

Abstract
Activation of host T cells that mediate allograft rejection is a 2-step process. The first occurs in secondary lymphoid organs where T cells encounter alloantigens presented by host DCs and differentiate to effectors. Antigen presentation at these sites occurs principally via transfer of intact, donor MHC-peptide complexes from graft cells to host DCs (cross-dressing) or by uptake and processing of donor antigens into allopeptides bound to self-MHC molecules (indirect presentation). The second step takes place in the graft, where effector T cells reengage with host DCs before causing rejection. How host DCs present alloantigens to T cells in the graft is not known. Using mouse islet and kidney transplantation models, imaging cytometry, and 2-photon intravital microscopy, we demonstrate extensive cross-dressing of intragraft host DCs with donor MHC-peptide complexes that occurred early after transplantation, whereas host DCs presenting donor antigen via the indirect pathway were rare. Cross-dressed DCs stably engaged TCR-transgenic effector CD8+ T cells that recognized donor antigen and were sufficient for sustaining acute rejection. In the chronic kidney rejection model, cross-dressing declined over time but was still conspicuous 8 weeks after transplantation. We conclude that cross-dressing of host DCs with donor MHC molecules is a major antigen presentation pathway driving effector T cell responses within allografts.
Keywords: Immunology, Transplantation
Keywords: Adaptive immunity, Antigen presentation, MHC class 1
Introduction
Allograft rejection is unique among immune responses in that it enlists 1%–10% of the total T cell repertoire, a proportion that is 100- to 1000-fold greater than that observed after microbial infection (1–3). The unusual number of T cells involved is attributed to the high frequency of T cells that recognize intact, allogeneic MHC molecules. This pathway of antigen recognition is referred to as “direct” to distinguish it from the “indirect” pathway in which T cells recognize foreign antigens as processed peptides bound to self-MHC molecules (4). Activation of T cells via the direct pathway was initially attributed exclusively to donor DCs that migrate from the graft to the host’s secondary lymphoid organs, as it was presumed that only donor DCs carry intact allogeneic MHC molecules (5, 6). Later studies in mouse heart, skin, and pancreatic islet transplantation, however, have shown that host DCs in secondary lymphoid organs are cross-dressed with intact donor MHC molecules and that they, not donor DCs, are the principal activators of directly alloreactive T cells (7–9).
Once activated in secondary lymphoid organs, alloreactive T cells proliferate and differentiate to effector cells that migrate to the graft. The mere arrival of effector T cells in the graft, however, is not sufficient for rejection. A second, cognate interaction with graft DCs is necessary (10, 11). Recent studies have shown that the graft DCs that engage effector T cells are of host origin, which is not surprising, considering that donor DCs that accompany the graft migrate out during the first days after surgery and have a limited life span or are recognized as nonself and killed rapidly by host immune cells (11–13). We hypothesized here that, as in secondary lymphoid organs, host DCs in the graft are cross-dressed with intact, donor MHC-peptide complexes that they then present to alloreactive, host effector T cells. Using islet and kidney transplantation in the mouse, we provide direct evidence in support of this hypothesis and demonstrate that the intragraft cross-dressing pathway is sufficient for sustaining rejection.
Results
Islet transplantation model.
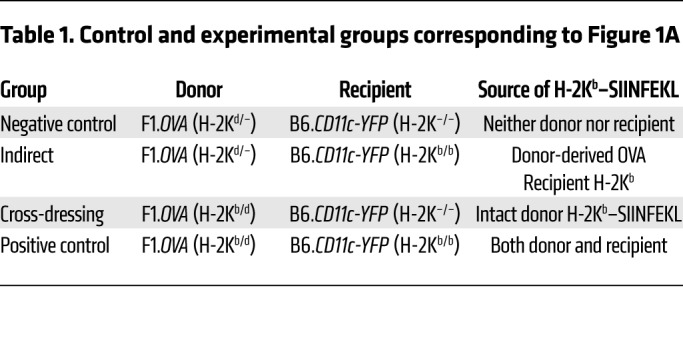
To characterize antigen presentation by host intragraft DCs, we transplanted (B6×BALB/c)F1.Act-mOVA (H-2b/d) islets, which express chicken OVA ubiquitously on surfaces of cells, under the kidney capsule of B6.CD11c-YFP (H-2b/b) recipients and transferred OVA-specific, DsRed+, TCR-transgenic OT-I effector CD8+ T cells 6 days later (Figure 1A). In this model, islet grafts are infiltrated with host, polyclonal T cells that recognize the semiallogeneic graft and OT-I cells that recognize the OVA257–264 SIINFEKL peptide bound to the H-2Kb MHC class I molecule (14). OT-I cells therefore serve as a tractable, antigen-specific T cell population for which antigen presentation can be manipulated. Moreover, the 6-day delay prior to OT-I transfer allows sufficient time for islet grafts to vascularize and host immune cells to replace donor immune cells (15). One day after OT-I transfer, grafts were analyzed by imaging flow cytometry to quantify H-2Kd molecules and H-2Kb–SIINFEKL complexes on host DCs and by 2-photon intravital microscopy (2P-IVM) to measure OT-I motility and interactions with host DCs. To restrict OVA antigen presentation to either the indirect or cross-dressing pathway, H-2Kb was eliminated from either the donor or recipient, respectively, by crossing the desired strain to B6 H-2Kb-deficient (H-2Kb–/–) mice (Table 1). To eliminate both OVA antigen presentation pathways simultaneously, we transplanted islets between donors and recipients lacking H-2Kb. These served as negative controls, while transplants between H-2Kb–sufficient mice, in which both OVA antigen presentation pathways are operational, served as positive controls (Table 1).
Figure 1. Host DCs are extensively cross-dressed with donor MHC class I–peptide complexes in islet allografts.

(A) F1.OVA H-2Kb–sufficient (H-2Kb/d) or F1.OVA H-2Kb-deficient (H-2Kd/–) islets were transplanted under the kidney capsule of B6.CD11c-YFP H-2Kb–sufficient (H-2Kb/b) or B6.CD11cYFP H-2Kb–deficient (H-2Kb–/–) recipients. 5 × 106 B6.Rag–/–.DsRed OT-I CD8+ effector T cells, which recognize the OVA peptide SIINFEKL bound to H-2Kb, were transferred i.v. 6 days later. One day after OT-I transfer, grafts were analyzed by imaging flow cytometry (B) and 2P-IVM (Figure 2). Control and experimental groups are shown in Table 1. (B) Leukocytes were isolated from transplanted allografts and analyzed by ImageStream. Intact H-2Kb–SIINFEKL complexes and donor H-2Kd molecules were identified as discreet spots on surface of host (CD11c-YFP) DCs. Representative images from each group are shown. (C) Proportion of host DCs positive for 1 or more spot of either H-2Kd or H-2Kb–SIINFEKL. In all groups, the majority of cells (~90%) carried only one spot, while the remainder had 2 to 5 spots (data not shown). The majority of DCs in the cross-dressing and control groups carried both MHC class I molecules. Each data point represents analysis of 1 transplanted animal. On average, 1071 (range = 100–3900) cells were analyzed per animal. **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way ANOVA with Tukey’s multiple comparison test.
Table 1. Control and experimental groups corresponding to Figure 1A.
Host DCs are extensively cross-dressed with donor MHC class I–peptide complexes in islet allografts.
Intact H-2Kd MHC class I molecules and H-2Kb–SIINFEKL complexes were identified as discreet spots on the surfaces of host (CD11c-YFP) intragraft DCs by imaging flow cytometry (Figure 1B), and the proportion of host DCs positive for 1 or more spot of either MHC molecule was quantified (Figure 1C). Donor H-2Kd molecules were equally present on host DCs in all groups despite B6 recipients lacking endogenous H-2Kd expression. This suggests that host DCs acquired donor MHC molecules from graft tissue. Likewise, H-2Kb–SIINFEKL complexes were present on H-2Kb–/– host DCs harvested from H-2Kb–sufficient F1.Act-mOVA grafts (cross-dressing group) at a level that was even greater than the positive control group, in which both donors and recipients expressed H-2Kb. This indicates that host DCs acquire intact, donor MHC class I–peptide complexes from the graft. In contrast, we did not detect H-2Kb–SIINFEKL complexes on H-2Kb–sufficient host DCs harvested from H-2Kb–/– F1.Act-mOVA grafts (indirect group) above the negative control group, suggesting that indirect OVA presentation occurs at very low levels. Finally, donor MHC class I molecule acquisition by host DCs was uniformly higher in the graft than the spleen (Figure 1C). Together, the data indicate that cross-dressing is a conspicuous phenomenon within the allograft early after transplantation.
Cross-dressed host DCs make stable contacts with antigen-specific, effector CD8+ T cells in islet allografts.
To investigate the function of cross-dressed DCs, we performed 2P-IVM on islet grafts 24 hours after transferring OT-I effector T cells to the mouse groups shown in Figure 1A. At the time of imaging, grafts were well vascularized and densely populated with host-derived, yellow fluorescent protein–positive (YFP+) DCs (Figure 2A). Equal numbers of OT-I effectors had infiltrated the tissue in and around the graft in all groups (Figure 2A and Supplemental Video 1; supplemental material available online with this article; https://doi.org/10.1172/JCI125773DS1), but significant differences in the dynamic behavior of these cells were observed (Figure 2, B and C, Supplemental Videos 1 and 2). To avoid pseudoreplication errors in the statistical analysis, we applied the stringent random intercept model to account for potentially correlated multiple observations within the imaged location, nested within the mouse, rather than the standard linear regression model, which ignores these correlations. As shown in Figure 2B, the frequency of OT-1 contacts with DCs (defined as contacts lasting more than 90 seconds) was significantly higher in the cross-dressed than the negative control group, while differences between the other groups did not reach statistical significance. However, the duration of these contacts was significantly greater in the cross-dressed than either the negative control or indirect groups, but was similar between the latter 2 groups (Figure 2B). Commensurate with these findings, OT-I effectors traveled much shorter distances, judged by track lengths (Figure 2, B and C), and had lower motility (speed) and higher arrest coefficient in the cross-dressing and positive control groups than in the indirect or negative control groups (Figure 2C). These data demonstrate that host DCs cross-dressed with donor MHC class I–peptide complexes make stable contacts with antigen-specific, CD8+ effector T cells in the islet graft, whereas host DCs presenting donor allopeptide via the indirect pathway were not sufficient for stabilizing the infiltrating T cells.
Figure 2. Cross-dressed host DCs make stable contacts with Ag-specific, CD8+ effector T cells in islet allografts.

2P-IVM was performed on islet grafts 24 hours after transferring OT-I effectors as outlined in Figure 1A. Three mice were imaged per group with multiple locations (between 2 and 6) imaged per graft. (A) Equal infiltration of allografts with OT-I effectors (red) in all groups. Allografts were well vascularized (magenta) at the time of imaging and had abundant host-derived YFP+ DCs (green) in and around islets (blue). Supplemental Video 1, corresponding to the micrographs, illustrates dynamic behavior of OT-I in the grafts. Each data point in the plot represents total OT-I counts in 1 graft location imaged. Ctrl, control. (B) OT-I paths over 5-minute imaging period (blue dragon tails) and contact with DCs (white surfaces) are shown in the photomicrographs and in Supplemental Video 2. Quantification of OT-I DC contacts per image volume and contact time are shown in the graphs. Each data point in the first plot represents total OT-I DC contacts counts in 1 graft location imaged. Each data point in the second plot represents contact time for each OT-I cell (all cells that made contact with DCs for more than 90 seconds were analyzed per location imaged; range = 259–1450 cells/location). (C) Arrest of OT-I effectors in the cross-dressing group is depicted by the shorter paths they took compared with the indirect group (photomicrographs with paths tracked over a 30-minute period) and by the quantitation of mean speed and arrest coefficients (graphs). The arrest coefficient is the proportion of time in which a cell moves less than 2 μm/min. Each data point in the plots represents a single OT-I cell (all cells were analyzed per location imaged; range = 987–2361 cells/location). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Random intercept model with the Bonferroni’s multiple comparison adjustment for pairwise comparisons between group means was applied to avoid pseudoreplication errors. Comparisons not shown in Figure 2, B and C, were not significant. Scale bars: 50 μm (A); 40 μm (B); 100 μm (C).
Cross-dressed host DCs are sufficient for sustaining acute islet allograft rejection by effector CD8+ T cells.
To investigate the biological consequence of cross-dressed DCs interaction with effector T cells, we studied islet allograft survival in a mouse model in which antigen presentation is restricted to the cross-dressing pathway, rejection is mediated by effector CD8+ T cells, and host DCs can be depleted by administration of diphtheria toxin (DT). We transplanted B6.Act-mOVA pancreatic islets to B6.H-2Kb–/– mice that had received either a B6.H-2Kb–/– or B6.H-2Kb–/–.CD11c-DTR bone marrow transplant and were rendered diabetic 1 week prior to transplantation. Recipient DCs in the latter group expressed human diphtheria toxin receptor (DTR) under control of the CD11c promoter such that DT administration depleted host DCs. DT was administered every other day starting on the day of transplantation to both mouse groups, and OT-I effectors were transferred 7 days after transplantation. Allograft survival was monitored by measuring blood glucose levels. In this model, allograft rejection is dependent on exogenous OT-I due to the extremely low frequency of endogenous T cells that react to OVA. The transfer of effector, instead of naive, OT-I allowed us to study the role of intragraft DCs, since effector T cells bypass secondary lymphoid organs and home directly to and interact with DCs in the graft (16). Finally, by transplanting H-2Kb–sufficient grafts into H-2Kb–/– recipients, antigen presentation was restricted to the cross-dressing pathway. As shown in Figure 3, all grafts were rejected by 26 days in the group without DTR, while only 1 of 5 grafts was rejected on day 55 in the DTR group. The other recipients in the DTR group remained euglycemic at time of death, which may have been caused by extended DT exposure (censored data points on days 46, 49, and 57), or at time of termination of the experiment (day 72). These data demonstrate that cross-dressing of host DCs in the graft with donor MHC class I–peptide complexes is sufficient for driving acute rejection by TCR-transgenic CD8+ effector T cells. Although we did not test whether indirect antigen presentation is sufficient for acute rejection in this study, our previous work had shown that the indirect pathway alone is insufficient for the entry and accumulation of OT-I effector T cells in B6.Act-mOVA islet grafts (15).
Figure 3. Cross-dressed host DCs are sufficient for acute islet allograft rejection by effector CD8+ T cells.

B6.Act-mOVA (B6.OVA) islets were transplanted to diabetic B6 bone marrow chimeras that had received either a B6.H-2Kb–/– (n = 7) or B6.H-2Kb–/–.CD11c-DTR (n = 5) bone marrow transplant. 1 × 105 OT-I effectors were transferred 7 days after transplantation. DT (5 ng/g) was administered every other day starting on the day of transplantation to both mouse groups. Allografts were deemed rejected when blood glucose level exceeded 300 mg/dl for 3 consecutive days. Mice that died with normal glucose levels before termination of experiment (day 72) were censored and are shown as white circles on the survival line. **P < 0.01, log-rank test.
Cross-dressing of host DCs with donor MHC class I–peptide complexes in kidney allografts.
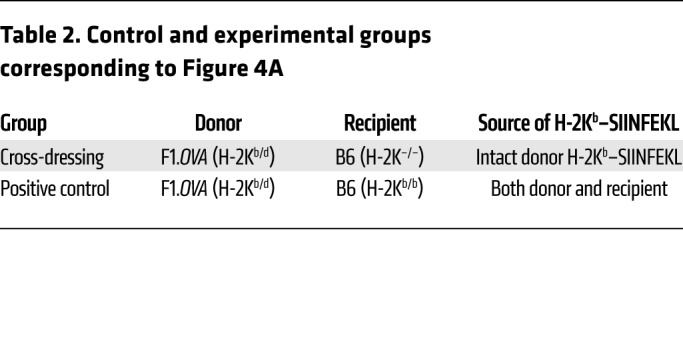
To investigate whether cross-dressing of host DCs also occurs in primarily vascularized allografts, we transplanted F1.Act-mOVA kidneys into B6 WT or H-2Kb–/– mice and transferred OT-I effectors 2 days later (Figure 4A and Table 2). In this model, acute histological rejection is observed in the first 2 weeks after transplantation, but the infiltrate declines and the grafts undergo slow chronic rejection. Allografts were harvested 10 or 60 days after transplantation and analyzed by cytometry and routine histology. As shown in Figure 4, B and C, the majority of host DCs (CD11c+) were cross-dressed with intact donor H-2Kd and H-2Kb–SIINFEKL complexes in both mouse groups at 10 days. Cross-dressed host DCs decreased significantly by 60 days, but remained quite conspicuous, representing approximately 5%–40% of the host DC population in the graft (Figure 4C). Histology and flow cytometry showed classical features of rejection with similar Banff grades and comparable infiltration with polyclonal CD4+ and CD8+ T cells in the 2 groups (Figure 4, D and E). Serum creatinine measurements at time of harvest were equivalent between groups (Figure 4D). We then examined the OT-I cells present in the grafts. We found that they constituted the majority of the T cell infiltrate at both time points and that their number and capacity to produce IFN-γ were comparable between H-2Kb–/– and WT recipients at the early time point (Figure 4E). However, significantly fewer OT-I cells were detected in the H-2Kb–/– group on day 60, although their capacity to produce IFN-γ remained equal to the WT group (Figure 4E). Together, these results indicate that cross-dressing of host DCs occurs extensively in mouse kidney allografts early after transplantation, is still present long-term in grafts, albeit at a lower level, and is sufficient for sustaining effector T cell accumulation and function in the graft. Diminished accumulation of OT-I cells in the cross-dressing group at the later time point, however, suggests that the indirect antigen presentation pathway may play an increasingly important role during chronic rejection.
Figure 4. Cross-dressing of host DCs with donor MHC class I–peptide complexes in kidney allografts.

(A) (B6×BALB/c)F1.Act-mOVA (F1.OVA) (H-2Kb/d) kidneys were transplanted to B6 H-2Kb–sufficient (H-2Kb/b) WT (n = 5–6) or B6 H-2Kb-deficient (H-2K–/–) (n = 4–6) recipients. 1 × 107 B6.Rag–/– OT-I CD8+ effector T cells, which recognize the OVA peptide SIINFEKL bound to H-2Kb, were transferred i.v. 2 days later. Grafts were analyzed 8 or 58 days after OT-I transfer. Control and experimental groups are shown in Table 2. (B) Leukocytes were isolated from transplanted allografts and analyzed by ImageStream. Intact H-2Kb–SIINFEKL complexes and donor H-2Kd molecules were identified as discreet spots on surface of host (CD11c+) DCs. Representative images from each group from day 10 grafts shown. On average, 426 (range = 153–784) and 3068 (range = 547–10366) cells were analyzed per animal on day 10 and day 60, respectively. (C) Proportion of host DCs positive for 1 or more spots of either H-2Kd or H-2Kb–SIINFEKL. Each data point represents analysis of 1 transplanted animal. (D) Histological analysis of grafts from cross-dressing and control groups removed on day 10 and day 60. Photomicrographs show examples of arteritis, tubulitis, intimal hyperplasia, and interstitial fibrosis and tubular atrophy (IFTA) in grafts from cross-dressed group. Scale bars: 50 μm. Bar graphs depict quantitation of Banff grades and serum creatinine on day 10 and day 60. Dashed line denotes lower detection limit for creatinine (<0.2 mg/dl). (E) Flow cytometric analysis of cellular infiltrate in allografts removed on day 10 and day 60 from cross-dressing and positive control groups. *P < 0.05; ***P < 0.001, 2-tailed unpaired t test.
Table 2. Control and experimental groups corresponding to Figure 4A.
Discussion
We have provided evidence in 2 transplantation models that cross-dressing of host DCs with donor MHC-peptide complexes is a major pathway by which donor alloantigens are presented to host effector T cells in the graft. We have also shown that this pathway is sufficient for allograft rejection. The significance of these findings is that they link alloantigen recognition in the graft to that in secondary lymphoid organs: directly alloreactive effector T cells, generated in response to intact donor allogeneic MHC-peptide complexes on host DCs in secondary lymphoid organs, recognize their cognate antigen on similarly cross-dressed host DCs in the graft. Since directly alloreactive T cells constitute the vast majority of host alloreactive T cells, targeting the cross-dressing pathway carries the promise of preventing or interrupting rejection mediated by effector or memory T cells that enter the graft.
Our findings also raise the possibility that cross-dressed DCs activate indirectly alloreactive T cells, which recognize allo-peptides bound to self-MHC molecules, if the donor and recipient share one or more MHC loci. In other words, a given MHC molecule common to the donor and recipient could efficiently present endogenous allopeptides to and activate indirectly alloreactive host T cells once transferred to host DCs. This would constitute a rapid mechanism of amplifying the indirect alloimmune response by bypassing the need for antigen uptake and processing by host DCs, akin to what has been observed after viral infection (17).
Our observation that cross-dressed host DCs are still present at a much later time point in kidney allografts underscores that the cross-dressing pathway is not restricted to the early posttransplantation period, but may also contribute to chronic rejection. It is unclear at this point whether host DCs that acquire donor MHC-peptide complexes immediately after transplantation persist long-term in the graft, or whether cross-dressing, for example via exosome release (18), is an ongoing, chronic process. It is also unclear what the relative contributions of cross-dressing versus the indirect pathway are to chronic rejection. Our observation that the number of graft-infiltrating, donor antigen-specific T cells diminishes with time in the absence of the indirect pathway suggests that this pathway becomes increasingly important over time. This concept is supported by older studies in the literature, but remains to be tested directly in our model (19, 20).
In this study, we adoptively transferred TCR-transgenic CD8+ T cells (OT-I) that recognize a model antigen (OVA) to lymphocyte-replete transplant recipients to investigate the role of cross-dressing in the alloimmune response. This approach was necessary for tracking donor antigen-specific T cells and for manipulating antigen presentation by eliminating a single MHC molecule (H-2Kb). Alloreactive polyclonal T cells cannot be tracked by present methodologies, and antigen presentation to these cells cannot be manipulated without global deletion of MHC class I and/or class II molecules, which would result in global deletion of target alloantigens in the graft or of CD8+ T cells, CD4+ T cells, or both in the host (21). We maximized the physiological relevance of our model by transplanting organs from F1.OVA mice that express allogeneic MHC molecules as well as the model antigen, thus eliciting both a polyclonal and a TCR-transgenic response in the host. We believe that our conclusions are applicable to polyclonal T cells because we did observe transfer of allogeneic H-2Kd molecules to which the polyclonal T cells react, because of the universality of TCR-MHC-peptide complex interactions at the molecular level, and because prior work has shown that TCR-transgenic T cells specific to a nonself (mismatched) MHC molecule are also activated via the cross-dressing pathway (8).
In summary, our findings imply that the recent shift in our understanding of which antigen presentation pathway is key to initiating alloimmune responses in secondary lymphoid organs also applies to events that occur in the transplanted organ. They also open avenues for therapeutic intervention aimed at interrupting effector or memory T cell activation and function in the graft.
Methods
Mice.
B6 (C57BL/6J; Thy1.2, CD45.2, H-2Kb), B6.Act-OVA (C57BL/6J-Tg[CAG-OVA]916Jen/J; CD45.2, H-2Kb), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1), OT-I (C57BL/6-Tg[TcraTcrb]1100Mjb/J; CD45.2, H-2Kb), CD11c-DTR/eGFP (B6.FVB-1700016L21RikTg(Itgax–DTR/EGFP)57Lan/J), DsRed (B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J), and B6.CD11c-YFP (B6.Cg-Tg(Itgax-Venus)1Mnz/J) mice were purchased from The Jackson Laboratory. B6.H-2Kb–/– (catalog 4216) mice were purchased from Taconic, and BALB/c mice (BALB/cAnNCrl; CD45.2, H-2Kd) from Charles River. B6.Act-OVA.H-2Kb–/– and B6.CD11c-YFP.H-2Kb–/– mice were generated in our animal facility by breeding B6.Act-OVA and B6.CD11c-YFP mice with B6 H-2Kb–/– mice, respectively. OT-I mice were bred onto a B6.RAG–/– (Thy1.2) background; then OT-I DsRed mice were generated by breeding B6.RAG–/–.OT-I mice with B6.DsRed mice. Flow cytometry of OT-I cells from these mice was performed on blood to ensure high-level DsRed expression. F1.Act-mOVA and F1.Act-mOVA.H-2Kb–/– mice were generated by breeding BALB/c mice with B6.Act-OVA and B6.Act-OVA.H-2Kb–/– mice, respectively. B6.H-2Kb–/–. CD11c-DTR.CD11c-YFP mice were generated by breeding B6.H-2Kb–/– with B6.CD11c-YFP mice, followed by crossing with B6.CD11c-DTR mice. B6.H-2Kb–/–.CD11c-YFP.CD11c-DTR and B6.H-2Kb–/–.CD11c-YFP bone marrow chimeras were generated by irradiating B6.H-2Kb–/– mice at 10 Gy followed by adoptive transfer of 1 × 107 cells i.v. taken from the femurs of B6.H-2Kb–/–.CD11c-YFP.CD11c-DTR and B6.H-2Kb–/– mice, respectively. These mice were given antibiotic (sulfatrim) in food for 2 weeks following irradiation. Reconstitution was confirmed 8 weeks after bone marrow transplantation. Mice were maintained under specific pathogen–free (SPF) conditions.
Reagents.
Anti-CD11c-biotin (clone HL3), andi-CD11b-biotin (M1/70), anti-CD16/32-biotin (2.4G2), anti-B220-biotin (RA3-6B2), and anti-CD45.1-FITC (A20) were purchased from BD Biosciences — Pharmingen; anti-CD4-BV421 (GK1.5) and anti-CD11b-PE-CF594 (M1/70) were from BD Horizon; anti-CD8-FITC (53-6.7), anti-IFN-γ-PE-Cy7 (XMG1.2), F4/80-biotin (BM8), anti-CD49b-biotin (DX5), anti-CD45.2-APC-eFluor780 (30-F11), streptavidin-APC, and anti-H-2Kb–SIINFEKL (eBio25-D1.16) were from eBioscience; anti-H-2Kd-PE-Cy7 (SF1-1.1) were from BioLegend; and Ter119-biotin (TER-119) and CellTracker Violet BMQC were from Invitrogen. From Sigma-Aldrich, we obtained Ficoll PM400, collagenase D, DNAse I, collagenase V, and DT. Dec205-OVA fusion antibody provided in-house. Anti-CD40 (FGK45) was purchased from BioXCell.
Islet transplantation.
Mouse pancreatic islets were isolated and transplanted as established by Bertera et al. (22). Briefly, pancreata were perfused with collagenase V injected into the common bile duct, harvested, and digested. Islets were separated over a Ficoll gradient, handpicked with a Pasteur pipette, and cultured overnight in complete RPMI medium supplemented with 10% FBS at 37°C and 5% CO2. For graft survival experiments, islets were injected immediately into the left renal subcapsular space of diabetic mice. For imaging experiments, cultured islets were labeled with 10 μM CellTracker Violet for 1 hour before transplantation. Diabetes was induced by injecting 200 mg/kg streptozocin in 0.1M sodium citrate buffer (pH 4.0) i.p. 4 days prior to transplantation and confirmed by elevated blood glucose levels (>300 mg/dl). Blood glucose of less than 150 mg/dl after transplantation was considered engraftment, and more than 300 mg/dl for 3 consecutive days was considered islet graft nonengraftment or rejection. For survival experiments, 5 ng/g body weight DT was injected i.p. every other day starting from the time of transplantation.
Kidney transplantation.
Mouse kidney transplants were performed as previously described (11) using (BALB/c × B6)F1.mAct-OVA donors and B6 recipients. Recipient native kidneys were removed at the time of transplantation. Allograft function was monitored by visual observation of recipients for signs of uremia. Serum creatinine was determined at time of harvest.
Generation and adoptive transfer of OT-I effector T cells.
Spleens from B6.RAG–/–.OT-I DsRed+ or Thy1.1+ (CD45.2) mice were processed to single-cell suspensions, and 1 to 5 × 105 splenocytes were transferred i.v. to B6 (CD45.1) mice. These recipient mice were then immunized with 25 μg of anti-Dec205-OVA antibody (Dec205-OVA) i.v. plus 50 μg of agonistic anti-CD40 (FGK45) i.p. to generate OT-1 effector cells (23). Spleens were harvested 5 days later,and CD45.2+DsRed+/Thy1.1+CD4–CD45.1– Lin (CD11c, CD11b, Ter119, CD16/32, B220, F4/80, DX5)– cells were sorted on a high-speed cell sorter (BD Aria). Sorted cells were washed and counted, and 5 × 106 cells were injected i.v. into transplanted mice for islet allograft imaging experiments, 1 × 105 for islet allograft survival studies, and 1 × 107 for kidney allograft studies. The vast majority of transferred OT-I cells were CD44hiCD62Llo (10).
2P-IVM.
2P-IVM was performed on islet grafts transplanted under the left kidney capsule. A Nikon A1 Multiphoton microscope equipped with a Chameleon femtosecond-pulsed laser (Coherent) tuned and mode-locked to 860 nm, available through the University of Pittsburgh Center for Biological Imaging, was used for all experiments. Mice were anesthetized with isoflurane and oxygen, and core body temperature was maintained at 37°C with a homeothermic controller (CWE Inc.). Blood vessels were visualized by injecting 75 μg Evans blue i.v. The kidney housing the transplanted islets was extraverted from its original location with intact vascular connection and immobilized in a custom cup mount (24). Z-stacks were acquired with a ×25 water immersion objective (NA: 1.05) 20–80 μm below the kidney capsule. Slices were acquired at a step size of 4.5 μm. Brightness and laser power were adjusted based on imaging depth and kept below phototoxic level. Images were acquired with a resonance scanner with 8× averaging at a resolution of 512 × 512 pixels. Approximately 36-second–long stacks were repeatedly scanned up to 60 times for a maximum imaging time of 36 minutes per location. Multiple different locations per islet graft were imaged. Spectral unmixing was performed on all video files offline using Nikon Elements software (version 4.x). All acquired videos were analyzed using Imaris software (Bitplane). Drift was corrected using YFP+ cells as a reference point. All DsRed+ cells detected during the imaging time were enumerated and tracked using Imaris.
Tissue digestion and flow cytometry.
After imaging, mice were euthanized and the kidney containing the graft was excised and placed immediately in cold PBS. The kidney capsule was then manually incised around the graft, and tissue containing the islets was scraped off with forceps using a stereoscope (Olympus). Islets were incubated in RPMI media containing 500 U/mL collagenase D, 0.02 mg/mL DNAse I, and 5% FBS for 30 minutes at 37°C. This suspension was filtered through a 70 μm filter and washed. The pellet was treated with RBC lysis buffer for 5 minutes at RT and washed again. Spleens were homogenized by crushing the organ through a 40 μm filter, followed by RBC lysis for 5 minutes at RT. Cells were then stained for viability, CD11b, H-2Kd, and H-2Kb–SIINFEKL and analyzed by imaging flow cytometry (Amnis ImageStreamX Mark II). Viable CD11c+(YFP+)CD11b+DsRed–CTV– cell expression characteristics, such as spot counting, were then analyzed offline using IDEAS software (BD). For renal allografts, kidney tissue was digested and cell suspension analyzed by imaging flow cytometry as described for islet grafts. Additional flow cytometry was performed to quantify CD4+, CD8+, and OT-I cells. IFN-γ production was measured by flow cytometry 16 hours after ex vivo stimulation with donor splenocytes as previously described (25).
Histological analysis.
Kidney allograft tissue was fixed in paraformaldehyde, paraffin embedded, sectioned, and stained with H&E, Masson trichrome, PAS, and Verhoeff–Van Giessen stain. All slides were scanned on a Zeiss Axioscan, with blinded reading by a renal pathologist and graded according to the Banff criteria (26). For quantification purposes, Banff grades were assigned the following numerical values: normal = 0, borderline = 0.5, IA = 1.0, IB = 1.5, IIA = 2.0, and IIB = 2.5.
Statistics.
Statistical analysis of allograft survival was calculated using the log-rank test. Data shown in Figure 2 were analyzed as follows: to assess statistical significance of differences in the contact count or OT-I number across groups (negative control, positive control, indirect, and cross-dressed), a random intercept linear model was fit in Stata (27) to account for potential within-mouse correlations across multiple videos. If the overall (Wald) test was statistically significant (P < 0.05), each of the 6 pair-wise contrasts was then tested using Bonferroni’s adjustment to account for multiple comparisons. For assessing differences in contact time, mean speed, or arrest coefficient, a random intercept model (and the same test of the overall model followed by tests of the pair-wise group contracts) was again fit except with a separate random intercept for each video to account for the multiple measurements and associated within-movie correlation.
All other experiments were analyzed using either 1-way ANOVA with Tukey’s multiple comparison test or the 2-tailed, nonparametric, unpaired t test. All statistical calculations were made using GraphPad Prism, version 8.12, or Stata software. P < 0.05 was considered significant.
Study approval.
All animal studies were approved by the University of Pittsburgh IACUC.
Author contributions
ADH, DZ, HD, KIAD, and RT performed experiments and data analysis. RR interpreted renal allograft pathology. ALW managed animal breeding and genotyping. ADH, MHO, AEM, and FGL planned experiments and wrote the manuscript. WDS participated in experimental design and edited the manuscript. DPL performed statistical analyses. MHO and FGL directly supervised the work.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI099465 and AI064343 (to FGL), HL130191 (to AEM), an ASN Merrill Grant in Transplantation (to MHO), the American Society of Transplantation TIRN predoctoral scholarship (to ADH), the American Society of Nephrology Ben J. Lipps Research Fellowship (to KIAD), and the Frank & Athena Sarris Chair in Transplantation Biology (to FGL). The Center for Biologic Imaging and the Flow Core at the University of Pittsburgh are funded by NIH grants 1S10RR028478 and 1S10OD019942, respectively.
Version 1. 11/25/2019
Electronic publication
Version 2. 01/02/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(1):287–294.https://doi.org/10.1172/JCI125773.
Contributor Information
Andrew D. Hughes, Email: Hughes.Andrew@medstudent.pitt.edu.
Daqiang Zhao, Email: DAQIANGZ@pitt.edu.
Hehua Dai, Email: daih@upmc.edu.
Khodor I. Abou-Daya, Email: kha17@pitt.edu.
Roger Tieu, Email: rot35@pitt.edu.
Rayan Rammal, Email: rr52@aub.edu.lb.
Amanda L. Williams, Email: williamsal@upmc.edu.
Douglas P. Landsittel, Email: dpl12@pitt.edu.
Warren D. Shlomchik, Email: warrens@pitt.edu.
Adrian E. Morelli, Email: morelli@pitt.edu.
Martin H. Oberbarnscheidt, Email: mho6@pitt.edu.
Fadi G. Lakkis, Email: lakkisf@upmc.edu.
References
- 1.Lombardi G, Sidhu S, Daly M, Batchelor JR, Makgoba W, Lechler RI. Are primary alloresponses truly primary? Int Immunol. 1990;2(1):9–13. doi: 10.1093/intimm/2.1.9. [DOI] [PubMed] [Google Scholar]
- 2.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166(2):973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 3.Macedo C, et al. Contribution of naïve and memory T-cell populations to the human alloimmune response. Am J Transplant. 2009;9(9):2057–2066. doi: 10.1111/j.1600-6143.2009.02742.x. [DOI] [PubMed] [Google Scholar]
- 4.Alegre ML, Lakkis FG, Morelli AE. Antigen presentation in transplantation. Trends Immunol. 2016;37(12):831–843. doi: 10.1016/j.it.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lafferty KJ, Prowse SJ, Simeonovic CJ, Warren HS. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu Rev Immunol. 1983;1:143–173. doi: 10.1146/annurev.iy.01.040183.001043. [DOI] [PubMed] [Google Scholar]
- 6.Larsen CP, Morris PJ, Austyn JM. Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med. 1990;171(1):307–314. doi: 10.1084/jem.171.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrera OB, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol. 2004;173(8):4828–4837. doi: 10.4049/jimmunol.173.8.4828. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q, et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J Clin Invest. 2016;126(8):2805–2820. doi: 10.1172/JCI84577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marino J, et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Sci Immunol. 2016;1(1):aaf8759. doi: 10.1126/sciimmunol.aaf8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walch JM, et al. Cognate antigen directs CD8+ T cell migration to vascularized transplants. J Clin Invest. 2013;123(6):2663–2671. doi: 10.1172/JCI66722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhuang Q, et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat Commun. 2016;7:12623. doi: 10.1038/ncomms12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrod KR, Liu FC, Forrest LE, Parker I, Kang SM, Cahalan MD. NK cell patrolling and elimination of donor-derived dendritic cells favor indirect alloreactivity. J Immunol. 2010;184(5):2329–2336. doi: 10.4049/jimmunol.0902748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laffont S, et al. CD8+ T-cell-mediated killing of donor dendritic cells prevents alloreactive T helper type-2 responses in vivo. Blood. 2006;108(7):2257–2264. doi: 10.1182/blood-2005-10-4059. [DOI] [PubMed] [Google Scholar]
- 14.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, et al. CD8+ effector T cell migration to pancreatic islet grafts is dependent on cognate antigen presentation by donor graft cells. J Immunol. 2016;197(4):1471–1476. doi: 10.4049/jimmunol.1600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci USA. 2002;99(9):6175–6180. doi: 10.1073/pnas.092596999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature. 2011;471(7340):629–632. doi: 10.1038/nature09863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montecalvo A, et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol. 2008;180(5):3081–3090. doi: 10.4049/jimmunol.180.5.3081. [DOI] [PubMed] [Google Scholar]
- 19.Vella JP, et al. Indirect allorecognition of major histocompatibility complex allopeptides in human renal transplant recipients with chronic graft dysfunction. Transplantation. 1997;64(6):795–800. doi: 10.1097/00007890-199709270-00001. [DOI] [PubMed] [Google Scholar]
- 20.Baker RJ, Hernandez-Fuentes MP, Brookes PA, Chaudhry AN, Cook HT, Lechler RI. Loss of direct and maintenance of indirect alloresponses in renal allograft recipients: implications for the pathogenesis of chronic allograft nephropathy. J Immunol. 2001;167(12):7199–7206. doi: 10.4049/jimmunol.167.12.7199. [DOI] [PubMed] [Google Scholar]
- 21.Grusby MJ, et al. Mice lacking major histocompatibility complex class I and class II molecules. Proc Natl Acad Sci USA. 1993;90(9):3913–3917. doi: 10.1073/pnas.90.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertera S, Balamurugan AN, Bottino R, He J, Trucco M. Increased yield and improved transplantation outcome of mouse islets with bovine serum albumin. J Transplant. 2012;2012:856386. doi: 10.1155/2012/856386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonifaz LC, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camirand G, et al. Multiphoton intravital microscopy of the transplanted mouse kidney. Am J Transplant. 2011;11(10):2067–2074. doi: 10.1111/j.1600-6143.2011.03671.x. [DOI] [PubMed] [Google Scholar]
- 25.Oberbarnscheidt MH, et al. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest. 2014;124(8):3579–3589. doi: 10.1172/JCI74370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solez K, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8(4):753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 27. Hardin JW, Hilbe JM. Generalized linear models extensions. College Station, TX; Stata Press: 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.