

Abstract
Recent occurrences of filoviruses and the arenavirus Lassa virus (LASV) in overlapping endemic areas of Africa highlight the need for a prophylactic vaccine that would confer protection against all of these viruses that cause lethal hemorrhagic fever (HF). We developed a quadrivalent formulation of VesiculoVax that contains recombinant vesicular stomatitis virus (rVSV) vectors expressing filovirus glycoproteins and that also contains a rVSV vector expressing the glycoprotein of a lineage IV strain of LASV. Cynomolgus macaques were vaccinated twice with the quadrivalent formulation, followed by challenge 28 days after the boost vaccination with each of the 3 corresponding filoviruses (Ebola, Sudan, Marburg) or a heterologous contemporary lineage II strain of LASV. Serum IgG and neutralizing antibody responses specific for all 4 glycoproteins were detected in all vaccinated animals. A modest and balanced cell-mediated immune response specific for the glycoproteins was also detected in most of the vaccinated macaques. Regardless of the level of total glycoprotein-specific immune response detected after vaccination, all immunized animals were protected from disease and death following lethal challenges. These findings indicate that vaccination with attenuated rVSV vectors each expressing a single HF virus glycoprotein may provide protection against those filoviruses and LASV most commonly responsible for outbreaks of severe HF in Africa.
Keywords: Vaccines, Virology
Keywords: Adaptive immunity

Introduction
Viral hemorrhagic fevers are a group of infectious diseases with symptoms that are similar among humans (1). The causative viruses affect the vascular system and can cause a hemorrhagic fever (HF) characterized by edema, hypotension, shock, and multiorgan failure. HF viruses include members of the Arenaviridae, Nairoviridae, Hantaviridae, Phenuiviridae, Filoviridae, and Flaviviridae families. Among these HF viruses, members of 2 genera of filoviruses, Ebolavirus and Marburgvirus, and the arenavirus Lassa virus (LASV), are among the deadliest emerging viruses affecting human health in Africa (2).
The 2013–2016 West African epidemic caused by Zaire ebolavirus (EBOV) was responsible for more than 28,000 cases and 11,000 deaths, primarily in Guinea, Sierra Leone, and Liberia (3). The current EBOV outbreak in the Democratic Republic of Congo, which began in August 2018, has caused 2108 cases and 1411 deaths to date and has spilled over into Uganda (4). LASV is constantly emerging in these and surrounding West African countries, with an estimate of more than 500,000 cases of Lassa fever and approximately 5000 deaths annually (5). A current LASV outbreak in Nigeria, which began in 2018, has caused 5821 suspected cases and 295 deaths in the 1187 confirmed cases (6).
In addition to causing morbidity and mortality as naturally acquired infections, filoviruses and LASV are also categorized as Category A priority pathogens by several US government agencies because of the concern for deliberate misuse and lack of effective medical countermeasures. Filoviruses and LASV were also recently listed on the World Health Organization’s (WHO) 2018 Blueprint list of priority pathogens (2). While there are no licensed filovirus or LASV vaccines for human use, several filovirus vaccines all directed against one specific filovirus, EBOV, were used in phase II/III clinical trials in Africa during the 2013–2016 EBOV epidemic (7, 8), and are being employed in the current EBOV outbreak in the Democratic Republic of Congo (9). However, there have been no phase II/III clinical trials of any vaccine for any other filovirus or LASV in endemic areas.
A number of vaccines expressing filovirus glycoproteins (GPs) have demonstrated protective efficacy in nonhuman primate (NHP) models, including recombinant human (rAd5, rAd36, and rAd35) and chimpanzee (rChAd3) adenoviruses, plasmid DNA (pDNA), modified vaccinia virus Ankara (MVA), virus-like particles (VLPs), alphavirus replicons, recombinant human parainfluenza virus 3 (rHPIV 3), recombinant rabies virus (rRV), and recombinant vesicular stomatitis virus (rVSV) (10–12). Of these vaccine vectors, recombinant adenovirus–, VLP-, and rVSV-based vaccines have demonstrated the ability to completely protect NHPs against Marburg virus (MARV) and multiple species of ebolaviruses, including both EBOV and Sudan ebolavirus (SUDV) (13–17). Two major rVSV vector designs have been explored and shown to be equally protective in filovirus vaccines: a rVSV vector where VSV G is directly replaced by the filovirus GP (rVSVΔG GP) (10–12), and a more attenuated rVSV vector that expresses the filovirus GP from an additional transcriptional unit and which is highly attenuated due to translocation of the N gene and truncation of the VSV G cytoplasmic tail (N4CT1) (18).
In contrast to filoviruses, there have been very few preclinical studies in NHPs assessing candidate vaccines against LASV. Three vaccines expressing the LASV glycoprotein precursor (GPC) have shown complete protection in NHPs against the prototype lineage IV Josiah strain of LASV. These include a recombinant vaccine based on the related Mopeia arenavirus (19), a DNA vaccine (20), and a rVSVΔG-based vaccine (21, 22). Importantly, no LASV vaccine has been evaluated in NHPs against the lineage II or III LASVs currently circulating in Nigeria or any other lineage of LASV.
Here, we describe the development of a new quadrivalent VesiculoVax vaccine consisting exclusively of highly attenuated rVSV vectors that express the glycoproteins of EBOV, SUDV, MARV, and LASV, respectively, and report the immunogenicity and protective efficacy of this vaccine formulation in NHPs challenged with all 4 viral pathogens.
Results
Quadrivalent vaccine design and study strategy.
Our quadrivalent VesiculoVax vaccine is made of 3 live, attenuated, replication-competent rVSV vectors that express the EBOV (Kikwit strain), SUDV (Boniface strain), and MARV (Angola strain) glycoproteins, respectively, plus 1 live, attenuated, replication-competent rVSV vector encoding the LASV (Josiah strain) surface glycoprotein (Figure 1, A and B). Both rVSV vector designs share a key attenuating feature, a translocation of the VSV N gene from the first to the fourth position in the VSV genome. The filovirus rVSV vaccine candidates also express VSV G containing a 28 amino acid truncation from the cytoplasmic tail, which further attenuates the vector. We previously demonstrated that the combination of these 2 attenuating changes, henceforth referred to as N4CT1, resulted in a well-tolerated and immunogenic vaccine vector against HIV-1 in phase I clinical testing (23). By contrast, our LASV rVSV vaccine candidate lacks VSV G completely, referred to as N4ΔG, and therefore the LASV GP becomes the only viral glycoprotein involved in viral entry and particle formation for this construct. This approach was chosen because stable expression of both VSV G CT1 and LASV GPC during virus amplification could not be achieved (data not shown). Compared with their native nucleotide sequence the genes expressing the filovirus glycoproteins were minimally altered to introduce silent mutations for cloning purposes and to avoid sequence similarities with motifs important for VSV transcription. The LASV GPC nucleotide sequence was mammalian codon-optimized to ensure adequate GPC expression for viral growth and immunogenicity within the context of the rVSV N4ΔG backbone. All glycoprotein genes were inserted in position 1 of the viral genome, immediately adjacent to the 3′ transcription promoter, in order to maximize glycoprotein expression. Expression of LASV glycoprotein was demonstrated by Western blot analysis (Figure 1C). Filovirus glycoprotein expression was demonstrated in a previous report (18). rVSVN4CT1 expressing HIV-1 gag protein was used as a nonspecific control vaccine. A total of 20 cynomolgus macaques were vaccinated by intramuscular (i.m.) injection with the quadrivalent rVSV N4CT1-EBOV / SUDV / MARV / rVSV N4ΔG LASV vaccine formulation containing 1 × 107 PFU of each vector, for a total dose of 4 × 107 PFU/macaque (Figure 1D). Animals were given a boost vaccination with the quadrivalent vaccine 56 days after the prime vaccination. Twelve macaques were vaccinated in parallel with an equivalent dose of rVSV N4CT1-HIVgag as a study control. All animals were divided into 4 challenge arms, each including 5 active (received quadrivalent vaccine) and 3 placebos (received control vaccine).
Figure 1. Quadrivalent vaccine design and study strategy.

(A) Genetic organization of attenuated rVSV quadrivalent vaccine vectors. Vectors of the rVSV N4CT1-FiloGP design contained a filovirus GP gene(s) in position 1, an N gene translocation (N4), and a truncated G gene (CT1). (B) The rVSV N4ΔG-LASVGPC vector design contained the LASV GPC gene in position 1, an N gene translocation (N4), and a deletion of the VSV G gene. (C) Expression of the LASV GP in virus-infected cell lysates by Western blot analysis. (D) Flow chart showing the days of vaccination (triangles) and days of sampling for immunogenicity study (arrows). Day 28 samples were only taken from SUDV and MARV arms. *Day of challenge.
Immunogenicity of quadrivalent vaccine in macaques.
Blood samples were drawn immediately before dosing from all 32 macaques on the day of prime vaccination, at days 10 and 28 after prime vaccination, immediately before dosing on the day of boost vaccination, and at day 10 after boost vaccination to assess cellular and/or humoral immune responses elicited against each GP protein. The humoral immune response elicited by each vector was assessed by measuring serum GP–specific IgG binding antibody by ELISA (Figure 2) and filovirus or LASV GP–specific neutralization activity by a 50% fluorescence cell reduction neutralization (FRNT50) test (Figure 3).
Figure 2. Filovirus and LASV GP–specific IgG ELISA titers in macaques immunized with the quadrivalent vaccine.

At study days 0 and 56, macaques were immunized i.m. with 4 × 107 PFU of the quadrivalent vaccine (black circles) or rVSV N4CT1-HIVgag as control (gray squares). A total of 20 vaccinated animals and 12 control animals are represented, except as noted. At study days 0, 10, 28 (only from SUDV and MARV challenge monkeys), 56, and 66, sera were collected and tested for (A) LASV GP–, (B) EBOV GP–, (C) SUDV GP–, and (D) MARV GP–specific IgG responses by ELISA. Data represent the geometric mean of log-transformed LASV and filovirus GP–specific IgG endpoint titer, with the 95% confidence interval. An unpaired 2-tailed t test was used to determine statistically significant differences (*P <0.05, **P < 0.005, ***P < 0.0005) between quadrivalent and control groups as indicated. Mean values and standard deviations are depicted in red.
Figure 3. Filovirus and LASV GP–specific neutralization antibody responses in macaques immunized with the quadrivalent vaccine.

At study days 0 and 56, macaques were immunized i.m. with 4 × 107 PFU of the quadrivalent vaccine (black circles) or rVSV N4CT1-HIVgag as control (gray squares). A total of 20 vaccinated animals and 12 control animals are represented, except as noted. At study days 0, 10, 28 (only from SUDV and MARV challenge monkeys), 56, and 66, sera were collected and tested for (A) LASV-, (B) EBOV-, (C) SUDV-, and (D) MARV-specific neutralization responses. Data represent the geometric mean of log-transformed LASV and filovirus neutralization titer (FRNT50), with the 95% confidence interval. An unpaired 2-tailed t test was used to determine statistically significant differences (**P < 0.005, ***P < 0.0005) between quadrivalent and control groups as indicated. Mean values and standard deviations are depicted in red.
For LASV GP–specific IgG, significant levels of IgG were detected after the second dose of quadrivalent vaccine immunization. The geomean endpoint titer on study day 66 (10 days after boost) was 3.5 log (Figure 2A). For EBOV GP– and SUDV GP–specific IgG, significant levels of IgG were already detected after a single dose of quadrivalent vaccine. The geomean endpoint titer at study day 10 was about 4 logs. The level of IgG was maintained well and was boosted an additional 2 log by the second dose of quadrivalent vaccine. The geomean endpoint titer on study day 66 (10 days after boost) was 6.3 log (Figure 2, B and C). For MARV GP–specific IgG, low but significant levels of IgG were elicited by a single quadrivalent vaccination. The geomean endpoint titer for MARV GP–specific IgG was 3.3 log at study day 10 and boosted for 2 log by the second dose of the vaccine (Figure 2D). No significant filovirus and LASV GP–specific IgG were observed in the control group animals.
Sera collected on study days 0, 10, 28, 56, and 66 were also tested for their neutralizing antibodies against LASV, EBOV, SUDV, and MARV using 4 rVSVΔG chimeric viruses expressing LASV or filovirus GPs, and additionally eGFP as a reporter (Figure 3). A statistically significant level of LASV-specific neutralizing antibody was detected after a single dose of the quadrivalent vaccine, with a FRNT50 of 1.8 log. This level of neutralizing antibody declined but boosted with the second dose of quadrivalent vaccine by over 1 log. The mean FRNT50 titer on study day 66 (10 days after boost) was 2.9 log (Figure 3A). Higher responses were observed with EBOV- and SUDV-specific neutralizing antibody after a single dose of vaccine and boosted over 1.5 to 2.0 log by the second dose of quadrivalent vaccine. The mean FRNT50 titers for EBOV and SUDV on study day 66 (10 days after boost) were 4.8 and 3.8 log, respectively (Figure 3, B and C). The MARV-specific neutralizing response was similar to that of LASV, with the titer being 0.5 log lower at day 10 after prime vaccine dose (Figure 3D). Neither LASV- nor filovirus-specific neutralizing antibodies were observed in control group animals.
PBMCs collected on study days 0, 10, 56, and 66 were tested for their IFNG responses against filovirus and LASV GPs. Figure 4 provides a graphic summary of the mean LASV GP–, EBOV GP–, SUDV GP–, and MARV GP–specific IFNG responses from both the quadrivalent vaccine and control groups over all time points. On study day 0, there were no detectable IFNG ELISPOT responses to any GP peptides. At days 10 and 56 after a single dose of the quadrivalent vaccine, low but detectable IFNG ELISPOT responses were observed in less than 25% animals. All 4 virus GP–specific IFNG ELISPOT responses were markedly enhanced by quadrivalent vaccine boosting. At study day 66 (10 days after the second dose), the quadrivalent group mean responses were enhanced from 10–20 SFC/million PBMCs (at study day 10) to over 90 SFC/million PBMCs for LASV, EBOV, and SUDV GPs, and 3–40 SFC/million PBMCs for MARV GP. The responder rates were also markedly enhanced. There were positive LASV GP–specific interferon gamma (IFNG) ELISPOT responses in 16 of 18 animals (~89%); positive EBOV and SUDV GP–specific IFNG ELISPOT responses in 15 of 18 animals (~83%); and positive MARV GP–specific IFNG ELISPOT responses in 9 of 18 animals (50%). Animals from the control group had no significant IFNG ELISPOT responses at any time point.
Figure 4. Filovirus and LASV GP–specific IFNG ELISPOT responses in macaques immunized with the quadrivalent vaccine.

At study days 0 and 56, macaques were immunized i.m. with 4 × 107 PFU of the quadrivalent vaccine (black circles) or rVSV N4CT1-HIVgag as control (gray squares). A total of 20 vaccinated animals and 12 control animals are represented, except as noted. At study days 0, 10, 56, and 66, PBMCs were collected and tested for LASV GP–, EBOV GP–, SUDV GP–, and MARV GP–peptide pool-specific IFNG secretion by ELISPOT assay. Data represent the average LASV GP–, EBOV GP–, SUDV GP–, or MARV GP–specific IFNG ELISPOT response, with the standard error of the mean indicated. The ratio number represents the responder rate of the animals with an antigen-specific IFNG ELISPOT response greater than or equal to 25 SFC/106 PBMCs. An unpaired 2-tailed t test was used to determine statistically significant differences (*P < 0.05, **P < 0.005) between quadrivalent and control groups, as indicated. Mean values and standard deviations are depicted in red.
Protective efficacy of the quadrivalent vaccine in macaques.
At day 28 after the boost vaccination (day 84 after prime vaccination), 4 groups of 8 macaques, each group containing 5 vaccinees and 3 controls, were challenged i.m. with 1000 PFU of low-passage-number LASV (strain 0043/LV/14), EBOV (strain Kikwit), SUDV (strain Gulu), or MARV (strain Angola). By days 6–9 after the challenge, control macaques in all 3 filovirus groups had succumbed with disease consistent with filovirus HF reported in previous studies, while all 3 of the LASV-infected controls succumbed between days 13–15 after virus challenge (Figure 5). All 3 LASV-infected animals also displayed clinical signs consistent with those shown in our previous work (21, 24, 25), and in contrast to filovirus-infected macaques, all of the LASV-infected animals showed neurologic disease as evidenced by intention tremors, ataxia, and/or seizures (Table 1). In contrast, all 20 macaques receiving the quadrivalent vaccine remained generally healthy and survived the lethal filovirus or LASV challenges (Figure 5 and Tables 1–4). In the animals immunized with the quadrivalent vaccine, low-grade transient fevers were noted in 1 animal challenged with EBOV at day 6, 1 animal challenged with MARV at day 6, and 1 animal challenged with SUDV at day 10. Mild and transient changes in clinical pathology were noted in a number of animals vaccinated with the quadrivalent vaccine for all 4 challenge groups, and these mostly included changes associated with activation of the innate immune response such as leukocytosis, granulocytosis, and elevated levels of CRP (Tables 1–4). A few of the animals that received the quadrivalent vaccine and were challenged with filoviruses showed small increases in liver-associated enzymes during days 6–14 after filovirus exposure. All 20 macaques that received the quadrivalent vaccine were completely healthy at the study endpoint 28 days after challenge and any animal that had altered clinical pathology values had returned to baseline levels.
Figure 5. Post-challenge survival of cynomolgus macaques immunized with the quadrivalent vaccine.

At study days 0 and 56, macaques were immunized i.m. with 4 × 107 PFU of the quadrivalent vaccine or rVSV N4CT1-HIVgag as control. At study day 84, macaques were challenged i.m. with 1000 PFU (A) LASV (strain 0043/LV/14), (B) EBOV (strain Kikwit), (C) SUDV (strain Gulu), or (D) MARV (strain Angola), and monitored up to 28 days.
Table 1. Clinical description and outcome of LASV-challenged NHPs.

Table 4. Clinical description and outcome of MARV-challenged NHPs.

Filovirus and LASV RNA and infectious virus.
The levels of post-challenge filovirus and LASV infectious virus present in plasma, and genomic RNA present in blood from macaques, were assessed by plaque assay (Figure 6, A–D) and real-time quantitative PCR (RT-qPCR) (Figure 6, E–H), respectively. There was no detectible infectious virus present in plasma from 19 of the 20 macaques receiving the quadrivalent vaccine at any time after challenge, contrasting with peak levels of infectious virus (>5–8 log10 PFU/mL) detected in plasma from sham-vaccinated macaques between days 6–13 after challenge (Figure 6, A–D). One macaque that received the quadrivalent vaccine and was challenged with LASV had a transient viremia at the day 10 sampling point only (Figure 6A).
Figure 6. Filovirus and LASV viremia in cynomolgus macaques challenged i.m. with LASV, EBOV, SUDV, or MARV.

Quadrivalent (Q-V) or control (HIVgag) rVSV vaccinated macaques were challenged i.m. with 1000 PFU (A and E) LASV (strain 0043/LV/14), (B and F) EBOV (strain Kikwit), (C and G) SUDV (strain Gulu), or (D and H) MARV (strain Angola). Post-challenge viremia was monitored by infectious plaque assay (A–D) or RT-qPCR (E and F). Limit of detection (LOD) for LASV, EBOV, SUDV, and MARV genome equivalent qRT-PCR is log10 4, log10 3, log10 3, and log10 4, respectively.
Peak levels of LASV genomic RNA (~7–9 log10 genome equivalents/mL) were detected in blood at day 10 after challenge in 3 of the 5 vaccinated macaques, remaining detectable until day 21 after challenge in one of these macaques. In contrast, higher levels of LASV genomic RNA (~10–11 log10 genome equivalents/mL) were detected in the 3 sham-vaccinated controls peaking between days 10 and 15 after exposure (Figure 6E). Peak levels of EBOV genomic RNA (~6–7 log10 genome equivalents/mL) were detected in blood at day 6 after infection in 2 of the 5 vaccinated animals, remaining detectable until day 21 after challenge in 1 of these macaques, whereas much higher levels of EBOV genomic RNA (~9.5–10.5 log10 genome equivalents) were detected in the sham-vaccinated controls between days 6–8 after infection (Figure 6F). For SUDV there was no detectable genomic RNA in any of the 5 vaccinated macaques at any time point after virus challenge, whereas low-level genomic RNA was detected in only 1 of 5 vaccinated animals challenged with MARV at the day 6 post-exposure time point only. In contrast, high levels of genomic RNA were detected in all 3 SUDV control animals (~10–11.5 log10 genome equivalents/mL) between days 6–9 after infection, and in all 3 MARV controls (~11.5–12.5 log10 genome equivalents/mL) (Figure 6, G and H).
Gross pathology and histopathology.
All 3 control cynomologus macaques challenged with LASV had lesions that were consistent with LASV infection at necropsy (24, 25). Significant lesions present in all LASV-infected controls included necrotizing hepatitis, splenomegaly, and lymphadenomegaly. Some of the animals also had petechial rashes, pleural effusion, ascities, interstitial pneumonia, and adrenomegaly. Significant lesions present in the 9 filovirus-infected controls had lesions consistent with filovirus infection at necropsy (26–28). Lesions in these filovirus-infected control macaques included necrotizing hepatitis, splenomegaly, and petechial rash. Some of the animals also had lymphadenomegaly, adrenomegaly, adrenal hemorrhage, interstitial pneumonia, and mucosal reddening of the proximal duodenum. In contrast to control macaques, all 20 specifically vaccinated cynomologus macaques that survived to the study endpoint at day 28 after challenge failed to display lesions that were consistent with the respective challenge virus.
All control macaques infected with LASV or filoviruses displayed histopathologic lesions that were consistent with the respective viruses (24–28). These lesions included lymphoid medullary histiocytosis, multifocal to diffuse necrotizing hepatitis and sinusoidal leukocytosis, splenitis with lymphocytosis and fibrin deposition, nephritis, adrenalitis, and interstitial pneumonia with pulmonary edema. Immunohistochemistry for the detection of specific anti–filovirus or anti–LASV antigen identified positive immunolabeling within the following cells: mononuclear cells within the lymph nodes (axillary and inguinal); mononuclear cells, sinusoidal lining cells, endothelium, and rarely hepatocytes in the liver (Figure 7); mononuclear cells and rarely endothelium within the spleen; mononuclear cells, endothelium, rarely tubular epithelium within the kidney; mononuclear cells, cortical cells, medullary cells, and endothelium in the adrenal gland; and mononuclear cells, endothelium, and rarely airway epithelium in the lung. All 20 specifically vaccinated macaques that survived to the study end at day 28 failed to display histologic lesions that were consistent with filovirus or LASV infection (Figure 7).
Figure 7. Immunohistochemistry.
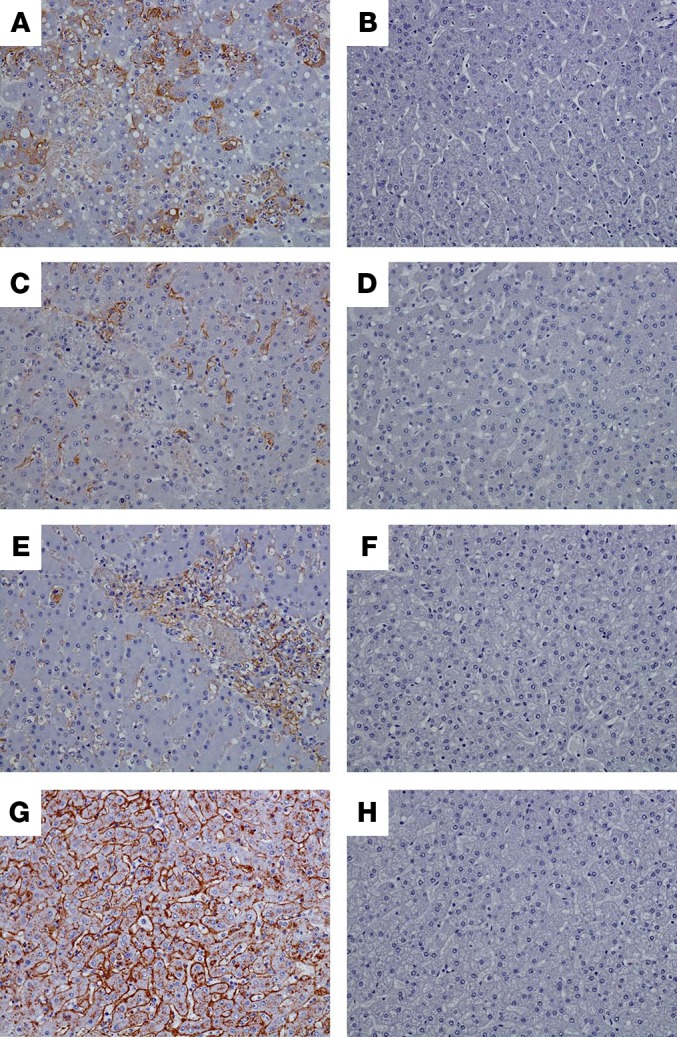
Images of liver of representative control macaques (A, C, E, and G) showing diffuse cytoplasmic immunolabeling (brown color) of sinusoidal lining cells, Kupffer cells, and rare hepatocytes of animals infected with LASV (A), EBOV (C), SUDV (E), and MARV (G). No immunolabeling was detected in liver of specifically vaccinated macaques that survived virus challenge, as shown by representative animals exposed to LASV (B), EBOV (D), SUDV (F), or MARV (H). All representative images taken at ×20.
Discussion
There are presently no approved active or passive interventions for exposure to filoviruses or LASV resulting from natural outbreak, laboratory accident, or deliberate misuse. Public health concern is based on both the emerging infectious disease status of these viruses and their potential use as biologic weapons. Effective prophylactic filovirus and LASV vaccines would find application in endemic areas, with medical personnel and close contacts during natural or deliberate outbreaks, and with laboratory workers engaged in research on these viruses. Due to the overlap in the endemic ranges of these viruses, the ideal prophylactic vaccine for use in Africa would confer protection against EBOV, SUDV, MARV, and LASV.
We previously demonstrated the protective efficacy of a highly attenuated rVSV N4CT1 trivalent VesiculoVax vaccine in cynomolgus monkeys against the filoviruses EBOV, SUDV, and MARV (18). However, additional protection from LASV disease by one vaccine presents an enormous challenge for several reasons. Not only is LASV genetically distinct from filoviruses because it is in the family Arenaviridae, but also, more importantly, there are 4 recognized lineages of LASV (29) and 2 newly proposed lineages (30, 31). These 4 to 6 LASV lineages are genetically distinct and there is significant concern that a vaccine that protects against one lineage may not necessarily protect against a different lineage. This further complicates the development of a multivalent vaccine that can confer protection against the major filoviruses and LASV. While we have shown that a monovalent rVSVΔG-based vaccine expressing the lineage IV Josiah strain LASV GPC can protect macaques against homologous challenge with isolates from the same lineage (21, 22), it is unknown if any LASV vaccine could protect NHPs against lethal challenge with an isolate from a heterologous lineage. A major advance shown in the current study is the finding that the new quadrivalent VesiculoVax vaccine based on a lineage IV LASV strain completely protected macaques against disease and death following high-dose challenge with a contemporary heterologous lineage II strain of LASV. These data strongly suggest that a single vaccine component may be able to provide protection against all major lineages of LASV. Future studies need to be conducted to test this vaccine against other lineages of LASV.
In this study, we demonstrated complete protection from lethal disease via 2 doses of the quadrivalent VesiculoVax vaccine. However, we have observed that antigen-specific immune responses, specifically antibody responses, were elicited with single dose vaccination, although at relatively low levels for LASV and MARV GPs. In previous studies, we showed that similar levels of immune responses after single dose trivalent filovirus vaccination protects macaques against lethal high-dose challenges of all 3 filoviruses (18), and a single dose of monovalent rVSVΔG-based LASV GPC vaccine can protect macaques against homologous lineage challenge (21, 22). These data suggest that we may be able to reach similar protection with a single dose of the quadrivalent VesiculoVax vaccine. While a single-dose vaccination strategy is clearly needed for rapid response to outbreaks in terms of a ring vaccination approach, the prime and boost regimen may be needed for long-term immunity. Further studies will be required to address these questions.
Our results show that a quadrivalent rVSV N4CT1-EBOV/SUDV/MARV/rVSV N4ΔG-LASV vaccine formulation protects NHPs from overt disease and death following high-dose challenge of any of the 3 filoviruses and LASV that are endemic in large overlapping regions of Africa. In addition to these preclinical results in the gold standard NHP models of viral HF, very promising preliminary safety data have been obtained from an ongoing phase I clinical study with an analogous monovalent rVSV N4CT1-EBOV vaccine (Profectus BioSciences; https://clinicaltrials.gov/ct2/show/NCT02718469). These results support the testing of a quadrivalent filovirus and LASV vaccine formulation in phase I clinical trials as the next step to identify and advance a vaccine that is safe and has broad-spectrum protective efficacy against this important group of viral HFs. Importantly, new filovirus vaccine targets, such as the Bundibugyo ebolavirus GP, could be incorporated into the attenuated rVSV backbone to combat new emerging filoviruses, if needed.
Methods
Study design.
The objectives of this work were to develop a new quadrivalent VesiculoVax formulation to protect against multiple species of filoviruses and heterologous lineages of LASV, and to assess the immunogenicity and protective efficacy of this new quadrivalent vaccine in cynomolgus monkeys. Studies were powered (n = 5 per vaccine group and n = 3 per control group) to detect large differences in protective efficacy. Animals were randomized into vaccine or control groups by Excel. The animal study was blinded to the research staff. All animal studies were approved by the University of Texas Medical Branch (UTMB) Institutional Animal Care and Use Committee and adhere to the NIH Guide for the Care and Use of Laboratory Animals.
Challenge viruses.
Zaire ebolavirus (EBOV) isolate 199510621 (strain Kikwit) originated from a 65-year-old female patient who died on May 5, 1995. The study challenge material was from the second Vero E6 passage of EBOV isolate 199510621. Briefly, the first passage at UTMB consisted of inoculating CDC 807223 (passage 1 of EBOV isolate 199510621) at an MOI of 0.001 onto Vero E6 cells (ATCC CRL-1586). The cell culture fluids were subsequently harvested at day 10 after infection and stored at –80°C as approximately 1-mL aliquots. Deep sequencing indicated the EBOV was greater than 98% 7U (consecutive stretch of 7 uridines). No detectable mycoplasma or endotoxin levels were measured (<0.5 endotoxin units (EU)/mL). Sudan ebolavirus (SUDV) isolate 200011676 (strain Gulu) originated from a 35-year-old male patient who died on October 16, 2000. The study challenge material was from the second Vero E6 cell passage of SUDV isolate 200011676. Briefly, the first passage at UTMB consisted of inoculating CDC 808892 (CDC passage 1 of SUDV isolate 200011676) at an MOI of 0.001 onto Vero E6 cells (ATCC CRL-1586). The cell supernatants were subsequently harvested at day 7 after infection and stored at –80°C as approximately 1-mL aliquots. No detectable mycoplasma or endotoxin levels were measured (<0.5 EU/mL). Marburg marburgvirus (MARV) Angola isolate 200501379 originated from human serum from an 8-month-old female patient in Uige, Angola. The serum was collected day 17 after onset, 1 day before death. The study challenge material was from the second Vero E6 cell passage of MARV Angola isolate 200501379. Briefly, the first passage at UTMB consisted of inoculating CDC 810820 at an MOI of 0.001 onto Vero E6 cells (ATCC CRL-1586). The cell supernatants were subsequently harvested at day 6 after infection and stored at –80°C as approximately 1-mL aliquots. No detectable mycoplasma or endotoxin levels were measured (<0.5 EU/mL). A lineage II LASV isolate number 0043/LV/14 originated from human serum collected from a 30-year-old male patient in Edo State, Nigeria who died in January of 2014. The study challenge material was from the first Vero 76 cell passage of this serum. Briefly, the serum was inoculated at 1:100 dilution on Vero 76 cells (ATCC CRL-1587) at UTMB. The cell supernatants were harvested at day 7 after infection and stored at –80°C as approximately 1-mL aliquots. No detectable mycoplasma or endotoxin levels were measured (<0.5 EU/mL).
Nonhuman primate vaccination and challenge.
Thirty-two healthy, filovirus- and LASV-naive, adult (~3 to 9.5 kg) cynomolgus macaques (Macaca fascicularis; PreLabs) were randomized into 4 groups of 5 experimental animals and 3 control animals each. The experimental animals were vaccinated by intramuscular (i.m.) injection of 4 × 107 PFU of the quadrivalent VesiculoVax vaccine, and the control animals were vaccinated by i.m. injection of 4 × 107 PFU of an irrelevant rVSV N4CT1-HIVgag vaccine. A boost vaccine was given 56 days after the prime vaccine. All of the macaques were challenged 4 weeks after the boost vaccination by i.m. injection with a target dose of 1000 PFU of EBOV strain Kikwit for group 1, 1000 PFU of SUDV strain Gulu for group 2, 1000 PFU of MARV strain Angola for group 3, and 1000 PFU of LASV lineage II strain Nigeria for group 4. All animals were given physical examinations, and blood was collected before prime vaccination, at day 10 after prime vaccination, at the time of boost vaccination, at day 10 after boost vaccination, at the time of virus challenge, and on days 3, 6, 10, 14, 21, and 28 after virus challenge. The macaques were monitored daily and scored for disease progression with an internal filovirus or LASV humane endpoint scoring sheet approved by the UTMB IACUC. UTMB facilities used in this work are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and adhere to principles specified in the eighth edition of the Guide for the Care and Use of Laboratory Animals, National Research Council. The scoring changes measured from baseline included posture and activity level, attitude and behavior, food intake, respiration, and disease manifestations, such as visible rash, hemorrhage, ecchymosis, or flushed skin. In addition, central nervous system abnormalities were scored in NHPs challenged with LASV. A score of 9 or more for filoviruses and 10 or higher for LASV indicated that an animal met the criteria for euthanasia.
NHP filovirus and LASV anti-GP IgG ELISA.
Immulon 2HB flat-bottom ELISA plates (Thermo Labsystem) were coated for 18 hours at 4°C with 30 ng/well recombinant filovirus glycoprotein (minus the transmembrane region: EBOV rGPdTM, catalog 0501-015; SUDV rGPdTM, catalog 0502-015; and MARV rGpTM, catalog 0506-015; Integrated Biotherapeutics) or 100 ng/well of gamma-irradiated Lassa virus (Josiah Strain NR-31822, BEI Resources) diluted in 0.1 mL DPBS. The plates were washed 3 times with 300 μL of 1× PBS containing 0.1% Tween 20. Monkey serum samples, diluted with 1× PBS containing 5% nonfat dry milk and 0.1% Tween-20 were added to the ELISA plates at a starting dilution of 1:100, and further diluted 3-fold across the plates. Plates were kept at 4°C overnight and washed afterwards 3 times with 300 μL of 1× PBS containing 0.1% Tween 20, followed by 1 hour at 37°C with 100 μL/well of streptavidin–horseradish peroxidase–conjugated goat anti-NHP IgG antibody (Fitzgerald, catalog 43R-IG020HRP) diluted 1:10,000 with 1× PBS supplemented with 0.1% Tween 20 and 5% nonfat dry milk. The plates were then washed 5 times with 300 μL of 1× PBS containing 0.1% Tween 20 and developed by the addition of 100 μL/well of TMB (3,3′, 5,5′-tetramethyl benzidine, MilliporeSigma). The reaction was stopped after 10 minutes by adding 100 μL/well of 1N sulfuric acid (VWR International LLC).
The OD values for each plate were captured using SoftMax Pro version 5.4 (Molecular Devices), the software that accompanies the VERSAmax plate reader. The SoftMax Pro software calculated the test sample ELISA endpoint titer, defined as the reciprocal of the highest serum sample dilution resulting in a background absorbance subtracted OD greater than 0.1 using a 4-parameter logistic equation. The background OD was calculated using the average OD values of blank wells.
Serum antibody neutralization assay.
Duplicate 3-fold serial dilutions of heat-inactivated NHP serum samples (days 0, 10, 28, 56, and 66) were incubated in 96-well tissue-culture plates with a fixed quantity of the rVSV N4ΔG-(filovirus or LASV GP)5-(eGFP)1 chimeric virus (500 fluorescence PFU per well) in DMEM 10 medium for 16–24 hours at 4°C with 5% human complement (lot no. PBS-154-1, Profectus). Following the incubation, the serum/virus mixture was transferred onto a monolayer of 2 × 104 Vero E6 cells (ATCC, CRL-1586) per well in a 96-well tissue culture plate and incubated at 37°C for 18–20 hours. Cell monolayers were then fixed with 50 μL of ice-cold 4% formaldehyde buffered for 20 minutes at room temperature. The plates were scanned and eGFP-expressing cells were counted using an S6 Universal Analyzer (Cellular Technology Ltd.). The 50% fluorescence cell reduction neutralization titer (FRNT50) is reported as the concentration of serum to reduce the number of GFP-expressing cells by 50% compared with the mean value relative to virus control wells in the absence of the serum.
Macaque IFNG ELISPOT assay.
Vaccine-elicited IFNG ELISPOT responses were determined using a T cell human IFNG ELISPOT kit (CTL HIFNG-10; Cellular Technology Ltd.). Briefly, 96-well flat-bottomed ELISPOT plates (Millipore) were coated overnight with anti-human IFNG capture antibody according the kit manual. Heparinized whole blood was collected on study days 0, 10, 56, and 66, and PBMCs were isolated from the whole blood by Ficoll-Hypaque density gradient centrifugation and cryopreserved. Cryopreserved macaque PBMCs were thawed and cultured overnight with complete R10 culture medium (RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 100U/mL penicillin, 100 μg/mL streptomycin sulfate, 1 mM sodium pyruvate, 1 mM HEPES, 100 mM nonessential amino acids) before the assay. The cells were then collected and counted. Samples with over 70% viability (19 of 20 animals at days 10 and 56 and 18 of 20 animals at day 66) were resuspended in complete R05 culture medium (same as R10 culture medium but supplemented with 5% FBS) containing either 5 μL/mL phytohemagglutinin mucoprotein (MilliporeSigma) or peptide pools (15 mers overlapping by 11 amino acids; final peptide concentration, 1 μM) spanning the EBOV GP (Mayinga strain), SUDV GP (Gulu strain), MARV GP (Angola strain), LASV GPC (Josiah strain), or medium alone. The input cell number was 3 × 105 PBMCs per well (3 × 106 PBMCs/mL), and the cells were assayed in duplicate wells. The cells were incubated for 18 to 24 hours at 37°C. Following development and drying, the plates were scanned and cells were counted with a CTL ImmunoSpot analyzer using the SmartCount data analysis module. Peptide-specific IFNG ELISPOT responses were considered positive if the responses (minus the medium background) were 3-fold above the medium response and greater than or equal to 25 SFC/106 PBMCs.
Viral titration by plaque assay.
Titration of the rVSV N4CT1 and N4ΔG vaccine vectors, confirmation of the challenge dose, and titration of EBOV, SUDV, MARV, or LASV in plasma after challenge were performed by conventional plaque assay on Vero E6 cell monolayers as previously described (18, 25). Briefly, serial 10-fold dilutions of the samples were adsorbed to Vero E6 monolayers (ATCC CRL-1586) in duplicate wells of 6-well plates (200 μL/well). Following adsorption, the inoculum was removed by aspiration, and the monolayers were overlaid with 3 mL of 0.8% SeaPlaque agarose in DMEM. The agarose was permitted to set at room temperature for 15 minutes, and the plates were then incubated at 32°C under 5% CO2 for 3–4 days (for rVSV N4CT1 vectors), 7–10 days for filoviruses, or 5 days for LASV to allow plaque development. The limit of detection was 25 PFU/mL.
Detection of viremia by RT-qPCR.
RNA was isolated from whole blood utilizing the Viral RNA mini-kit (Qiagen) using 100 μL of blood added to 600 μL of the viral lysis buffer. Primers and probe targeting the NP gene of LASV, the VP30 gene of EBOV, the L gene of SUDV, and the NP gene of MARV were used for RT-qPCR with the following probes: LASV, 6-carboxyfluorescein (FAM)-5′TGCAGCTTGACCCAAGTGCCAA 3′-6 carboxytetramethylrhodamine (TAMRA); EBOV, FAM-5′CCGTCAATCAAGGAGCGCCTC 3′-TAMRA; SUDV, FAM-5′CATCCAATCAAAGACATTGCGA 3′-TAMRA; and MARV, FAM-5′CCCATAAGGTCACCCTCTTC 3′-TAMRA (Life Technologies). Viral RNA was detected using the CFX96 detection system (Bio-Rad Laboratories) in 1-step probe RT-qPCR kits (Qiagen) with the following cycle conditions: LASV, 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 53°C for 30 seconds; EBOV, 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 57°C for 30 seconds; SUDV and MARV, 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 59°C for 30 seconds. Threshold cycle (CT) values representing viral genomes were analyzed with CFX Manager software, and the data are shown as genome equivalents (GEq) per milliliter. To create the GEq standard, RNA from viral stocks was extracted, and the number of strain-specific genomes was calculated using Avogadro’s number and the molecular weight of each viral genome.
Hematology and serum biochemistry.
Total white blood cell counts, white blood cell differentials, red blood cell counts, platelet counts, hematocrit values, total hemoglobin concentrations, mean cell volumes, mean corpuscular volumes, and mean corpuscular hemoglobin concentrations were analyzed from blood collected in tubes containing EDTA using a laser-based hematologic analyzer (Beckman Coulter). Serum samples were tested for concentrations of alanine aminotransferase (ALT), albumin, alkaline phosphatase (ALP), amylase, aspartate aminotransferase (AST), C-reactive protein (CRP), calcium, creatinine, gammaglutamyltransferase (GGT), glucose, total protein, blood urea nitrogen (BUN), and uric acid, and by using a Piccolo point-of-care analyzer and Biochemistry Panel Plus analyzer discs (Abaxis).
Histopathology and immunohistochemistry.
A partial necropsy was performed on all subjects. Tissue samples of major organs were collected for histopathologic and immunohistochemical examination, immersion-fixed in 10% neutral buffered formalin, and processed for histopathology as previously described (24–27). For immunohistochemistry, specific anti-LASV immunoreactivity was detected using anti-LASV NP protein rabbit primary antibody (Zalgen Labs, catalog CLD4) at a 1:1000 dilution. Anti-EBOV immunoreactivity was detected using an anti-EBOV VP40 protein rabbit primary antibody (Integrated Biotherapeutics, catalog 0301-010) at a 1:4000 dilution; anti-SUDV immunoreactivity was detected using an anti-SUDV VP40 protein rabbit primary antibody (Integrated Biotherapeutics, catalog 0302-001) at a 1:4000 dilution; and anti-MARV immunoreactivity was detected using an anti-MARV NP protein mouse primary antibody (provided by USAMRIID, catalog BB06-BB01 [ref. 32]) at a 1:4000 dilution. Tissue sections were processed for immunohistochemistry using the Dako Autostainer. The secondary antibody used was biotinylated goat anti-rabbit IgG (Vector Laboratories) or biotinylated goat anti-mouse IgG (Vector Laboratories) at 1:200 followed by Dako LSAB2 streptavidin-HRP (Dako). Slides were developed with Dako DAB chromagen (Dako) and counterstained with hematoxylin. Nonimmune rabbit IgG was used as a negative control.
Statistical analysis.
Comparisons of log-transformed serum IgG and neutralizing antibody titers in the different vaccine groups at each time point were performed with an unpaired 2-tailed Student’s t test using GraphPad Prism version 5.02. P values less than 0.05 indicated that the tests were statistically significant.
Study approval.
The animal studies were performed at the Galveston National Laboratory, University of Texas Medical Branch at Galveston and were approved by the University of Texas Medical Branch Animal Care and Use Committee.
Author contributions
RWC, RX, JHE, and TWG conceived and designed the animal challenge experiments. DM and SH conceived and designed the vaccine vectors and together with TEL, CSG, RMN, and SEW, generated and characterized all vaccine candidates. RWC, JBG, DJD, CEM, and TWG performed the animal procedures and conducted clinical observations. KNA performed the clinical pathology assays and PCR assays. JBG performed the filovirus and LASV infectivity assays. AL and LS performed the ELISA assays. AOS and LS performed the virus neutralization assays. AOS, AL, and LS performed the ELISPOT assays. RWC, RX, AOS, JBG, CH, KNA, HF, JHE, and TWG analyzed the data. KAF performed gross pathologic, histologic and immunohistochemical analysis of the data. RWC, RX, and TWG wrote the paper. HF and JHE edited the paper. All authors had access to all of the data and approved the final version of the manuscript. The authorship order among co–first authors was determined alphabetically.
Table 3. Clinical description and outcome of SUDV-challenged NHPs.

Table 2. Clinical description and outcome of EBOV-challenged NHPs.

Acknowledgments
The authors thank Kimberly Schuenke for administrative assistance, Viktoriya Borisevich for assistance with clinical pathology assays, Natalie Dobias for assistance with tissue preparations, and the UTMB Animal Resource Center for animal husbandry support. We also thank Pardis Sabeti, Robert Garry, and Luis Branco for assistance with obtaining the LASV isolate used in this study. This work was supported by Department of Health and Human Services/National Institute of Allergy and Infectious Diseases (NIAID)/NIH contract number HHSN272201700077C (to JHE) and NIAID/NIH grant number UC7AI094660 (to UTMB for BSL-4 operations support of the Galveston National Laboratory). Some samples used to optimize assays were made available by NIAID/NIH grant number U19AI142785 (to TWG). Partial funding was provided through the NIAID/NIH Intramural Research Program (to HF). Support was also received from the National Human Genome Research Institute (NHGRI), NIH Department of Health and Human Services, under grant numbers U01HG007480 and U54HG007480 (to CH). World Bank project ACE019 supports CH’s research at Redeemer’s University, Ede, Nigeria. The opinions, interpretations, conclusions, and recommendations contained here are not necessarily endorsed by UTMB at Galveston or the NIH.
Version 1. 10/22/2019
In-Press Preview
Version 2. 12/09/2019
Electronic publication
Version 3. 01/02/2020
Print issue publication
Footnotes
Conflict of interest: RX, DM, SH, TEL, CSG, RMN, AOS, AL, SEW, LS, and JHE are paid employees of Profectus Biosciences, Inc. Profectus Biosciences in-licenses a portfolio of patents (US patent 7,875,446; U.S. patent 8,287,878) previously obtained from Wyeth to support the development of VesiculoVax vectors as vaccines against hemorrhagic fever viruses. HF and TWG are named on patents for different filovirus and/or arenavirus-based vaccines (US patent 8012489B2 [HF]; US patents 7,635,485; 8,017,130; 8,796,013 [TWG]) but have no intellectual property regarding the VesiculoVax vectors used in this project.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(1):539–551.https://doi.org/10.1172/JCI131958.
Contributor Information
Robert W. Cross, Email: rwcross@utmb.edu.
Rong Xu, Email: RXu@profectusbiosciences.com.
Demetrius Matassov, Email: dmatassov@profectusbiosciences.com.
Stefan Hamm, Email: shamm@profectusbiosciences.com.
Theresa E. Latham, Email: tlatham@profectusbiosciences.com.
Cheryl S. Gerardi, Email: cgerardi@profectusbiosciences.com.
Rebecca M. Nowak, Email: bnowak@profectusbiosciences.com.
Joan B. Geisbert, Email: jbgeisbe@utmb.edu.
Ayuko Ota-Setlik, Email: ayuko.ota-setlik@pfizer.com.
Krystle N. Agans, Email: knagans@utmb.edu.
Amara Luckay, Email: luckay@profectusbiosciences.com.
Susan E. Witko, Email: Susan.E.Witko@pfizer.com.
Lena Soukieh, Email: lsoukieh@profectusbiosciences.com.
Daniel J. Deer, Email: djdeer@utmb.edu.
Chad E. Mire, Email: chmire@UTMB.EDU.
Heinz Feldmann, Email: feldmannh@mail.nih.gov.
Christian Happi, Email: happic@run.edu.ng.
Karla A. Fenton, Email: kafenton@UTMB.EDU.
John H. Eldridge, Email: eldridge@profectusbiosciences.com.
Thomas W. Geisbert, Email: twgeisbe@utmb.edu.
References
- 1.Paessler S, Walker DH. Pathogenesis of the viral hemorrhagic fevers. Annu Rev Pathol. 2013;8:411–440. doi: 10.1146/annurev-pathol-020712-164041. [DOI] [PubMed] [Google Scholar]
- 2.Mehand MS, Al-Shorbaji F, Millett P, Murgue B. The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antiviral Res. 2018;159:63–67. doi: 10.1016/j.antiviral.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. WHO. Ebola data and statistics. Situation summary. Latest available situation summary, 11 May 2016. http://apps.who.int/gho/data/view.ebola-sitrep.ebola-summary-latest?lang=en Accessed October 7, 2019.
- 4. WHO. Ebola Virus Disease. Democratic Republic of the Congo. External Situation Report 51. https://www.who.int/publications-detail/ebola-virus-disease-democratic-republic-of-congo-external-situation-report-51-2019.
- 5.McCormick JB, Webb PA, Krebs JW, Johnson KM, Smith ES. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis. 1987;155(3):437–444. doi: 10.1093/infdis/155.3.437. [DOI] [PubMed] [Google Scholar]
- 6. Nigeria Centre for Disease Control. An update of Lassa fever outbreak in Nigeria. http://ncdc.gov.ng/diseases/sitreps/?cat=5&name=An%20update%20of%20Lassa%20fever%20outbreak%20in%20Nigeria Accessed October 7, 2019.
- 7.Henao-Restrepo AM, et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!) Lancet. 2017;389(10068):505–518. doi: 10.1016/S0140-6736(16)32621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy SB, et al. Phase 2 placebo-controlled trial of two vaccines to prevent Ebola in Liberia. N Engl J Med. 2017;377(15):1438–1447. doi: 10.1056/NEJMoa1614067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. WHO. Preliminary results on the efficacy of rVSV-ZEBOV-GP Ebola vaccine using the ring vaccination strategy in the control of an Ebola outbreak in the Democratic Republic of the Congo: an example of integration of research into epidemic response. https://www.who.int/csr/resources/publications/ebola/ebola-ring-vaccination-results-12-april-2019.pdf Accessed October 7, 2019.
- 10.Kozak RA, Kobinger GP. Vaccines against ‘the other’ Ebolavirus species. Expert Rev Vaccines. 2016;15(9):1093–1100. doi: 10.1586/14760584.2016.1170597. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds P, Marzi A. Ebola and Marburg virus vaccines. Virus Genes. 2017;53(4):501–515. doi: 10.1007/s11262-017-1455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cross RW, Mire CE, Feldmann H, Geisbert TW. Post-exposure treatments for Ebola and Marburg virus infections. Nat Rev Drug Discov. 2018;17(6):413–434. doi: 10.1038/nrd.2017.251. [DOI] [PubMed] [Google Scholar]
- 13.Swenson DL, et al. Vaccine to confer to nonhuman primates complete protection against multistrain Ebola and Marburg virus infections. Clin Vaccine Immunol. 2008;15(3):460–467. doi: 10.1128/CVI.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geisbert TW, et al. Single-injection vaccine protects nonhuman primates against infection with marburg virus and three species of ebola virus. J Virol. 2009;83(14):7296–7304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hensley LE, et al. Demonstration of cross-protective vaccine immunity against an emerging pathogenic Ebolavirus Species. PLoS Pathog. 2010;6(5):e1000904. doi: 10.1371/journal.ppat.1000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mire CE, Geisbert JB, Marzi A, Agans KN, Feldmann H, Geisbert TW. Vesicular stomatitis virus-based vaccines protect nonhuman primates against Bundibugyo ebolavirus. PLoS Negl Trop Dis. 2013;7(12):e2600. doi: 10.1371/journal.pntd.0002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warfield KL, et al. Homologous and heterologous protection of nonhuman primates by Ebola and Sudan virus-like particles. PLoS ONE. 2015;10(3):e0118881. doi: 10.1371/journal.pone.0118881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matassov D, et al. Single-dose trivalent VesiculoVax vaccine protects macaques from lethal Ebolavirus and Marburgvirus challenge. J Virol. 2018;92(3):e01190-17. doi: 10.1128/JVI.01190-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carnec X, et al. A vaccine platform against arenaviruses based on a recombinant hyperattenuated mopeia virus expressing heterologous glycoproteins. J Virol. 2018;92(12):e02230-17. doi: 10.1128/JVI.02230-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cashman KA, et al. A DNA vaccine delivered by dermal electroporation fully protects cynomolgus macaques against Lassa fever. Hum Vaccin Immunother. 2017;13(12):2902–2911. doi: 10.1080/21645515.2017.1356500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geisbert TW, et al. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005;2(6):e183. doi: 10.1371/journal.pmed.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Safronetz D, et al. A recombinant vesicular stomatitis virus-based Lassa fever vaccine protects guinea pigs and macaques against challenge with geographically and genetically distinct Lassa viruses. PLoS Negl Trop Dis. 2015;9(4):e0003736. doi: 10.1371/journal.pntd.0003736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchs JD, et al. First-in-human evaluation of the safety and immunogenicity of a recombinant vesicular stomatitis virus human immunodeficiency virus-1 gag vaccine (HVTN 090) Open Forum Infect Dis. 2015;2(3):ofv082. doi: 10.1093/ofid/ofv082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hensley LE, et al. Pathogenesis of Lassa fever in cynomolgus macaques. Virol J. 2011;8:205. doi: 10.1186/1743-422X-8-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mire CE, et al. Human-monoclonal-antibody therapy protects nonhuman primates against advanced Lassa fever. Nat Med. 2017;23(10):1146–1149. doi: 10.1038/nm.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geisbert TW, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003;163(6):2347–2370. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hensley LE, et al. Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J Infect Dis. 2011;204 Suppl 3:S1021–S1031. doi: 10.1093/infdis/jir339. [DOI] [PubMed] [Google Scholar]
- 28. Feldmann H, Sanchez A, Geisbert TW. Filoviridae: Marburg and Ebola Viruses. In: Knipe DM, Howley PM, eds. Fields Virology. Philadelphia, Pennsylvania, USA: Lippincott Williams & Wilkins; 2013:923–956. [Google Scholar]
- 29.Bowen MD, et al. Genetic diversity among Lassa virus strains. J Virol. 2000;74(15):6992–7004. doi: 10.1128/JVI.74.15.6992-7004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manning JT, Forrester N, Paessler S. Lassa virus isolates from Mali and the Ivory Coast represent an emerging fifth lineage. Front Microbiol. 2015;6:1037. doi: 10.3389/fmicb.2015.01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitmer SLM, et al. New lineage of Lassa virus, Togo, 2016. Emerging Infect Dis. 2018;24(3):599–602. doi: 10.3201/eid2403.171905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geisbert TW, Jahrling PB. Differentiation of filoviruses by electron microscopy. Virus Res. 1995;39(2-3):129–150. doi: 10.1016/0168-1702(95)00080-1. [DOI] [PubMed] [Google Scholar]