

Abstract
X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia (XMEN) disease are caused by deficiency of the magnesium transporter 1 (MAGT1) gene. We studied 23 patients with XMEN, 8 of whom were EBV naive. We observed lymphadenopathy (LAD), cytopenias, liver disease, cavum septum pellucidum (CSP), and increased CD4–CD8–B220–TCRαβ+ T cells (αβDNTs), in addition to the previously described features of an inverted CD4/CD8 ratio, CD4+ T lymphocytopenia, increased B cells, dysgammaglobulinemia, and decreased expression of the natural killer group 2, member D (NKG2D) receptor. EBV-associated B cell malignancies occurred frequently in EBV-infected patients. We studied patients with XMEN and patients with autoimmune lymphoproliferative syndrome (ALPS) by deep immunophenotyping (32 immune markers) using time-of-flight mass cytometry (CyTOF). Our analysis revealed that the abundance of 2 populations of naive B cells (CD20+CD27–CD22+IgM+HLA-DR+CXCR5+CXCR4++CD10+CD38+ and CD20+CD27–CD22+IgM+HLA-DR+CXCR5+CXCR4+CD10–CD38–) could differentially classify XMEN, ALPS, and healthy individuals. We also performed glycoproteomics analysis on T lymphocytes and show that XMEN disease is a congenital disorder of glycosylation that affects a restricted subset of glycoproteins. Transfection of MAGT1 mRNA enabled us to rescue proteins with defective glycosylation. Together, these data provide new clinical and pathophysiological foundations with important ramifications for the diagnosis and treatment of XMEN disease.
Keywords: Immunology
Keywords: Genetic diseases, Glycobiology, Proteoglycans

Introduction
X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia (XMEN) disease is a rare primary immunodeficiency caused by hemizygous loss-of-function (LOF) mutations in the X-linked magnesium transporter 1 (MAGT1) gene in males (1–3). Heterozygous females are healthy carriers with lyonization skewed in their hematopoietic cells to the X chromosome bearing the normal allele (1). MAGT1 loss was first shown to affect intracellular magnesium (Mg2+) homeostasis, leading to defective T cell immune responses and uncontrolled EBV infection with increased susceptibility to EBV+ lymphoma. Patients had an inverted CD4/CD8 ratio, increased B cells, and decreased surface expression of the activator receptor natural killer group 2, member D (NKG2D) on NK and CD8+ T cells (1–3). NKG2D loss predisposes individuals to EBV-driven lymphoproliferative disease (LPD) and lymphoma (4). In certain patients, features resembling autoimmune lymphoproliferative syndrome (ALPS), a disease of lymphocyte homeostasis due to defective FAS-mediated apoptosis, became apparent (5–9). Patients with ALPS have enlarged secondary lymphoid tissue and an expansion of αβ T cells lacking both CD4 and CD8 coreceptors (αβ double-negative T cells [αβDNTs]) but expressing the CD45R isoform B220 (10). The full scope of XMEN disease manifestations and their pathogenic cause compared with ALPS have not yet been described.
Protein glycosylation is a posttranslational modification critical for normal immune function (11). MAGT1 has high amino acid sequence homology with the human tumor suppressor candidate 3 protein (TUSC3) and the yeast oligosaccharyl transferase 3/6 (OST3/6) proteins that participate in the enzymatic complex that performs asparagine N-linked glycosylation (NLG) in the endoplasmic reticulum (ER) (12–14). Each OST complex has 1 catalytic subunit, either STT3A or STT3B, and multiple noncatalytic subunits creating distinct but complementary NLG enzyme complexes (15, 16). Although there is substantial overlap in the peptides glycosylated by the 2 OST complexes, STT3A primarily glycosylates substrate peptides cotranslationally, whereas STT3B is involved in either cotranslational or posttranslational glycosylation of peptides skipped by STT3A (16, 17). STT3A preferentially glycosylates acceptor sites in cysteine-rich regions and the amino terminus of multipass transmembrane (TM) proteins (18). Conversely, STT3B favors sequons that would be difficult to glycosylate cotranslationally, including those in the terminal 50–55 amino acids of the carboxyl tail and short loops between TM regions (17, 18). MAGT1 can associate with the STT3B-containing OST complex and promote NLG of STT3B-dependent glycoproteins in human tumor cell lines (14, 19). Genetic diseases affecting protein glycosylation, congenital disorders of glycosylation (CDG), can involve genes that add glycans to proteins in the ER (type I) or further process protein-bound glycans in the Golgi apparatus (type II) (20, 21). The clinical manifestations and severity of CDG are heterogeneous depending on the specific genetic and molecular defects. More recently, a different clinical phenotype manifested by intellectual and developmental disability was described for 2 patients with MAGT1 mutations. These individuals had abnormal glycosylation as determined by serum transferrin isoelectric focusing (sTf IEF) and hypoglycosylated STT3B–dependent substrates in patient-derived cell lines (22). However, the extent of the glycosylation defect and an in-depth analysis of the glycopeptides affected by loss of MAGT1 in human lymphocytes have not been described.
Here, we report new aspects of the largest cohort of EBV-naive and EBV-infected patients with XMEN. We use deep immunophenotyping of PBMCs by mass cytometry combined with a new machine learning algorithm and cluster analysis of multidimensional data to delineate lymphocyte subsets that distinguish patients with XMEN, patients with ALPS, and healthy controls (HCs). We performed global glycoproteomics analysis of T lymphocytes, which revealed a selective NLG defect in XMEN disease affecting multiple immune proteins. Finally, we show that MAGT1 mRNA transfection reversed defective glycosylation in peripheral lymphocytes. Together, our data show that XMEN disease has previously unidentified features, some of which may be attributable to MAGT1 as a newly recognized facilitator of NLG.
Results
Patient mutations and demographics.
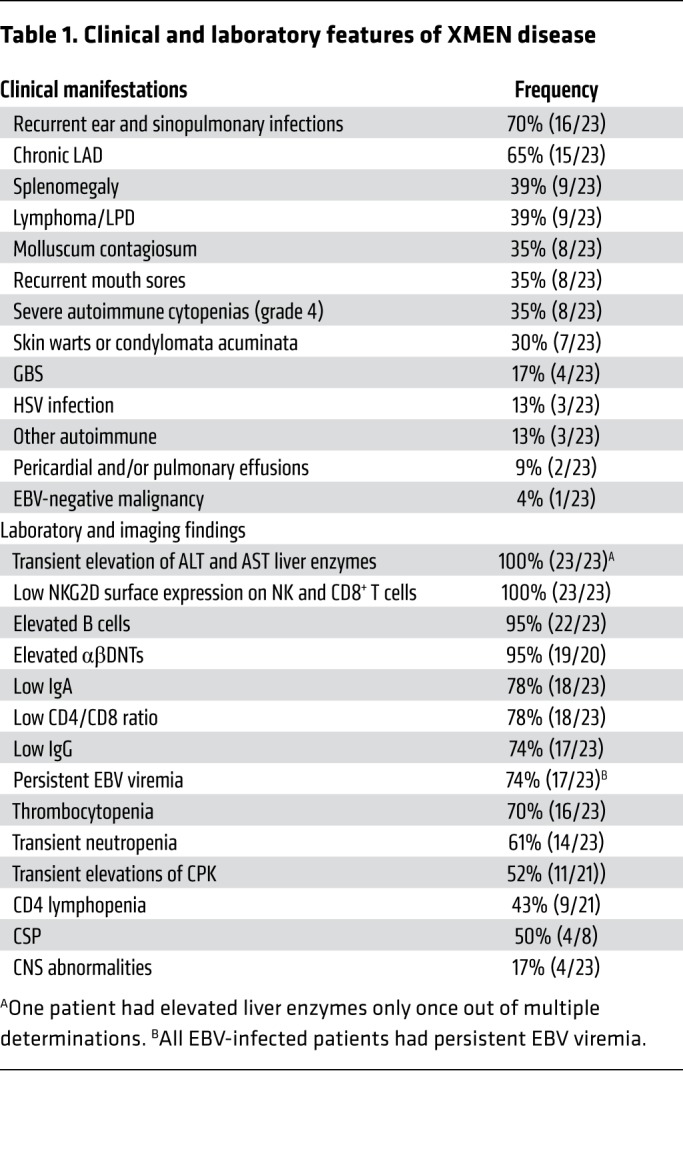
We reviewed the records of 23 patients from 17 unrelated families (A, B, and D–R) with LOF MAGT1 mutations. We observed that XMEN is a multisystem disease that is more complex than previously appreciated (3, 23–26). (Figure 1, A and B, Table 1, and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI131116DS1). The cohort was 70% white, non-Hispanic, 13% black, 13% multirace, and 4% Hispanic. All patients were males, consistent with the X-linked inheritance. Eight individuals (aged 5–17 years) were EBV naive, whereas 15 (aged 9–50 years) had chronic EBV infection (Supplemental Table 1 and Supplemental Table 2). Two of the EBV-naive patients subsequently developed EBV infection.
Figure 1. Clinical, laboratory, and genetic findings in XMEN disease.

Clinical manifestations (A) and laboratory findings (B) in XMEN disease. AHA, autoimmune hemolytic anemia (AHA); ITP, immune thrombocytopenic purpura. (C) Immunoblot showing MAGT1 and β-tubulin proteins in T cell blasts from HCs (HC 1 and HC 2) and patients with XMEN with the indicated mutations. (D) NKG2D expression on CD8+ T cells and NK cells from HCs (blue), patients with XMEN (red), and an isotype control (gray).
Table 1. Clinical and laboratory features of XMEN disease.
We identified 14 unique molecular alterations of MAGT1: missense mutations in 10 families, nucleotide deletions in 4, nucleotide insertions in 2, and large deletions in 2 (Supplemental Table 3). The most common mutation (c.409C>T, p.Arg137X) appeared in 4 unrelated families. The 409 C residue occurs in a CpG dinucleotide, which has a propensity for deamination and conversion of cytosine to thymine, thereby producing the mutation. All mutations tested abolished MAGT1 protein expression (Figure 1C). The most consistent diagnostic finding was decreased NKG2D surface expression on both CD8+ T cells and NK cells (P < 0.0001), making it a hallmark of the disease whether or not EBV infection was present (Figure 1D and Supplemental Figure 1A). Interestingly, we observed that in 5 families with 2 male nonidentical siblings and no other children, the frequency of coincidence of disease in both males (4 of 5 families) was much higher than expected by chance (P = 0.04).
Multisystem abnormalities.
Our examination of this larger cohort of EBV-infected and EBV-naive patients revealed clinical abnormalities in other organ systems. We observed mild thrombocytopenia (70%) (Figure 2A), transient neutropenia (61%) sometimes associated with mouth sores (Figure 2B), and antibody-mediated cytopenias (35%) (Table 1, and Supplemental Table 1 and Supplemental Table 2). Bone marrow biopsies done to evaluate cytopenias in patients without malignancies showed normal trilineage hematopoiesis. We also found noninfectious liver abnormalities including transient elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with preserved hepatic function (Figure 2C, Supplemental Table 2). Transient elastography was abnormal in 3 of 7 patients. Liver ultrasound showed increased echogenicity (steatosis) in some patients (Figure 2D). Liver biopsies from both EBV-naive and EBV-infected patients and pathology of the liver from a patient who died from EBV-associated LPD revealed variable degrees of periportal inflammatory infiltrates and sinusoidal fibrosis, hepatosteatosis, mild-to-moderate iron deposits, and diffuse glycogenosis (Figure 2, E–G, Supplemental Figure 1, I–N, and Supplemental Table 4). All liver specimens from EBV-infected patients were EBV-encoded small RNA (EBER) negative. Conventional autoantibodies associated with autoimmune hepatitis (AH) were negative, except for 1 previously reported patient for whom we reviewed the liver biopsy and found no histopathological evidence of AH (25). Transient and asymptomatic elevations of serum muscle creatine phosphokinase (CPK) were detected (52%). Acute immune-mediated polyneuropathy or Guillain-Barré syndrome (GBS) was seen in 1 EBV-naive and 3 EBV-positive patients. Three patients had seizures. Eight patients had brain imaging (MRI or CT scan) and the most serious finding was atrophy of the cerebrum, cerebellum, brainstem, and spinal cord, with calcifications in the basal ganglia and thalami of a 30-year-old male with progressive neurological and cognitive decline (Figure 2H, Supplemental Figure 1G). Two patients, aged 45 and 48 years, had brain atrophy greater than expected for their age, and 2 patients had white matter abnormalities consistent with leukoencephalopathy (Figure 2I). Interestingly, 50% of the patients who underwent brain imaging had cavum septum pellucidum (CSP), a developmental ventricular change of uncertain significance that is found in only approximately 15% of the general population (Figure 2J). We found no evidence of intellectual disability and/or facial dysmorphism in our XMEN cohort. Taken together, these findings suggest a much broader disease phenotype outside of the immune system than previously suspected.
Figure 2. Multisystem abnormalities in XMEN disease.

Platelet (A) and absolute neutrophil (B) counts for an EBV-naive (red) patient and an EBV-positive (blue) patient with a normal range (gray) and patients’ age. (C) ALT and AST levels in EBV-naive (black), EBV-infected (red), and EBV-naive patients who became EBV infected (blue) with a normal range (light blue) and patients’ identities (shown on the x axis). (D) Increased hepatic echogenicity in liver ultrasound from an EBV-naive patient. (E and F) H&E-stained (HE) liver biopsy from an EBV-naive patient showing mild focal portal chronic inflammatory infiltrates (black arrow) without interface hepatitis and hepatocytes with pale cytoplasm. (G) Masson’s trichrome–stained image of liver biopsy sample from the same EBV-naive patient shows periportal fibrosis (black arrow). (H) T1-weighted MRI demonstrating severe diffuse atrophy of the cerebrum, cerebellum, brainstem, and spinal cord (yellow arrows). (I) T2 fluid-attenuated inversion recovery (FLAIR) brain MRI showing occipital leukomalacia (yellow arrow). (J) T2-weighted brain MRI demonstrating CSP (yellow arrow). (K) CT scan of paranasal sinuses showing opacification of the maxillary sinuses (yellow arrows) in a patient with bacterial sinusitis. (L) CT scan showing extensive tree-in-bud nodular infiltrates in the right lung (yellow circle) in a patient with longstanding hypogammaglobulinemia. (E–G) Scale bars: 50 μm (gray) and 100 μm (black).
Hypogammaglobulinemia, lymphoproliferation, and malignancy.
Most patients had decreased IgG and IgA with normal IgM associated with recurrent ear and sinus infections (Figure 2K, Table 1, Supplemental Table 1, and Supplemental Table 2). Upper and lower respiratory infections due to longstanding hypogammaglobulinemia sometimes led to bronchiectasis (Figure 2L). We observed molluscum contagiosum in 35% of patients (Supplemental Figure 1B). We observed flat warts predominantly affecting the palms and soles in 30% of the patients, and 1 young adult developed large perineal condylomata acuminata. These findings suggested a broader immune deficiency than previously noted (1–3).
Many EBV-naive and EBV-infected patients had chronic lymphoadenopathy (LAD) (65%) (Figure 3, A–D, Supplemental Figure 1, C–F, Table 1, and Supplemental Table 1 and Supplemental Table 5). We detected splenomegaly and lymphoma in half of the patients with persistent EBV viremia, whereas only 1 EBV-naive patient had splenomegaly (Figure 3C). A lymph node (LN) biopsy from an EBV-naive child showed Castleman-like changes (Figure 3E), whereas a tissue biopsy of a large mediastinal mass in another EBV-naive patient revealed a diffuse, predominantly IgD+ B lymphoid proliferation without underlying discernible follicular or nodal architecture and no evidence of clonal expansion (Figure 3F). A LN biopsy from a patient with persistent EBV viremia revealed florid reactive lymphoid hyperplasia and increased EBER-positive cells (Figure 3G). We also observed EBV-LPD with a characteristic increase in CD20+ and CD30+ cells in a patient with recent EBV infection (Figure 3H). Lymphoma occurred frequently at a young age in some EBV-infected patients but not in the EBV-naive patients (Supplemental Table 6). The most common neoplasia was Hodgkin lymphoma (HL) (n = 4). A young adult with a history of HL in complete remission developed EBV-negative liposarcoma (Supplemental Figure 1H).
Figure 3. Lymphoproliferation in XMEN disease.

(A) CT scan of chest, abdomen, and pelvis depicting bilateral axillary, mesenteric, and inguinal LAD in an EBV-infected patient (yellow arrows). (B) Contrast-enhanced CT scan of the chest showing axillary LAD (yellow arrow) in an EBV-naive patient. (C) Contrast-enhanced CT scan of the abdomen depicting splenomegaly (red arrow) and mesenteric LAD (yellow arrows) in an EBV-infected patient. (D) FDG-PET scan of a patient with XMEN and EBV-positive LPD showing hypermetabolic splenomegaly and bilateral axillary LAD (yellow arrows). (E) HE stain of cervical LN biopsy from an EBV-naive patient showing reactive lymphoid hyperplasia with Castleman-like changes (black arrows) and negative EBER ISH staining. (F) Image of biopsy sample from a large mediastinal mass showing lymphoid tissue obliterating normal LN architecture and negative EBER staining. (G) H&E-stained sections of an inguinal LN biopsy from an EBV-positive patient showing reactive lymphoid hyperplasia and positive EBER staining. (H) HE, EBER, CD20, and CD30 staining of cervical LN biopsy tissue from a patient with EBV LPD. (I) Percentage relative to CD3+ DNTs, αβDNTs, and B220+ αβDNTs from age-matched HCs (n = 10), EBV-naive patients (EBV–, n = 6), EBV-infected patients (EBV+, n = 10), and ALPS patients (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, by ordinary 1-way ANOVA with Tukey’s multiple comparisons test (α = 0.05). (J) Percentage of T cell death after treatment with different concentrations of the agonistic anti-FAS antibody APO-1.3 or anti-CD3 antibody for HCs, patients with ALPS (ALPS-1 and ALPS-2), and patients with XMEN (A.3, Q.1, and H.1). Results are representative of all XMEN patients tested (n = 13) in 3 independent experiments. (E–H) Scale bars: 50 μm (gray), 100 μm (black), and 200 μm (white).
The immune phenotype of EBV-infected and EBV-naive XMEN patients was very similar (Supplemental Figure 2, and Supplemental Table 1 and Supplemental Table 2). Characteristic peripheral blood abnormalities included increased B cells (95%) and αβDNT cells (95%). Most patients (78%) had low (<1.1) CD4/CD8 ratios (range, 0.17–1.9) (Supplemental Figure 2 and Supplemental Table 2). CD4 T cell lymphopenia was noted in 43% of patients, although opportunistic infections were rarely observed. The NK and NKT cell counts were normal for most patients. Recent CD31+ thymic emigrants were also normal (n = 13). The LPD, lymphoma predisposition, and increased double-negative T cells (DNTs) in patients with XMEN disease were reminiscent of ALPS, raising a question as to how these disorders can be distinguished by peripheral blood lymphocyte populations (5–9).
Patients with XMEN share some features of ALPS including elevated αβDNTs, defective apoptosis, and secondary lymphoid expansion.
Most patients with XMEN (95%) had elevated αβDNT cells warranting further investigation (Figure 3I and Supplemental Table 2). Because ALPS αβDNTs have been well documented to express an unusual CD45 glycosylation form similar to mouse B220, we stained PBMCs from HCs, patients with ALPS-FAS, and patients with XMEN for the CD4 and CD8 coreceptors, the αβ T cell receptor (αβTCR), and the CD45R (B220) (10, 27). By contrast to ALPS, most αβDNTs from patients with XMEN were negative for CD45R (B220), whether the patients were EBV infected or EBV naive (Figure 3I). Notably, patients with XMEN did not have marked elevations of the serum biomarkers soluble FasL, IL–10, IL-18, or vitamin B12, which can be dramatically elevated in ALPS-FAS (Supplemental Figure 3) (25). We then investigated FAS-induced apoptosis and anti-CD3 restimulation-induced cell death (RICD). The latter is a process by which previously activated T lymphocytes undergo clonal depletion upon TCR reengagement (28). We found that all XMEN patients tested (n = 9) had normal FAS-induced apoptosis but that RICD was defective (Figure 3J). By contrast, ALPS cells showed the expected prominent defect in FAS-induced apoptosis and a minor RICD defect (Figure 3J). These data suggest that abnormal RICD may contribute to the expansion of secondary lymphoid tissue in XMEN.
Leukocyte surface marker clusters distinguish XMEN, ALPS, and HCs.
To more precisely define the lymphocyte population differences between patients with XMEN and those with ALPS, we hypothesized that high-dimensional phenotyping of PBMCs could reveal novel differences on the basis of surface marker combinations that would not have previously been suspected. If true, this would provide a new and powerful approach for exploring more subtle and complex cellular alterations in immunological diseases that is not possible by conventional flow cytometric analysis. Hence, we stained PBMCs from age-matched XMEN and ALPS patients with a custom-designed antibody panel against 32 surface markers for in-depth immune phenotyping by time-of-flight mass cytometry (CyTOF) (Figure 4, Supplemental Figure 4, and Supplemental Table 7). We analyzed PBMCs from 18 patients with XMEN, 11 patients with ALPS-FAS, and 24 HCs and performed hierarchical agglomerative learning (HAL-x), a new machine learning algorithm to automatically identify clusters of differentiation (CoD) of leukocytes on the basis of distinctive expression levels of surface markers (Figure 4, A–C). HAL-x enabled us to execute an unsupervised random forest classification of machine learning attributes with a tunable threshold to self-consistently identify and validate 69 CoD (29, 30). We found that XMEN and ALPS patients had 6 and 2 CoD, respectively, that were statistically significantly different from those of HCs (Supplemental Figure 5). Specifically, patients with XMEN had a remarkably subtle and distinct immunological signature due to multiple elevated populations of naive B cells (Figure 4C). We then ranked the CoD to classify the samples and found that 2 populations of naive B cells, CoD 32 (CD20+CD27–CD22+IgM+HLA-DR+CXCR5+CXCR4++CD10+CD38+) and CoD 34 (CD20+CD27–CD22+IgM+HLA-DR+CXCR5+CXCR4+CD10–CD38–), were sufficient to generate receiver operating characteristics (ROCs) that achieved a near-perfect classification of our samples as either XMEN, ALPS, or HC (Figure 4D, Supplemental Figure 6, A and B). The specific cell markers for statistically different CoD are shown in Supplemental Figure 6B and Supplemental Figure 7, A and B. To compare and validate the findings from our novel pipeline, we also performed high-dimensional cluster analysis of the CyTOF data using Phenograph (Supplemental Figure 8, A–C) (31). Ranking of the phenograph-generated CoD (PG-CoD) revealed 1 population of naive B cells, PG-CoD 0 (CD20+CD27–CD22+IgM+HLA-DR+CCR6+CXCR5+CXCR4++CD10+CD38+) and 1 population of cytotoxic T cells, PG-CoD 22 (CD3+CD8+CD27+CD45RA+PD-1+CD38+CD57+NKG2D+) as the lymphocyte subsets with the most divergent abundance among HCs, patients with XMEN, and patients with ALPS (Supplemental Figure 9 and Supplemental Figure 10, A and B). Our Phenograph analysis also showed 7 additional lymphocyte subsets (PG-CoD 2, 6, 10, 13, 14, 18, and 19) that distinguished XMEN from both HC and ALPS lymphocytes (Supplemental Figure 9 and Supplemental Figure 11). Thus, the mass cytometric analysis of cells from XMEN and ALPS patients uncovered both global as well as remarkably subtle but characteristic CoD patterns of peripheral blood leukocytes in these diseases.
Figure 4. Deep immunophenotyping of PBMCs shows distinctive immune subsets for XMEN compared with HCs and ALPS.

HAL-x on CyTOF data acquired from PBMCs from patients with XMEN (n = 18), patients with ALPS (n = 11), and HCs (n = 24) identified 69 CoD. (A) 2D projection of the identified CoD as visualized by t-distributed stochastic neighbor embedding (t-SNE). (B) Dendrogram showing these CoD based on their abundance of surface epitopes. (C) Dendrogram of the frequencies of these CoD based on unsupervised grouping showing the clustering of patients with XMEN (blue), patients with ALPS (magenta), and HCs (green). (D) ROCs for the random forest classification of XMEN, ALPS, and HCs based on the frequency of their CoD. The AUC value is the mean AUC taken after a 4-fold cross-validation. Each fold of the cross-validation represents a different separation (of the patients) into training and testing sets. 95% CIs are shown (gray).
Restricted NLG defect in patients with XMEN.
Because of the broader scope of the clinical findings, we sought additional pathogenetic mechanisms besides the alteration of Mg2+ transport. Our bioinformatics analysis and other data showed that MAGT1 is the human homolog of the yeast OST3/6 subunit of the OST complex and functions in NLG (15, 16, 19, 22). The recently reported structure of the yeast OST complex showed an integral association of 2 TM helices of OST3 with TM segments of the major catalytic STT3 subunit (32) (Figure 5A). We hypothesized that the selective reduction of NKG2D but not that of other surface proteins such as CD5 in XMEN was due to abnormal glycosylation (Figure 5B). NKG2D is a dimeric single-pass TM glycoprotein of the C-type lectin-like family of receptors (2, 33, 34). NKG2D forms a tetrameric complex with 2 copies of the signaling protein DAP10 that is obligatory for cell-surface expression (Supplemental Figure 12A). Silencing the expression of human NKG2D or DAP10 decreases the cytotoxic effector function of CD8+ T cells and NK cells (34). Human NKG2D has 3 predicted asparagine-X-serine/threonine (NXS/T) motif NLG sites, where X is any amino acid except proline, Asn131 and Asn163, which are conserved, and Asn202, which differs in location compared with murine NKG2D (35) (Supplemental Figure 12A). Consistent with previous results, we observed that the full-length NKG2D glycosylated species that was observed as a broad band between 42 and 50 kDa (Figure 5C, lane 1, band 3) in the HC sample was replaced in the XMEN sample, with a smaller, less intense band migrating at 25 kDa and several faint indistinct bands ranging up to 28 kDa (Figure 5C, lane 3, band 1), which we hypothesized were partially glycosylated forms in samples from patients with XMEN. Treatment with N-glycosidase F (PNGase F), which removes almost all N-linked oligosaccharides from glycoproteins, caused these bands to collapse into the presumptive unglycosylated polypeptide of 23 kDa expected for a 216–amino acid protein (Figure 5C, lane 2, band 0). This unglycosylated polypeptide was markedly less abundant in samples from patients with XMEN, presumably due to instability of the partially glycosylated NKG2D protein (Figure 5C, lane 4, band 0). As expected, MAGT1 protein was absent from the patient lysates compared with those from HCs (Figure 5C, middle panel, lanes 3 and 4). These data raised the question of whether defective NLG accounted for decreased surface NKG2D expression. Therefore, we treated T cells from HCs with tunicamycin, an inhibitor of NLG, for 24 hours and found that this dramatically reduced NKG2D surface expression but only mildly affected CD5 expression (Figure 5D). Biochemical analysis showed that the fully glycosylated NKG2D species (Figure 5E, lane 1, band 1) and the nonglycosylated polypeptide revealed by PNGase F treatment (Figure 5E, lane 2, band 2) were diminished by tunicamycin treatment (Figure 5E, lane 3, band 1 and lane 4, band 2, respectively). Taken together, these data show that direct impairment of glycosylation causes instability and loss of the NKG2D protein, thereby reducing cell-surface expression. Hence, this provides a paradigm of how defective glycosylation could interfere with the proper expression of NKG2D, leading to impaired EBV antiviral immunity.
Figure 5. Selective N-glycosylation defects in XMEN disease.

(A) Model of MAGT1 as a facilitator of STT3B-dependent transference of oligosaccharides (blue and pale pink) to glycosites (red) of nascent peptides (gray) in the ER (13, 15, 16, 19). (B) Flow cytometric histogram and MFI quantification relative to HCs of NKG2D and CD5 in CD8+T cells from HCs (blue) and patients with XMEN (red), with an unstained control (gray, n = 6). (C) Immunoblot of NKG2D, MAGT1, and β-tubulin in T cells from HCs and patients with XMEN, with (+) or without (–) PNGase F treatment. Unglycosylated (0), partially glycosylated (1), and fully glycosylated (3) NKG2D bands and nonspecific (2) and PNGase F (*) bands. (D) Flow cytometric histogram and MFI quantification relative to HCs of NKG2D and CD5 in CD8+ T cells treated for 24 hours with DMSO vehicle (blue) or tunicamycin (red) with unstained control (gray). PE, fluorophore R-phycoerythrin. (E) Immunoblot of NKG2D and β-tubulin in T cells with (+) or without (–) PNGase F treatment after incubation with DMSO (Veh) or 10 μg/mL tunicamycin (Tun) for 18 hours. Fully glycosylated (1) and unglycosylated (0) NKG2D and PNGase F (*) bands. (F) Heatmap depicting significantly different glycopeptides between HCs (n = 3) and patients with XMEN (n = 3), with increased (red) and decreased (black) glycosylation shown. Numbers on the left for immunoblots indicate kDa standards. Data are representative of 6 (C) and 3 (E) replicates, respectively. Data represent the mean ± SEM. ****P < 0.0001, by 1-sample t test with μ = 1 (B and D) and 2-sample t test (F).
To further investigate the NLG defect due to the absence of MAGT1, we performed glycoproteomic analysis on T cells from 3 patients with XMEN and from 3 HCs using filter-aided, mixed-lectin glycopeptide capture followed by peptide identification by liquid chromatography and tandem mass spectrometry (LC-MS/MS) (36). In total, we obtained sequence data for 2481 peptides from 1421 proteins, essentially all of which contained the canonical NXS/T sequon (Supplemental Figure 12, B and C). Comparing the glycoacceptor sites, we found that the abnormal XMEN subset had a serine/threonine (S/T) ratio of 1.56, whereas the total data set had a ratio of 0.75 (Supplemental Figure 12B). Hence, proteins perturbed by the loss of MAGT1 predominantly contained the NXS sequon, with a smaller fraction containing the NXT sequon, consistent with observations in the yeast Δost3Δost6 knockout (37, 38). Although the proteins had a broad cellular distribution, over 50% were derived from the ER or the cell membrane (Supplemental Figure 12D). Unsupervised hierarchical clustering of the glycopeptide abundances readily sorted the samples into 2 distinct groups that corresponded to the MAGT1-deficient XMEN patients and the HCs that were not immediately obvious in comparing all peptides (Supplemental Figure 12E). However, with a more refined heatmap of the differences, we found 105 specific glycopeptides encoded by 73 unique genes having 1 or more glycopeptide in the samples from patients with XMEN (Figure 5F and Supplemental data file 1). Most of the NLG alterations that had a significant P value in a t test comparison of XMEN with HC samples exhibited lower glycosylation, although a few had higher glycosylation (Supplemental Figure 12F, red dots, and Supplemental data file 1). Since the lack of glycoproteins could result from decreased protein abundance, we also examined the complete LC-MS/MS proteome. This did not reveal major peptide abundance differences between the XMEN and HC lysates, except for the marked reduction of MAGT1 protein in all the XMEN samples, confirming that the gene mutations in these individuals abrogated protein expression (Supplemental Figure 12G). Because both XMEN and HC samples displayed incomplete glycosylation, we compared both using a scatter plot of the log (glycosite/protein), which showed that XMEN samples had more peptides with lower glycosylation site occupancy compared with HC samples, supporting the conclusion that MAGT1 deficiency leads to a selective NLG defect (Supplemental Figure 12H, red dots).
More detailed examination of the data set showed, as expected, that peptides from NKG2D containing the Asn131 and Asn202 residues had reduced glycosylation; no peptide with Asn163 was detected (Supplemental data file 1). We also found underglycosylation of other key immune regulatory molecules including the costimulatory molecule CD28, CD70 (or TNF [ligand] superfamily member 7), the MHC protein HLA-DRB1, the T cell receptor α chain (TCRα), the ceramide synthase 2 (CERS2) protein, and the solute carrier family 4 member 7 (SLC4A7) protein, all of which might contribute to the vulnerability of XMEN patients to EBV and other infectious agents (Figure 6, A and B, Supplemental data file 1). Notably, using glycoproteomics MS, we detected reduced NLG of the T cell receptor α chain (TCRα), but we also detected defective glycosylation of the T cell receptor β chain (TCRβ) by Western blotting. Since CD28, CD70, HLA-DRB1, and TCRβ must be expressed on the cell surface to execute their immune function, we used flow cytometry to test whether reduced NLG affected surface expression on T cells from patients with XMEN (Figure 6A). CD28, CD70, and HLA-DRB1 showed a marked reduction in surface expression. Surprisingly, it appeared that TCRβ surface immunolabeling on T cells was increased in XMEN. We surmised that this was due to less steric interference with the staining antibody binding due to underglycosylation because the level of protein was mildly reduced rather than increased in the XMEN samples by Western blotting (Figure 6B). We therefore treated T cells from HCs with tunicamycin for 48 hours and found that, despite a relative decrease in total TCRβ expression in the tunicamycin-treated samples as determined by immunoblotting (Supplemental Figure 12I), its surface immunolabeling by flow cytometry appeared to be increased, especially at lower concentrations of tunicamycin, consistent with the hypothesis of steric interference with antibody binding by glycosylation (Supplemental Figure 12J).
Figure 6. Impaired glycosylation of immune proteins and CDT pattern in XMEN disease.

(A) Flow cytometric histogram and MFI quantification relative to HCs of CD28, CD70, HLA-DR, and TCRβ in T cells from HCs (blue) and patients with XMEN (red), with an unstained control (gray, n = 6). (B) Immunoblot of CD70, HLA-DR, TCRβ, CERS2, SLC4A7, and β-tubulin in T cells from HCs and patients with XMEN with (+) or without (–) PNGase F treatment. Unglycosylated (0), partially glycosylated (1), and fully glycosylated (2) bands. (C) Immunoblot of CD28 and HSP90 in T cells from HCs (n = 2) and patients with XMEN (n = 12) with the indicated mutations. Glycosylated (1) and unglycosylated (2) CD28 bands. (D) Immunoblot of CD28 and HSP90 in T cells treated with DMSO (0) or tunicamycin for 48 hours. Glycosylated (1) and unglycosylated (2) CD28 bands. The numbers on the left for immunoblots indicate kDa standards. (E) Pie chart showing the biological function of abnormally glycosylated proteins in XMEN. (F) Mass spectrometric traces of a CDT test for HCs (blue) and patients with XMEN (red). Mass and intensity are expressed in daltons (Da) and counts per second (cps), respectively. Data are representative of 5 (B), 2 (C), and 3 (D) replicates. Data represent the mean ± SEM. *P < 0.05 and **P < 0.01, by 1-sample t test with μ = 1.
We also examined other immunologically important molecules and observed that impaired glycosylation of CD28 in T cells from patients with XMEN also correlated with its decreased protein expression by Western blotting (Figure 6C), and tunicamycin treatment of T cells from HCs showed a dose-dependent decrease in expression of both the total and fully glycosylated CD28 as well as an increase in the presumptive unglycosylated form (Figure 6D). Reduced glycosylation was verified by Western blotting for CD28, CD70, HLA-DRB1, TCRβ, CERS2, and SLC4A7 proteins, for which we saw a reduction in fully or partly glycosylated species (Figure 6B, bands 1 or 2; and Figure 6C, band 1) to underglycosylated or nonglycosylated species in the XMEN samples compared with the HC samples (Figure 6B, band 0 or 1). The high-molecular-weight species of CD70 and TCRβ are likely due to modification of the oligosaccharide core in the Golgi (39). We also assessed the surface protein expression and glycosylation status of ICAM-3 by flow cytometry and Western blotting, respectively, and confirmed that this protein is representative of overglycosylated proteins in XMEN disease (Supplemental Figure 12, K and L). Overall, we found that the affected 73 proteins fall into at least 6 categories: neural function, glycosylation, transport, adhesion, immunity, and lipid metabolism, with 18 proteins being uncategorized (Figure 6E). A large number of these proteins are associated with disease states affecting different organ systems, especially the hemopoietic system, when they are genetically deficient (Supplemental Table 8). Thus, although most proteins in XMEN cells appeared to be represented at levels equivalent to those in normal cells, a small, select group, affecting a diverse set of cellular processes, showed abnormal glycosylation in MAGT1-deficient cells. Certain of these, though not necessarily all, may contribute for the broader phenotype we observed, depending on the specific functional effect of the glycosylation defect on the role of the individual protein in its cellular pathway.
Because of the clear glycosylation defects in patients with XMEN, we investigated both the carbohydrate-deficient transferrin (CDT) test and the apolipoprotein CIII (Apo-CIII) isoforms as clinical adjuvants for the screening of XMEN disease (20). Apo-CIII is a single core 1 mucin type O-glycosylated protein that carries a single O-glycan at Thr-94 and is not N-glycosylated (40). All XMEN patients tested (n = 10) had a mild but distinctly abnormal glycosylation pattern on MS resembling a CDG type I, but displayed a unique combined defect in both CDT and Apo-CIII glycosylation. Most of the transferrin molecules from XMEN patients’ cells had fully glycosylated asparagine sites (Figure 6F, pattern 3), but some were defective for the initial transfer of the oligosaccharide precursor from the dolichol carrier leaving unoccupied asparagine glycosylation sites (Figure 6F, pattern 1, and Supplemental Table 9). Transferrin from XMEN patients’ cells also exhibited a mild defect in final processing of oligosaccharides with sialic acid (Figure 6F, pattern 2). The Apo-CIII O-linked glycosylation (OLG) that we observed for some patients could be a secondary effect of defective NLG of enzymes/proteins important for OLG (Supplemental Table 9). Thus, these tests reveal the nature and extent of the glycosylation defect and provide useful additional clinical diagnostic tools for XMEN disease.
Affected N-glycosylation sites are close to TM regions.
To investigate whether the N-glycosylation sites affected in XMEN disease are STT3A or STT3B dependent, we performed a prediction analysis of our glycoproteomic site occupancy data set and found that only 36% and 17% of the differentially glycosylated peptides mapped to STT3B- and STT3A-predicted motifs, respectively (Supplemental data file 1). Interestingly, we found that the majority (57%) of the affected sequons were within 60 amino acids of a TM region, and these peptides accounted for most (83%) of the STT3A-dependent peptides (Figure 7A and Supplemental data file 1). We also observed that most glycosylation sequons that mapped to a described protein region were hypoglycosylated (Figure 7B).
Figure 7. Motifs of glycopeptides affected in XMEN disease.

(A) Venn diagram shows the number of differentially glycosylated peptides in XMEN that mapped to regions on a protein that were within the last 55 amino acids of the C-terminus (C-Term), in short loops between 2 TM regions (Loop), in cysteine-rich regions of cysteine-rich proteins (Cys-Rich), in the N-terminus domain of multi-TM proteins (N-Term), and/or within 60 amino acids of TM regions. (B) Bar plot shows the counts of hypoglycosylated (blue) and hyperglycosylated (red) peptides in XMEN that mapped to the above regions.
Defective glycosylation can be corrected by RNA replacement.
Finally, we explored the hypothesis that restoring MAGT1 gene expression might reverse the glycosylation defects in XMEN. We transfected synthetic mRNAs encoding MAGT1 or a GFP control into XMEN patients’ PBMCs ex vivo using the Maxcyte transfection device. This procedure achieved greater than 90% transfection efficiency with high cell viability. Forty-eight to seventy-two hours after transfection, the cells were analyzed by flow cytometry, which showed that surface expression of key glycoproteins including NKG2D and CD70 was restored (Figure 8A). We further examined the specific glycosylated protein forms using Western blotting and found that the fully glycosylated forms of NKG2D, CD70, CERS2, SLC4A7, and TCRβ proteins were mostly or completely rescued by mRNA transfection–mediated expression of MAGT1 (Figure 8B).
Figure 8. Rescue of N-glycosylation by MAGT1 mRNA transfection in XMEN T cells.

(A) Flow cytometric histograms of NKG2D, CD70, and CD5 in T cells from a HC (blue line) and a patient with XMEN, 48 hours after transfection of mRNA encoding GFP (red line) or MAGT1 (light blue fill). (B) Immunoblot of NKG2D, CD70, CERS2, TCRβ, SLC4A7, and β-actin in T cells from a HC and a patient with XMEN, 48 hours after GFP or MAGT1 mRNA transfection. Fully glycosylated (2), partially glycosylated (1), and unglycosylated (0) bands are indicated. Data in A and B are representative of 3 independent experiments.
Discussion
In a larger group of patients, we found new features that were not manifested or recognized in the earlier cases (1–3). Here, we report a comprehensive clinical description of the largest cohort of patients with XMEN yet described, to our knowledge. We found that XMEN disease is a complex multisystem disorder. Although all patients had MAGT1 mutations that cause complete protein loss, the disease has variable expressivity, even among individuals harboring the same mutation. Classic XMEN disease can now be recognized as affecting males with recurrent ear and sinopulmonary infections, LAD with or without splenomegaly, autoimmune cytopenias, and/or LPD (Table 1). Remarkably, EBV-naive and EBV-infected XMEN patients are clinically similar except for lymphoma and EBV-LPD, which occur exclusively in the latter. Mild thrombocytopenia, neutropenia, elevated liver enzymes, and increased CPK levels are more variable in occurrence, and normal results do not rule out XMEN disease. Other common findings are increased B cells, an inverted CD4/CD8 ratio, elevated αβDNTs, low serum IgG and IgA, and CD4+ lymphopenia. Chronic EBV viremia was characteristic of patients with XMEN who were EBV infected, confirming our previous observations (1). The expanded set of clinical features and diagnostic tests should allow clinicians to make a positive presumptive diagnosis that can then be confirmed by molecular testing.
An important new insight that emerged from exploring the molecular details of MAGT1 as a component of OST complexes involved in NLG was that MAGT1 controls a specific pattern of glycoproteins that are important for proper immune function. Our mass spectrometric glycoproteomics analysis of T lymphocytes was able to detect a surprisingly small and distinctive set of glycosylation changes between patients and controls. We confirmed that there were 73 unique proteins whose glycosylation was affected, and we demonstrated that several of the hypoglycosylated proteins important for immunity had secondarily decreased expression. However, the MAGT1-dependent glycoproteins are a tiny subset of the thousands of proteins that undergo posttranslational NLG.
Our glycoproteomics analysis also revealed that defective glycosylation in XMEN disease preferentially affects NxS sequons. We found that most affected glycosites were located within 60 amino acids of TM domains, including both acceptor sites predicted to be STT3A and STT3B dependent, which represents a novel class of MAGT1-facilitated sites. We show that MAGT1 deficiency caused a more immune-restricted and less-severe clinical presentation than does STT3B deficiency or other CDG, in which brain and neurodevelopmental abnormalities, intellectual disability, failure to thrive, severe liver disease, and hypotonia are commonly seen (20, 21, 41). In contrast to a recent report, we did not find intellectual disability or dysmorphic features in our cohort of patients, including 2 patients (only 1 reported here) with the same c.991C>T (p.R331X) MAGT1 mutation (22). This suggests that loss of MAGT1 expression alone does not lead to intellectual disability. We did observe varying degrees of brain, cerebellar, and/or spinal cord atrophy in 3 patients. Because cerebellar atrophy has been described in STT3B deficiency, defective glycosylation may play a role in these CNS findings (42). Liver disease is commonly seen in CDG, and we noted intermittent elevations of liver enzymes (AST and ALT) in the patients with XMEN. However, liver biopsies did not reveal a specific pattern of injury. Liver disease in EBV-naive patients showed that EBV was not the cause, so it may be related to impaired glycosylation.
We show that the glycosylation of transferrin as detected by a clinical mass spectrometric assay was clearly abnormal in all XMEN patients tested. Although it was recently reported that MAGT1 deficiency results in a type 1 sTf IEF pattern, we now show that sTfs from patients with XMEN also have a mild defect in final processing of the glycans (22). Furthermore, and in contrast with most CDG type I in which Apo-CIII is not affected, patients with XMEN may also have an OLG defect in Apo-CIII, which could be secondary to defective NLG of components of the OLG enzymatic apparatus. Nevertheless, the limited but broader range of glycosylation defects we describe here compared with our earlier reports may offer a window for further investigation into the expanded phenotype of patients with XMEN. We now propose that XMEN is a restricted CDG. The presence of a CDT and Apo-CIII isoform pattern similar to the one observed in this study (Supplemental Table 9) in a male patient with a suspected CDG should prompt further immune and genetic testing for XMEN disease. Since we know very little about how specific amino acid residues in the MAGT1 protein carry out their function, having a simple straightforward clinical diagnostic test is useful. Both staining for NKG2D (which may or may not be available in different hospitals) as well as the CDT test can help solidify the diagnosis.
Interestingly, we found defective glycosylation of certain ion transporters in XMEN samples, and Mg2+ deficiency can affect glycosylation (14). The MAGT1 gene is conserved in unicellular eukaryotes such as yeast, indicating an ancient phylogenetic connection between glycosylation and Mg2+ (12). Previously, abnormalities of intracellular levels and transport of Mg2+ have been found in patients with XMEN (1, 2). However, in our study, both total and ionized serum magnesium concentrations were normal and hence were not diagnostic of XMEN (Supplemental Table 2). Intracellular Mg2+ determination or TCR-induced Mg2+fluxes have been extensively tested and not proven useful for clinical diagnostic tests. These assays are based on fluorescent dyes that bind Mg2+ but that also have low affinity for calcium Ca2+ and other ions (our unpublished observations). Thus, results can be compromised by fluctuations in Ca2+ concentrations and intracellular dye compartmentalization, among other variables, and are therefore unreliable. The previously reported NKG2D surface expression deficiency remains the best flow cytometry diagnostic test, and this can now be augmented with the CDT test (1, 2).
Another important clinical insight derived from our study is the recognition that XMEN has certain clinical similarities to ALPS and should be considered in the differential diagnosis for the latter disease. LAD, previously attributed to uncontrolled EBV infection in XMEN, was detected in both EBV-naive and EBV-infected patients. Interestingly, both ALPS and XMEN have apoptotic effects that may contribute to secondary lymphoid expansion, but in ALPS, this primarily involves defective FAS-induced apoptosis, whereas in XMEN, it is due to defective RICD. αβDNTs are a shared abnormal cell population, but in ALPS, this population characteristically expresses high CD45R (B220), whereas we show that in XMEN, it does not. In addition, these 2 diseases have other distinct clinical features and a genetic difference, all of which will aid proper diagnosis and treatment (Supplemental Table 10). In order to define the differences between these diseases and healthy humans, we undertook deep immunophenotyping with a multidimensional CyTOF procedure that yielded 69 different CoD groups. Using a new machine learning pipeline (HAL-x), we defined 2 rare naive B cell populations that accurately discriminate between XMEN, ALPS, and HCs. We also used a classical high-dimensional clustering method (Phenograph) to validate and expand our CyTOF analyses. It will be of great interest to examine these newly defined cell populations functionally and determine whether and how they contribute to disease pathogenesis.
Finally, we discovered that CD70, whose genetic deficiency was recently shown to predispose individuals to uncontrolled EBV infection and hypogammaglobulinemia, is underglycosylated and has reduced surface expression on XMEN lymphocytes (43). We also now demonstrate that reduced surface expression of NKG2D occurs when glycosylation is abrogated. Therefore, there are 2 key molecules whose restoration by improved glycosylation may enhance antiviral immunity against EBV, which is a strongly lymphotrophic and oncogenic virus. We also show that mRNA transfection can achieve restoration of these and other glycoproteins. This provides evidence that gene transfer approaches to restoration of MAGT1 expression in patients with XMEN disease might rescue critical cellular functions. However, restoration of MAGT1 in blood cells may not be sufficient to correct abnormalities in all organ systems.
Methods
Patients.
Twenty patients with XMEN were seen at the NIAID and 3 elsewhere, with samples evaluated at the NIH.
Cells.
PBMCs were isolated by Ficoll-Paque PLUS (GE Healthcare) gradient density centrifugation and ACK lysis (Quality Biological). PBMCs were cryopreserved in liquid nitrogen in dialyzed FBS containing 10% DMSO.
Activation and expansion of human T lymphocytes.
Cryopreserved PBMCs were rapidly thawed and washed in prewarmed complete RPMI-1640 medium (Lonza) containing 10% FBS, 2 mM glutamine, and penicillin and streptomycin (100 U/mL each; Invitrogen, Thermo Fisher Scientific). Cells at 1 × 106/mL were stimulated with anti-CD3 (HIT3a, BioLegend) and anti-CD28 (CD28.8, BioLegend) at 1 μg/mL each in complete RPMI medium. After 3 days, cells were supplemented with 100 U/mL recombinant human IL-2 (rhIL-2) (R&D Systems).
Tunicamycin treatment.
Tunicamycin (MilliporeSigma) was dissolved in DMSO to generate a concentrated stock solution. T cells from healthy donors were activated and cultured for 7–14 days prior to tunicamycin treatment. Cells (1 × 106 to 1 × 107) were treated with 10 μg/mL tunicamycin or DMSO as a vehicle control at a cell density of 5 × 105 cells/mL in complete RPMI-1640 medium containing 10% FBS, 2 mM glutamine, penicillin and streptomycin (100 U/mL each) (Invitrogen, Thermo Fisher Scientific), and 100 U/mL rhIL-2 (R&D Systems) for the specified duration at 37°C in a 5% CO2 ambient atmosphere. Lower tunicamycin concentrations at 1 and 3 μg/mL were also used for the determination of TCRβ surface expression by flow cytometry.
Deglycosylation.
Cells (5 × 106) were lysed in 100 μL RIPA lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4) with protease inhibitor (Roche). The lysates were then centrifuged at 18,000 × g at 4°C for 10 minutes to remove insoluble material and treated with PNGase F (New England BioLabs) according to the manufacturer’s instruction. Briefly, a stock of 10× denaturation buffer (5% SDS and 400 mM DTT) was added to the samples to a final concentration of 1×. The samples were denatured at 100°C for 10 minutes and then chilled on ice. Denatured samples were mixed with 2 μL 10× Glyco Buffer 2 and 2 μL 10% NP-40 and then diluted to 20 μL with water. Twenty microliters of the mixture was then transferred into 2 separate tubes (i.e., 40 μL total). One tube was treated with 1 μL PNGase F (500,000 units/mL, New England BioLabs) and the other with ddH2O vehicle at 37 °C for 1 hour.
FAS receptor–mediated apoptosis and RICD assays.
For FAS receptor–mediated apoptosis (FAS kill) and RICD assays, cultured T lymphocytes were washed and resuspended in RPMI without IL-2. Cells (1 × 105 cells/0.2 mL media/well) were plated in triplicate in 96-well round-bottomed plates and treated with varying concentrations of agonistic anti-FAS antibody (APO-1-3, Enzo Life Sciences) plus protein A (MilliporeSigma) at twice the concentration of the anti-FAS antibody or anti–human CD3 antibody (HIT3a, BioLegend) for 24 hours. Nonviable cells were stained with 5 μg/mL propidium iodine (MilliporeSigma). Cell death was quantified by flow cytometry using an LSR II (BD Biosciences), analyzed with FlowJo (Tree Star), and plotted in GraphPad Prism (GraphPad Software).
Plasma biomarkers.
Plasma biomarkers were quantified with Quantikine Kits (R&D Systems) following the manufacturer’s instructions (44). A mixed-effects statistical model with a likelihood ratio test was used to analyze the data using R, version 3.3.2 (45).
Flow cytometry.
Cryopreserved PBMCs were thawed and incubated with human Fc receptor blocking solution (BioLegend) and Zombie Green Viability Dye (Biolegend) in PBS at 25°C for 20 minutes. Cells were stained in ice-cold FACS buffer (Dulbecco’s PBS, 2% FCS, 1% sodium azide) with fluorochrome-conjugated antibodies on ice for 30 minutes. T cell blasts were processed similarly but without incubation with human Fc receptor blocker and viability dye. The following antibodies were used: anti-CD3 (UCHT1, clone HIT3a or SK7, Invitrogen, Thermo Fisher Scientific), CD4 (clone OKT4 or RPA-T4, BioLegend), CD5 (L17F12, BioLegend), CD8 (clone RPA-T8 or SK1, BioLegend), CD16 (3G8, BioLegend), CD20 (clone 2H7, BioLegend), CD45R/B220 (RA36B2, BioLegend) or rat IgG2aκ isotype control (clone RTK2758, BioLegend), CD28 (clone CD28.2, BioLegend), CD50/ICAM-3 (clone CBR-IC3/1, BioLegend), CD56 (clone MEM-188, BioLegend), CD70 (clone 113-16, BioLegend), CD314/NKG2D (clone 1D11, BioLegend) or mouse IgG1κ isotype control (clone MOPC-21, BioLegend), HLA-DR (clone Tü36, BioLegend), TCRαβ (clone T10B9 or IP26, BD Biosciences), and TCRγδ (B1, BioLegend). Cells were washed in FACS buffer prior to acquisition on a Fortessa or LSR II instrument (BD Biosciences) and analyzed with FlowJo software. Gating strategies are summarized in Supplemental Figure 13. Statistical tests were conducted using GraphPad Prism, version 8.0.2 (GraphPad Software), with α = 0.05.
Immunoblotting.
Cells were lysed in RIPA buffer supplemented with a protease and phosphatase inhibitor cocktail (Roche). Standard Western blotting procedures were followed. A rabbit monoclonal antibody against human MAGT1 (N-terminal sequence: FINFPAKGKPKRGDTYELQVRGFS) was produced (Epitomics). The following antibodies were used for immunoprobing of specific glycoproteins: anti-NKG2D (clone D-20, Santa Cruz Biotechnology), anti-CD70 (catalog ab96323, Abcam), anti-CD28 (clone D2Z4E, Cell Signaling Technology), anti- HLA-DR (clone TAL 1B5, Abcam), anti–TCRβ (clone H-197, Santa Cruz Biotechnology), anti-CERS2 (A303-193A, Bethyl Laboratories), anti-SLC4A7 (clone L-15, Santa Cruz Biotechnology), and anti-ICAM-3 (A304-334A, Bethyl Laboratories). Anti–β-tubulin antibody (clone AA2, MilliporeSigma), anti–β-actin (ab20272, Abcam), and anti-Hsp90αβ (clone 3H3C27, BioLegend) were used as loading controls.
Determination of transferrin and Apo-CIII isoforms.
Transferrin and Apo-CIII isoforms in serum were identified by immunoaffinity liquid chromatography and electrospray MS as previously described (46). In brief, a 10-μL diluted sample was injected into a 200-μL/min flow of pH 7.4 PBS and retained on an immunoaffinity column composed of 60% anti–human Apo-CIII antibody and 40% anti–human transferrin antibody–bound POROS 20 AL Media (Applied Biosystems, Thermo Fisher Scientific). The immunoaffinity column was then washed for 2 minutes with PBS. Following the 2-minute wash, captured proteins were eluted to a C4 column at 200 μL/min with 100 mM glycine (pH 2.5) and 2% acetic acid buffer. The C4 column was then washed with water/methanol/glacial acetic acid (97:2:1) to remove signal-suppressing salts. Next, the proteins were eluted from the C4 column with methanol/water/glacial acetic acid/trifluoroacetic acid (94.5:5:0.5:0.04) and introduced into an API 4000 LC-MS/MS System (SCIEX) equipped with a Turbo V source configured for electrospray ionization. The mass spectrometer was operated in positive Q1 scan mode with 2 scan ranges: m/z 1090–2000 for Apo-CIII and m/z 2000–3000 for transferrin.
Mass cytometry.
Cryopreserved PBMCs from 18 patients with XMEN (patients A.1, A.2, A.3, D.1, E.1, G.1, G.2, H.1, I.2, J.1, J.2, K.1, K.2, L.1, L.2, N.1, P.1, and Q.1), 11 patients with ALPS, and 24 healthy volunteers were rapidly thawed at 37°C and washed in prewarmed complete RPMI immediately prior to cell-surface staining and acquisition. All reagents were from Fluidigm unless otherwise noted. Cells were first incubated with a 5-μM solution of cisplatin in PBS to mark dead cells. The cells were then washed and resuspended in Maxpar Cell Staining Buffer (MCSB) (Fluidigm). Human Fc receptor blocking solution (BioLegend) was added to each sample and incubated for 10 minutes at room temperature. All metal-labeled antibodies were purchased from Fluidigm. Biotin anti–human CD70 (BioLegend) was detected with Qdot-streptavidin conjugate (Thermo Fisher Scientific). The antibody panel for deep immunophenotyping by CyTOF is summarized in Supplemental Table 7. Antibodies were diluted in MCSB at concentrations validated for minimal channel spillover, added to each sample, and incubated for 30 minutes at room temperature. Cells were washed twice with MCSB and then fixed for 15 minutes in a 1.6% solution of paraformaldehyde (MilliporeSigma) in PBS. Cell intercalation solution was prepared by adding Cell-ID Intercalator-Ir into Maxpar Fix and Perm Buffer (both from Fluidigm) to a final concentration of 125 nM. Cells were incubated with cell intercalation solution overnight at 4°C and then washed once with MCSB and Maxpar Water twice. Cells were finally resuspended in a water solution containing EQ Four Element Calibration Beads (Fluidigm) immediately prior to CyTOF data acquisition at a cell concentration of 106 cells/mL and filtered into cell trainer cap tubes. Data were acquired on a Helios Mass Cytometer (Fluidigm) as previously described (47).
Computational methods and statistical analysis of mass cytometric data.
Raw mass cytometric data were normalized with the Normalizer algorithm as recommended by the software developers (48). Data were then analyzed using a pipeline custom programmed in Python (fast density clustering; https://github.com/alexandreday/fast_density_clustering). Data were first gated as live singlet cells using [EQ4–cisplatin–] and [CD45+DNA(2n)] manual gates (Supplemental Figure 13D). Automatic identification of CoD based on surface marker expression was performed using a new pipeline termed HAL-x (https://pypi.org/project/hal-x). In brief, HAL-x performs data clustering through a modified hierarchical agglomerative learning algorithm, assigns a phenotype to each cluster across all models, and applies stochastic selection and random forest classification to identify defining features (e.g., frequencies or expression levels) for the cell clusters of each group of individuals (XMEN, ALPS, or healthy donor). This custom-programmed algorithm relies on cross-validated identification of high-density regions, followed by rapid random forest classification in the 32-dimensional space of surface markers. HAL-x then labeled all the cells in all samples (>15 million cells from 53 individuals) and computed the inverse hyperbolic sine and normalized frequencies for each leukocyte population. A random forest algorithm used the population frequencies to determine whether samples were taken from XMEN, ALPS, or healthy tissues. A similar analysis was performed using the previously described Python-based Phenograph application, for which a 1-million-cell downsampled subset of all the cells was randomly chosen across all samples for computation (31).
N-linked glycoproteome analysis.
Protein digestion, lectin enrichment, and deglycosylation were performed as previously described (36, 49). In brief, following protein digestion, tryptic peptides were incubated for 60 minutes with a lectin mixture (concanavalin A [Con A], wheat germ agglutinin [WGA], and ricinus communis agglutinin [RCA]) on filtration units. Given the large size of the glycopeptide-lectin complexes, they were retained on the filter after washing away nonglycosylated peptides. This was followed by treatment of the samples with PNGase F in H218O, which leaves a characteristic mass shift on the previously glycosylated site. Deglycosylated peptides were then eluted and measured using LC-MS/MS.
LC-MS/MS analysis.
Both enriched deglycosylated peptides (glycoproteome analysis) and total proteome peptides were separated by a nanoflow HPLC system (Proxeon Biosystems, now Thermo Fisher Scientific) coupled to a Quadrupole-Orbitrap Mass Spectrometer (Q Exactive, Thermo Fisher Scientific) with a nanoelectrospray ion source (Proxeon Biosystems). MaxQuant software was used to analyze the mass spectrometric raw data. We searched the MS/MS spectra against the Uniprot database using the Andromeda search engine incorporated into the MaxQuant framework. For the glycoproteome, cysteine carbamidomethylation was set as a fixed modification, and N-terminal acetylation, methionine oxidation, and deamidation in H218O were set as variable modifications. For the proteome, cysteine carbamidomethylation was set as a fixed modification. N-terminal acetylation and methionine oxidation were set as variable modifications. A FDR of 0.01 was required for proteins, peptides, and sites. Enzyme specificity was set to trypsin, allowing N-terminal cleavage to proline. A minimum of 7 amino acids per identified peptide were required, and 2 miscleavages were allowed. The initial allowed mass deviation of the precursor ion was up to 6 parts per million (ppm), and for the fragment masses it was up to 20 ppm. Mass accuracy of the precursor ions was improved by time-dependent recalibration algorithms of MaxQuant. The “match between runs” option was enabled to match identifications across different replicates. For both the proteome and glycoproteome, label-free quantification was performed using MaxQuant with standard settings. We analyzed the MaxQuant output data with the Perseus tools, which was also available in the MaxQuant environment.
Unsupervised hierarchical clustering and t test analysis of glycoproteomics data.
A filter was initially applied to remove contaminants and anomalous data. A subsequent filter was then applied to identify canonical class I NxS/T sequons using a localization probability of greater than 0.75 and a score difference of more than 5. The values were then converted to a log2 scale of intensity. The site table was then expanded and a 50% valid value filter applied (which filtered for 9 valid values in total). Unsupervised hierarchical clustering (Euclidean distance) was then performed using automated data analysis tools and the data presented as a heatmap. A dendrogram was added to depict the relative similarity between samples. For subsequent identification of sites differently glycosylated and expressed between groups A and B, we set the FDR at 0.05 and the S0 (artificial within groups variance) at 0.25. The t test values of the data were then presented as both a volcano plot (t test difference versus –log t test P value) and a heatmap. The results of our mass spectrometric analysis have been deposited in the Mass Spectrometry Interactive Virtual Environment (MassIVE) data set (https://doi.org/doi:10.25345/C5MH21).
Glycopeptide mapping.
Glycosites were categorized as previously described (18). In brief, the FASTA sequence for each significantly affected protein was downloaded from UniProt (https://www.uniprot.org/). The affected glycosite was mapped onto the sequence, as were TM domains predicted by the TMHMM Server, version 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). We categorized glycosites as follows: C-term, within 55 amino acids of the C-terminus of the protein; near TM, within 60 amino acids of a predicted TM domain; loop, in short loops between TM domains; Cys-rich, in cysteine-rich domains of cysteine rich proteins; and/or N-term, before the first TM domain of a multi-TM protein. Short loops were defined as fewer than 100 amino acids between the TM regions. Cysteine-rich proteins were defined as having greater than 4.0% cysteine as calculated by ExPASy’s ProtParam tool (https://web.expasy.org/protparam/). Glyosites in cysteine-rich proteins were defined as being in cysteine-rich domains if they fit at least 1 of the following criteria: (a) either the +1 or +2 sites were a cysteine (i.e., NCS/T or NXC); (b) there were at least 3 cysteines between the –9 and +10 positions surrounding the glycosite; or (c) the glycosite fell within a cysteine-rich domain predicted by the ExPASy PROSITE (https://prosite.expasy.org/prosite.html).
mRNA electroporation.
PBMCs were activated and cultured in complete medium with IL-2 for 2 weeks (described above). Then, cells were counted and resuspended in electroporation buffer (MaxCyte System) at 2 × 107 cells/mL. RNA (100 μg/mL) was added as indicated and electroporated according to the manufacturer’s specifications. After electroporation, cells were incubated at 37°C for 20 minutes before transfer to complete medium for culturing.
Statistics.
Unless otherwise stated, statistical analysis was conducted using GraphPad Prism, version 8.1.2 (GraphPad Software), P values of less than 0.05 were considered statistically significant, and data are shown as the mean ± SEM. For multiple comparisons, an ordinary 1-way ANOVA with Tukey’s multiple comparisons correction for all pairwise comparisons was used. To compare test samples relative to controls, the controls were normalized to 1, and a 1-sample t test was conducted on the experimental samples, with μ = 1.
Study approval.
All subjects provided written informed consent to participate in the study, in accordance with IRB-approved protocols at their respective institutions and with Declaration of Helsinki principles, and written informed consent was provided for photographs appearing in this report.
Author contributions
JCR, MML, GAB, and MJL designed and supervised the research. JCR, S. Price, KB, AP, DK, HM, PA, CMTV, JLF, JOA, SGH, NCP, and EU collected data. JCR, MML, JZ, MB, SJD, PJ, AM, GN, GAB, BS, LZ, ABG, SDC, TH, RM, KR, and SSDR performed research. JCR, MML, JZ, MB, SJD, HCS, CK, JTA, SDC, EM, GAB, MM, and MJL analyzed and interpreted the data. S. Pittaluga, AP, and DEK conducted pathology evaluations. JCR, MML, SJD, SDC, SH, and MM performed statistical analysis. AGRD and PM developed the data clustering algorithm. JCR, S. Price, KB, DK, MG, JIC, HCS, VKR, GU, JB, LW, CT, WG, LRF, JSO, IKC, CMTV, JLF, JOA, SGH, NCP, TP, EU, MK, SSDR, and HLM provided clinical insight. JCR, MML, MJL, and other authors wrote the manuscript. All authors reviewed and edited the manuscript. JCR and MML contributed equally to this work; JCR led the clinical study and is thus listed first.
Supplementary Material
Acknowledgments
This work was supported by the Division of Intramural Research, NIAID, NIH; the Intramural Research Program of the National Human Genome Research Institute, NIH; the Japan Society for the Promotion of Science (JSPS) and NIH grant (71403); and by cofunding through the Office of Disease Prevention, NIH. MML was supported by a Uehara Research Fellowship from the Uehara Memorial Foundation (201330032). We also acknowledge the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute for their grant (UM1 HG006542) to the Baylor-Hopkins Center for Mendelian Genomics; the NIAID for their grant (NIH R01-AI120989) to JSO; and COLCIENCIAS for their grant (1115.569.34430) to the Universidad de Antioquia. This project was also supported by federal funds from the NCI under contract HHSN261200800001E. The CyTOF Facility at the Center for Human Immunology is supported by NIH intramural funding (AI001226-01). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The authors thank Ryan Kissinger for designing the diagrams, Brigit S. Sullivan for reviewing the manuscript, and Hudson H. Freeze for important insights and advice. We thank James R Lupski, Richard Gibbs, and Zeynep Coban-Akdemir for their contribution in the genetic diagnosis of one of our patients. We thank Maxcyte Inc. for providing the MAGT1 and GFP mRNAs and their collaboration. Finally, we thank all patients described in this manuscript and their families for facilitating this work.
Version 1. 11/05/2019
In-Press Preview
Version 2. 12/09/2019
Electronic publication
Version 3. 01/02/2020
Print issue publication
Footnotes
MML’s present address is: Division of Experimental Immunology, Institute of Advanced Medical Sciences, University of Tokushima, Tokushima, Japan.
MB’s present address is: New York University Langone Medical Center, New York, New York, USA.
GN’s present address is: Department of Cell Biology, Harvard Medical School, Boston, Massachusetts, USA.
JSO’s present address is: Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
EM’s present address is: University of California San Diego School of Medicine, San Diego, California, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(1):507–522.https://doi.org/10.1172/JCI131116.
See the related Commentary at XMEN: welcome to the glycosphere.
Contributor Information
Juan C. Ravell, Email: juan.ravell@nih.gov.
Mami Matsuda-Lennikov, Email: mmlennikov@gmail.com.
Samuel D. Chauvin, Email: samuel.chauvin@nih.gov.
Juan Zou, Email: JUZOU@augusta.edu.
Matthew Biancalana, Email: matt.biancalana@gmail.com.
Sally J. Deeb, Email: sally.deeb@genedata.com.
Susan Price, Email: pricesu@niaid.nih.gov.
Helen C. Su, Email: Hsu@niaid.nih.gov.
Giulia Notarangelo, Email: giulia_notarangelo@g.harvard.edu.
Ping Jiang, Email: ping.jiang@nih.gov.
Aaron Morawski, Email: aaron.morawski@nih.gov.
Chrysi Kanellopoulou, Email: chrysi.kanellopoulou@gmail.com.
Ratnadeep Mukherjee, Email: ratnadeep.mukherjee@nih.gov.
James T. Anibal, Email: jta54@case.edu.
Brian Sellers, Email: brian.sellers@nih.gov.
Lixin Zheng, Email: LZHENG@niaid.nih.gov.
Tingyan He, Email: Hetingyan2017@outlook.com.
Stefania Pittaluga, Email: stefpitt@mail.nih.gov.
Astin Powers, Email: apowers88md@gmail.com.
David E. Kleiner, Email: kleinerd@mail.nih.gov.
Devika Kapuria, Email: devika.kapuria@gmail.com.
Marc Ghany, Email: MarcG@intra.niddk.nih.gov.
Sally Hunsberger, Email: sallyh@niaid.nih.gov.
Jeffrey I. Cohen, Email: jcohen@niaid.nih.gov.
Gulbu Uzel, Email: guzel@niaid.nih.gov.
Jenna Bergerson, Email: Jenna.Bergerson@nih.gov.
Lynne Wolfe, Email: lynne.wolfe@nih.gov.
Camilo Toro, Email: toroc@mail.nih.gov.
William Gahl, Email: gahlw@mail.nih.gov.
Les R. Folio, Email: les.folio@nih.gov.
Helen Matthews, Email: helen.matthews@nih.gov.
Pam Angelus, Email: pam.angelus@nih.gov.
Ivan K. Chinn, Email: ivan.chinn@bcm.edu.
Jordan S. Orange, Email: jso2121@cumc.columbia.edu.
Claudia M. Trujillo-Vargas, Email: claudia.trujillo@udea.edu.co.
Jose Luis Franco, Email: jose.franco@udea.edu.co.
Julio Orrego-Arango, Email: julio.orrego@udea.edu.co.
Sebastian Gutiérrez-Hincapié, Email: sebasghincapie@gmail.com.
Niraj Chandrakant Patel, Email: Niraj.Patel@atriumhealth.org.
Kimiyo Raymond, Email: raymond.kimiyo@mayo.edu.
Turkan Patiroglu, Email: turkanp@erciyes.edu.tr.
Ekrem Unal, Email: drekremunal@yahoo.com.tr.
Musa Karakukcu, Email: mkkukcu@yahoo.com.
Alexandre G.R. Day, Email: agrday@bu.edu.
Pankaj Mehta, Email: pankajm@bu.edu.
Evan Masutani, Email: evmasuta@gmail.com.
Suk S. De Ravin, Email: sderavin@niaid.nih.gov.
Harry L. Malech, Email: hmalech@nih.gov.
Grégoire Altan-Bonnet, Email: gregoire.altan-bonnet@nih.gov.
V. Koneti Rao, Email: korao@niaid.nih.gov.
Matthias Mann, Email: mmann@biochem.mpg.de.
Michael J. Lenardo, Email: mlenardo@niaid.nih.gov.
References
- 1.Li FY, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475(7357):471–476. doi: 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaigne-Delalande B, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. 2013;341(6142):186–191. doi: 10.1126/science.1240094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. 2014;123(14):2148–2152. doi: 10.1182/blood-2013-11-538686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravell J, Chaigne-Delalande B, Lenardo M. X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia disease: a combined immune deficiency with magnesium defect. Curr Opin Pediatr. 2014;26(6):713–719. doi: 10.1097/MOP.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fisher GH, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 6.Rieux-Laucat F, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 7.Sneller MC, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90(2):334–341. doi: 10.1172/JCI115867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130(7):591–601. doi: 10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 9.Price S, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123(13):1989–1999. doi: 10.1182/blood-2013-10-535393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bleesing JJ, et al. TcR-alpha/beta(+) CD4(-)CD8(-) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol. 2001;100(3):314–324. doi: 10.1006/clim.2001.5069. [DOI] [PubMed] [Google Scholar]
- 11.Lee HS, Qi Y, Im W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci Rep. 2015;5:8926. doi: 10.1038/srep08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, Clapham DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A. 2009;106(37):15750–15755. doi: 10.1073/pnas.0908332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohorko E, Owen RL, Malojčić G, Brozzo MS, Aebi M, Glockshuber R. Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure. 2014;22(4):590–601. doi: 10.1016/j.str.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda-Lennikov M, et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J Biol Chem. 2019;294(37):13638–13656. doi: 10.1074/jbc.RA119.008903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16(4):47R–62R. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136(2):272–283. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shrimal S, Trueman SF, Gilmore R. Extreme C-terminal sites are posttranslocationally glycosylated by the STT3B isoform of the OST. J Cell Biol. 2013;201(1):81–95. doi: 10.1083/jcb.201301031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cherepanova NA, Venev SV, Leszyk JD, Shaffer SA, Gilmore R. Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J Cell Biol. 2019;218(8):2782–2796. doi: 10.1083/jcb.201904004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cherepanova NA, Shrimal S, Gilmore R. Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J Cell Biol. 2014;206(4):525–539. doi: 10.1083/jcb.201404083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7(4):389–396. doi: 10.2174/156652407780831548. [DOI] [PubMed] [Google Scholar]
- 21.Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 22.Blommaert E, et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype. Proc Natl Acad Sci U S A. 2019;116(20):9865–9870. doi: 10.1073/pnas.1817815116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhalla F, et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol. 2015;35(2):112–118. doi: 10.1007/s10875-014-0116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brigida I, et al. Large deletion of MAGT1 gene in a patient with classic Kaposi sarcoma, CD4 lymphopenia, and EBV infection. J Clin Immunol. 2017;37(1):32–35. doi: 10.1007/s10875-016-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patiroglu T, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol. 2015;159(1):58–62. doi: 10.1016/j.clim.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 26.He TY, Xia Y, Li CG, Li CR, Qi ZX, Yang J. [X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia: report of a family and literature review] Zhonghua Er Ke Za Zhi. 2018;56(1):48–52. doi: 10.3760/cma.j.issn.0578-1310.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Coffman RL, Weissman IL. B220: a B cell-specific member of th T200 glycoprotein family. Nature. 1981;289(5799):681–683. doi: 10.1038/289681a0. [DOI] [PubMed] [Google Scholar]
- 28.Zheng L, Li J, Lenardo M. Restimulation-induced cell death: new medical and research perspectives. Immunol Rev. 2017;277(1):44–60. doi: 10.1111/imr.12535. [DOI] [PubMed] [Google Scholar]
- 29.van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 30. doi: 10.1016/j.physrep.2019.03.001. Mehta P, et al. A high-bias, low-variance introduction to machine learning for physicists. ArXiv e-prints. http://www.arXiv:1803.08823v3 Submitted March 23,2018 REvised May 27, 2019. Access November 11, 2019. [DOI] [PMC free article] [PubMed]
- 31.Levine JH, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162(1):184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wild R, Kowal J, Eyring J, Ngwa EM, Aebi M, Locher KP. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science. 2018;359(6375):545–550. doi: 10.1126/science.aar5140. [DOI] [PubMed] [Google Scholar]
- 33.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227(1):150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karimi M, Cao TM, Baker JA, Verneris MR, Soares L, Negrin RS. Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J Immunol. 2005;175(12):7819–7828. doi: 10.4049/jimmunol.175.12.7819. [DOI] [PubMed] [Google Scholar]
- 35.Ho EL, Heusel JW, Brown MG, Matsumoto K, Scalzo AA, Yokoyama WM. Murine Nkg2d and Cd94 are clustered within the natural killer complex and are expressed independently in natural killer cells. Proc Natl Acad Sci U S A. 1998;95(11):6320–6325. doi: 10.1073/pnas.95.11.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zielinska DF, Gnad F, Wiśniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141(5):897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Schulz BL, et al. Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc Natl Acad Sci U S A. 2009;106(27):11061–11066. doi: 10.1073/pnas.0812515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jamaluddin MF, Bailey UM, Schulz BL. Oligosaccharyltransferase subunits bind polypeptide substrate to locally enhance N-glycosylation. Mol Cell Proteomics. 2014;13(12):3286–3293. doi: 10.1074/mcp.M114.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karaoglu D, Kelleher DJ, Gilmore R. Functional characterization of Ost3p. Loss of the 34-kD subunit of the Saccharomyces cerevisiae oligosaccharyltransferase results in biased underglycosylation of acceptor substrates. J Cell Biol. 1995;130(3):567–577. doi: 10.1083/jcb.130.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaith P, Assmann G, Uhlenbruck G. Characterization of the oligosaccharide side chain of apolipoprotein C-III from human plasma very low density lipoproteins. Biochim Biophys Acta. 1978;541(2):234–240. doi: 10.1016/0304-4165(78)90396-3. [DOI] [PubMed] [Google Scholar]
- 41.Al Teneiji A, et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol Genet Metab. 2017;120(3):235–242. doi: 10.1016/j.ymgme.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 42.Shrimal S, Ng BG, Losfeld ME, Gilmore R, Freeze HH. Mutations in STT3A and STT3B cause two congenital disorders of glycosylation. Hum Mol Genet. 2013;22(22):4638–4645. doi: 10.1093/hmg/ddt312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abolhassani H, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. 2017;214(1):91–106. doi: 10.1084/jem.20160849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caminha I, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125(4):946–949.e6. doi: 10.1016/j.jaci.2009.12.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pinheiro JC, Bates D. Mixed-Effects Models in S and S-PLUS. New York, NY: Springer-Verlag; 2000. [Google Scholar]
- 46.Lacey JM, Bergen HR, Magera MJ, Naylor S, O’Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001;47(3):513–518. [PubMed] [Google Scholar]
- 47.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finck R, et al. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83(5):483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deeb SJ, Cox J, Schmidt-Supprian M, Mann M. N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol Cell Proteomics. 2014;13(1):240–251. doi: 10.1074/mcp.M113.033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.