

Abstract
We report an efficient synthesis of protected (2S,3R)-3-hydroxy-3-methylproline that proceeds in three steps with complete stereoselectivity. This route represents a significant improvement over previous approaches to this noncanonical amino acid. Key to this success is the development of a one-pot chemoenzymatic procedure for the preparation of (2S,3S)-3- methylproline from L-isoleucine. This work lays the foundation for future chemoenzymatic syntheses of polyoxypeptin A and FR225659.
Keywords: biocatalysis, depsipeptide, chemoenzymatic synthesis, hydroxylation
Graphical Abstract
1. Introduction
Isolated from Streptomyces sp. MK498–98 F14, polyoxypeptin A (1) is a unique hexadepsipeptide with potent cytotoxicity against apoptosis-resistant human pancreatic adenocarcinoma AsPC-1 cells (Figure 1).1 Specifically, 1 was shown to induce early cell death, nuclear fragmentation, and internucleosomal DNA scission with an ED50 value of < 0.1 μg/mL.2 In contrast, known anticancer agents adriamycin and vinblastine are not capable of inducing apoptosis in AsPC-1 cells even at 30 μg/mL, suggesting that 1 could warrant further development as treatment for recalcitrant cancer. Structurally, 1 consists of several highly unusual building blocks, including two N-hydroxylated amino acids, (2S,3R)-3-hydroxy-3- methylproline (2), 5-hydroxypiperazic acid, and a complex C15 acyl side chain bearing five stereocenters. While several synthetic studies have been attempted, no total synthesis of 1 has been disclosed to date. Most of these synthetic studies have focused on the preparation of 2, an exceedingly rare fragment in peptide natural products. To date, 2 has only been found in polyoxypeptins A and B, as well as in FR225659 (3), a fungal peptide natural product with potent inhibitory effects on glucagon-induced gluconeogenesis.3,4
Figure 1.

The chemical structures of polyoxypeptin A (1) and FR225659 (3), two natural products containing (2S,3R)-3-hydroxy-3-methylproline motif (2).
One of the earliest routes to access 2 was developed by Kobayashi and co-workers in 2001 (Figure 2A).5 This approach proceeded from geraniol in 12 steps and featured the use of Sharpless asymmetric epoxidation as a stereogenerating step, and the use of Pd-catalyzed N- allylation to furnish the desired pyrrolidine ring. Two routes starting from protected glycine were reported in 2005.6,7 However, they required either the use of lipase-catalyzed kinetic resolution that proceeded with sub-optimal enantioselectivity, or the use of expensive (S)-BINOL as a stoichiometric chiral auxiliary. Finally, Ye and co-worker have also developed an 11-step approach towards 2 starting from Boc-Thr-OH in 2005.8 Given the shortcomings of previously developed syntheses, we believe that a more efficient strategy to prepare 2 is warranted. Our synthesis design leveraged the use of biocatalytic retrosynthetic logic and drew inspiration from the proposed biosynthesis of 2 (Figure 2B). A seminal isotope-labelling work by Umezawa and co-workers established that 2 arises in nature through sequential C5 and C3 functionalizations of L-isoleucine (5).9 The biosynthetic gene cluster of 1 was identified in 2014,10 and two iron- and α-ketogluratate-dependent enzymes (Fe/αKGs), PlyO and PlyP, were proposed to take part in the production of 2. However, no biochemical characterization was performed to confirm the functions of these two enzymes.
Figure 2.

A. Prior synthetic approaches to 2. B. Our biocatalytic retrosynthetic analysis of 2.
While characterization of PlyO and PlyP merited serious considerations, we were cognizant of two recently-characterized Fe/αKGs that could offer potential solutions for selective C3 and C5 functionalizations of 4 and 5, respectively. GetF is a pipecolic acid 3-hydroxylase from the GE81112 biosynthesis pathway that has been reported to exhibit promiscuous hydroxylation activity towards 4.11 However, at >$100/g from commercial vendors, 4 is not a viable starting material from an economic standpoint. While a Hofmann-Loffler-Freytag approach to 4 has previously been developed,12 adaptation of this strategy to the synthesis of 5 will necessitate unnecessary amino acid protection/deprotection sequence. Recent elucidation of the biogenesis of UCS1025A by Tang and co-workers13 identified the role of an Fe/αKG enzyme, UcsF, in the construction of its pyrrolizidine motif via initial C5 oxidation of 5. Encouragingly, in vitro analysis by Tang and co-workers suggested that UcsF is capable of performing iterative C5 oxidation on 5 to afford the corresponding cyclic imine. Given previous precedent from our laboratory in leveraging such reactivity for the construction of 4-methylproline derivatives,14 we hypothesized that biocatalytic oxidation of 5 with UcsF will provide a more efficient solution for large-scale preparation of 4.
2. Results and discussion
We first set out to determine the efficiency of isoleucine oxidation with UcsF. To this end, we carried out extensive optimization of enzyme loading and αKG equivalents in the biotransformation, monitoring the reaction conversion with 1H NMR analysis (Table 1). While we found that 0.05 mol% catalyst loading of UcsF is sufficient to fully process 5 to its C5- hydroxylated counterpart (6), complete conversion to the desired cyclic imine 7 could not be achieved even with 0.3 mol% enzyme loading. In fact, for reasons that are unclear at this stage, the reaction conversion seemed to plateau at ca. 1:1 ratio of 6:7. Addition of catalase to the reaction to suppress unwanted reactive oxygen species or change in reaction headspace volume failed to elicit further improvement in reaction conversion. Further temperature screening also revealed that the reaction proceeds optimally at 20 °C. Despite the sub-optimal conversion, we reasoned that this ratio is sufficient to provide ample quantity of 7 for further manipulations towards 2.
Table 1.
Optimization of reaction parameters for the oxidation of 5 with UcsF.a
 | |||
|---|---|---|---|
| % mol UcsF | αKG equiv. | 6 (%) | 7 (%) |
| 0.01 | 3.0 | 23 | 0 |
| 0.025 | 3.0 | 53 | 0 |
| 0.05 | 3.0 | 98 | 2 |
| 0.1 | 4.0 | 76 | 24 |
| 0.2 | 5.0 | 56 | 44 |
| 0.2* | 5.0 | 55 | 45 |
| 0.2^ | 5.0 | 78 | 22 |
| 0.2# | 5.0 | 100 | 0 |
| 0.3 | 5.0 | 53 | 47 |
Reaction conditions: L-isoleucine (20 mM, 1.0 equiv), αKG (60–100 mM, 3.0–5.0 equiv), L- ascorbic acid (10 mM, 0.50 equiv), FeSO4 (1 mM, 0.050 equiv), kPi buffer (pH = 8.0, 50 mM),12 h at 20 °C. Percent conversions were assessed by 1H NMR analyses at the end of the reactions.
Reaction was run in the presence of bovine liver catalase (0.1 mol%).
Reaction was conducted at 30 °C.
Reaction was conducted at 37 °C.
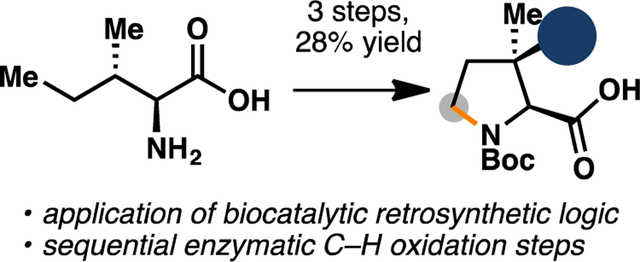
To streamline the workflow for large-scale reaction, we next tested the use of crude lysate for the oxidation as it would allow us to bypass laborious protein purification steps. Gratifyingly, similar levels of conversion (ca. 1:1 mixture of 6 and 7, full consumption of 5) could also be observed when the biotransfomation was conducted with clarified lysate of E. coli expressing UcsF (pre- lysis OD600 = 40). Similar to our previous work in the one-pot chemoenzymatic synthesis of 4- methylproline derivatives, we observed that the imine moiety of 7 could undergo facile and complete reduction upon treatment with NH3•BH3 (Scheme 1).15 Purification of the crude reaction with ion-exchange chromatography yielded a 1:1 inseparable mixture of 4 and 6 with excellent mass recovery. Without further separation, this mixture was carried forward to biocatalytic hydroxylation with GetF. In agreement with previous report, GetF was observed to exhibit excellent hydroxylation activity on 4. Biocatalytic hydroxylation of 4 could be performed with clarified lysate of E. coli co-expressing GetF and GroES/GroEL16,17 (pre-lysis OD600 = 30) to afford complete conversion to 2 without any detrimental effects from the presence of 6 during the reaction. At this stage, the crude product mixture was subjected to N-Boc protection, during which Boc-protected 6 underwent intramolecular cyclization to the six-membered lactone, thereby facilitating purification of Boc-protected (2S,3R)-3-hydroxy-3-methylproline (8). Overall, this strategy allows 8 to be obtained in 28% yield over 3 steps starting from L-isoleucine (5). To highlight its scalability and practicality, this route has been performed on up to 300 mg scale, without any significant loss in isolated yield of the desired product.
Scheme 1.

Chemoenzymatic synthesis of Boc-protected (2S,3R)-3-hydroxy-3-methylproline (8) featuring two enzymatic C–H oxidation events.
3. Conclusion
This work describes the development of a biocatalytic approach towards (2S,3R)-3-hydroxy-3- methylproline by employing two key enzymatic C–H hydroxylation steps that both proceed with exquisite regio- and stereocontrol and good yields. By enlisting the use of biocatalytic C–H oxidation logic,18 we successfully streamlined the preparation of a traditionally-challenging amino acid motif and minimized the use of concession steps and protecting group manipulations. In contrast to prior approaches, our synthesis of 2 commenced from inexpensive starting material (isoleucine) and relied only on the use of green and environmentally benign oxidation biocatalysts. This work lays the initial foundation for the total syntheses of polyoxypeptin A and FR225659.
4. Experimental
4.1. General materials and methods
Unless otherwise noted, all chemicals and reagents for chemical reactions were purchased at the highest commercial quality and used without further purification. Reactions were monitored by thin layer chromatography (TLC) and liquid chromatography/mass spectrometry (LC/MS). TLC was performed with 0.25 mm E. Merck silica plates (60F-254) using short-wave UV light as the visualizing agent, and ninhydrin, KMnO4, or phosphomolybdic acid and heat as developing agents. LC/MS was performed with Agilent 1260 Infinity System equipped with Poroshell 120 EC-C18 column (3.0 × 50 mm, 2.7 micron). Flash column chromatography was performed using a Biotage® Isolera One automated purification system loaded with Zip KP-Sil cartridges filled with SilicaFlash® P60 silica gel (230–400 mesh). NMR spectra were recorded on a Bruker spetrometer and calibrated using residual undeuterated solvent. Optical rotations were measured on Autopol IV polarimeter (Rudolph Research Analytical). Expression and purification of Fe/αKG enzymes were performed by following previously reported protocols.14,16
4.2. Enzyme expression of UcsF
An overnight culture of BL21(DE3) E. coli cells harboring pET-28a(+)-UcsF was used to inoculate 2 × 500 mL TB media (in 2 × 2 L non-beveled Erlenmeyer flasks) containing 50 μg/mL kanamycin. The cultures were shaken at 250 rpm at 37 °C for roughly 3 h or until an optical density (OD600) of 0.7–1.0 was reached. Cultures were cooled on ice (20 min) and then induced by adding IPTG to a final concentration of 0.025 mM. The cultures were allowed to shake at 250 rpm for another 16 hours at 20 °C. The cells were harvested by centrifugation (4 °C, 15 min, 4,000xg). For enzyme purification, the cell pellet was stored at –20 °C or below for at least 2 h. For reaction with clarified lysate, the pellet was resuspended in 50 mM phosphate buffer (pH = 8.0) to OD600 = 40 (for 1000 mL expression culture, this usually requires ca. 240 mL of buffer). Cells were lysed by sonication (3 min, cycle = 1 s on/4 s off, 45% amplitude). The lysate was centrifuged at 10,000xg for 15 min at 4 °C to remove cell debris and split into 2 batches.
4.3. Protein purification of UcsF
Purification was performed with an AKTA pure FPLC system (GE Healthcare). For the purification of 6XHis tagged enzymes, the thawed cell pellet was resuspended in Ni-NTA buffer A (25 mM Tris.HCl, 200 mM NaCl, 25 mM imidazole, pH 8.0, 4 mL/g of cell wet weight) and lysed by sonication (3 min, cycle = 1 s on/4 s off, 50% amplitude). The lysate was centrifuged at 15,000xg for 30 min at 4 °C to remove cell debris. The collected supernatant was subjected to a Ni-NTA chromatography step using a Ni Sepharose column (HisTrap-HP, GE healthcare, Piscataway, NJ). The protein was eluted from the Ni Sepharose column using 25 mM Tris.HCl, 200 mM NaCl, 300 mM imidazole, pH 8.0. Ni-purified protein was buffer exchanged into 50 mM phosphate buffer (pH = 8.0) using a 10 kDa MW cut-off centrifugal filter. Protein concentrations were determined by A280 with calculated extinction coefficients (UcsF – 38,390 M−1cm−1). For storage, proteins were portioned into 100 μL aliquots, flash frozen on liquid N2, and stored at –80 °C.
4.4. In vitro hydroxylation of isoleucine with purified UcsF
A 20 mL scintillation vial was charged with the L-isoleucine (60 μmol, 1.0 equiv, 20 mM final concentration), L-ascorbic acid (30 μmol, 0.5 equiv, 10 mM final concentration), and α- ketoglutaric acid (disodium salt dihydrate, 150 μmol, 3.0–5.0 equiv, 60–100 mM final concentration). 50 mM kPi buffer was added to the vial (pH 8.0, 3.0 mL), followed by 15 μL of FeSO4 solution in H2O (200 mM, 0.05 equiv, 1 mM final concentration). The reaction was started by the addition of UcsF stock solution (final concentration = 2.0–60 μM, 0.0001–0.003 equiv) and shaken for 12 h at 20 °C, 250 rpm under air. After quenching with 300 μL of 1 M HCl, the crude reaction mixture was centrifuged (15,000 rpm, 5 min) and lyophilized. The crude mixture was resuspended in D2O and directly submitted to 1 H NMR analysis to determine the ratio of 5:6:7.
4.5. Chemoenzymatic synthesis of 4 using clarified lysate of E. coli expressing UcsF
A 1 L Erlenmeyer flask was charged with isoleucine (314 mg, 2.40 mmol, 1.0 equiv, 20 mM final concentration), L-ascorbic acid (211 mg, 1.20 mmol, 0.5 equiv, 10 mM final concentration), and α-ketoglutaric acid (disodium salt dihydrate, 1356 mg, 6.00 mmol, 2.5 equiv, 50 mM final concentration). 120 mL of clarified lysate of E. coli expressing UcsF from section 4.2 was added to the flask, followed by FeSO4•7H2O (53 mg, 0.192 mmol, 0.08 equiv, 1.6 mM final concentration). The reaction was shaken for 4 h at 20 °C, 200 rpm under air. At this point, L- ascorbic acid (211 mg, 1.20 mmol, 0.5 equiv), α-ketoglutaric acid (disodium salt dihydrate, 1356 mg, 6.00 mmol, 2.5 equiv) and FeSO4•7H2O (53 mg, 0.192 mmol, 0.08 equiv)were added, followed by the remaining 120 mL of clarified lysate. The reaction was shaken for another 4 h, and treated with NH3•BH3 (223 mg, 7.20 mmol). After shaking for 30 min, the reaction was quenched with 35 mL of 1 M HCl (final pH = 2). The suspension was centrifuged at 10,000xg for 15 min at 4 °C to remove protein debris. Dowex 50WX8 resin (50 g) was slurry-packed with 1 M NH4OH in a flash chromatography column then washed with H2O until pH = 7. Next, the resin was washed with 1 M HCl until pH = 1, and then with H2O until pH = 7. The reaction supernatant was loaded directly onto the column, washing with H2O (ca. 600 mL), and with 1 M NH4OH (ca. 500 mL) until product no longer eluted from the column. Amino acid containing fractions were pooled, concentrated in vacuo, and then lyophilized. 1H NMR analysis of the crude product mixture at this stage showed approximately 1:1 mixture of 4:6, with no detectable presence of 5. Compound 4 was further characterized as its N-Boc counterpart.19 Spectroscopic data are in agreement with the structure and the 1H NMR data and 13C NMR data are consistent with previously reported values.20
4.6. Enzyme expression of GetF
An overnight culture of BL21(DE3) E. coli cells harboring pET-28a(+)-GetF and pGro7 plasmids was used to inoculate 500 mL TB media (in 2 L non-beveled Erlenmeyer flasks) containing 50 μg/mL kanamycin and 25 μg/mL chloramphenicol. The cultures were shaken at 250 rpm at 37 °C for roughly 3 h or until an optical density (OD600) of 0.7–1.0 was reached. Cultures were cooled on ice (20 min) and then induced by adding IPTG and L-arabinose to a final concentration of 0.025 mM and 1 mg/mL respectively. The cultures were allowed to shake at 250 rpm for another 16 hours at 23 °C. The cells were harvested by centrifugation (4 °C, 15 min, 4,000xg) and resuspended in 50 mM phosphate butter (pH = 7.0) to OD600 = 30 (for 500 mL expression culture, this usually requires ca. 200 mL of buffer). Cells were lysed by sonification (3 min, cycle = 1 s on/4 s off, 45% amplitude). The lysate was centrifuged at 10,000xg for 15 min at 4 °C to remove cell debris.
4.7. Hydroxylation of 4 using clarified lysate of E. coli expressing GetF
A 500 mL Erlenmeyer flask was charged with the crude product from section 4.5 (containing approximately 1.20 mmol 4, 1.0 equiv, 20 mM final concentration), L-ascorbic acid (106 mg, 0.60 mmol, 0.50 equiv, 10 mM final concentration), and α-ketoglutaric acid (disodium salt dihydrate, 678 mg, 3.00 mmol, 2.5 equiv, 50 mM final concentration). 60 mL of clarified lysate of E. coli expressing GetF was added to the flask, followed by FeSO4•7H2O (27 mg, 96 μmol, 0.08 equiv, 1.6 mM final concentration). The reaction was shaken for 12 h at 20 °C, 200 rpm under air, then quenched with 9 mL of 1 M HCl (final pH = 2). The suspension was centrifuged at 10,000xg for 15 min at 4 °C to remove protein debris. Dowex 50WX8 resin (50 g) was slurry- packed with 1 M NH4OH in a flash chromatography column, and then washed with H2O until pH = 7. Next, the resin was washed with 1 M HCl until pH = 1, and then with H2O until pH = 7. The reaction supernatant was loaded directly onto the column, washing with H2O (ca. 600 mL), and with 1 M NH4OH (ca. 500 mL) until product no longer eluted from the column. Amino acid containing fractions were pooled, concentrated in vacuo, and then lyophilized. 1H NMR analysis of the crude product mixture at this stage showed approximately 1:1 mixture of 2:6, with no detectable presence of 4.
4.8. Synthesis of Boc-protected (2S,3R)-3-hydroxy-3-methylproline (8)
The crude product from section 4.7 (containing approximately 1.20 mmol 2 and 1.20 mmol 6, 1.0 equiv) was dissolved in a mixture of water (5 mL) and 1,4-dioxane (5 mL). The resulting solution was cooled to 0 °C and treated with NaHCO3 (605 mg, 7.20 mmol, 3.0 equiv) and Boc2O (1.30 g, 6.00 mmol, 2.5 equiv). The reaction mixture was slowly warmed to room temperature and allowed to stir for 24 hours. At this stage, LCMS analysis showed that all starting material had reacted. The reaction was acidified with 1N HCl (final pH = 2), and then extracted with CH2Cl2 (2 × 15 mL). The organic layer was washed with water, followed by brine, and then dried over anhydrous Na2SO4. 0.15 mL of formic acid was added and the organic layer was allowed to stir overnight. Column chromatography purification (0.2% formic acid, methylene chloride:methanol 50:1 to 10:1) provided 8 (165 mg, 0.672 mmol, 28% yield) as colorless foam.
Rf = 0.2 (CH2Cl2:MeOH 9:1)
1H NMR (400 MHz, methanol-d4, 20 °C) a mixture of rotamers: δ 4.00 and 3.98 (s, 1H), 3.65 – 3.54 (m, 1H), 3.49 – 3.39 (m, 1H), 2.09 – 2.00 (m, 1H), 1.91 – 1.82 (m, 1H), 1.47 – 1.41 (m, 12H).
13C NMR (101 MHz, methanol-d4, 20 °C) a mixture of rotamers: δ 173.8 and 173.5, 156.2 and 156.1, 81.6 and 81.3, 79.2 and 78.3, 70.4 and 69.8, 46.0 and 45.5, 39.5 and 39.0, 28.7 and 28.5, 27.2 and 27.0.
1H NMR (400 MHz, DMSO-d6, 80 °C) δ 3.81 (s, 1H), 3.45 (ddd, J = 10.4, 7.9, 6.1 Hz, 1H), 3.31 (dt, J = 10.4, 7.0 Hz, 1H), 1.99 – 1.88 (m, 1H), 1.74 (ddd, J = 12.1, 7.6, 6.2 Hz, 1H), 1.41 – 1.34 (m, 12H).
HRMS (ESI): calcd for C11H18NO 5− (M-H)− 244.1190, found: 244.1205 [α]25D = –15.0 (c = 1.0, MeOH).
Supplementary Material
Acknowledgement
We thank the Roush, Shen, and Kodadek labs for access to their instrumentations. We acknowledge Prof. Yi Tang (UCLA) for providing a plasmid encoding for UcsF. Financial support for this work is generously provided by The Scripps Research Institute (TSRI) and the National Institutes of Health (grant GM128895).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Umezawa K; Nakazawa K; Uemura T; Ikeda Y; Kondo S; Naganawa H; Kinoshita N; Hashizume H; Hamada M; Takeuchi T; Ohba S Tetrahedron Lett 1998, 39, 1389–1392. [Google Scholar]
- 2.Umezawa K; Nakazawa K; Ikeda Y; Naganawa H; Kondo SJ Org. Chem 1999, 64, 3034–3038. [DOI] [PubMed] [Google Scholar]
- 3.Ohtsu Y; Sasamura H; Tsurumi Y; Yoshimura S; Takase S; Hashimoto M; Shibata T; Hino M; Fujii TJ Antibiot 2003, 56, 682–688. [DOI] [PubMed] [Google Scholar]
- 4.Hatori H; Zenkoh T; Kobayashi M; Ohtsu Y; Shigematsu N; Setoi H; Hino M; Handa MJ Antibiot 2004, 57, 456–461. [DOI] [PubMed] [Google Scholar]
- 5.Noguchi Y; Uchiro H; Yamada T; Kobayashi S Tetrahedron Lett 2001, 42, 5253–5256. [Google Scholar]
- 6.Haddad M; Larcheveque M Tetrahedron: Asymm 2005, 16, 2243–2247. [Google Scholar]
- 7.Makino K; Nagata E; Hamada Y Tetrahedron Lett 2005, 46, 8159–8162. [Google Scholar]
- 8.Chen Z; Ye T Synlett 2005, 2781–2785. [Google Scholar]
- 9.Umezawa K; Ikeda Y; Kawase O; Naganawa H; Kondo SJ Chem. Soc., Perkin Trans 1, 2001, 1550–1553. [Google Scholar]
- 10.Du Y; Wang Y; Huang T; Tao M; Deng Z; Lin S BMC Microbiol 2014, 14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattay J; Hüttel W ChemBioChem 2017, 18, 1523–1528. [DOI] [PubMed] [Google Scholar]
- 12.Reddy LR; Reddy BVS; Corey EJ Org. Lett 2006, 8, 2819–2821. [DOI] [PubMed] [Google Scholar]
- 13.Li L; Tang M-C; Tang S; Gao S; Soliman S; Hang L; Xu W; Ye T; Watanabe K; Tang YJ Am. Chem. Soc 2018, 140, 2067–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zwick CR III; Renata HJ Am. Chem. Soc 2018, 140, 1165–1169. [DOI] [PubMed] [Google Scholar]
- 15.Ghislieri D; Green AP; Pontini M; Willies SC; Rowles I; Frank A; Grogan G Turner NJ J. Am. Chem. Soc 2013, 135, 10863–10869. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X; King-Smith E; Renata H Angew. Chem. Int. Ed 2018, 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
- 17.Thomas JG; Ayling A; Baneyx F Appl. Biochem. Biotechnol 1997, 66, 197–238. [DOI] [PubMed] [Google Scholar]
- 18.Li F; Zhang X; Renata H Curr. Opin. Chem. Biol 2019, 49, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jurica EA et al. J. Med. Chem 2017, 60, 1417–1431. [DOI] [PubMed] [Google Scholar]
- 20.Sato H; Yoshida M; Murase H; Nakagawa H; Doi T ACS Comb. Sci 2016, 18, 590–595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.