

Abstract
Immunotherapy has transformed the treatment landscape for a wide range of human cancers. Immune checkpoint inhibitors (ICIs), monoclonal antibodies that block the immune-regulatory “checkpoint” receptors CTLA-4, PD-1, or its ligand PD-L1, can produce durable responses in some patients. However, coupled with their success, these treatments commonly evoke a wide range of immune-related adverse events (irAEs) that can affect any organ system and can be treatment-limiting and life-threatening, such as diabetic ketoacidosis, which appears to be more frequent than initially described. The majority of irAEs from checkpoint blockade involve either barrier tissues (e.g., gastrointestinal mucosa or skin) or endocrine organs, although any organ system can be affected. Often, irAEs resemble spontaneous autoimmune diseases, such as inflammatory bowel disease, autoimmune thyroid disease, type 1 diabetes mellitus (T1D), and autoimmune pancreatitis. Yet whether similar molecular or pathologic mechanisms underlie these apparent autoimmune adverse events and classical autoimmune diseases is presently unknown. Interestingly, evidence links HLA alleles associated with high risk for autoimmune disease with ICI-induced T1D and colitis. Understanding the genetic risks and immunologic mechanisms driving ICI-mediated inflammatory toxicities may not only identify therapeutic targets useful for managing irAEs, but may also provide new insights into the pathoetiology and treatment of autoimmune diseases.
Introduction
The 2018 Nobel Prize in Physiology or Medicine was awarded to James P. Allison and Tasuku Honjo for their work on the role of inhibitory immune “checkpoint” receptors in the regulation of antitumor immunity. Their work and the work of others in the field laid the foundation for a revolution in cancer treatment, unleashing the immune system to attack cancer. Allison’s early intuition was correct: “What we needed to do was to release the brakes of the immune system to fight cancer.” Allison demonstrated that cytotoxic T lymphocyte antigen 4 (CTLA-4), a protein expressed by activated T cells, acts as an important check on immune activation, in particular inhibiting the response to cancer. Subsequent research has focused on other immune checkpoints, most prominently the receptor programmed cell death protein 1 (PD-1) and its ligand (PD-L1) (1–3). Immune checkpoint inhibitors (ICIs) targeting these proteins are now approved by the US FDA to treat a variety of types of cancers, such as melanoma, lung, kidney, bladder, gastric, and liver tumors. Physiologically, CTLA-4, PD-1, and PD-L1 play critical roles in peripheral tolerance, as clearly demonstrated both in animal models and in patients with haploinsufficiency in CTLA-4. Not surprisingly, pharmacologic disruption of these checkpoints leads to a wide range of inflammatory toxicities, collectively referred to as immune-related adverse events (irAEs) (ref. 4 and Table 1). These toxicities can affect any organ system of the body, although most occur either at barrier organs (e.g., the gastrointestinal or pulmonary mucosa) or in endocrine glands (4, 5). Many of these irAEs are mild, yet they can carry considerable morbidity, and in rare cases these toxicities can be fatal, particularly when recognized late (6).
Table 1. Immune-related adverse events from cancer immunotherapy.

Elucidating the mechanisms underlying irAEs caused by CTLA-4 and PD-1 pathway inhibition may provide essential clues to understanding the pathogenesis of autoimmune diseases, potentially leading to the identification of novel treatments. In support of this concept, CTLA-4 haploinsufficiency, a rare genetic disorder, severely impairs the normal regulation of the immune system, resulting in inflammatory intestinal disease, multilineage autoimmune cytopenias, and respiratory infections (7). Furthermore, conditional deletion of Ctla-4 on regulatory T cells (Tregs) during adulthood leads to resistance to experimental autoimmune encephalomyelitis, the mouse model of multiple sclerosis (8); this suggests that peripheral Treg expansion and/or increased Treg activation as a result of CTLA-4 blockade could prevent autoimmune disease. Finally, the CTLA-4 splice variant li-CTLA-4, a ligand-independent isoform, reduces diabetes incidence and insulitis in nonobese diabetic (NOD) mice, when expressed at physiologic levels in CTLA-4–sufficient animals (9). li-CTLA-4 is expressed in naive and activated T cells and can modify T cell signaling despite its lack of a B7 binding domain. Here, we will explore how these and other mechanistic insights into irAEs provide the groundwork for understanding how to limit the toxicity of immunotherapy as well as treat autoimmune disease.
Mechanisms of CTLA-4 and PD-1/PD-L1 signaling
CTLA-4 plays a critical role in the chain of events leading to T cell activation and regulation. CTLA-4 upregulation occurs following T cell receptor (TCR) engagement (signal 1 of T cell activation) and reduces TCR signaling by competing with the costimulatory molecule CD28 for the B7 ligands B7-1 (CD80) and B7-2 (CD86), for which CTLA-4 has higher avidity and affinity (Figure 1 and Figure 2A) (10–12). B7-1 and B7-2 binding leads to positive costimulatory signals through CD28, and competitive inhibition of both molecules by CTLA-4 is essential to produce a negative effect on T cell activation (13).
Figure 1. Schematic representation of CTLA-4 and PD-1 blockade of T cell activation and attenuation.
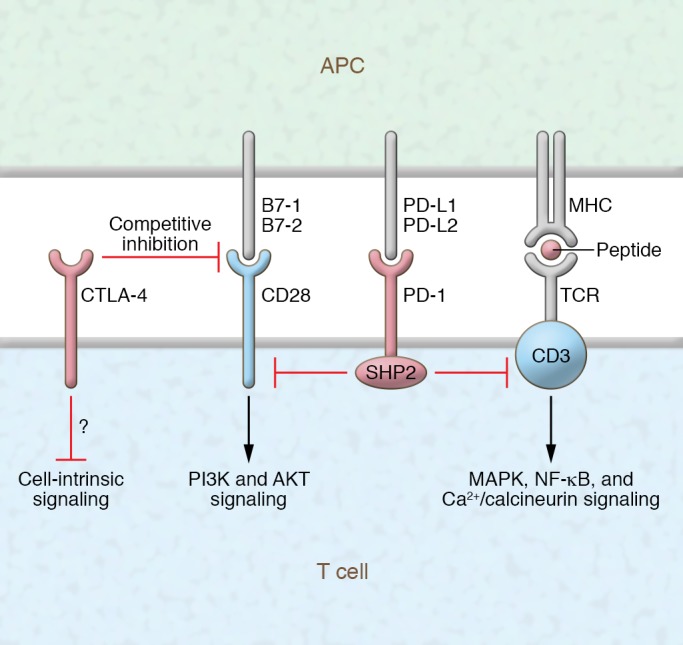
Molecular interactions and downstream signaling as a result of ligation of CTLA-4 and PD-1 with their corresponding ligands.
Figure 2. Immune checkpoint inhibitor mechanisms and design.

(A) Mechanisms by which T cell activation by CTLA-4 and PD-1 blockade therapy may cause pituitary and pancreatic β cell damage. CTLA-4 is expressed by normal pituitary cells. Following CTLA-4 blockade (i.e., ipilimumab), the classic complement pathway is activated, resulting in severe inflammation (hypophysitis) and destruction of pituitary cells (23). T cell activation by PD-1 blockade (i.e., nivolumab) can cause pancreatic β cell destruction. Interestingly, PD-L1 is specifically upregulated on pancreatic β cells of patients with T1D, and it is induced by both type I and II interferons via IRF1 (74). Several additional mechanisms are thought to contribute to the efficacy of anti–CTLA-4 and anti–PD-1 therapy (right). These include antibody-mediated depletion of Tregs, enhancement of T cell–positive costimulation within the tumor microenvironment, blockade of host-derived PD-L1 signals from nontumor cells in the microenvironment, and blockade of interactions between PD-L1 and B7-1 (2). Some of these additional mechanisms theoretically play a role in the development of specific organ inflammatory toxicities related to anti–CTLA-4 and anti–PD-1 immunotherapy. (B) Therapeutic mAbs targeting CTLA-4, PD-1, or PD-L1. Left: IgG1 is the isotype of the majority of approved mAb immunotherapies, such as anti–CTLA-4 or anti-CD20 (rituximab). This mAb drives potent antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC), engaging both cellular and humoral immune responses. Center: IgG1-induced ADCC can be increased by defucosylation of the glycan sequences (IgG1fut). This modification, obtained using a specific CHO cell line, enhances mAb binding to FcγRIIIa/CD16. The approved anti-CD20 obinutuzumab is engineered with reduced fucose content. Right: IgG4S228P is an engineered isotype of IgG4 that displays reduced ADCC and ADCP and no CDC. A serine-to-proline substitution at position 228 (S228P) in the hinge region prevents Fab arm exchanges that frequently occur between IgG4 molecules. IgG4S228P mAbs, such as anti–PD-1 nivolumab, are mainly blocking agents.
The upregulation of CTLA-4 is not the only mechanism regulating T cell activation. CTLA-4 is present in intracellular vesicles that are recruited into the immunologic synapse (2). The degree of CTLA-4 recruitment to the immunologic synapse correlates with TCR signal strength, and it is stabilized by B7 ligand binding and outcompetes CD28 (14). Through this mechanism, CTLA-4 dampens positive costimulation by CD28 and in turn decreases CD28 downstream signaling that is primarily mediated by PI3K and AKT (15, 16). CTLA-4 regulation of T cell activity takes place not only in lymphoid organs but also in peripheral tissues, given that B7 ligands are expressed (to a certain extent) by antigen-presenting cells within tissues, including tumors, and can also be expressed by activated T cells.
The primary biological function of the PD-1/PD-L1 pathway is to maintain peripheral tolerance in the setting of chronic inflammation. PD-1, like CTLA-4, is expressed after T cell activation, but its expression increases upon repeated stimulation (17). PD-1 has also been found on several other cell types, including B cells (17). PD-L1 is upregulated by inflammatory cytokines such as IFN-γ on a wide variety of tissues, including many tumors (18–20). A second ligand for PD-1, PD-L2, is expressed primarily on cells of hematologic origin. PD-1 and its ligand PD-L1 exhibit a critical role in tumor progression and appear to play a central role in mediating tumor immune escape. PD-1 regulates T cell activation through interaction with PD-L1 and PD-L2 (Figure 1 and Figure 2A) (18–20). Upon engagement with PD-L1 and PD-L2, PD-1 delivers a negative costimulatory signal through the tyrosine phosphatase SHP2, leading to diminished activation. The recruitment of SHP2 directly attenuates TCR signaling through dephosphorylation of proximal signaling elements (Figure 1). Recently it has been shown that CD28 is an important target for PD-1–induced attenuation of T cell signaling (21).
Structure of anti–CTLA-4 and anti–PD-1 antibodies
At present, ipilimumab is the only approved antibody targeting CTLA-4, while three antibodies against PD-1 (nivolumab, pembrolizumab, and cemiplimab) have been approved for various indications, as have three antibodies against PD-L1 (avelumab, durvalumab, and atezolizumab). The success of ICI antibodies in cancer immunotherapy has substantially improved the mechanistic understanding of the regulation of T cell responses, although ICIs targeting negative regulatory molecules are able to elicit durable immune responses only in a subgroup of cancer patients (22). ICIs can cause a variety of cell toxicities, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (23, 24).
Ipilimumab is a human IgG1 monoclonal antibody (mAb) that targets CTLA-4. Ipilimumab induces ADCC, ADCP, and CDC in vitro, and in turn elicits both humoral and cellular responses. IgG1-induced ADCC can be enhanced by defucosylation of the Fc region’s glycan sequences (IgG1fut) (Figure 2B). The IgG4 immunotherapeutic antibodies nivolumab and pembrolizumab are both IgG4S228P. This variant of IgG4, an engineered isotype with a serine-to-proline substitution at position 228 (S228P) in the hinge region, prevents Fab arm exchanges that otherwise frequently occur between IgG4 molecules. In in vitro assays, IgG4 shows reduced ADCC and ADCP and no CDC (25). Thus, these IgG4S228P mAbs mainly block the activation of PD-1 (Figure 2B). Glycosylation of the Fc region of IgGs has major implications for the safety and clinical efficacy of therapeutic antibodies. For example, the enhancement of ADCC is attributed to the increased affinity of non-fucosylated IgG for FcγRIIIa expressed on natural killer (NK) cells (26–28).
CTLA-4 and PD-1 pathways contribute to barrier tolerance
Inflammation at barrier organs (skin, pulmonary epithelium, and gastrointestinal mucosa) is a common toxicity in patients treated with antibodies against CTLA-4 or PD-1/PD-L1 (5). However, differences in the frequency and severity of this inflammation indicate important differences in the regulatory roles of CTLA-4 and PD-1/PD-L1 (5). The majority of irAEs caused by antibodies against CTLA-4 or PD-1/PD-L1 are mild to moderate in severity, such as autoimmune thyroid disease. More rarely these irAEs are serious or occasionally life-threatening, such as severe colitis, pneumonitis, encephalitis, toxic epidermal necrolysis, myocarditis, autoimmune type 1 diabetes (T1D) presenting in diabetic ketoacidosis (DKA), and primary adrenal insufficiency caused by autoimmune adrenalitis (6).
CTLA-4 has a critical role for maintaining immune homeostasis in the gut. Blockade of CTLA-4 with ipilimumab induces inflammation in the colon (colitis) and/or small intestine (enteritis) in a high frequency of patients. Approximately 10% of patients treated with ipilimumab will develop severe, even life-threatening colitis, often within the first two or three cycles of treatment (5, 29–35). The frequency and severity of colitis is dose-dependent, suggesting that we have not yet seen the full effect of complete CTLA-4 inhibition. Once initiated, colitis can rapidly progress over a period of days if left untreated, causing severe dehydration and risking colonic perforation (5, 29–31, 34, 35).
In contrast, while PD-1 and PD-L1 blockade produces mild colonic inflammation at a reasonably high frequency, inflammation that escalates to the point of requiring intervention is uncommon. Severe enterocolitis occurs in fewer than 2% of patients in most clinical trials (5, 32, 33, 35–43). The difference in frequency and severity demonstrates that gastrointestinal mucosal tolerance is dominated by CTLA-4, with PD-1 playing a relatively minor role (5, 32, 33, 36–43). In addition, many patients with mild PD-1–induced colitis can have stable or slowly escalating symptoms over a period of weeks or months, even with continued immunotherapy treatment (5). This period of stable, smoldering inflammation suggests that other regulatory mechanisms compensate to prevent complete loss of mucosal tolerance in these patients, a clear distinction from the rapid evolution of colitis driven by CTLA-4 blockade (5).
Not surprisingly, the homeostatic roles for PD-1 and CTLA-4 do not precisely overlap, as blocking both pathways induces more frequent and more severe enterocolitis than is seen with blockade of either pathway alone (5, 44). The effect is also somewhat synergistic (more than additive), an indication that CTLA-4 and PD-1 have some role in compensating for each other’s absence in the setting of single pathway inhibition, though this degree of synergy is far less than is seen for some other organs, such as the liver (5, 44, 45).
Although CTLA-4 clearly plays a more important role in gut homeostasis than does PD-1/PD-L1, the opposite appears to be true in the lungs. Pneumonitis is more common in patients treated with antibodies that block PD-1 or PD-L1 than in patients treated with ipilimumab (29, 31, 36–41, 46). Differences in the patient populations treated with these drugs likely account for some, though not all, of this difference. PD-1/PD-L1 blockade is approved for the treatment of lung cancer, which has a higher incidence of pneumonitis than other malignancies, while ipilimumab is not. Whether this distinction relates to important differences in the immune inflammatory mechanisms or to the antigenic targets in these two tissues remains to be determined.
Inciting events and antigenic targets
One of the most valuable aspects of studying the mechanisms driving the inflammatory toxicities of ICIs is that the timing of the immune perturbation is well defined. However, presently we have a paucity of data on disease-associated biomarkers following ICI treatment, and still have a rudimentary understanding of the association between irAEs and cancer outcome. Skin reactions appear to be more frequent among patients treated with ICIs who have preexisting rheumatoid factor, whereas thyroid dysfunction seems to be more frequent among patients with preexisting anti-thyroid autoantibodies (47, 48). Hence, organ-specific autoantibodies developing under ICI treatment could be potential biomarkers of ICI toxicity and efficacy, though further data will be required before we can determine whether this is the case.
Patients have a clear start date on therapy, may have biological samples available before treatment, and often have samples during treatment but before emergence of clinical toxicity. This is in direct distinction to patients with spontaneous autoimmune disease, for whom the initial immune perturbation is unknown and may have occurred years or decades before the onset of clinically apparent disease (5). Without a clearly defined disease duration, determining which disease-associated immune changes are driving inflammation and which are consequences of the underlying pathology can be extremely difficult.
ICI-related autoimmune/inflammatory responses
Despite the theoretical advantages of studying irAEs, at present too little is known about the underlying immune pathogenesis of these toxicities to draw mechanistic conclusions about the inciting events. The rapid onset of colitis after CTLA-4 blockade suggests that cells capable of driving colonic inflammation may be present at baseline in many individuals, or at the least that triggers are common and commonly encountered. In contrast, the delayed onset of PD-1 blockade–associated colitis suggests that triggers beyond PD-1 inhibition may play an important role in most patients (5, 32, 33, 49). Colonic infections, alterations in the microbiome, dietary changes, and toxic injuries are all potential factors that could break tolerance and precipitate colitis in patients who initially maintained it in the setting of PD-1 blockade.
The antigenic targets in checkpoint colitis are similarly unclear at present. Based on our current understanding of the mechanism of action of CTLA-4 and PD-1/PD-L1 blockade, antigen-specific T cells are presumed to be the key orchestrators of colitis as they are in driving antitumor responses. Although in theory, self-proteins could be recognized in ICI-induced colitis, the microbial and dietary antigen diversity along the gastrointestinal mucosa provides a prolific source of non-self proteins that could be recognized as well, and these are the more plausible antigenic targets. Preliminary evidence indicates difference in the baseline microbiomes of patients who develop colitis on ICIs compared with those who do not, and in animal models, histopathologic changes associated with CTLA-4 treatment are influenced by microbiota, with Bacteroides and Burkholderiales associated with preserved intestinal architecture and improved antitumor responses (50, 51). This does not directly implicate specific microbial organisms as the targets of T cell–mediated immunity, however, as these differences may reflect differences in dietary or other environmental exposures, or differences in baseline inflammatory states induced by microbial products. Further implicating the microbiome in ICI-colitis, fecal microbiota transplant has been reported to be effective in two patients with colitis refractory to standard treatment (52).
ICI-related autoimmune endocrinopathies
Autoimmune endocrinopathies, including T1D, hypopituitarism, hypothyroidism, hypogonadism, and hypoadrenalism, have emerged as frequent and clinically meaningful adverse events (53, 54). The most common of the reported endocrinopathies with ipilimumab has been thyroid dysfunction (55).
In several studies and in large phase III clinical trials, thyroid dysfunction (hypothyroidism, hyperthyroidism, and thyroiditis) was reported in 6% to 20% of patients (55–57). Myxedema crisis has also been described (58). The majority of patients affected by ICI-induced thyroid dysfunction are asymptomatic, and conservative therapy during the thyrotoxic phase of thyroiditis is usually sufficient. Anti–CTLA-4 and anti–PD-1 usually leads to permanent hypothyroidism after an average of 1 month following the thyrotoxic phase of the disease and 3 months from initiation of immunotherapy, and lifelong thyroid hormone replacement is necessary (54, 58). Interestingly, the risk for thyroid dysfunction may be related to the existence of preexisting autoantibodies (47, 48). Graves’ disease is a rare cause of immunotherapy-related hyperthyroidism generally following the use of ipilimumab. Graves’ ophthalmopathy may also occur (59, 60). High-dose steroids and antithyroid drugs are used successfully to treat this condition (54, 61).
Autoimmune hypophysitis is on the rise, particularly after initiation of ipilimumab treatment (53). An interesting mechanism has been proposed in which CTLA-4 is expressed by normal pituitary cells, and administration of an anti–CTLA-4 mAb activates classic complement pathway, resulting in destruction of pituitary cells (Figure 2A) (23, 53).
Primary adrenal insufficiency caused by autoimmune adrenalitis predominantly occurs in patients affected by melanoma treated with anti–PD-1 immunotherapy (62). The majority of these patients recovered clinically on hydrocortisone replacement therapy and on fludrocortisone if necessary (63). In ICI-induced autoimmune adrenalitis, once therapy with hydrocortisone and fludrocortisone is initiated and the symptoms have resolved, patients should continue maintenance therapy (54, 64, 65). This recommendation is also clearly indicated in recent guidelines for the management of endocrine complications of ICIs (66). The mechanism of ICI-induced autoimmune adrenalitis is unknown.
In an original report of five subjects with diabetes mellitus, aged 55–83 years, their presentation ranged from symptoms of hyperglycemia, such as polyuria, to DKA requiring treatment in the intensive care unit (67). Remarkably, a marked increase of ICI-related autoimmune diabetes was reported, with over 50% of cases reported in 2017. Overall, half of the patients with ICI-related diabetes presented in DKA (50.2%) (56). Although archetypal T1D develops years or even decades after the appearance of islet autoantibodies, autoimmune diabetes induced by ICIs presents weeks to months after initiation of ICI therapy. Islet autoantibodies are frequently present in these patients, though with a prevalence lower than in T1D, and their association with HLA-DQ/DR alleles conferring T1D risk appears to be stronger than in T1D. In ICI-induced T1D, cytotoxic T lymphocytes responding to islet peptides have also been described (68).
Furthermore, there have been several reports describing rapid-onset as well as fulminant T1D, the latter usually characterized by rapid onset and absence of diabetes-related antibodies in patients treated with anti–PD-1/PD-L1 antibodies or, rarely, with ipilimumab (54, 69–73).
A recent study indicates that PD-L1 is specifically upregulated on pancreatic β cells from patients with T1D, and it is induced by both type I and II interferons via interferon regulatory factor 1 (IRF1) (74). This PD-L1 upregulation was correlated with the presence of CD8+ T cells within the islets. A provocative hypothesis is that β cells displaying high PD-L1 expression may resist T cell–mediated apoptosis over a longer period of time and persist despite sustained islet T cell responses, similarly to PD-L1–expressing cancer cells (Figure 2A) (75). Interestingly, in NOD mice, long-term-surviving β cells in the setting of ongoing immune responses express high levels of PD-L1, and blockade of PD-L1 precipitates rapid onset of fulminant diabetes (75). Stamatouli et al. described 27 patients with ICI-induced diabetes. This syndrome has similarities and differences compared with classic T1D (76). There was a predominance of HLA-DR4, which was present in 76% of patients, whereas other HLA alleles associated with high risk of spontaneous T1D were not overrepresented, including HLA-DR3, -DQ2, and -DQ8. Approximately 40% of the patients described by Stamatouli et al. exhibited islet autoantibodies that are found in spontaneous T1D, a prevalence lower than that of islet autoantibodies found in spontaneous T1D (76). A possible explanation of the latter finding is that ICI-induced T cell and autoantibody responses may recognize as-yet unidentified islet autoantigen(s). In the Stamatouli et al. report, more detailed clinical information (serology and HLA-DQ and -DR haplotypes) should have been provided regarding a group of patients with T1D not treated with ICIs. Furthermore, in ICI-induced T1D, random glucagon levels were not reduced, suggesting that α cells were still functioning.
T1D was diagnosed in one patient who developed autoimmune syndrome type 2 (including Addison’s disease and hypophysitis) after treatment with atezolizumab (77). The HLA genotype was DRB1*04 and DQB1*03 haplotype, typically associated with T1D, whereas islet cell antibodies were not detected. Other cases of polyendocrinopathies resulting from ICI treatment have been reported (65, 78). Interestingly, PD-1 inhibitors can induce worsening of preexisting type 2 diabetes (69), and an increase in hemoglobin A1c in nondiabetic patients (34).
Immune-related pancreatic exocrine insufficiency was described in a case series of patients treated with ICIs (79). Interestingly, immune-related pancreatic exocrine insufficiency caused by pancreatitis was linked to pembrolizumab or nivolumab for metastatic melanoma, and a meta-analysis has shown that CTLA-4 inhibitors alone as well as combination treatment of nivolumab and ipilimumab are associated with increased risk of amylase or lipase elevation (80, 81). Overall, these observations suggest an opportunity to identify those at highest risk of ICI-induced autoimmune diseases, which may yield insights into spontaneous autoimmune disease, and improve our ability to treat these irAEs.
Restoration of immune homeostasis
For most irAEs, with the notable exception of the endocrine toxicities, corticosteroids are first-line treatments, including for ICI-induced colitis (5, 32, 33, 82, 83). In the majority of patients, corticosteroids are sufficient to resolve symptoms, although a substantial fraction of patients will require further immune suppression (32, 33, 84). Because of its frequency and severity, treatment and restoration of immune homeostasis is best understood for ICI-induced colitis, though even for this irAE few mechanistic details are known. For patients with ICI-induced colitis who do not respond to corticosteroids, the TNF-α–blocking antibody infliximab is highly effective, implicating TNF-α in the molecular pathogenesis of ICI-induced colitis (5, 32–34, 82, 83, 85). The presumption is that other anti–TNF-α therapies would also be effective, but these drugs have not been evaluated directly. Most patients require one to three doses of infliximab to resolve colitis, underscoring the importance of TNF-α in ICI-induced colitis. Intriguingly, TNF-α may play a role in some cases of ICI-induced diabetes (86). A recent case report described a patient who simultaneously developed ICI-induced colitis and diabetes, and whose diabetes improved after treatment of the colitis with infliximab (86).
The antibody vedolizumab, which blocks the gut-homing integrin α4β7, has also shown efficacy as an alternative to infliximab, or for patients who fail to respond to TNF-α blockade (87, 88). This finding suggests that trafficking of new immune cells from the circulation into the gut is required to perpetuate the inflammatory response in ICI-induced colitis, a finding that is consistent with our understanding of colitis more generally. Additional therapies for refractory ICI-induced colitis have been reported, including immune suppression directed at T cells and borrowed from the transplant experience, such as mycophenolate mofetil and tacrolimus (5).
Recurrence is rare for most irAEs, including colitis, after resolution of the initial inflammation, and chronic inflammatory syndromes do not typically develop, demonstrating that immune homeostasis can be restored in most patients after transient immune receptor disruption (5). Whether long-term changes in immune regulation occur after ICI therapy is not clear, as subtle changes in the risk for spontaneous autoimmune disease would be too difficult to detect with the small populations of treated patients and the relatively short periods of follow-up to date. We also do not know the extent to which immune-suppressive medications such as corticosteroids are necessary in order to achieve restoration of immune tolerance. Patients with severe inflammation are always treated, and many with less severe inflammation, who may present with grade 1 symptoms, are never definitively diagnosed.
Relationship to primary immunodeficiency and inflammatory bowel disease
Inflammatory bowel disease (IBD) is a relatively common spontaneous autoinflammatory disease of the gastrointestinal mucosa that resembles ICI-induced colitis in many aspects (5, 89, 90). Phenotypically, IBD is divided into two diseases: ulcerative colitis (UC) and Crohn’s disease (CD) (5, 89, 90). UC is a mucosal inflammatory disease that affects the colon in a continuous region spreading proximally from the anus. UC does not involve the small intestines (5, 89, 90). CD, in contrast, can involve any part of the gastrointestinal mucosa from the mouth to the anus, and often occurs as discrete segments of inflammation surrounded by tissue that is spared. Inflammation extends from the mucosa to the serosa, leading to complications that are unique to CD, such as deep ulcerations, luminal strictures, and fistulas, which can involve other organs such as the bladder, vagina, or perianal region, and skin (5, 89, 90).
Much like UC, ICI-induced colitis typically involves the gastrointestinal mucosa, and complications such as fistulas are not seen. In addition, inflammation is typically observed in a continuous pattern, with pancolitis as the most common presentation (5, 89, 90). In contrast to sporadic UC, however, involvement of the small intestines is common in ICI-induced colitis (5, 35, 89, 90). The rapid onset of ICI-induced colitis from CTLA-4 blockade is also more reminiscent of an infectious colitis than of either form of IBD. IBD is more often an indolent disease with periods of symptom flares, but it is unusual for a patient to present within the first few days of symptom onset with life-threatening inflammation (5, 89, 90). PD-1/PD-L1 blockade–induced colitis is much more like IBD in this respect. ICI-induced colitis is almost always a monophasic disease, in sharp distinction to IBD, which is nearly uniformly a chronic disease with relapses and remissions (5, 89, 90). Histopathologically, the chronic nature of IBD is associated with architectural distortion in the colonic epithelium, a feature rarely seen in ICI-induced colitis (5, 32, 33, 35, 89, 90).
The importance of CTLA-4, PD-1, and PD-L1 in the immune pathophysiology of IBD itself is presently unclear. A multicenter retrospective cohort of patients including more than 100 patients with underlying IBD who were treated with CTLA-4 or PD-(L)1 blockade was recently reported, adding to several smaller case series (91–94). In a cohort that included both patients with UC and with CD, the gastrointestinal adverse event rate was 41% compared with 11% in a control cohort, implicating these immune checkpoints in maintenance of IBD remission (94). The risk of a flare was unrelated to the type of underlying IBD (CD versus UC), but showed a trend toward an increased risk associated with blockade of CTLA-4. All of the currently reported studies are retrospective, however, and based on the data available, the patients were disproportionately likely to have quiescent disease, with many off of all therapy and without symptoms (94). Indeed, several of the patients with UC had had prior colectomies, a functional cure (91–94).
Although neither PD-1 nor PD-L1 genetic deficiency has been reported in patients, CTLA-4 haploinsufficiency is now well described. Patients with monoallelic loss of function of CTLA-4 develop a multiorgan inflammatory syndrome called CTLA-4 haploinsufficiency with autoimmune infiltration (CHAI) (95–97). This rare disease impairs normal regulation of the immune system and results in excessive numbers of lymphocytes and autoimmunity (95–97). Mucosal inflammation is common in CHAI patients, with variable severity (95–97). CHAI can be treated with immune suppression, but it responds extremely well to CTLA-4 Ig or abatacept (95–97). Abatacept is likely to reverse many, if not all, of the ongoing inflammation observed in patients treated with CTLA-4 blockade (95–97). While potentially useful in cases of life-threatening and refractory inflammation induced by CTLA-4 blockade, abatacept is also likely to reverse the therapeutic antitumor effect, presenting a powerful downside to its use.
Effects of immune checkpoint blockade on tumor cell microenvironment
ICIs have dramatically improved treatment outcomes of a variety of aggressive tumors. Although approximately 50% of cancer patients benefit from immune checkpoint blockade, a subset develop autoimmune disorders that can be life-threatening.
Mounting evidence indicates that elevated PD-L1 expression may be associated with response to immunotherapy for many malignancies. In particular, in patients with advanced non–small cell lung cancer, PD-L1 expression in at least 50% of tumor cells correlated with improved efficacy of pembrolizumab (36). High levels of PD-L1 expression by tumor cells suggests immune evasion by the tumor, and, in essence, this leaves tumor cells vulnerable to PD-1/PD-L1 blockade (Figure 3). Interestingly, PD-L1 appears to be specifically upregulated on pancreatic β cells from patients with T1D, and it is induced by both type I and II interferons via interferon regulatory factor 1 (IRF1) (Figure 3) (74, 98, 99). Thus, the enhancement of effector T cell function with PD-1/PD-L1 blockade results in tumor cell killing and response to immunotherapy, as well as hypothetically causing irreversible bystander β cell damage in pancreatic β cells overexpressing PD-L1, and possibly neoantigens (100), microbial antigens, or antigens shared by tumor cells and normal cells (101). These mechanisms may apply to all inflammatory disorders precipitated by immunotherapy.
Figure 3. Neoantigens and dendritic cell and CD8+ T cell activation at the tumor site following checkpoint blockade immunotherapy.

CD8+ T cells are the primary effectors of antitumor immune responses, though other immune cell types (i.e., CD4+ T cells) are also involved. Middle: Dendritic cells (DCs) are activated by neoantigens from the tumor. Dead and dying tumor cells release damage-associated molecular patterns (DAMPs; e.g., heat shock proteins, ATP, nucleic acids) that can also activate DCs. Left: The activated DCs travel to lymph nodes, whereby they present MHC class I–bound neoantigens to naive CD8+ T cells. HLA class I genotype can influence cancer response to checkpoint blockade immunotherapy (107). TCRs binding to the MHC class I–bound neoantigen along with B7-CD28 binding results in the activation of CD8+ T cells specific for the neoantigen. Right: Cytotoxic CD8+ T cells traffic to the tumor site following a chemokine signal (e.g., CXCL9/10 secretion binding to CXCR3 on the T cells). At the tumor site, TCR binding to MHC class I–bound neoantigens to tumor cells has two outcomes: First, it induces IFN-γ secretion, which is bound by IFN-γ receptors in nearby tumor and normal cells, leading to upregulation of MHC class I antigen presentation in those cells. In tumor cells, this facilitates further TCR engagement and cytotoxic activity. Concurrently, IFN-γ also induces PD-L1 expression. Second, it leads to T cell activation and tumor killing through Fas/FasL apoptotic signaling, granzyme and perforin secretion, and direct cell membrane lysis.
Many immunotherapeutic approaches to cancer take advantage of the effects of tumor neoantigens. Nonetheless, their role in the generation of organ-specific autoimmune responses triggered by ICI remains to be elucidated. Tumor neoantigens derived from gene fusions are considered highly immunogenic and can mediate robust responses to immunotherapy (102). These findings highlight an essential class of tumor-specific antigens and have implications for targeting gene fusion events in cancers that would otherwise be less poised for response to immunotherapy (Table 2). Furthermore, the higher likelihood of tumors with DNA mismatch repair deficiency to respond to immunotherapy is thought to be due to their higher tumor mutation burden (neoantigens) (Table 3) (103).
Table 2. Major ICI-induced irAEs.

Table 3. Tumor neoantigens and mechanisms/biomarkers of immune responses in general, and response to immunotherapy.
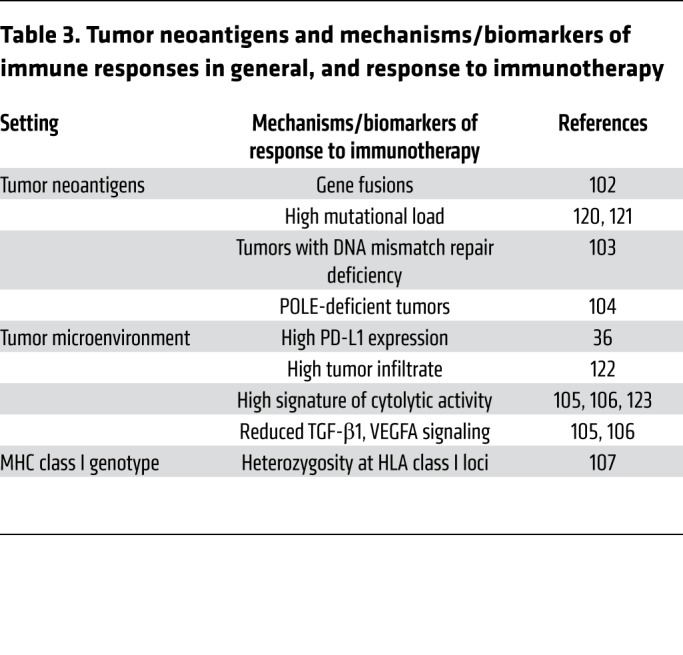
Mutations in POLE, a polymerase involved in DNA replication, lead to even higher mutational burden in tumors and have been associated with excellent response to immunotherapy (104). Other described mechanisms/markers associated with response to immunotherapy include downregulation of TGF-β1 signaling (105), VEGFA secretion (106), and a specific HLA class I genotype (102, 107).
Concluding remarks
IrAEs represent a clinical challenge that can reduce the benefits of antitumor immunotherapy by limiting the treatment dose and duration, and in some cases by preventing safe treatment as a result of underlying autoimmunity or transplant status. But these irAEs also provide, on a more fundamental level, important insights into basic immunobiology. The irAEs represent the phenotype of loss of function of checkpoint receptors in humans and thus reveal the regulatory role of the targeted receptors in maintaining immune homeostasis, an important complement to true loss-of-function mutations in patients. In genetic loss of function, effects on immune system development are difficult to disentangle from effects on mature immune cells, while irAEs specifically teach us about the roles of these receptors in immune homeostasis after otherwise normal development.
The lessons learned from a detailed understanding of irAEs will likely provide important clues into autoimmune diseases and potentially critical insights toward the development of new treatments. Spontaneous autoimmune diseases progress over an unclear period of time, with rarely identified initial events. This means that we often study the manifestations of autoimmune disease without knowing which of the observed changes are the key drivers of the response, and which are the consequences of the true immune pathology. The irAEs induced by ICIs clarify the relationship between the manifestations of the disease and the immune perturbation. We know precisely what the initial disruption in immune homeostasis was, and when that disruption began.
This may well lead to the identification of new treatment targets, in addition to deepening our understanding of autoimmunity. Potential treatment targets include immune populations; signaling pathways of various ICIs targeting negative immune receptors (108); and secreted factors that are expanded/amplified at the initiation of the inflammatory toxicity and diminish during disease resolution, but that may not be apparent in the tumor microenvironment.
The treatment of most autoimmune diseases must balance immune suppression with the need to provide protective immunity from pathogenic microorganisms. Similarly, immune-suppressive treatment for irAEs must balance the effects of these suppressive treatments on antitumor immunity. Yet currently, our understanding of the relationship between effective antitumor immunity and the mechanisms driving irAEs is rudimentary. We have evidence that productive antitumor responses can occur in patients treated with corticosteroids, but we also have evidence that corticosteroids may reduce the full effectiveness of checkpoint blockade (109–111). Mounting evidence suggests that irAEs may correlate with more effective antitumor responses, yet treatment of these irAEs may limit the scope of that benefit (109, 112–114). Innate inflammatory cytokines such as TNF-α are clear drivers of some irAEs, and evidence has implicated these factors in tumor promotion, making these cytokines potentially attractive treatment targets, though definitive evidence that such an approach will be beneficial in humans is lacking (115, 116).
Checkpoint blockade targeting CTLA-4 and PD-1/PD-L1 has already taught us critical lessons about the regulation of immunity and autoimmunity in humans. These immunotherapies are only the first in a probable long line of conceptually similar medications that will target the full array of regulatory pathways that modulate immune responses, and we stand to learn equally important lessons from the irAEs induced by these next-generation therapeutics (22). This will include agents targeting alternate immune checkpoints such as TIM-3 or LAG3, but it will also include blockade of innate immune-regulatory pathways such as CD47 and drugs targeting specific regulatory cells such as Tregs or myeloid-derived suppressor cells (22). The development of these next-generation agents, including combination treatments, will undoubtedly pose a considerable clinical challenge for irAE management, but at the same time will further expand our understanding of immune regulation and may deepen our understanding of autoimmunity.
Acknowledgments
This work was supported by the McNair Medical Institute at the Robert and Janice McNair Foundation (to MP), the American Gastroenterological Association Research Scholars Award (to MD), and NIH/National Institute of Diabetes and Digestive and Kidney Diseases grant 1K08 DK114563-01 (to MD).
Version 1. 01/02/2020
Print issue publication
Footnotes
Conflict of interest: MD receives research funding from Novartis and consulting fees from Genentech-Roche, Tillotts, and Partner Therapeutics.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(1):51–61.https://doi.org/10.1172/JCI131194.
Contributor Information
Michael Dougan, Email: Michael_Dougan@dfci.harvard.edu.
Massimo Pietropaolo, Email: Massimo.Pietropaolo@bcm.edu.
References
- 1.Wei SC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 3.Wei SC, et al. Negative co-stimulation constrains T cell differentiation by imposing boundaries on possible cell states. Immunity. 2019;50(4):1084–1098.e10. doi: 10.1016/j.immuni.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young A, Quandt Z, Bluestone JA. The balancing act between cancer immunity and autoimmunity in response to immunotherapy. Cancer Immunol Res. 2018;6(12):1445–1452. doi: 10.1158/2326-6066.CIR-18-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dougan M. Checkpoint blockade toxicity and immune homeostasis in the gastrointestinal tract. Front Immunol. 2017;8:1547. doi: 10.3389/fimmu.2017.01547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang DY, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018;4(12):1721–1728. doi: 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rieux-Laucat F, Casanova JL. Immunology. Autoimmunity by haploinsufficiency. Science. 2014;345(6204):1560–1561. doi: 10.1126/science.1260791. [DOI] [PubMed] [Google Scholar]
- 8.Paterson AM, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med. 2015;212(10):1603–1621. doi: 10.1084/jem.20141030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stumpf M, Zhou X, Bluestone JA. The B7-independent isoform of CTLA-4 functions to regulate autoimmune diabetes. J Immunol. 2013;190(3):961–969. doi: 10.4049/jimmunol.1201362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1(9):793–801. doi: 10.1016/S1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 12.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185(3):393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lanier LL, et al. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154(1):97–105. [PubMed] [Google Scholar]
- 14.Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21(3):401–413. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 15.Pagès F, et al. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369(6478):327–329. doi: 10.1038/369327a0. [DOI] [PubMed] [Google Scholar]
- 16.Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2(1):37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- 17.Agata Y, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 18.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 19.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 21.Hui E, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dougan M, Dranoff G, Dougan SK. Cancer immunotherapy: beyond checkpoint blockade. Annu Rev Cancer Biol. 2019;3:55–75. doi: 10.1146/annurev-cancerbio-030518-055552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Transl Med. 2014;6(230):230ra45. doi: 10.1126/scitranslmed.3008002. [DOI] [PubMed] [Google Scholar]
- 24.Romano E, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112(19):6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846–856. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 26.Kanda Y, et al. Establishment of a GDP-mannose 4,6-dehydratase (GMD) knockout host cell line: a new strategy for generating completely non-fucosylated recombinant therapeutics. J Biotechnol. 2007;130(3):300–310. doi: 10.1016/j.jbiotec.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 27.Mori K, et al. Non-fucosylated therapeutic antibodies: the next generation of therapeutic antibodies. Cytotechnology. 2007;55(2–3):109–114. doi: 10.1007/s10616-007-9103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamane-Ohnuki N, Satoh M. Production of therapeutic antibodies with controlled fucosylation. MAbs. 2009;1(3):230–236. doi: 10.4161/mabs.1.3.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 31.Eggermont AM, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845–1855. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geukes Foppen MH, et al. Immune checkpoint inhibition-related colitis: symptoms, endoscopic features, histology and response to management. ESMO Open. 2018;3(1):e000278. doi: 10.1136/esmoopen-2017-000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, et al. Endoscopic and histologic features of immune checkpoint inhibitor-related colitis. Inflamm Bowel Dis. 2018;24(8):1695–1705. doi: 10.1093/ibd/izy104. [DOI] [PubMed] [Google Scholar]
- 34.Gauci ML, et al. Checkpoint inhibitor treatment induces an increase in HbA1c in nondiabetic patients. Melanoma Res. 2019;29(3):328–332. doi: 10.1097/CMR.0000000000000585. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ML, Neyaz A, Patil D, Chen J, Dougan M, Deshpande V. Immune-related adverse events in the gastrointestinal tract: diagnostic utility of upper gastrointestinal biopsies. Histopathology. doi: 10.1111/his.13963. doi: 10.1111/his.13963. [published online ahead of print July 30, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 37.Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 38.Robert C, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 39.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carbone DP, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415–2426. doi: 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 42.Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 43.Socinski MA, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–2301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 44.Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds K, Thomas M, Dougan M. Diagnosis and management of hepatitis in patients on checkpoint blockade. Oncologist. 2018;23(9):991–997. doi: 10.1634/theoncologist.2018-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eggermont AMM, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378(19):1789–1801. doi: 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- 47.Toi Y, et al. Profiling preexisting antibodies in patients treated with anti-PD-1 therapy for advanced non-small cell lung cancer. JAMA Oncol. 2019;5(3):376–383. doi: 10.1001/jamaoncol.2018.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Moel EC, et al. Autoantibody development under treatment with immune-checkpoint inhibitors. Cancer Immunol Res. 2019;7(1):6–11. doi: 10.1158/2326-6066.CIR-18-0245. [DOI] [PubMed] [Google Scholar]
- 49.Wang DY, et al. Clinical characterization of colitis arising from anti-PD-1 based therapy. Oncoimmunology. 2019;8(1):e1524695. doi: 10.1080/2162402X.2018.1524695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dubin K, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391. doi: 10.1038/ncomms10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis [published correction appears in Nat Med. 2019;25(1):188]. Nat Med. 2018;24(12):1804–1808. doi: 10.1038/s41591-018-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy — immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13(4):195–207. doi: 10.1038/nrendo.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan MH, et al. Spectrum of immune checkpoint inhibitors-induced endocrinopathies in cancer patients: a scoping review of case reports. Clin Diabetes Endocrinol. 2019;5:1. doi: 10.1186/s40842-018-0073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puzanov I, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5(1):95. doi: 10.1186/s40425-017-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright JJ, et al. Increased reporting of immune checkpoint inhibitor-associated diabetes. Diabetes Care. 2018;41(12):e150–e151. doi: 10.2337/dc18-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51(3):145–156. doi: 10.1055/a-0843-3366. [DOI] [PubMed] [Google Scholar]
- 58.Khan U, Rizvi H, Sano D, Chiu J, Hadid T. Nivolumab induced myxedema crisis. J Immunother Cancer. 2017;5:13. doi: 10.1186/s40425-017-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borodic G, Hinkle DM, Cia Y. Drug-induced graves disease from CTLA-4 receptor suppression. Ophthalmic Plast Reconstr Surg. 2011;27(4):e87–e88. doi: 10.1097/IOP.0b013e3181ef72a1. [DOI] [PubMed] [Google Scholar]
- 60.Sagiv O, et al. Extraocular muscle enlargement and thyroid eye disease-like orbital inflammation associated with immune checkpoint inhibitor therapy in cancer patients. Ophthalmic Plast Reconstr Surg. 2019;35(1):50–52. doi: 10.1097/IOP.0000000000001161. [DOI] [PubMed] [Google Scholar]
- 61.Dadu R, Zobniw C, Diab A. Managing adverse events with immune checkpoint agents. Cancer J. 2016;22(2):121–129. doi: 10.1097/PPO.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 62.Hanna RM, et al. Acute kidney injury after pembrolizumab-induced adrenalitis and adrenal insufficiency. Case Rep Nephrol Dial. 2018;8(2):171–177. doi: 10.1159/000491631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Min L, Ibrahim N. Ipilimumab-induced autoimmune adrenalitis. Lancet Diabetes Endocrinol. 2013;1(3):e15. doi: 10.1016/S2213-8587(13)70031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trainer H, Hulse P, Higham CE, Trainer P, Lorigan P. Hyponatraemia secondary to nivolumab-induced primary adrenal failure. Endocrinol Diabetes Metab Case Rep. 2016;2016:16-0108. doi: 10.1530/EDM-16-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paepegaey AC, et al. Polyendocrinopathy resulting from pembrolizumab in a patient with a malignant melanoma. J Endocr Soc. 2017;1(6):646–649. doi: 10.1210/js.2017-00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Higham CE, et al. Society for Endocrinology Endocrine Emergency Guidance: acute management of the endocrine complications of checkpoint inhibitor therapy. Endocr Connect. 2018;7(7):G1–G7. doi: 10.1530/EC-18-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hughes J, et al. Precipitation of autoimmune diabetes with anti-PD-1 immunotherapy. Diabetes Care. 2015;38(4):e55–e57. doi: 10.2337/dc14-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akturk HK, Michels AW. Adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(12):1163–1164. doi: 10.1056/NEJMc1801663. [DOI] [PubMed] [Google Scholar]
- 69.Kotwal A, Haddox C, Block M, Kudva YC. Immune checkpoint inhibitors: an emerging cause of insulin-dependent diabetes. BMJ Open Diabetes Res Care. 2019;7(1):e000591. doi: 10.1136/bmjdrc-2018-000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shiba M, et al. Fulminant type 1 diabetes mellitus accompanied by positive conversion of anti-insulin antibody after the administration of anti-CTLA-4 antibody following the discontinuation of anti-PD-1 antibody. Intern Med. 2018;57(14):2029–2034. doi: 10.2169/internalmedicine.9518-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, Michels AW. Immune checkpoint inhibitor-induced type 1 diabetes: a systematic review and meta-analysis. Diabet Med. 2019;36(9):1075–1081. doi: 10.1111/dme.14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. Osaka IDDM Study Group. N Engl J Med. 2000;342(5):301–307. doi: 10.1056/NEJM200002033420501. [DOI] [PubMed] [Google Scholar]
- 73.Tsiogka A, Jansky GL, Bauer JW, Koelblinger P. Fulminant type 1 diabetes after adjuvant ipilimumab therapy in cutaneous melanoma. Melanoma Res. 2017;27(5):524–525. doi: 10.1097/CMR.0000000000000384. [DOI] [PubMed] [Google Scholar]
- 74.Colli ML, et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and -γ via IRF1 induction. EBioMedicine. 2018;36:367–375. doi: 10.1016/j.ebiom.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β Cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab. 2017;25(3):727–738. doi: 10.1016/j.cmet.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stamatouli AM, et al. Collateral damage: insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes. 2018;67(8):1471–1480. doi: 10.2337/dbi18-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lanzolla G, Coppelli A, Cosottini M, Del Prato S, Marcocci C, Lupi I. Immune checkpoint blockade anti-PD-L1 as a trigger for autoimmune polyendocrine syndrome. J Endocr Soc. 2019;3(2):496–503. doi: 10.1210/js.2018-00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akturk HK, Alkanani A, Zhao Z, Yu L, Michels AW. PD-1 inhibitor immune-related adverse events in patients with preexisting endocrine autoimmunity. J Clin Endocrinol Metab. 2018;103(10):3589–3592. doi: 10.1210/jc.2018-01430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eshet Y, et al. Clinical significance of pancreatic atrophy induced by immune-checkpoint inhibitors: a case-control study. Cancer Immunol Res. 2018;6(12):1453–1458. doi: 10.1158/2326-6066.CIR-17-0659. [DOI] [PubMed] [Google Scholar]
- 80.Su Q, Zhang XC, Zhang CG, Hou YL, Yao YX, Cao BW. Risk of immune-related pancreatitis in patients with solid tumors treated with immune checkpoint inhibitors: systematic assessment with meta-analysis. J Immunol Res. 2018;2018:1027323. doi: 10.1155/2018/1027323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hofmann L, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190–209. doi: 10.1016/j.ejca.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 82.Haanen JBAG, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl 4):iv119–iv142. doi: 10.1093/annonc/mdx225. [DOI] [PubMed] [Google Scholar]
- 83.Brahmer JR, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;36(17):1714–1768. doi: 10.1200/JCO.2017.77.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beck KE, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283–2289. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Badran YR, Cohen JV, Brastianos PK, Parikh AR, Hong TS, Dougan M. Concurrent therapy with immune checkpoint inhibitors and TNFα blockade in patients with gastrointestinal immune-related adverse events. J Immunother Cancer. 2019;7(1):226. doi: 10.1186/s40425-019-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trinh B, Donath MY, Läubli H. Successful treatment of immune checkpoint inhibitor-induced diabetes with infliximab. Diabetes Care. 2019;42(9):e153–e154. doi: 10.2337/dc19-0908. [DOI] [PubMed] [Google Scholar]
- 87.Abu-Sbeih H, et al. Outcomes of vedolizumab therapy in patients with immune checkpoint inhibitor-induced colitis: a multi-center study. J Immunother Cancer. 2018;6(1):142. doi: 10.1186/s40425-018-0461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergqvist V, et al. Vedolizumab treatment for immune checkpoint inhibitor-induced enterocolitis. Cancer Immunol Immunother. 2017;66(5):581–592. doi: 10.1007/s00262-017-1962-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361(21):2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62(10):1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnson DB, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol. 2016;2(2):234–240. doi: 10.1001/jamaoncol.2015.4368. [DOI] [PubMed] [Google Scholar]
- 92.Leonardi GC, et al. Safety of programmed death-1 pathway inhibitors among patients with non-small-cell lung cancer and preexisting autoimmune disorders. J Clin Oncol. 2018;36(19):1905–1912. doi: 10.1200/JCO.2017.77.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Menzies AM, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28(2):368–376. doi: 10.1093/annonc/mdw443. [DOI] [PubMed] [Google Scholar]
- 94.Hamzah AS, et al. Immune checkpoint inhibitor therapy in patients with preexisting inflammatory bowel disease [published online December 4, 2019] J Clin Oncol. 2019;37 doi: 10.1200/JCO.19.01674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeissig S, et al. Early-onset Crohn’s disease and autoimmunity associated with a variant in CTLA-4. Gut. 2015;64(12):1889–1897. doi: 10.1136/gutjnl-2014-308541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–1627. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Atkinson MA, et al. How does type 1 diabetes develop? The notion of homicide or β-cell suicide revisited. Diabetes. 2011;60(5):1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Newby BN, Mathews CE. Type I interferon is a catastrophic feature of the diabetic islet microenvironment. Front Endocrinol (Lausanne) 2017;8:232. doi: 10.3389/fendo.2017.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.James EA, Pietropaolo M, Mamula MJ. Immune recognition of β-cells: neoepitopes as key players in the loss of tolerance. Diabetes. 2018;67(6):1035–1042. doi: 10.2337/dbi17-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Friday RP, Pietropaolo M. Oncogenic autoimmunity. Eisenbarth GS, ed. Molecular Mechanisms of Endocrine and Organ-Specific Autoimmunity. Georgetown, Texas, USA: R.G. Landes Co.; 1999:63–84. [Google Scholar]
- 102.Yang W, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25(5):767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johanns TM, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6(11):1230–1236. doi: 10.1158/2159-8290.CD-16-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mariathasan S, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen PL, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chowell D, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davar D, Zarour HM. Immunological targets for immunotherapy: inhibitory T cell receptors. Methods Mol Biol. 2020;2055:23–60. doi: 10.1007/978-1-4939-9773-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Faje AT, et al. High-dose glucocorticoids for the treatment of ipilimumab-induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer. 2018;124(18):3706–3714. doi: 10.1002/cncr.31629. [DOI] [PubMed] [Google Scholar]
- 110.Horvat TZ, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33(28):3193–3198. doi: 10.1200/JCO.2015.60.8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arbour KC, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36(28):2872–2878. doi: 10.1200/JCO.2018.79.0006. [DOI] [PubMed] [Google Scholar]
- 112.Wang Y, et al. Immune-checkpoint inhibitor-induced diarrhea and colitis in patients with advanced malignancies: retrospective review at MD Anderson. J Immunother Cancer. 2018;6(1):37. doi: 10.1186/s40425-018-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Teulings HE, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33(7):773–781. doi: 10.1200/JCO.2014.57.4756. [DOI] [PubMed] [Google Scholar]
- 114.Molina GE, et al. Prognostic implications of co-occurring dermatologic and gastrointestinal toxicity from immune checkpoint inhibition therapy for advanced malignancies: a retrospective cohort study. J Am Acad Dermatol. doi: 10.1016/j.jaad.2019.07.049. doi: 10.1016/j.jaad.2019.07.049. [published online ahead of print July 23, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ridker PM, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 116.Bertrand F, et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. 2017;8(1):2256. doi: 10.1038/s41467-017-02358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arnaud-Coffin P, et al. A systematic review of adverse events in randomized trials assessing immune checkpoint inhibitors. Int J Cancer. 2019;145(3):639–648. doi: 10.1002/ijc.32132. [DOI] [PubMed] [Google Scholar]
- 118.Mahmood SS, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. 2018;71(16):1755–1764. doi: 10.1016/j.jacc.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wolchok JD, Chiarion-Sileni V, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]