement in phylogeny.
To calculate depth of coverage, a custom database was created by species containing all viral contigs generated in this study in addition to the 45S rDNA sequence assembled from Ae. aegypti. Reads from each mosquito species were competitively aligned to this database using Bowtie2 under default settings (Langmead and Salzberg, 2012). The resulting SAM file was converted into BAM format, and depth of coverage at each nucleotide position was calculated using SAMtools -depth (Li et al., 2009). Read identification for field collect mosquitoes was determined using the same methodology described above. Novel Narna-Levi virus sequences were aligned as described above, and pairwise nucleotide identity was calculated in Geneious.
2.9. Data availability
All sequencing data has been deposited to the SRA database under PRJNA505498. Novel virus genomes are listed in GenBank with accession numbers MK2835331-MK285338.
3. Results
3.1. Approach
3.1.1. DNase treatment of total RNA
Note: Starting material should be total RNA isolated using Trizol or kit-based extraction methods.
Clean all surfaces and pipettes thoroughly with RNase AWAY (ThermoFisher) or similar.
Prepare a 50 μL DNase reaction by mixing 5 μL of 10x TURBO DNase buffer, 1 μL of TURBO DNase and the appropriate amount of sample (no more than 10 μg of RNA). Bring the solution up to 50 μL in molecular grade water.
- Incubate at 37 °C for 30 min.
- All steps in a thermal cycler should have the heated lid set to 105 °C.
- During the incubation, remove the RNAClean XP beads from 4 °C and allow to warm to room temperature. Thoroughly mix the RNAClean XP beads before use.
Remove the tubes from the thermal cycler and add 1.4 volumes of RNAClean XP beads (70 μL) to each sample, mix well by pipetting 10 times. Incubate at room temperature for 5 min.
Apply the tubes to a magnet and remove the supernatant, taking care not to remove the beads.
Wash 3 times with 70% Ethanol and then let air dry for ten minutes following the final wash.
Elute in 20 μL molecular grade water.
3.1.2. RT/RNase H and DNase treatment
Note: A master mix can be prepared for all reaction mixes.
Prepare a 16.5 μL RT reaction by mixing 2 μL 10 mM dNTPs, 0.5 μL 20 mg/mL BSA, 5 μL of the pooled 100 μM probe mix, 1 μL 5 M betaine and 8 μL of sample RNA.
Denature the RNA in a thermal cycler by heating to 95 °C for 2 min followed by a slow cool to 50 °C at 0.1 °C/s. Hold the RNA at 50 °C for 5 min.
While the RNA is denaturing, prepare the enzyme mix by combining 2 μL AMV RT buffer, 0.5 μL RNase inhibitor and 1 μL AMV RT.
Add 3.5 μL of the enzyme mix to each sample following the 5-min incubation period.
Allow the reaction to incubate for 2 h at 50 °C.
After two hours, prepare the RNase H mix by combining 0.5 μL RNase H, 1.5 μL 10x RNase H Buffer and 8 μL molecular grade water.
Add 10 μL of this mix to the RT reaction from step 5. Incubate at 37 °C for 30 min.
Prepare DNase mix by adding 2 μL DNase, 4 μL 10x DNase buffer and 14 μL molecular grade water.
Add 20 μL of this mix to the reaction from step 7. Incubate at 37 °C for 30 min.
Purify this reaction with 1.4x RNAClean XP beads as described above.
Assess depletion efficiency by a qPCR of pre- and post-depleted samples for rRNA.
3.2. Reverse-transcriptase (RT) mediated ribosomal RNA (rRNA) depletion is effective for mosquitoes from three medically relevant genera
The workflow for our proposed ribosomal depletion method is outlined in Fig. 1 and detailed in the approach section of the results. Briefly, DNase treated RNA was reverse-transcribed using DNA probes that are in the reverse-complement orientation to the sequences for mosquito sequences for the 18S, 28S and 5.8S cellular rRNA and the 12S and 16S mitochondrial rRNA sequences. In order to design probes that work against the majority of mosquitoes, we aligned sequences from several mosquito genera obtained from the SILVA rRNA database project (Quast et al., 2013). The probes were designed specifically to regions of high sequence homology and to have a melting temperature around 65 °C, thereby giving them high specificity while maintaining binding to all genera. The probe sequences are presented in Supplemental Table 1 and a schematic showing the probes aligned to the Aedes albopictus 45 S rRNA sequence is presented in Supplemental Fig. 1. For the depletion, the RNA was heat denatured in the presence of the probes and slowly cooled to favor specific binding of the probes to the RNA. cDNA was then synthesized using a variety of reverse transcriptases (RT) (all in triplicate). It was determined that AMV RT was superior to other RTs tested in depleting 18S and 28S from An. gambiae mosquitoes (Fig. 2A–B). While MMLV RT was also able to significantly reduce rRNA, it also depleted WNV RNA, while AMV did not, suggesting that the depletion was highly specific (Fig. 2C). AMV and SSIII RT were the only RTs tested that significantly reduced the amount of 18S and 28S rRNA while maintaining the same amount of WNV RNA (p < 0.0001 for 18S and 28S and p = 0.9990 for WNV RNA all when comparing with and without probes and by One-Way ANOVA with Tukey comparison). We continued with AMV RT because the reduction in rRNA was more dramatic and because it is less expensive than SSIII. Following AMV RT, we treated samples with RNase H and finally DNase I to degrade the RNA in the DNA: RNA hybrid and any DNA present, respectively. Using qRT-PCR, we saw a significant reduction in 18S and 28S rRNA from An. gambiae only when the RT and RNase/DNase steps were included and not when any steps were omitted, suggesting that the reverse transcription and RNaseH/DNase I treatment are all required for specific depletion (Fig. 2D–E, all p < 0.0001 by One-Way ANOVA with Tukey comparison as compared to the non-depleted group). The reduction from the DNase treated RNA to the samples not treated with RT or depletion probes is likely due to the removal of small fragments during RNAClean bead purification.
Fig. 1. Workflow for reverse-transcriptase mediated ribosomal depletion from total RNA.

To perform ribosomal RNA (rRNA) depletion, total RNA is first extracted, DNase treated and subsequently purified with RNAClean XP Beads (Agencourt). DNA-free RNA is then bound to oligonucleotide probes designed to bind to rRNA from mosquito species in Aedes, Culex and Anopheles genera that are in the reverse complement orientation to both the long and short ribosomal subunit and 12S and 16S mitochondrial rRNA. The RNA with bound oligos is then subjected to reverse transcription using Avian Myeloblastosis Virus (AMV) Reverse Transcriptase (NEB). RNA that is reverse transcribed to cDNA is then digested using RNase H, which selectively destroys RNA in an RNA:DNA hybrid. Remaining DNA is then digested using DNase I (NEB), leaving mostly non-ribosomal RNA which is then used for library preparation.
Fig. 2. Reverse Transcriptase mediated ribosomal RNA (rRNA) depletion is most effective with AMV RT and requires all steps to be effective.

Nucleic acids were eluted from FTA cards with midgut contents of An. gambiae that had been exposed to a bloodmeal containing West Nile virus (WNV) placed on them. RNA and DNA was then extracted to obtain total nucleic acid. The nucleic acid was then treated with DNase I and then purified to obtain total RNA. This RNA was then subjected to cDNA synthesis with a panel of reverse transcriptases (n = 3 for each treatment) in the presence (+probes) or absence (- probes) of DNA probes specific to rRNA. The RTs tested were Tth DNA polymerase, Superscript III (SSIII), Superscript IV (SSIV), AMV and MMLV. All the samples were then treated with RNase H and then DNase I to remove the RNA present in an RNA:DNA hybrid and cDNA, respectively. The samples were then purified and subjected to qRT-PCR with primer probe combinations specific for 18S rRNA (A), 28S rRNA (B) or WNV (C). Further tests were performed exclusively with AMV RT. Panels D and E show the results of qRT-PCR for samples that underwent the process of depletion but omitting some step or reagent. 18S (D) and 28S (E) rRNA was quantified in the input RNA, RNA with no RT added, RNA with no depletion probes added and RNA treated with RT with depletion probes. All statistical tests were performed by One-Way ANOVA with Tukey test for multiple comparisons. ****Indicates p-value < 0.0001. All stars without an accompanying bar are statistical comparisons between the DNase Treated RNA and the other group. The other comparisons with a bar are statistical comparisons between the two groups that are below the corresponding bar.
We next sought to determine if the ribosomal depletion protocol was effective for mosquito species from three distinct medically relevant genera; Anopheles, Aedes and Culex. Total RNA was extracted from pools (n = 10) of whole mosquitoes and the rRNA was depleted as previously described, with the exception that additional probes were added to the mixture that targeted non-depleted rRNA sequences identified in preliminary NGS analysis (data not shown). We then subjected the input RNA, depleted RNA (RT - with probes) and RNA that went through the depletion process without probes (RT - no probes) to qRT-PCR analysis. For all species tested, the input RNA had significantly higher 18 S (Fig. 3A) and 28S (Fig. 3B) rRNA levels when compared to the depleted group (p < 0.0001 Two-Way ANOVA with Tukey comparison). We also subjected both the input RNA and the depleted RNA to electrophoretic analysis using a Bioanalyzer 2100. For all three species tested, the peak for rRNA (both 18S and 28S typically appear at ~ 2000 nt) is inapparent following the depletion protocol (Fig. 3C-E). In contrast, the input RNA has a prominent peak for rRNA. The traces for the depleted and input RNA are overlaid on the same graph to facilitate comparison.
Fig. 3. Reverse Transcriptase mediated ribosomal RNA (rRNA) depletion is effective against mosquitoes from three distinct medically relevant genera.

Total RNA was extracted from three distinct pools of whole mosquitoes from three medically relevant genera; Culex (Cx.) quinquefasciatus, Aedes (Ae.) aegypti and Anopheles (An.) gambiae. The RNA was treated with DNase I and then purified; this will now be called input RNA. An aliquot was then taken, and reverse transcribed to cDNA using AMV reverse transcriptase (RT) and DNA probes specific for mosquito ribosomal RNA (RT – with Probes) or in the absence of probes (RT – no probes). The samples were then treated with RNase H and DNase I to remove the RNA present in an RNA:DNA hybrid and cDNA, respectively. The samples were then purified and subjected to qRT-PCR with primer probe combinations specific for 18S or 28S rRNA (A and B). The input RNA and RT – with Probes were then assessed using a Bioanalyzer. Panels C-E show a representative trace for each of the three mosquito species tested, Cx. quinquefasciatus (C), Ae. aegypti (D), An. gambiae (E). The blue trace for each panel shows the input RNA and the red trace shows the RT – with Probes treated RNA. The peak present at roughly 40 s in each trace is the peak for both 18S and 28S rRNA. All statistical tests were performed by Two-Way ANOVA with Tukey test for multiple comparisons. ****Indicates p-value < 0.0001. All stars without an accompanying bar are statistical comparisons between the input RNA and the other group. The other comparisons with a bar are statistical comparisons between the two groups that are below the corresponding bar.
3.3. RT mediated rRNA depletion increases sequencing reads to viruses and mRNA
Samples to test depletion efficacy were prepared using a method termed Xenosurveillance, prepared as described previously (Fauver et al., 2018). Briefly, An. gambiae mosquitoes were exposed to an infectious bloodmeal containing WNV (which doesn’t replicate in these mosquitoes) and then midguts containing the partially digested blood were collected the next day on FTA cards. The nucleic acids were eluted and extracted as previously described and then the samples were depleted with AMV RT and depletion probes (RT - with probes). We also tested the input RNA and samples that underwent the depletion protocol with the omission of probes (RT - no probes). Following depletion, the RNA underwent Illumina library prep and was sequenced using the MiSeq platform (Illumina). The reads were then trimmed and then each file was normalized to contain 1.5 million reads. Normalization to the number of reads allows for direct comparison between each file. We next removed PCR duplicates and compared the number of unique reads between each group after deduplication (Fig. 4A). When compared to either the input RNA or RT – no probes the number of unique reads in the depleted samples approached significance (p = 0.0504 and 0.0765, respectively, by One-Way ANOVA with Tukey comparison). The unique reads were then mapped to either 18S (Fig. 4B), 28 S (Fig. 4C) or mitochondrial 16S (Fig. 4D) rRNA. For 18S rRNA, significantly fewer reads mapped to the depleted samples when compared to either the input RNA or RT – no probes (p = 0.0056 and 0.0097, respectively, by One-Way ANOVA with Tukey comparison). The same was observed for 16S rRNA when the number of reads aligning in the depleted samples was compared to either the input RNA or RT – no probes (p = 0.0014 and 0.0089, respectively, by One-Way ANOVA with Tukey comparison). The differences were not significant for any comparisons for 28S rRNA when using just mapped reads. However, when we further normalized our reads to a highly abundant host mRNA in the same manner as previously described (Kumar et al., 2012) the differences became clearer. After normalization to a highly expressed gene, lipophorin (AGAP001826), all statistical comparisons between the depleted samples and the other two groups for 18 S (Fig. 4E) and 28S rRNA (Fig. 4F) became highly statistically different (all comparisons p < 0.0001 by One-Way ANOVA with Tukey comparison). Before lipophorin normalization, we observed a 69.3% and 18.7% decrease in the number of unique reads aligning to 18S and 28S rRNA for the depleted samples, respectively, as compared to the input RNA. After lipophorin normalization we observed a 96.8% and 89.1% reduction in 18S and 28S rRNA, respectively between the depleted samples and the input RNA. We used several different genes to normalize, including actin, with similar results (data not shown).
Fig. 4. Reverse Transcriptase mediated ribosomal RNA (rRNA) depletion increases target-specific coverage while reducing the number of rRNA reads in next-generation sequencing.

Anopheles gambiae mosquitoes were exposed to an infectious bloodmeal containing 107 PFU of West Nile virus strain NY99. The following day, midguts were dissected and the residual bloodmeal was spread onto a CloneSaver FTA card (GE Healthcare, USA) and then soaked in RNAlater solution to stabilize the nucleic acid and facilitate dispersion. Total nucleic acid was then extracted, and DNase treated. This is considered the input RNA. DNase-free RNA was then reverse transcribed using either ribosomal RNA specific probes (RT – with Probes) or without probes (RT – no Probes). The samples were then treated with RNase H and DNase I and purified. The samples were then subjected to library preparation and sequenced on an Illumina MiSeq. Reads were then demultiplexed and subsequently trimmed using BBDuk. Each sample was then normalized to contain 1.5 million reads to allow for direct comparisons. Duplicate reads were removed using Clumpify and then unique reads were mapped using BBSplit to the appropriate reference sequence, unique reads after duplicate removal (A), 18S rRNA (B), 28S rRNA (C) and mitochondrial 16S rRNA (D). Reads for 18S (E) and 28S (F) rRNA were then normalized to host gene AGAP001826-RA and then compared. All statistical tests were performed by One-Way ANOVA with Tukey test for multiple comparisons. The p-values are indicated as numbers. All p-values without an accompanying bar are statistical comparisons between the input RNA and the other group. The other comparisons with a bar are statistical comparisons between the two groups that are below the corresponding bar.
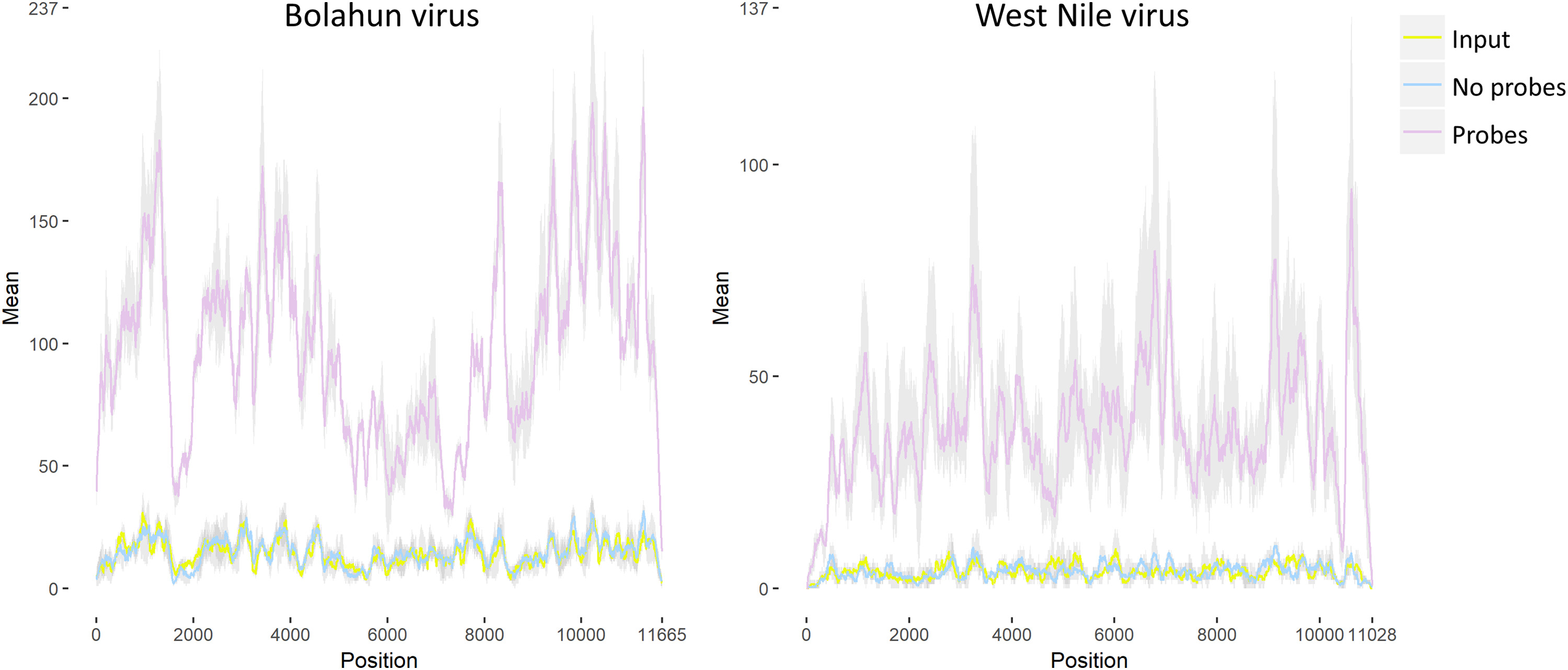
We next sought to determine the effect of depletion on read counts for potentially interesting species of RNA. Specifically, we aligned the unique reads to the transcriptome of Anopheles gambiae and found significantly more reads in the depleted samples compared to either the input RNA or RT – no probes (Fig. 5A, p = 0.0031 and 0.0027, respectively, by One-Way ANOVA with Tukey comparison). This resulted in a significantly increased number of genes with greater than 20 reads aligning in the depleted samples as compared to the other two groups (Fig. 5B, p = 0.0041 and 0.0040, respectively, by One-Way ANOVA with Tukey comparison). A table that lists genes, their read counts and the fold-difference to the input RNA is presented in Supplemental Table 4. We next compared viral sequences, namely, WNV (Fig. 5C) or Bolahun virus (BOLV, Fig. 5D) sequences. BOLV is known to persistently infect these mosquitoes (Fauver et al., 2016). A significantly increased number of reads aligned to both viruses (One-Way ANOVA with Tukey comparison, all p < 0.005, all depleted samples when compared to either input RNA or RT – no probes). We observed an 838.1% and 562.0% increase in the number of reads for WNV and BOLV, respectively in the depleted samples as compared to the input RNA. Coverage plots from input, depleted and non-depleted RNA samples are presented in Supplemental Fig. 2 for both BOLV and WNV. Finally, we assessed the ability to analyze intrahost viral variation in BOLV by calling variants with LoFreq. A significantly greater number of minority variants could be called in the depleted RNA when compared to the input RNA or RT - no probes group (Fig. 5E, One-Way ANOVA with Tukey comparison, p < 0.05 for depleted compared to either input to depleted and RT - no probes).
Fig. 5. Percent of reads to bacterial rRNA and host transcripts are increased in depleted samples.

Anopheles gambiae mosquitoes were exposed to an infectious bloodmeal containing 107 PFU of West Nile virus strain NY99. The following day, midguts were dissected and the residual bloodmeal was spread onto a CloneSaver FTA card (GE Healthcare, USA) and then soaked in RNAlater solution to stabilize the nucleic acid and facilitate dispersion. Total nucleic acid was then extracted, and DNase treated. This is considered the input RNA. DNase-free RNA was then reverse transcribed using either ribosomal RNA specific probes (RT – with Probes) or without probes (RT – no Probes). The samples were then treated with RNAse H and DNase I and purified. The samples were then subjected to library preparation and sequenced on an Illumina MiSeq. Reads were then demultiplexed and subsequently trimmed using BBDuk. Each sample was then normalized to contain 1.5 million reads to allow for direct comparisons. Duplicate reads were removed using Clumpify and then unique reads were mapped using either BBMap or BBSplit to the appropriate reference sequence, An. gambiae transcriptome (A and B), West Nile virus (C) or Bolahun virus (D). The reads called for each gene was determined using BBMap with output flag rpkm (B). Variants detected in Bolahun virus were called using LoFreq (F). Only variants present at greater than 5% were used for analysis. All statistical tests were performed by One-Way ANOVA with Tukey test for multiple comparisons. The p-values are indicated as numbers. All p-values without an accompanying bar are statistical comparisons between the input RNA and the other group. The other comparisons with a bar are statistical comparisons between the two groups that are below the corresponding bar.
We also aligned these sequences to bacterial 16S and 23S rRNA, since the bacterial midgut is known to contain bacteria that can influence vector competence of different pathogens (Beier et al., 1994; Dennison et al., 2014). We observed a significantly increased number of reads in the depleted samples as compared to the input RNA or RT - no probes group for 16 S (Fig. 6A, p = 0.0223 and 0.0235, respectively, One-Way ANOVA with Tukey comparison). The differences approached significance for 23S rRNA (Fig. 6B, p = 0.0564 and 0.0570, respectively, One-Way ANOVA with Tukey comparison). We also assessed the effect of depletion on the percentage of reads aligning to the transcriptome of a bacterial midgut resident. We chose to focus on Elizabethkingia anophelis, a known member of the Anopheles gambiae microbiome (Kämpfer et al., 2011). More reads aligned in the depleted samples as compared to the input RNA or RT - no probes groups (Fig. 6C, p = 0.0285 and 0.0267, respectively, One-Way ANOVA with Tukey comparison). A significantly increased number of genes with at least 20 reads aligning could be detected in the depleted samples, as compared to the input RNA or RT - no probes groups (Fig. 6D, both p = 0.037 by One-Way ANOVA with Tukey comparison). A table that lists the genes, their read counts and the fold-difference to the input RNA is presented in Supplemental Table 5. Additionally, we observed a significant increase in the percentage of reads aligning to a reference sequence containing all bacterial genomes in the depleted samples as compared to either the input RNA or RT- no probes (Fig. 6E and Supp. Table 6, p = 0.0217 and 0.0218, respectively, by One-Way ANOVA with Tukey comparison).
Fig. 6. Percent of reads to bacterial rRNA and host transcripts are increased in depleted samples.

Anopheles gambiae mosquitoes were exposed to an infectious bloodmeal containing 107 PFU of West Nile virus strain NY99. The following day, midguts were dissected and the residual bloodmeal was spread onto a CloneSaver FTA card (GE Healthcare, USA) and then soaked in RNAlater solution to stabilize the nucleic acid and facilitate dispersion. Total nucleic acid was then extracted, and DNase treated. This is considered the input RNA. DNase-free RNA was then reverse transcribed using either ribosomal RNA specific probes (RT – with Probes) or without probes (RT – no Probes). The samples were then treated with RNAse H and DNase I and purified. The samples were then subjected to library preparation and sequenced on an Illumina MiSeq. Reads were then demultiplexed and subsequently trimmed using BBDuk. Each sample was then normalized to contain 1.5 million reads to allow for direct comparisons. Duplicate reads were removed using Clumpify and then unique reads were mapped using either BBSplit to the appropriate reference sequence, 16S bacterial rRNA (A), 23S bacterial rRNA (B), Elizabethkingia anophelis transcriptome (C and D) and the NCBI bacterial genome database (E). The reads called for each gene was determined using BBMap with output flag rpkm (D). All statistical tests were performed by One-Way ANOVA with Tukey test for multiple comparisons. The p-values are indicated as numbers. All p-values without an accompanying bar are statistical comparisons between the input RNA and the other group. The other comparisons with a bar are statistical comparisons between the two groups that are below the corresponding bar.
3.4. Mosquito collections and sequencing summary
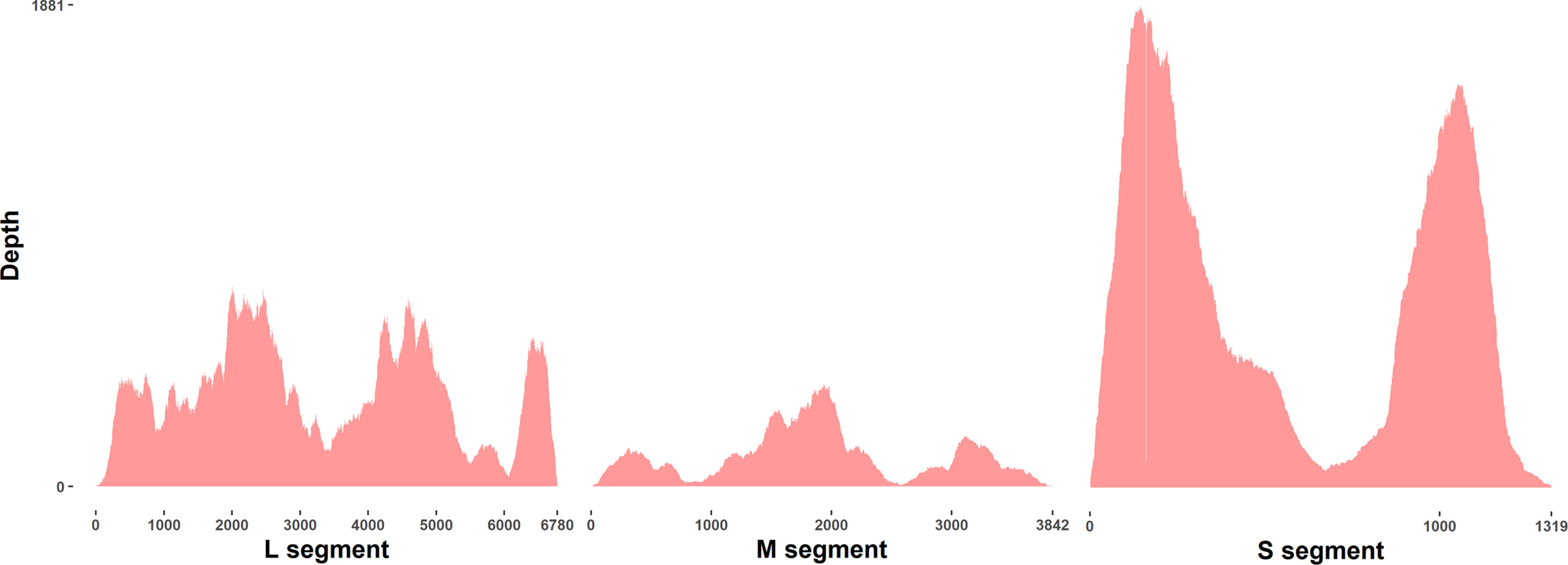
A total of 978 adult field-collected mosquitoes from 10 species were pooled for analysis by NGS (Supplemental Table 2). The most abundant species collected (242) was Coquillettidia venezuelensis, followed by Ae. albopictus (238), Psorophora albipes (110), Ps. varipes (101), Ae. angustivittatus (91), Cx. nigripalpus (87), Ae. aegypti (72), Ae. taeniorhynchus (33), Ae. serratus (2), and Ps. ferox (2). All species collected in this study have previously been reported from Chiapas state (Bond et al., 2014; Heinemann and Belkin, 1977). A single MiSeq run following quality filtering and removal of duplicate reads yielded 25.9 million total reads, resulting in 3.8 Gb of paired-end data. The total percentage of reads mapping to rRNA in the field samples was in line with what we observed after depletion in our colony mosquitoes (Supplemental Fig. 3).
3.5. Virus sequences identified in field collected mosquitoes following rRNA depletion
Each mosquito species sequenced, except a single pool of 2 Ps. ferox mosquitoes, produced contigs aligning to known viral sequences (Fig. 7, Supplemental Table 3). Based off amino acid similarity and phylogenetic placement, 8 major clades as well as multiple families of RNA viruses were represented across all samples. Amino acid similarities spanned anywhere from 28% (Reovirus contig from Ae. angustivittatus) to 99% (Phasi Charoen-like phasivirus RDRP from multiple Aedes species). Multiple previously described viruses were identified, based on a > 95% pairwise nucleotide identity, including Phasi Charoen-like phasivirus (PCLV) in Ae. aegypti, Ae. angustivittatus, and Ps. varipes. A complete genome of PCLV was assembled from pools of both male and female Ae. aegypti mosquitoes (Supplemental Fig. 4). This PCLV genome aligned to Phasi Charoen-like phasivirus strain 2b (Accession: MH237598) with ~ 98% pairwise nucleotide identity. PCLV sequences from Ae. angustivittatus and Ps. varipes aligned only to a portion of the RDRP. Partial sequences aligning to both the RDRP and capsid proteins of Humaita-Tubiacanga (HTV) virus were identified from female Ae. aegypti and male Ae. albopictus mosquitoes. Sequences aligned to HTV with 98.5% and 97.5% pairwise nucleotide identity, respectively.
Fig. 7. Viral sequences belonging to diverse clades of RNA viruses identified in field-collected mosquitoes following rRNA depletion.

Individual reads from each mosquito species sequenced were mapped back to all virus contigs identified in this study. Virus clade is inferred by amino acid similarity to other closely related sequences. The total number of reads aligning to each virus clade was graphed as a proportion of the total number of viral reads produced from each mosquito species.
Short flavivirus sequences (100−250) were found in 7 of 8 mosquito species sequenced aligning to the same portion of the WNV genome. Based on the sequence similarity between species, its presence in nearly all groups, and our frequent use of WNV in our laboratory, it is likely these sequences are the result of laboratory contamination during library preparation opposed to an authentic infection in these mosquito samples.
While numerous contigs were generated that distantly resembled known viral sequences, indicating the presence of divergent viruses in these species, we chose to further analyze only contigs that produced coding complete viral genomes (Ladner et al., 2014). Our computational approached generated 7 novel viral genomes, including a novel strain of a previously described Negevirus (Fig. 8A), 5 Levi-Narnaviruses (Fig. 9A-E), and 1 Luteo-Sobemo virus (Fig. 10A).
Fig. 8. Description of a novel variant of the negevirus Cordoba virus from Culex nigripalpus.

A- Virus cartoon depicting the genomic structure and depth of coverage Cordoba virus Cx. nigripalpus variant. The large boxes represent predicted ORFs and the small boxes represent areas of protein homology to viral methyltransferase (pfam01660), FtsJ-like methyltransferase (pfam01728), viral RNA helicase (pfam01443), and viral RNA-dependent RNA polymerases (cd1699). B. Phylogenetic placement of multiple strains of Cordoba virus highlighted in blue. Phylogenies were created using 1234 A.A. residues across the complete ORF. Tree is midpoint rooted. Phylogenetic trees were generated in FigTree. C. Expansion of phylogenetic tree containing the sequenced strains of Cordoba virus. The strain sequenced in this study is highlighted in blue. Bootstrap proportions are shown for each node. Phylogenetic trees were viewed in FigTree.
Fig. 9. Multiple, unique narnaviruses described from multiple mosquito species.

A–E cartoons depicting the simple genomic structure and depth of coverage to newly described narnaviruses. The large boxes represent predicted ORFs and the small boxes represent protein homology to viral RNA-dependent RNA polymerases (cd1699). A- Coquillettidia venezuelensis narnavirus 1 (CVNV1), B-Coquillettidia venezuelensis narnavirus 2 (CVNV2), C-Psorophora varipes narnavirus (PVNV), D- Aedes taeniorhynchus narnavirus (ATNV), E- Aedes angustivittatus narnavirus (AANV). F- Pairwise identify of 295 amino acid residues across the predicted RDRP between the newly described narnaviruses. The darker the color indicates a higher level of pairwise nucleotide identity. H- Phylogenetic placement of novel narnaviruses highlighted in blue. Tree based on alignments of RDRP from multiple narnavirus and is midpoint rooted. Bootstrap proportions are shown for each node. Phylogenetic trees were viewed in FigTree.
Fig. 10. Description of a novel Luteo-Sobemo like virus from Aedes aegypti mosquitoes.

A- Cartoon depicting the predicted bipartite genomic structure of RENV. Large boxes represent ORFs, small boxes represent areas of protein homology to Trypsin-like serine protease (cd00190), viral RNA-dependent RNA polymerase (cd1699), and capsid protein (cd00205). B- Phylogenetic placement of RENV. Phylogeny was created using a 289 amino acid portion of the RDRP. Trees are midpoint rooted. Bootstrap proportions are shown for each node. Phylogenetic trees were viewed in FigTree.
A total of 4 contigs identified in Cx. nigripalpus mosquitoes aligned to the CoB_37B strain of Cordoba virus with estimated gaps of 188, 72, and 55 nucleotides. The assembly of these contigs produced a final sequence approximately 7300 nucleotides long that contained a single ORF predicted to code for 4 proteins (Fig. 8A). These proteins include a viral methyltransferase (pfam01660), FtsJ-like methyltransferase (pfam01728), Viral RNA helicase (pfam01443), and RDRP (cd01699) (Fig. 8A). Both the type of proteins encoded and synteny of the genome are in agreement with representative +ssRNA viruses from the Nelorpivirus group of Negeviruses (Nunes et al., 2017). Phylogenetic placement and high pairwise nucleotide identity (78.8–93.6%, depending on strain) indicated this genome to be a novel strain of Cordoba virus, a negevirus described from a variety of mosquito species, including Cx. nigripalpus, from Nepal, the U.S., and Colombia (Nunes et al., 2017) (Fig. 8B, C).
Multiple sequences related to viruses in the +ssRNA Narna-Levi clade were identified from Ae. angustivittatus, Ae. taeniorhynchus, Cq. venezuelensis, and Ps. varipes. Two distinct contigs were generated from Cq. venezuelensis mosquitoes. These sequences were found to be approximately 2 kb in length and contain a single ORF that encodes for RDRP (cd01699) (Fig. 9 A–E). Pairwise amino acid identity was approximately 72–80% between 4 of the virus sequences, while a sequence from pools of Ae. angustivittatus mosquitoes varied substantially (30–33%) compared to other sequences described in this study (Fig. 9F). The 4 more similar genomes grouped with other narnavirus-like sequences described from mosquitoes, where the sequence from Ae. angustivittatus mosquitoes grouped with narnavirus-like sequences from crustaceans (Fig. 9G). These virus genomes have provisionally been designated Aedes angustivittatus narnavirus (AANV), Aedes taeniorhynchus narnavirus (ATNV), Coquillettidia venezuelensis narnavirus 1 & 2 (CVNV1, CVNV2), and Psorophora varipes narnavirus (PVNV).
Two sequences related to +ssRNA Luteo-Sobemo like viruses, 2718 and 1131 nucleotides in length, were identified in pools of both male and female Ae. aegypti mosquitoes. The longer sequence is predicted to encode for two proteins, a Trypsin-like serine protease (cd00190) and RDRP (cd01699), respectively, in two separate ORFs (Fig. 10A). These ORFs overlap and appear to be on the same segment indicating the reading frame difference is likely the result of frameshift mutation, which is common in Luteo-Sobemo viruses (Barry and Miller, 2002). The identified “slippery sequence”, a conserved heptanucleotide sequence that causes the ribosome to shift reading frames, in Sobemoviruses is “UUUAAAC”(Mäkinen et al., 1995). This specific sequence was not identified, however, as these viruses are divergent and not well characterized, it is possible a non-canonical heptanucleotide sequence could exist. A sequence 24 base pairs upstream of the second ORF reads “GGGCCCG”, which deviates slightly from the typical slippery sequence construct of “XXXYYYZ” (Plant, 2012). It remains to be determined whether this sequence is responsible for ribosomal frameshifting in this virus. The smaller sequence contains a single ORF encoding the predicted viral coat protein (pfam00729). This virus sequence was predicted to contain a bipartite genome based on 1) homology to the most similar virus currently described, Hubei mosquito virus (Shi et al., 2016), 2) the identification of two contigs with complete ORFs, 3) similar depth of coverage across viral segments, and 4) the co-occurrence of each segment in the same libraries. This sequence, provisionally named Renna virus (RENV), groups phylogenetically with viruses identified from a variety of ticks and insects, including mosquitoes (Fig. 10B). Both segments had a high average depth of coverage, 650 and 1351, respectively in Ae. aegypti females. RENV from male and female Ae. aegypti mosquito pools shared a > 99% pairwise nucleotide identity.
As all field-caught mosquito samples underwent rRNA depletion, no non-depleted samples exist for comparison purposes. In lieu of this comparison, we quantified the number of reads aligning to rRNA as well as virus sequences from each species of mosquitoes (Supplemental Fig. 3). The ratio of virus to rRNA reads was highest in Aedes aegypti mosquitoes, which were included as a template for probe design. The number of reads aligning to virus sequences and rRNA sequences varied substantially between mosquito species. The percentage of reads mapping to viruses was relatively high, particularly for Ae. aegypti. In addition, we classified reads from each mosquito species sequenced to broad taxonomic categories (Supplemental Table 7).
3.6. Transcriptome and bacterial analysis from field-caught mosquitoes
In addition to viral sequences, researchers may be interested in identifying host transcripts or bacterial reads. We therefore aligned the normalized reads from Ae. aegypti and Ae. albopictus to their respective transcriptome reference file and generated a table containing the number of reads aligning to each gene (Supp. Tables 8 and 9). We were able to identify many genes in each sample for each species and sex. In fact, we identified the gene “female-specific chymotrypsin” in both Ae. aegypti and Ae. albopictus that was present 156 and 121 times, respectively in the female pools. Only 12 and 30 reads were counted in the male pools, respectively, highlighting our ability to identify sex-specific transcripts from field-caught mosquitoes. We also present the percentage of reads that align to the transcriptome for all mosquito species tested in Supplemental Table 7. For bacteria, we identified a high percentage of reads aligning to the 23 S rRNA gene and to a lesser extent the 16S rRNA.
4. Discussion
Studies involving sequencing viral RNA; such as viral metagenomics, intrahost viral dynamics, transcriptomics and virus discovery require target reads to be at sufficient levels to perform meaningful analysis. These analyses are often hampered by the high percentage of ribosomal RNA (rRNA) present in total RNA, which can reach greater than 80–90% of the total sample (Eun, 1996). Since these reads are rarely used, this represents significant waste of both financial and computational resources and limits the amount of multiplexing that can be performed. While procedures such as selection of polyadenylated transcripts can be used to enrich RNA preparations for mRNA, this is not relevant to RNA viruses that lack polyadenylation. Furthermore, other methods like amplicon sequencing or probe capture are sequence specific, and thus unknown pathogens sequences will not be enriched for. Therefore, selective depletion of highly abundant rRNA is beneficial. Several methods and commercial kits are available to do this, but most are designed to work specifically for human or mouse samples. Here, we describe a novel method that utilizes specific reverse transcription of rRNA using small DNA probes for depletion along with RNase H. This allowed us to design depletion probes that could simultaneously deplete rRNA from mosquitoes of highly diverse genetic backgrounds. Using this method, we show that specific depletion of rRNA results in increased reads to meaningful RNA, such as viruses and host mRNA. In addition, we detected more intrahost variants using this depletion method. Although we subjected all field-collected mosquito pools to rRNA depletion, thus no non-depleted libraries were generated for comparison, we were able to detect novel virus genomes from a single, highly multiplexed (64 libraries), MiSeq run of nearly 1000 diverse field-collected mosquito samples that underwent rRNA depletion. Taken together, these findings suggest that RT-mediated rRNA depletion can facilitate sequencing of mosquito samples both from the lab and field.
To our knowledge, only two other studies have aimed to assess rRNA depletion strategies from insect species. The first used a commercial kit designed for mammalian rRNA, Epicentre’s Ribo-Zero rRNA, to deplete rRNA from Drosophila flies. While the approach seemed to effectively remove rRNA and enrich mRNA transcripts, it suffers from being high-cost and also doesn’t allow for additional targets to be added (Kumar et al., 2012). Kumar et al. observed a 66% decrease in the raw number of 18S rRNA reads and a 6.2-fold increase in reads to actin. We observed a 69% decrease in 18S rRNA reads and a 9.4- and 6.6-fold decrease in WNV and BOLV reads, respectively. In addition, when we normalized the number of rRNA reads to a highly-expressed gene (AGAP001826-RA) in the same manner as Kumar et al. we found a 96.8% and 89.1% decrease in 18S and 28S rRNA, respectively. This suggests that our approach is as good, if not better, than the Ribo-Zero kit that they assessed for insect samples. Another study showed by bioanalyzer effective removal of rRNA from mosquito midguts using RNA probes to the rRNA (Kukutla et al., 2013). However, this technique required large amounts of input RNA (50 pooled midguts), uses unstable RNA probes and expensive streptavidin beads. Furthermore, it’s unclear if this technique works for other species or just An. gambiae. Accordingly, we devised a novel method for depleting rRNA using RNase H depletion that was based on the method described by Morlan et al. (Morlan et al., 2012) with the exception that it uses shorter probes and incorporates a reverse transcription (RT) step. The shorter probes allow highly conserved regions to be targeted, thus making it possible to simultaneously deplete rRNA from divergent species or even genera. The RT step extends the bound DNA probes to produce cDNA complementary to the rRNA that is destroyed following both a RNase H and DNase I digestion.
First, we assessed the efficacy of several RTs to convert rRNA to cDNA and subsequently be degraded by RNase H. We found AMV to be the optimal enzyme, depleting a significant amount of rRNA with no off-target effects. While M-MLV RT depleted as much or more rRNA as AMV, it also depleted WNV RNA, suggesting it was converting non-target RNA species to cDNA as well (Fig. 2C). It’s unclear why M-MLV RT would have off-target effects and not AMV, especially as there has been evidence of the opposite occurring in a previous publication (Agranovsky, 1992). This and other publications have shown primer-independent cDNA synthesis for both AMV and M-MLV RTs, which could explain the high level of non-specific depletion in M-MLV but not AMV observed here (Freeh and Peterhans, 1994). Agranovsky et al. presented evidence that a tRNA contaminant in the AMV RT preparation tested at that time was responsible for this primer-independent cDNA synthesis. We cannot rule out the possibility that the M-MLV obtained from NEB contained some contaminant that could effectively primer non-target RNA species such as WNV. There may also be small RNAs present in our samples that could have primed cDNA synthesis particularly well for M-MLV. Both Superscript (SS) III and IV, mutants of M-MLV, were effective at depleting 18S rRNA with no off-target effects. While SSIII also depleted 28S rRNA, SSIV did not effectively deplete this RNA species. Finally, Tth DNA polymerase, which shows RT activity in the presence of manganese, did not effectively deplete rRNA, even in the presence of specific DNA probes. This may be related to the fact that Tth and SSIV lack functional RNase H domains (Myers and Gelfand, 1991), suggesting that this intrinsic activity is important for the mechanism of depletion with this technique, even if RNase H is added after the RT step. Next, we assessed whether the RT step was necessary for depletion in the workflow, as Morlan et al. had previously shown efficient depletion in the absence of this step (Morlan et al., 2012). The RT step was critical to the depletion observed and the specific depletion probes were necessary, as samples treated with DNA probes in the absence of RT had only a modest depletion effect. However, in the presence of RT and specific depletion probes, 18S and 28S rRNA were depleted roughly 100- and 1000-fold, respectively.
Depletion was then tested on RNA from three medically important mosquito species representing three distinct genera; Ae. aegypti, An. gambiae and Cx. quinquefasciatus. These species transmit a significant proportion of vector-borne pathogens; including dengue virus, Zika virus, chikungunya virus, malaria parasites and WNV, among others. We found by qRT-PCR and bioanalyzer, depletion in the presence of rRNA probes was associated with a significant reduction in 18S and 28S rRNA from all three species tested. Despite the almost complete removal of the peak for rRNA in the bioanalyzer traces, we were still able to detect rRNA sequences by both qRT-PCR and NGS. This might be a result of incomplete digestion of the RNA by RNase H due to incomplete activity or RNA that hadn’t been reverse transcribed. It’s possible that the secondary structure of rRNA prevents the complete synthesis of cDNA from RNA and that this is not degraded by the RNase H. Different methods to increase the efficiency of cDNA synthesis or adding additional DNA probes may be beneficial in future iterations of this protocol. This result suggested that this protocol could be used for a wide array of mosquito species, as Aedes and Culex are significantly divergent from Anopheles mosquitoes, having separated likely over 200 million years ago (Reidenbach et al., 2009). In fact, we have seen rRNA depletion by NGS in virus stocks prepared in mammalian cells as well, suggesting a broad range of cross-reactivity to rRNA from different species.
We then depleted rRNA from midguts isolated from An. gambiae mosquitoes that were fed a bloodmeal containing WNV. This RNA was then subjected to Illumina deep-sequencing and the resulting reads were aligned to several sequences. We observed significant depletion of rRNA while increasing the number of normalized reads to host and bacterial mRNA, WNV and the insect-specific virus BOLV (Fauver et al., 2016). We were also able to identify significantly more minority variants present in BOLV, suggesting intrahost virus population analyses are facilitated following depletion. It has been shown that high levels of sequencing coverage are necessary to perform intrahost virus analysis, which can be difficult to achieve without depletion or enrichment (McCrone and Lauring, 2016). We were also able to identify a significantly greater number of genes with 20 or more reads aligning in our transcriptomic analysis of the depleted samples as compared to the input RNA. This was true for both Anopheles gambiae and its bacterial symbiont, Elizabethkingia anophelis. This would suggest that non-model organism and microbiome transcriptomic analysis is possible with this depletion method. A considerable proportion of reads also aligned to the transcriptome in field-caught mosquitoes, which allowed us to identify several reads, specifically the female-specific chymotrypsin gene that was present at higher coverage in Ae. aegypti and Ae. albopictus females than males.
Furthermore, we found more reads aligned to bacterial 16S and 23S rRNA sequences in the depleted samples as compared to input RNA. While we did not intend this depletion technique for metagenomics studies, this suggests that this technique can be used for insect or other non-model organism studies of this kind. We did not originally test our probes for off-target hits on bacterial rRNA and as such results would be expected to improve if any probe that binds to this RNA is removed. Furthermore, if bacterial rRNA reads are not informative for the desired analysis, one could design probes specific to these sequences to be depleted. Additionally, we detected a significant number of genes from the Anopheles bacterial symbiont Elizabethkingia anophelis, which could potentially allow researchers to perform transcriptomic analysis from bacterial residents of insects or other non-model organisms. The number of genes with 20 or more reads aligning was significantly increased in the depleted samples.
As second and third generation sequencing based approaches for the detection and analysis of vector-borne pathogens from field-collected mosquitoes are becoming commonplace, techniques that increase reads to target sequences in complex samples will be sorely needed. Accordingly, we employed our rRNA depletion method to a diverse group of field-collected mosquitoes and subjected them to NGS with the goal of identifying both human-infecting and insect-specific viruses. While we did not identify arbovirus sequences from these pools of mosquitoes, we were able to identify partial and coding complete genomic sequences of a variety of presumed insect specific viruses. As all pools were subjected to rRNA depletion, we do not have non-depleted libraries to compare the efficacy of rRNA depletion to. However, the total number of reads aligning to rRNA from these samples was congruent with what we observed in our laboratory studies. In fact, in libraries constructed from Ae. aegypti females, more reads competitively aligned to virus sequences than to 28S or 18S rRNA sequences, although the number of reads aligning to both viruses and rRNA sequences varied widely between divergent genera. Using our bioinformatic approach, 7 novel coding complete viral genomes were identified, in addition to the previously described insect specific viruses PCLV and HTV. Complete PCLV genomes were assembled from pools of both male and female Ae. aegypti mosquitoes at a relatively high depth of coverage and pairwise nucleotide identity. PCLV has been identified mosquito cell culture and in numerous populations of Ae. aegypti mosquitoes from across the globe (Chandler et al., 2014; Di Giallonardo et al., 2018; Yamao et al., 2009; Zhang et al., 2018). In addition to PCLV, we identified large contigs with > 97% nucleotide identity to HTV in both female Ae. aegypti and male Ae. albopictus mosquitoes (Aguiar et al., 2015; Zakrzewski et al., 2018). A total of 5 coding complete narnavirus genome sequences were identified from 4 species of mosquitoes collected in this study. Of the 5 virus genomes described here, 4 group closely together and with other Narnaviruses described from mosquitoes. While multiple Narnaviruses have been identified by metagenomic sequencing of whole mosquito samples, it remains to be determined if these represent infections of fungi in the normal microbiota, or bona fide infections of mosquitoes (Chandler et al., 2015; Cook et al., 2013; Shi et al., 2016). A novel strain of Cordoba virus, a negevirus described previously from mosquitoes, was identified in Cx. nigripalpus mosquitoes (Nunes et al., 2017). We were also able to assemble the coding complete genome of RENV, a virus that groups with Luteo-Sobemo viruses identified in mosquitoes (Shi et al., 2016). Based on the phylogenetic placement of these sequences, all the viruses described in this study are presumed to be insect specific, however this is yet to be validated. As well, the effect these viruses may have on mosquito biology or vector competence remains to be determined. It is highly probable that these viruses would have been detected if we did not perform rRNA depletion, but based on the NGS data from laboratory experiments, our depletion method likely aided in discovery and characterization by allowing more unique, non-rRNA sequences to be identified. Although the amount of viral RNA, or other microbial RNA, from any given mosquito depends upon individual infection status and the amount of replication occurring, we were able to identify and assemble multiple viral genomes from a highly multiplexed sequencing run on a comparatively low-output sequencing platform. As well, we were able to assign reads from each mosquito species to broad taxonomic levels. Increasing reads to target sequences of interest (e.g. viruses) by depleting uninformative rRNA sequences from complex, field-collected mosquito samples has the potential to improve the efficacy and feasibility of using metagenomic sequencing for mosquito-borne disease surveillance.
In conclusion, we have developed an effective approach for depleting unwanted sequences such as rRNA from complex RNA samples. While we specifically targeted depletion of rRNA from mosquitoes in this report, we have effectively used this approach for tick, human and mouse RNA as well (data not shown). Furthermore, since probes can be designed to any RNA species, one can deplete any target gene that is not necessary for downstream analysis. This approach can be adapted for a range of non-model pathogen: host systems, making it highly versatile.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to acknowledge Nunya Chotiwan, Rushika Pereira, Karla Saavedra, Ildefonso Fernández-Salas and all the employees at the Centro Regional de Investigación en Salud Pública for assistance in collecting mosquitoes in Tapachula, Chiapas, Mexico. We also acknowledge the funding sources for providing the resources to perform this work; notably NIH grant AI067380 (NIAID).
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.virol.2018.12.020
References
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. , 2013. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agranovsky AA, 1992. Exogenous primer-independent cDNA synthesis with commercial reverse transcriptase preparations on plant virus RNA templates. Anal. Biochem. 203, 163–165. [DOI] [PubMed] [Google Scholar]
- Aguiar ERGR, Olmo RP, Paro S, Ferreira FV, de Faria I.J. da S., Todjro YMH, Lobo FP, Kroon EG, Meignin C, Gatherer D, et al. , 2015. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 43, 6191–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, 1990. Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Aubry M, Teissier A, Huart M, Merceron S, Vanhomwegen J, Roche C, Vial A-L, Teururai S, Sicard S, Paulous S, et al. , 2017. Zika virus Seroprevalence, French polynesia, 2014–2015. Emerg. Infect. Dis 23, 669–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JK, Miller WA, 2002. A- 1 ribosomal frameshift element that requires base pairing across four kilobases suggests a mechanism of regulating ribosome and replicase traffic on a viral RNA. Proc. Natl. Acad. Sci 99, 11133–11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier MS, Pumpuni CB, Beier JC, Davis JR, 1994. Effects of para-aminobenzoic acid, insulin, and gentamicin on Plasmodium falciparum development in anopheline mosquitoes (Diptera: culicidae). J. Med. Entomol 31, 561–565. [DOI] [PubMed] [Google Scholar]
- Bond JG, Casas-Martínez M, Quiroz-Martínez H, Novelo-Gutiérrez R, Marina CF, Ulloa A, Orozco-Bonilla A, Muñoz M, Williams T, 2014. Diversity of mosquitoes and the aquatic insects associated with their oviposition sites along the Pacific coast of Mexico. Parasit. Vectors 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T, 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler JA, Thongsripong P, Green A, Kittayapong P, Wilcox BA, Schroth GP, Kapan DD, Bennett SN, 2014. Metagenomic shotgun sequencing of a Bunyavirus in wild-caught Aedes aegypti from Thailand informs the evolutionary and genomic history of the Phleboviruses. Virology 464–465, 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler JA, Liu RM, Bennett SN, 2015. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front. Microbiol 6, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S, Chung BY-W, Bass D, Moureau G, Tang S, McAlister E, Culverwell CL, Glücksman E, Wang H, Brown TDK, et al. , 2013. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One 8, e80720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross ST, Kapuscinski ML, Perino J, Maertens BL, Weger-Lucarelli J, Ebel GD, Stenglein MD, 2018. Co-infection patterns in individual Ixodes scapularis ticks reveal associations between viral, eukaryotic and bacterial microorganisms. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darsie RF, Ward RA, 2005. Identification and Geographical Distribution of the Mosquitoes of North America, North of Mexico: : University Press of Florida Google Scholar, Gainesville. [Google Scholar]
- Dennison NJ, Jupatanakul N, Dimopoulos G, 2014. The mosquito microbiota influences vector competence for human pathogens. Curr. Opin. Insect Sci 3, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giallonardo F, Audsley MD, Shi M, Young PR, McGraw EA, Holmes EC, 2018. Complete genome of Aedes aegypti anphevirus in the Aag2 mosquito cell line. J. Gen. Virol 99, 832–836. [DOI] [PubMed] [Google Scholar]
- Eun H-M, 1996. Enzymology Primer for Recombinant DNA Technology. Elsevier. [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M, 2016. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver JR, Grubaugh ND, Krajacich BJ, Weger-Lucarelli J, Lakin SM, Fakoli LS III, Bolay FK, Diclaro JW II, Dabiré KR, Foy BD, et al. , 2016. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 498, 288–299. [DOI] [PubMed] [Google Scholar]
- Fauver JR, Gendernalik A, Weger-Lucarelli J, Grubaugh ND, Brackney DE, Foy BD, Ebel GD, 2017. The use of xenosurveillance to detect human bacteria, parasites, and viruses in mosquito bloodmeals. Am. J. Trop. Med. Hyg 97, 324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver JR, Weger-Lucarelli J, Fakoli LS 3rd, Bolay K, Bolay FK, Diclaro JW 2nd, Brackney DE, Foy BD, Stenglein MD, Ebel GD, 2018. Xenosurveillance reflects traditional sampling techniques for the identification of human pathogens: a comparative study in West Africa. PLoS Negl. Trop. Dis 12, e0006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni D, Filippi G, Cagliani R, De Gioia L, Pozzoli U, Al-Daghri N, Clerici M, Sironi M, 2015. The heptad repeat region is a major selection target in MERS-CoV and related coronaviruses. Sci. Rep 5, 14480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeh B, Peterhans E, 1994. RT-PCR: “background priming” during reverse transcription. Nucleic Acids Res. 22, 4342–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh ND, Sharma S, Krajacich BJ, Fakoli LS III, Bolay FK, Diclaro JW II, Johnson WE, Ebel GD, Foy BD, Brackney DE, 2015. Xenosurveillance: a novel mosquito-based approach for examining the human-pathogen landscape. PLoS Negl. Trop. Dis 9, e0003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh ND, Weger-Lucarelli J, Murrieta RA, Fauver JR, Garcia-Luna SM, Prasad AN, Black WC 4th, Ebel GD, 2016. Genetic drift during systemic arbovirus infection of mosquito vectors leads to decreased relative fitness during host switching. Cell Host Microbe 19, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O, 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Heinemann SJ, Belkin JN, 1977. Collection records of the project “Mosquitoes of Middle America” 9. Mexico (MEX, MF, MT, MX). Mosq. Syst 9, 483–535. [Google Scholar]
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P, 2008. Global trends in emerging infectious diseases. Nature 451, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM, 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. , 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukutla P, Steritz M, Xu J, 2013. Depletion of ribosomal RNA for mosquito gut metagenomic RNA-seq. J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Creasy T, Sun Y, Flowers M, Tallon LJ, Dunning Hotopp JC, 2012. Efficient subtraction of insect rRNA prior to transcriptome analysis of Wolbachia-Drosophila lateral gene transfer. BMC Res. Notes 5, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner JT, Beitzel B, Chain PSG, Davenport MG, Donaldson EF, Frieman M, Kugelman JR, Kuhn JH, O’Rear J, Sabeti PC, et al. , 2014. Standards for sequencing viral genomes in the era of high-throughput sequencing. MBio 5 (e01360–14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, Komar N, Panella NA, Allen BC, Volpe KE, et al. , 2000. Rapid detection of west nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J. Clin. Microbiol 38, 4066–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A, 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The sequence alignment/Map format and SAM tools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen K, Tamm T, Næss V, Truve E, Puurand Ü, Munthe T, Saarma M, 1995. Characterization of cocksfoot mottle sobemovirus genomic RNA and sequence comparison with related viruses. J. Gen. Virol 76, 2817–2825. [DOI] [PubMed] [Google Scholar]
- Matranga CB, Gladden-Young A, Qu J, Winnicki S, Nosamiefan D, Levin JZ, Sabeti PC, 2016. Unbiased deep sequencing of RNA viruses from clinical samples. J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrone JT, Lauring AS, 2016. Measurements of intrahost viral diversity are extremely sensitive to systematic errors in variant calling. J. Virol 90, 6884–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsky HC, Matranga CB, Wohl S, Schaffner SF, Freije CA, Winnicki SM, West K, Qu J, Baniecki ML, Gladden-Young A, et al. , 2017. Zika virus evolution and spread in the Americas. Nature 546, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moratorio G, Henningsson R, Barbezange C, Carrau L, Bordería AV, Blanc H, Beaucourt S, Poirier EZ, Vallet T, Boussier J, et al. , 2017. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol 2, 17088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlan JD, Qu K, Sinicropi DV, 2012. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One 7, e42882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy RM, Meola MA, Morin L-LL, Ebel GD, Kramer LD, 2007. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am. J. Trop. Med. Hyg 77, 365–370. [PubMed] [Google Scholar]
- Myers TW, Gelfand DH, 1991. Reverse transcription and DNA amplification by a Thermus thermophilus DNA polymerase. Biochemistry 30, 7661–7666. [DOI] [PubMed] [Google Scholar]
- Nunes MRT, Contreras-Gutierrez MA, Guzman H, Martins LC, Barbirato MF, Savit C, Balta V, Uribe S, Vivero R, Suaza JD, et al. , 2017. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 504, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte A, Sauter M, Daxer MA, McHardy AC, Klingel K, Gabriel G, 2015. Adaptive mutations that occurred during circulation in humans of H1N1 influenza virus in the 2009 pandemic enhance virulence in mice. J. Virol 89, 7329–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP, 2012. Ribosomal frameshift signals in viral genomes In: Garcia M (Ed.), Viral Genomes - Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus Interactions. InTech. [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO, 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidenbach KR, Cook S, Bertone MA, Harbach RE, Wiegmann BM, Besansky NJ, 2009. Phylogenetic analysis and temporal diversification of mosquitoes (Diptera: culicidae) based on nuclear genes and morphology. BMC Evol. Biol 9, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Lin X-D, Tian J-H, Chen L-J, Chen X, Li C-X, Qin X-C, Li J, Cao J-P, Eden J-S, et al. , 2016. Redefining the invertebrate RNA virosphere. Nature. [DOI] [PubMed] [Google Scholar]
- Shi P-Y, Tilgner M, Lo MK, Kent KA, Bernard KA, 2002. Infectious cDNA clone of the epidemic west nile virus from New York City. J. Virol 76, 5847–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S, 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 2018. 2018 annual review of the Blueprint list of priority diseases. [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N, 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamao T, Eshita Y, Kihara Y, Satho T, Kuroda M, Sekizuka T, Nishimura M, Sakai K, Watanabe S, Akashi H, et al. , 2009. Novel virus discovery in field-collected mosquito larvae using an improved system for rapid determination of viral RNA sequences (RDV ver4.0). Arch. Virol 154, 153–158. [DOI] [PubMed] [Google Scholar]
- Zakrzewski M, Rašić G, Darbro J, Krause L, Poo YS, Filipović I, Parry R, Asgari S, Devine G, Suhrbier A, 2018. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep 8, 4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang S, Jin T, Lin P, Huang Y, Wu C, Peng B, Wei L, Chu H, Wang M, et al. , 2018. Discovery and high prevalence of Phasi Charoen-like virus in fieldcaptured Aedes aegypti in South China. Virology 523, 35–40. [DOI] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V, 2018. A completely reimplemented MPI bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol 430, 2237–2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data has been deposited to the SRA database under PRJNA505498. Novel virus genomes are listed in GenBank with accession numbers MK2835331-MK285338.