

Abstract
The complex interplay between cancer cells and their microenvironment remains a major challenge in the design and optimization of treatment strategies for pancreatic ductal adenocarcinoma (PDAC). Recent investigations have demonstrated that mechanistically distinct combination therapies hold promise for treatment of PDAC, but effective clinical translation requires more accurate models that account for the abundant tumor-stroma and its influence on cancer growth, metabolism and treatment insensitivity. In this study, a modular 3D culture model that comprised PDAC cells and patient-derived cancer-associated fibroblasts (CAFs) was developed to assess the effects of PDAC-CAF interactions on treatment efficacies. Using newly-developed high-throughput imaging and image analysis tools, it was found that CAFs imparted a notable and statistically significant resistance to oxaliplatin chemotherapy and benzoporphyrin derivative-mediated photodynamic therapy, which associated with increased levels of basal oxidative metabolism. Increased treatment resistance and redox states were similarly observed in an orthotopic xenograft model of PDAC in which cancer cells and CAFs were co-implanted in mice. Combination therapies of oxaliplatin and PDT with the mitochondrial complex I inhibitor metformin overcame CAF-induced treatment resistance. The findings underscore that heterotypic microtumor culture models recapitulate metabolic alterations stemming from tumor-stroma interactions. The presented infrastructure can be adapted with disease-specific cell types and is compatible with patient-derived tissues to enable personalized screening and optimization of new metabolism-targeted treatment regimens for pancreatic cancer.
Keywords: Cancer organoids, Cancer metabolism, Redox homeostasis, Antioxidant and redox signaling, Mitochondrial respiration, Photo-chemotherapy combinations
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy that is projected to become a major cause of cancer deaths, as the late diagnosis and the limited efficacy of available treatment options leads to dismal prognoses [1]. Palliative chemotherapeutic strategies such as FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, and oxaliplatin), nanoparticular paclitaxel, and nanoparticular irinotecan have shown extended survival for patients with non-resectable pancreatic cancer, yet long-term survival rates remain poor and have not substantially improved over the last decades [2–4]. In the evaluation of new, more effective therapies for PDAC, photodynamic therapy (PDT) has demonstrated significant clinical potential [5]. This therapy comprises the pharmaceutical photosensitization of tumor masses, resulting in the local generation of cytotoxic reactive molecular species (RMS) upon irradiation with high-intensity visible light. These RMS trigger cell death as a result of direct and indirect cytotoxic mechanisms that include severe oxidative damage, hypoxia, hyponutrition, and an anti-tumor immune response [6–8]. As it is mechanistically distinct from chemotherapeutics, PDT synergizes well with various clinically used chemotherapies in preclinical models of pancreatic cancer [9–11].
Effective management of PDAC remains challenging, which is partially due to the failure of many preclinically validated therapeutics to succeed in clinical trials. This is attributed to the intrinsic biology of PDAC that is not completely recapitulated in most preclinical models. Clinical PDAC is characterized by excessive desmoplasia, which is associated with encapsulation of the tumor by cancer-associated fibroblasts (CAFs) and fibroblast-like pancreatic stellate cells [12]. The CAFs form a dense barrier that contributes to a harsh tumor microenvironment with limited blood flow and nutrient availability [13]. Recent reports have demonstrated that tumor survival and growth in these resource-limited environments is partly mediated by the aberrant metabolic properties of PDAC cells, high degrees of metabolic plasticity, and metabolite exchange between cancer cells and stromal partners [14]. Therapeutic strategies aiming at depleting the PDAC stroma have seen promising preclinical results but achieved ambiguous or dis-appointing results in clinical trials due to unforeseen biological effects and suboptimal dosimetry [15,16]. Therefore, finding effective therapies for PDAC requires a balanced and well-informed therapeutic approach [17], for which preclinical models that accurately mimic the interactions between cancer cells and stromal cells are a critical necessity [18]. To expedite the development of new therapies, cancer organoids are emerging as a promising preclinical platform [17,19]. The inclusion of CAFs in such models are critical to prevent inaccurate assessments of treatment response and biological effects [12,20,21].
To inform and expedite the development of new therapies, this study aimed to establish customizable, microtumor culture models of PDAC that account for the biological effects of CAFs on treatment susceptibility. The models were specifically designed to be compatible with recently developed high-content imaging assays and image analysis tools for redox states and treatment response [22–24]. The models demonstrated that CAFs influence microtumor development, redox state, and treatment susceptibility. These findings inspired combination therapies of oxaliplatin and PDT with metformin, in which inhibition of oxidative metabolism overcame CAF-induced treatment resistance. Taken together, these heterotypic microtumors represent a modular platform that can be optimized for specific cancer types and with patient-derived tissues to investigate the interplay between cancer development, metabolism, and treatment screening.
2. Materials and methods
2.1. Cell culture
MIA PaCa-2 (MP2) and AsPC-1 human pancreatic cancer cells were acquired from the American Type Culture Collection (ATCC, Manassas VA). MP2 cells were maintained in 5 mM L-glutamine and 4.5 mM glucose containing DMEM (Corning, Tewksbury MA) supplemented with 10% FBS (Corning) and 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, ThermoFisher, Waltham MA). AsPC-1 cells were grown in RPMI1640 supplemented with 5 mM glutamine, 10% FBS, and 100 U/mL penicillin, 100 μg/mL streptomycin. The culture media of both cell lines will hereafter be referred to as complete culture medium. Cells were typically passaged twice per week to a 1:10 ratio, and cultures were discarded at passage number 30.
Normal human skin samples and samples of squamous cell carcinoma were obtained at the Department of Dermatology, Massachusetts General Hospital (MGH), as discarded tissues not needed for diagnosis. All samples were processed as approved by the MGH Institutional Review Board. The age/stage, sex and gender identity of the subjects was not available. Healthy dermal fibroblasts (HDF1 and HDF2 primary dermal fibroblasts) were freshly isolated from healthy human abdominal skin tissue as described previously [25,26]. Cancer associated fibroblasts (CAF1 and CAF6 lines) were freshly isolated from clinically resected squamous cell carcinoma patients as described before [26]. Cells were maintained on complete DMEM culture medium and passaged weekly at a 1:4 ratio (HDFs) or 1:2 ratio (CAFs). Substantial expansion of the CAF lines was necessary to obtain sufficient cell numbers for 3D culture (~2.5 × 105 cells/75 cm2 culture flask), and were therefore typically discarded at passage number 20. No aberrations in proliferation rates, differentiation, or morphology were detected during these passages. For 3D co-cultures composed of AsPC-1+CAF6, the culture media of AsPC-1 was exchanged for fully supplemented DMEM for at least 2 passages prior to co-culture experiments.
All cell types were tested for mycoplasma contamination using the MycoAlert Plus mycoplasma detection kit (Lonza, Portsmouth NH), and underwent treatment with plasmocin (Mycozap, Lonza) when tested positively. All lines were confirmed mycoplasma negative prior to experimentation (Table S1).
2.2. Fibroblast characterization
For flow cytometry, cells were harvested as described above and aliquoted at 5 × 104 cells in separate centrifuge tubes. Cells were centrifuged at 500×g for 5 min, after which the supernatant was removed and the cell pellet was dissolved in 300 μL phosphate buffered saline (PBS). Cells were subsequently fixed in 700 μL 96% ice-cold ethanol, which was added in a drop-wise fashion under constant swerving of the cells. Prior to immunostaining, cells were washed in PBS and re-suspended in 100 μL staining solution (DAKO, Agilent, Lexington, MA) that was supplemented with mouse-anti-human α-smooth muscle actin (α-SMA) Cy3 (1:200, clone 1A4, Sigma-Aldrich, St Louis, MO) and polyclonal sheep-anti-human fibroblast activation protein (FAP) allophycocyanin (1:200, R&D Systems, Minneapolis, MN). Flow cytometry was performed using a FACS Aria cytometer (Becton Dickinson, Franklin Lakes, NJ) with which Cy3 and allophycocyanin fluorescence intensities were detected at λex 488 nm/λem 585 ± 42 nm and λex 633 nm/λem 660 ± 20 nm, respectively.
Fibroblasts were seeded on glass-bottom 24-well Sensoplates (Corning, Tewksbury MA) at a density of 2.5 × 104 cells/well. Cells were incubated overnight, after which they were fixed in 4% paraformaldehyde and 2% sucrose. The cells were subsequently stained with mouse-anti-human vimentin-Alexa488 (R&D Systems, Minneapolis MN) at a 1:50 dilution in staining solution for 1 h at room temperature. Cells were washed with staining buffer and submerged in with DAPI. After 30 min of incubation at room temperature in the dark, the cultures were imaged on an Olympus FV1000 confocal laser scanning microscope (DAPI: λex 405 nm/λem 440 ± 20 nm, Alexafluor488: λex 488 nm/λem 520 ± 40 nm), through a 0.65 NA 20 × air objective).
2.3. Adherent 3D cultures of pancreatic cancer microtumors
A 250 μL layer of 4 °C Matrigel (Corning) was solidified in each well of black walled, glass-bottom, 24-well Sensoplates (Corning), during a 20 min incubation at 37 °C, onto which 7500 MP2 cells were seeded on day 1. Matrigel lot numbers used throughout this study were 5173009 and 36819 and contained 9.2 mg/mL protein and < 1.5 U/mL endotoxin. Culture medium for 3D cultures was supplemented with 2% Matrigel throughout all experimental procedures. Cultures received fresh medium on culture day 5 and day 14. When indicated, 5 × 104 fibroblasts were added to the cultures on culture day 8, roughly corresponding to a cancer cell:fibroblast ratio of 2:1 [11]. This ratio was chosen as it was the lowest ratio at which substantial morphological alterations of the individual nodules were observed, whereas at higher ratios of CAFs resulted in extensive fibrosis to a degree that disabled further investigations. Cultures were maintained until day 15, during which medium was refreshed every 3 days. From here on, we will refer to these 3D cultures as cancer microtumors.
2.4. In vitro PDT and chemotherapy
PDT was performed on the microtumor cultures on day 11. Cultures were photosensitized during a 1 h incubation with 0.25 μM benzoporphyrin derivative (BPD, U.S. Pharmacopeia) in complete culture medium. Subsequently, medium was refreshed and microtumors were irradiated with 690 nm light (Intense Ltd., North Brunswick, NJ) at a fluence rate of 150 mW/cm2 and a total radiant exposure ranging from 1 to 50 J/cm2 as indicated. Oxaliplatin (SelleckChem, ThermoFisher) was provided on day 11 at concentrations ranging from 1 to 1000 μM as indicated (72 h exposure). Cultures received fresh oxaliplatin-free medium on day 14. When indicated, cells received fresh medium supplemented with 10 mM metformin hydrochloride (Sigma Aldrich) on day 9, day 11, and day 14.
2.5. Comprehensive assessment of treatment effects
Effects of treatment on microtumor viability was assessed using in situ live/dead staining with 2 μM Calcein AM (ThermoFisher), and 3 μM propidium iodide (PI, Sigma Aldrich) in PBS for 30 min at culture conditions, and subsequent confocal laser scanning fluorescence microscopy (Olympus FV1000). Calcein and PI fluorescence intensities were detected at λex 488 nm/λem 520 ± 20 nm and λex 559 nm/λem 630 ± 20 nm, respectively (0.16 NA 4 × air objective). Subsequent image acquisition and image analysis was performed using the CALYPSO methodology as developed and described previously [22]. Normalized dose-response curves were plotted in which the curve fits were constrained to 100% and 0% as the top and bottom plateaus, respectively, allowing us to determine the IC50 values with relative accuracy.
2.6. Label-free tracking of redox metabolism
The optical redox ratio (ORR) was determined using two-photon microscopy of reduced nicotinamide dinucleotide (NAD(P)H) and flavin adenine dinucleotide (FAD) autofluorescence intensity as described previously [23,24,31]. At the indicated growth and treatment conditions, cultures were imaged using an Olympus FV1000 laser scanning microscope. The excitation wavelength was set at 750 nm (Mai Tai DeepSee, Santa Clara, CA), and emission was collected at 480 ± 40 nm and 540 ± 20 nm for NAD(P)H and FAD, respectively. Image acquisition was performed through a 0.4 NA 10 × air objective, and consisted of stacks of 30 images taken at 0.1 μm intervals on the z-axis to prevent potential photothermal effects. The ORR was calculated on a pixel-by-pixel basis, using a custom built image analysis workflow in Matlab (Mathworks, Natick, MA).
2.7. Immunoblotting
Protein samples from the organoid cultures were obtained using Cell Recovery Solution (BD Bioscience), and lysis in RIPA buffer (Thermo Fisher) supplemented with protease inhibitor cocktail (Calbiochem) and phosphatase inhibitor cocktails I, II, and III (Sigma Aldrich). Lysates (1–25 μg protein) were separated using SDS-PAGE on 4–20% acrylamide gradient gels (BioRad) at 80 V for 1–2 h. The gels were blotted on PVDF membranes for 2 h at 150 A. Membranes were blocked for 1 h in 5% BSA (Sigma Aldrich) in TBST (Tris-buffered saline, Boston Bioproducts) supplemented with 0.1% Tween (Sigma-Aldrich). Membranes were incubated for 1 h in primary antibody in TBST with 5% BSA, washed excessively in TBST, and incubated for 1 h with secondary antibody labeled with horseradish peroxidase in TBST with 5% non-fat dry milk). After excessive washing in TBST, protein detection was performed using the ECL plus kit (Thermo Scientific) and CL-X Posure radiographic films (Thermo Fisher).
Antibodies and dilutions were: anti-heme oxygenase 1 (HO-1, clone A3, Santa Cruz, 1:1000), anti-cyclooxygenase 2 (COX-2, clone D5H5, Cell Signaling, 1:2000), and anti-β-actin (clone A-74, Sigma Aldrich, 1:20.000). Secondary antibodies conjugated to horseradish peroxidase were either goat-anti-mouse or goat-anti-rabbit (Cell Signaling, 1:2000). Immunoblots were quantified as described previously [27].
2.8. Extracellular metabolite analysis
Lactate concentrations in cell culture media were determined using the Lactate Plus lactate meter (Nova Biomedical, Waltham, MA). Culture medium was collected from the microtumor cultures and 5 μL of culture medium was diluted in 45 μL of milliQ grade water prior to measurement. Concentrations of alanine and aspartate in culture medium were determined using liquid chromatography tandem-mass spectrometry (LCMS/MS, Agilent). The instrumentation consisted of a 1290 Infinity solvent system and autosampler coupled to a 6430 Triple Quad LCMS (Agilent). Media contents were separated on a Zorbax Eclipse Plus C18 column (2.1 × 5 mm2, 1.8 μm, Agilent) using a mobile phase consisting of a NH4HCO2/acetonitrile gradient (0.4 mL/min). Amino acid detection was performed as described previously [28]. Concentrations were corrected for an internal standard of 15 μg/mL N-(1-naphthyl)ethylenediamine dihydrochloride (Sigma-Aldrich), and final concentrations were determined using a standard curve of alanine and aspartate (Sigma-Aldrich) in fully supplemented DMEM.
2.9. Evaluation of PDT efficacy in vivo
All animal experiments were performed in agreement with the IACUC of Massachusetts General Hospital, and procedures have been described previously (4). Male Swiss nu/nu mice received 1 × 106 MP2 cells or a mixture of 1 × 106 MP2 cells and 1 × 106 CAF6 cells in 50% growth factor-reduced Matrigel (200 μL injection volume) by injection in the pancreas under ketamine/xylazine anesthesia. Nine days post-implantation, mice were treated with PDT; 0.25 mg/kg Visudyne (QLT Phototherapeutics, Vancouver, Canada) via a 50 μL tail-vein injection, 1 h drug-light interval, and 690 nm irradiation (HPD/Intense, North Brunswick, NJ) at 100 mW/cm2 and a radiant exposure of 50 J/cm2. PDT was performed under ketamine/xylazine anesthesia, and PDT was delivered directly on the surgically exposed pancreas. Tumor size was monitored at 3/4-day intervals using ultrasound (Vevo LAZR, VisualSonics, Toronto, Canada) as described previously [9]. After 30 days (N = 3/group) and 60 days (N = 3/group), animals were sacrificed and tumor tissues were excised, embedded in Tissue-TEK Optimal Cutting Temperature compound (VWR, Franklin, MA), and frozen at −80 °C. Tissues were sliced in a cryomicrotome at a thickness of 10 μm and were placed on microscope slides. Frozen slides were thawed during a gentle wash in distilled water and were subsequently dried for 20 min at room temperature (in accordance with [29]). Immediately thereafter, slides were imaged using two-photon fluorescence microscopy to quantify NAD(P)H and FAD autofluorescence intensities as described previously [29–31].
2.10. Statistical analysis
Statistical analyses were performed in Graphpad Prism 7.0 (La Jolla, CA). Data was tested for normality. Normally distributed data sets were compared using a student’s t-test or one-way ANOVA and Tukey’s post-hoc test for multiple comparisons. Non-gaussian data sets were statistically compared using a Mann-Whitney U test or a Kruskal-Wallis test combined with a Dunn’s post-hoc test for multiple comparisons. Dose response curves were fitted using non-linear regression (least-squares fit), and fitted IC50 values were compared using an extra-sum-of-squares F-test.
3. Results
3.1. Formation of distinct cancer microtumor morphologies by healthy dermal fibroblasts and cancer associated fibroblasts
Various methods exist for establishing 3D culture models of cancer, including non-adherent cultures of spheroids or non-spherical cell aggregates, as well as matrix-adherent organotypic cultures. The latter has particular relevance in the screening for new therapies as large quantities of microtumors are cultured simultaneously, representing a heterogeneous mix of tumor nodules that can be individually analyzed to assess treatment susceptibility [11,22]. In this study, we leveraged a well-described protocol utilizing the liquid-overlay technique to establish adherent microtumor cultures that are compatible with recently developed methodologies for high-throughput assessment of treatment effects [11,22]. To account for the effects of stromal fibroblasts on the treatment of the PDAC microtumors, we explored the use of various types of fibroblasts to be used in these 3D co-culture models. As pancreatic CAFs can be typically difficult to obtain, we explored the use of primary CAFs isolated from squamous cell carcinoma as a more available resource.
Recent investigations demonstrated that dermal CAFs undergo both molecular and (epi)genetic alterations that are either pre-existing or occur upon interaction with malignant cells [25,26,32], and exhibit features that are common to CAFs found in pancreatic cancer specimens [26,33–35]. Thus, surgically resected squamous cell carcinoma specimens potentially represent a valuable and accessible resource for the frequent isolation of fresh CAFs to investigate cancer-stroma interactions. To evaluate the influence of CAFs on the development of PDAC microtumors, we first performed a basic molecular characterization of two individual CAF lines in comparison to HDFs from a similar origin. All fibroblast lines tested negative for the endothelial marker cytokeratin 8 (CK8, Fig. S1A), but did express high levels of FAP, with the highest levels observed in the HDF lines (Fig. S1B). CAF1 and CAF6 had significantly higher expression levels α-SMA (Fig. S1C) and vimentin (Fig. S1D) compared to HDF1 and HDF2 (Figs. S1C–D). Microscopy images additionally indicate that the morphology of the HDFs was largely spindle shaped, whereas the CAFs were larger and had a more stellate morphology (Fig. S1E). Taken together, the CAF1 and CAF6 cells exhibited characteristics typical of pancreatic CAFs, whereas the HDFs demonstrated features of both quiescent and wound-healing associated fibroblasts [33,34].
Based on previous studies in which microtumor cultures were established from various PDAC cell lines [11,36], an experimental timeline was designed to model desmoplastic PDAC and perform treatment screening (Fig. 1A). The PDAC cell lines MIA PaCa-2 (MP2) and AsPC-1 were selected as they represent well-characterized and widely-available cell types with different baseline treatment susceptibilities [9,11]. In these models, desmoplasia following the onset of PDAC was simulated by the addition of various types of fibroblasts to 8 day-old PDAC microtumors. The models were exposed to cancer therapies on day 11, after which treatment effects were assessed on culture day 15 (Fig. 1A).
Fig. 1. Experiment timeline of the 3D co-culture models.

(A) Adherent 3D cultures were initiated on day 1 by seeding 7.5 × 103 PDAC cells (MP2 cell line) on solidified Matrigel in each well of a 24-wells plate. Fibroblasts, either HDFs or CAFs, were added on day 8 at a density of 5 × 104, corresponding to a 2:1 cancer cell:fibroblast ratio [11]. Cultures were maintained until day 11, after which treatments were initiated. Experiments were terminated on day 15, after which live/dead staining and image analyses were performed. (B) Cancer microtumor development in absence and presence of the various fibroblasts. In absence of fibroblasts, the PDAC cultures develop into relatively homogeneously dispersed adherent spheroids. In presence of HDFs, large interconnected networks of microtumors are formed. In presence of CAFs, multiple individual cancer spheroids are condensed into large microtumor masses.
After addition of HDFs or CAFs to 8 day-old MP2 microtumor cultures, the fibroblast phenotype influenced the morphology of the heterotypic microtumors (Figs.1B and S1F). In the absence of fibroblasts, MP2 cells developed into a relatively monodispersed mix of individual oval microtumors (mean ± SEM = 3.3 × 104± 1.2 × 103 μm2, corresponding to an approximate diameter of 171.6 μm (Fig. 1B, S1G-H). The addition of HDFs resulted in the formation of interconnected networks between the individual MP2 nodules, resulting in a heterogeneous distribution in the size and shape of the microtumors (e.g., MP2+HDF1 mean ± SEM = 6.7 × 104 ± 5.3 × 103 μm2). CAFs displayed similar behavior (e.g., MP2+CAF1 mean ± SEM = 1.0 × 105 ± 1.6 × 104 μm2), but rapidly (< 24 h) condensed the connected microtumor networks into larger tumor masses, with CAFs residing in the center of these clusters (Figs.1B and S1F). Live/Dead staining on day 15 in the absence of treatment revealed the presence of viable cancer microtumors with necrotic cores (Fig. 1B). The effects of CAF6 fibroblasts were recapitulated in AsPC-1-based microtumor cultures, resulting in a significantly increased overall microtumor size (Figs. S1I–K). None of the heterotypic microtumor cultures displayed additional morphologic alterations with prolonged 3D culturing (Figs. S1G–J).
3.2. CAFs induce resistance to oxaliplatin and photodynamic therapy in heterotypic PDAC microtumors
Various investigations have implicated stromal CAFs in the development of therapy resistance, although the exact causal factors remain the subject of further study [33,37–40]. Potential mechanisms underlying CAF-induced resistance include increased extracellular matrix deposition, heterotypic cell-cell interactions, and metabolic reprogramming [33]. To ascertain whether the HDFs and CAFs were capable of imparting treatment resistance in our organotypic culture models, and thus whether these models could be used to investigate mechanisms underlying treatment insensitivity, the PDAC microtumor cultures were subjected to clinically relevant therapies.
PDAC patients may receive a variety of chemotherapies, including FOLFIRINOX which remains one of the most successful regimens [2]. The platinum-based chemotherapeutic oxaliplatin constitutes an integral component of this multi-line chemotherapy. Given the profound therapeutic recalcitrance associated with PDAC coupled with the high rates of relapse and progression, it has been postulated that combinations of mechanistically distinct adjuvant therapies, in which individual components of the regimen synergistically interact, may be required to achieve improved outcomes. One treatment that is mechanistically distinct of chemotherapies is PDT, which was demonstrated to be feasible and safe in a phase I/II clinical trial, with promising preliminary results [5]. Moreover, by leveraging 3D culture models, BPD-PDT was found to synergize with various chemotherapeutics, including platinum-agents carboplatin and oxaliplatin [11,41,42]. These findings are supported by other reports in which (neo)adjuvant PDT was found to improve the efficacy of various immuno- and chemotherapies [9,10,43], underscoring the translational value of PDT in the management of cancer. However, beneficial treatment outcomes from combinations of PDT and chemotherapeutics are strongly dependent on the selected photosensitizer and chemo agent [44]. As sensitivities to these treatments may be further influenced by cancer metabolism [45,46] and tumor-stroma interactions, responses to individual and combination therapies should be carefully characterized in appropriate preclinical models.
In this context, we evaluated the effects of oxaliplatin and BPD-PDT separately using the heterotypic PDAC microtumor models. Treatment efficacies were first evaluated on MP2-based microtumor cultures, and key conditions were tested on AsPC-1-based microtumor cultures. To evaluate treatment responses in heterotypic cultures with complex asymmetrical structures, we applied conventional live/dead staining in combination with a recently developed image analysis platform for high-throughput therapy screening that is agnostic to microtumor shape and size [22]. It should be noted that this assay does not evaluate viabilities of PDAC cells and CAFs separately, but rather considers individual microtumors as single entities containing both cell types. Dose-escalation studies of oxaliplatin revealed that the MP2 microtumors cultured with HDF1 and CAF6 had significantly higher viability compared to MP2 microtumors devoid of fibroblasts (Fig. 2A, 2C-D). Calculation of the oxaliplatin IC50 from the fitted dose-response curves of the fibroblast-containing microtumors indeed revealed a significant degree of treatment resistance (Fig. 2D) in comparison to the IC50 for MP2 microtumors (627.1 ± 28.9 μM). There was a 1.6-fold increase in IC50 for the MP2+HDF1 microtumors (992.6 ± 60.7 μM, p < 0.001), and a 2.4-fold increase in IC50 for the MP2+CAF6 microtumors (1492 ± 140.5 μM, p < 0.001). Additionally, there was a significant difference in oxaliplatin susceptibility between MP2+HDF1 and MP2+CAF6 microtumors (p < 0.001). Oxaliplatin resistance was also observed for the MP2+CAF1 microtumors, and to a lesser extent in MP2+HDF2 microtumors, as compared to MP2 without fibroblasts (Figs. S2A and S2B). Resistance to oxaliplatin occurred as well in 2D monolayer cultures in which MP2 cells were co-cultured with CAF6 (Fig. S2C). Lastly, these findings were corroborated in AsPC-1-based microtumors that were co-cultured with CAF6, demonstrating a significantly higher resistance to 500 μM oxaliplatin (Fig. 2E). It should be noted that AsPC-1 microtumors are inherently less susceptible to oxaliplatin chemotherapy compared to MP2-based microtumors [11].
Fig. 2. MP2 and AsPC-1 microtumors co-cultured with CAFs exhibit reduced treatment sensitivity to BPD-PDT and oxaliplatin chemotherapy.
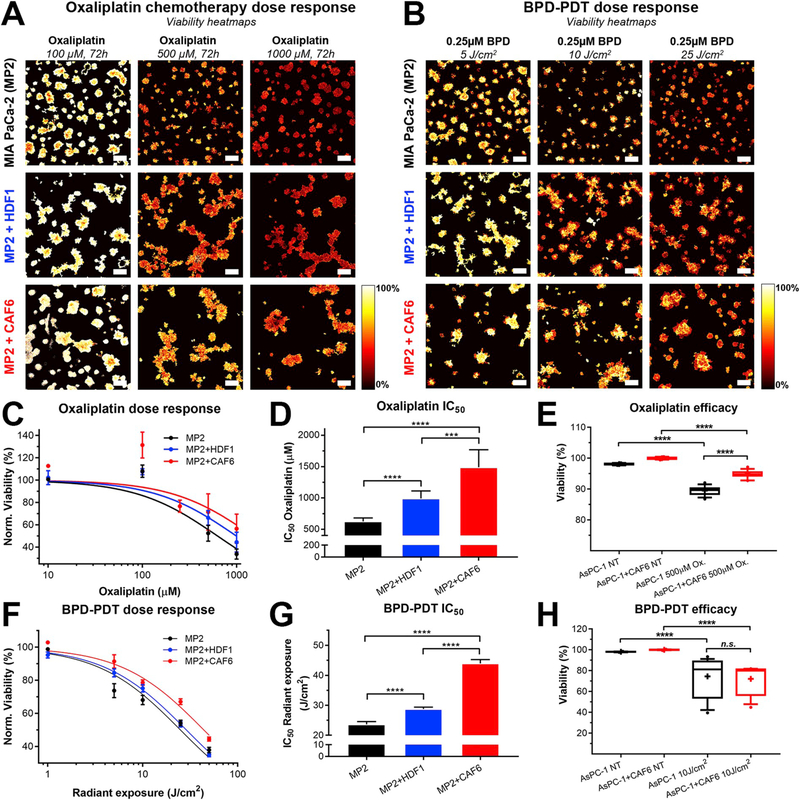
Depicted are representative viability heatmaps of the MP2, MP2+HDF1, and MP2+CAF6 microtumor co-cultures subjected to oxaliplatin dose-escalations (A) and BPD-PDT radiant exposure dose-escalations (B). Scalebar = 400 μm (C) Oxaliplatin dose-response curves based on normalized median viability. Depicted are the mean ± SD of N = 12–60 measurements. (D) Comparison of the fitted oxaliplatin IC50 doses between MP2 only (black), MP2+HDF1 (blue), and MP2+CAF6 (red) cultures, extracted from the dose-response data in panel (C), N = 151–206 values. (E) Box-whisker plot of 500 μM efficacy in AsPC-1-based microtumor cultures (median, 25th and 75th percentile, and the 90% confidence interval (CI), N = 16). (F) BPD-PDT radiant exposure dose-response curves based on normalized median viability. Depicted are the mean ± SD of N = 12–60. (G) Comparison of the fitted radiant exposure IC50 doses between MP only (black), MP2+HDF1 (blue), and MP2+CAF6 (red) cultures, extracted from the dose-response data in panel (F), N = 151–206 values. (H) Box-whisker plot of 10 J/cm2 BPD-PDT efficacy in AsPC-1-based microtumor cultures (median, 25th and 75th percentile, and the 90% CI, N = 16). Data depicted in panels C, D, F, and G was obtained from 3 to 6 technical repeats, containing 3 biological replicates for each condition containing ~700 individual microtumors in 4 images. Data depicted in panels E and H was obtained from 2 to 4 technical repeats, containing 3 biological replicates for each condition containing ~900 individual microtumors in 4 images.
With respect to BPD-PDT, the MP2+HDF1 and MP2+CAF6 microtumors exhibited higher resistance compared to MP2 microtumors (Fig. 2B & 1F). Compared to the IC50 values for BPD-PDT radiant exposure in the MP2 microtumors (23.65 ± 1.68 J/cm2), there was significant resistance in the MP2+HDF1 and MP2+CAF6 microtumor cultures (28.79 ± 1.47 J/cm2 and 43.97 ± 2.63 J/cm2, respectively) (Fig. 2G). Reduced BPD-PDT susceptibility was also observed for the MP2+CAF1 microtumors, but not in MP2+HDF2 microtumors, as compared to MP2 microtumors without fibroblasts (Figs. S2D and S2E). Reduced BPD-PDT sensitivity was similarly found for 2D monolayer cultures in which MP2 cells were co-cultured in presence of CAF6 (Fig. S2F). In contrast, no additional resistance to PDT was observed in As-PC1+CAF6 microtumors compared to AsPC-1 microtumors without fibroblasts (Fig. 2H).
The large size differences between the heterotypic microtumors prompted us to further investigate the correlation between microtumor size and treatment susceptibility (Fig. S3). In absence of treatment, there was a significant inverse relationship between microtumor size and viability, which can be explained by the presence of hypoxic/necrotic cores in the larger nodules. Treatment responses were heterogeneous, and there appeared to be no universal correlation between microtumor size and response to oxaliplatin or BPD-PDT treatment. We did note a significant positive correlation between MP2+HDF1 microtumor size and viability following PDT, indicating that larger nodules may have reduced sensitivity to treatment in these cultures. However, for the MP2+CAF6 cultures, the increased microtumor sizes did not account for the reduced treatment susceptibilities.
These dose-escalation experiments demonstrate that CAFs reduced the susceptibility of PDAC microtumors to both oxaliplatin chemotherapy and BPD-PDT in our models. Furthermore, while the highest degree of treatment resistance was observed for the PDAC co-cultures containing CAFs, a lesser degree of resistance was observed following co-culture with the HDF lines. The data suggests that the activation status of the fibroblast line used for co-culture impacts the degree of treatment resistance that is ultimately observed, and that the interactions between CAFs and cancer cells do indeed enhance the ability of heterotypic microtumors to escape treatment. Taken together, these heterotypic PDAC microtumor models recapitulate cancer-associated stroma induced treatment resistance as observed in various studies (e.g. Refs. [33,47–49]), and thus constitute a platform in which the potential mechanisms of treatment escape by cancer associated fibroblasts can be further investigated.
3.3. Cancer-associated fibroblasts affect PDAC microtumor metabolism and redox state
It is becoming increasingly clear that changes in the metabolic state of cancer cells can occur as a result of interactions with cancer associated stroma, and that these changes may comprise a key determinant of treatment resistance [50]. In separate studies, the oxidative state of PDAC has been linked to treatment resistance [51], whereas metabolic support of pancreatic CAFs to PDAC cells has been shown to promote oxidative metabolism [14]. We therefore set out to determine whether CAFs may promote treatment resistance through modulating oxidative metabolism in PDAC cells.
We utilized established methods for redox imaging based on the NAD(P)H and FAD autofluorescence in cells and tissues [23,30], and found that the redox states of MP2-based microtumors were significantly elevated in the presence of fibroblasts; the highest oxidative states were observed in the presence of CAF6 and CAF1 (Fig. 3A/C). We next evaluated whether the increased redox states correlated to the upregulation of oxidative stress-induced survival proteins (Fig. S4). Heme oxygenase 1 (HO-1) was selected as a marker of the nuclear factor E2-related factor 2 (NRF2)-mediated antioxidant stress response, and which relates to PDT-and chemoresistance [6,51,52]. Microtumor cultures containing CAF1 and CAF6 indeed overexpressed HO-1 compared to MP2 or MP2+HDF1 cultures, yet HO-1 was also highly expressed by MP2+HDF2 microtumors (Fig. S4B). Secondly, we investigated cyclooxygenase 2 (COX2) expression, which is induced by ROS-activated nuclear factor-κB (NF-κB) and also associates to both PDT and chemotherapy resistance [6,53]. Our findings clearly show that COX2 was overexpressed exclusively by MP2+CAF cultures (Fig. S4C).
Fig. 3. PDAC microtumors co-cultured with cancer-associated fibroblasts exhibit increased redox states and altered metabolism.

Brightfield images and their corresponding redox ratio heatmaps of (A) MP2-based microtumor (co-)cultures and (B). AsPC-1-based microtumor (co-)cultures. Scalebar = 200 μm. (C) Quantification of redox states of MP2 (black), MP2+CAF1 (orange), and MP2+CAF6 microtumors (red)on culture day 11, following a 48 h exposure to 10 mM metformin (median ± 95% CI of N = 56–252 from 3 to 6 technical repeats). (D–F) Levels of lactate (D), alanine (E), and aspartate (F) detected in the culture media of MP2 (black), MP2+CAF1 (orange), and MP2+CAF6 microtumors (red) upon no treatment (solid bars) or metformin (Metf) treatment (semi-opaque) (mean ± SD of N = 6 from 2 technical repeats). (G) Quantification of redox states of AsPC-1 microtumors (black) and AsPC-1+CAF6 microtumors (red) following a 48 h exposure to 10 mM metformin (mean ± SD from N = 46–64 from three technical repeats). (H–J) Levels of lactate (H), alanine (I), and aspartate (J) detected in the culture media of AsPC-1 (black) and AsPC-1+CAF6 microtumors (red) upon no treatment (solid bars) or Metf treatment (semi-opaque) (mean ± SD of N = 6 from two technical repeats). (For a better interpretation of the redox ratio heatmaps, the reader is referred to the full color renderings in the Web version of this article.)
To confirm that the increased oxidative state observed following co-culture with CAFs was due to an increase in oxidative phosphorylation, metformin was administered to the microtumor models. Metformin is a known inhibitor of mitochondrial complex I and is currently being evaluated clinically for PDAC as an inhibitor of intratumoral oxidative phosphorylation [54]. A 48 h incubation of the MP2+CAF co-cultures with metformin mediated a significant reduction in redox states, (Fig. 3C), without induction of toxicity (Figs. S5E–F). Interestingly, glycolysis was not correspondingly increased following treatment with metformin as lactate levels in the culture medium remained unchanged (Fig. 3D). These findings suggest that the MP2-CAF microtumors may exploit alternative metabolic pathways as sources of energy following inhibited mitochondrial respiration. In the MP2 cultures devoid of CAFs, the addition of metformin resulted in significantly elevated redox states, potentially as a result from non-lethal mitochondrial uncoupling and oxidative stress in these more treatment-sensitive models [55].
KRAS-mutated pancreatic cancers exhibit strong glutamine addiction [56], and this metabolic feature is retained by MP2 and AsPC-1 cells in culture [57]. Since glutaminolysis is accompanied by a build-up of intracellular nitrogen, cells secrete the excess nitrogen in the form of either urea, alanine (Ala), or to a lesser extent aspartate (Asp) [58]. Indeed, MP2 microtumors without fibroblasts secrete up to 300 μg/mL Ala and 4 μg/mL Asp in the culture medium, suggesting glutaminolytic activity in these microtumors (Fig. 3E and F). However, extracellular Ala and Asp levels drop in the presence of CAFs, indicating either increased consumption or reduced excretion of these amino acids by the microtumors. Increased Ala consumption by PDAC cells in the presence of stromal partners and under nutrient-starved conditions has indeed been reported previously [14]. Since blocking OxPhos with metformin also impairs oxidative glutamine catabolism resulting from decreased TCA cycle activity [59], the reduced levels of Ala and Asp upon metformin treatment may have resulted from impaired oxidative glutaminolysis [54,58]. Interestingly, as lactate levels were neither affected by the presence of CAFs, nor by metformin treatment in the MP2-based microtumors, the findings suggest that these microtumors utilize alternative pathways to meet their energy demands. One potential pathway is glutamine-dependent reductive carboxylation, an OxPhos-independent pathway in which glutamine-derived α-ketoglutarate acts as a source for lipogenesis and various other metabolites [54,60,61]. Although the exact metabolic activity in these models warrants further elucidation, the results unequivocally show that CAFs influence MP2 microtumor metabolism that is characterized by increased OxPhos activity and reduced Ala secretion. A schematic of the basal and adaptive metabolism of MP2-based microtumor cultures, as observed in this study is depicted in Fig. S6A.
Compared to the MP2 cell line that was derived from a primary PDAC, the AsPC-1 cell line originates from the ascites of a PDAC patient, thus constituting a metastatic cell line that has been previously identified to have a distinct metabolic phenotype compared to MP2 cells [62,63]. In contrast to MP2-based microtumors, the redox states of AsPC-1 microtumors were not affected following co-culture with CAF6 (Fig. 3B/G). However, basal redox states were substantially higher in the AsPC-1 microtumors compared to MP2 microtumors. This is consistent with established differences in their metabolic phenotypes: whereas MP2 cells have been characterized as highly glycolytic, AsPC-1 cells appear to rely more on OxPhos [62]. Indeed, inhibition of OxPhos with metformin in the AsPC-1 microtumors resulted in significantly elevated lactate excretion levels, reaching similar extents of lactate excretion as untreated MP2 microtumors. AsPC-1-cells thus display metabolic plasticity to maintain their energetic demands when confronted with a specific metabolic restriction (Fig. 3H). Similar to the MP2-based microtumors, AsPC-1 microtumors secreted Ala and Asp in the culture medium (Fig. 3I/J), suggesting that the basal metabolism of AsPC-1 microtumors involves both OxPhos and glutaminolysis [58,60]. In the presence of CAF6, secretion of Ala, but not Asp, was mildly reduced. Metformin facilitated a significant decrease in Ala and Asp secretion, suggesting that oxidative glutaminolysis is partially responsible for the amino acid secretion. Taken together, and in contrast to the MP2-based microtumors, CAFs exert little influence on the metabolism of AsPC-1-based microtumors that already possess intrinsic metabolic plasticity. As OxPhos activity has been linked to chemotherapy resistance [45], the increased basal levels of OxPhos activity of AsPC-1 microtumors compared to MP2-based microtumors may partially explain their treatment resistant nature. A schematic of the basal and adaptive metabolism of AsPC-1-based microtumor cultures as observed in this study is depicted in Fig. S6B.
The findings demonstrated that cancer-associated stromal partner cells promote OxPhos activity in glycolytic MP2-based microtumors, and give rise to treatment resistance. These results thus reiterate various accounts in which elevated OxPhos activity and oxidative states were found to be causal factors of treatment resistance in acute myeloid leukemia, triple-negative breast cancer, large B-cell lymphoma, and melanoma [45,64–67]. Regardless of the exact metabolic origin, increased redox states in PDAC have also been observed in various investigations, and such perturbations to the metabolic state have been linked to reduced treatment susceptibility [51,68]. The organotypic PDAC-CAF co-cultures described in this study at least partially recapitulate these features in vitro, and thus represent a promising platform for evaluating new cancer therapies.
3.4. CAF-induced elevated redox states and treatment resistance are recapitulated in vivo
As the complexity and heterogeneity of PDAC metabolism has become the subject of many investigations [13], a variety of studies have demonstrated that PDAC cells and tissues retain much of their metabolic features in mouse models of cancer [14,56,62]. To evaluate whether the observed alterations in redox state and treatment efficacies could be recapitulated in vivo, we established an orthotopic mouse model of PDAC in which MP2 and CAF6 cells were co-injected in the murine pancreas.
Analysis of tumor redox states ex vivo on day 10 post-implantation revealed that there was a significantly elevated redox state in tumors comprised of MP2+CAF6 cells compared to tumors comprised of MP2 cells only (Fig. 4A, E-F). Compared to orthotopic tumors of MP2 cells only, tumor growth was significantly delayed in the presence of CAF6 (Fig. 4B–D), corroborating the compressive nature of the CAFs in 3D co-cultures (Fig. 1B). Furthermore, whereas suboptimal BPD-PDT (0.25 mg/kg BPD, 60 min drug-light interval, 50 J/cm2, 100 mW/cm2 [9]) was capable of decreasing tumor growth rates in MP2-only xenografts (Fig. 4C), there was no significant reduction in tumor growth rates in the MP2+CAF6 xenografts (Fig. 4D). Together, these findings indicate that the compression of cancer tissues by CAFs, the CAF-related increased redox states, and CAF-induced treatment resistance observed in the heterotypic microtumor models in vitro are partially recapitulated in vivo, underscoring the capacity of these heterotypic microtumor cultures to bridge the gap between in vitro and in vivo research.
Fig. 4. Increased CAF6-induced redox states and treatment resistance is recapitulated in and orthotopic model of pancreatic cancer in vivo.

(A) Tumor redox states were quantified at day 10 post-implantation (N = 3 tumors/group). (B) Tumor size as quantified at day 24 post-implantation in the absence of treatment (N = 6 animals/group). (C–D) Linear regression analysis of tumor growth of MP2 (C) and MP2+CAF6 orthotopic xenografts (D) was quantified in the linear range in absence of treatment (solid lines) or following 50 J/cm2 BPD-PDT (0.25 mg/kg, 60 min drug-light interval) (dashed lines). (E–F) Representative hematoxylin & eosin (H&E) stained slides and stitched redox ratio heatmaps of MP2 (E) and MP2+CAF6 xenografts (F), which were harvested on day 10 post-implantation. (For a better interpretation of the redox ratio heatmaps, the reader is referred to the full color renderings in the Web version of this article.)
3.5. Metabolic restriction by metformin overcomes CAF-induced treatment resistance in MIA PaCa-2 microtumors
It is hypothesized that metabolic plasticity allows PDAC cells to survive in harsh and nutrient-poor microenvironments, while concomitantly rendering them less susceptible to treatments [13]. The findings of this study seem to support this notion, in which the normally glycolytic and glutaminolytic MP2 microtumors transition to auxiliary OxPhos in the presence of CAFs (Fig. 3C). Given its ability to impair OxPhos, metformin is a promising therapeutic agent to overcome treatment insensitivity by metabolic plasticity [69–71]. Metformin is a core agent in the treatment of type II diabetes [72,73], and is currently being explored in various clinical trials as a cancer therapeutic likely because of its metabolic modulatory effects [13,74]. Despite its demonstrated ability to modulate OxPhos and oxidative glutamine catabolism in MP2+CAF microtumors, metformin exerted no notable toxicity as a stand-alone therapeutic agent in MP2 or AsPC-1 based heterotypic microtumors (Fig. S5), a finding also observed in patient-derived PDAC xenografts [75]. We therefore explored whether metformin could be combined with oxaliplatin chemotherapy and BPD-PDT to yield beneficial treatment outcomes. To achieve this, the treatment regimen described in Fig. 1A was adapted to include the continuous exposure to 10 mM metformin from culture day 9–15, whereas oxaliplatin and PDT were initiated on culture day 11. The 10 mM metformin dose was based on previous investigations, as well as preliminary findings (Fig. S5).
Adjuvant treatment with metformin significantly enhanced the efficacy of oxaliplatin and BPD-PDT in the highly resistant MP2+CAF6 heterotypic microtumors (Fig. 5A and B). Similarly, a significant enhancement of efficacy was observed for the 10 J/cm2 BPD-PDT/500 μM oxaliplatin combination, (referred to as photochemotherapy) (Fig. 5C). In the MP2 microtumor cultures without CAFs, metformin was similarly capable of enhancing the efficacy of oxaliplatin and PDT-oxaliplatin combination, but not for BPD-PDT as a monotherapy (Fig. 5D–F). Although the MP2+CAF6 microtumors remained significantly less susceptible to oxaliplatin compared to the MP2 microtumors even following metformin treatment, the metformin still facilitated a 3.5-fold decrease in the differential between oxaliplatin IC50 in these cultures (Fig. 5G). Likewise, whereas BPD-PDT exhibited a 2-fold higher radiant exposure IC50 in the MP2+CAF6 microtumors without metformin pretreatment, the pretreatment with metformin yielded similar, non-significantly different, BPD-PDT efficacy between the MP2+CAF6 vs the MP2 alone microtumors (Fig. 5H). These results suggested that metformin pretreatment was sufficient to overcome CAF induced BPD-PDT resistance in the heterotypic microtumors. With respect to the PDT-oxaliplatin combination in absence of metformin pretreatment (10 J/cm2 BPD-PDT + 500 μM oxaliplatin), the MP2+CAF6 microtumors were less susceptible compared to the MP2 microtumors, albeit to a non-significant extent (p = 0.14). Upon adjuvant treatment of metformin, there was a significant reduction in viability for both cultures, a more homogeneous response, and an equal treatment susceptibility (p > 0.99) between the two culture types (Fig. 5I). Lastly, we demonstrate that inhibition of OxPhos with metformin also significantly improves the susceptibility of AsPC-1+CAF6 microtumors to oxaliplatin, albeit to a minor extent (Figs. S7A–C). Contrastingly, neither the efficacy of BPD-PDT alone (Fig. S7D), nor that of a PDT-oxaliplatin combination was improved by metformin (Fig. S7E), indicating that OxPhos alone may be less vital as a mechanism for treatment resistance in AsPC-1-based microtumors.
Fig. 5. Inhibition of oxidative phosphorylation with metformin overcomes CAF6-induced treatment resistance in MP2-based microtumors.

(A–C) Dose-response effects of oxaliplatin (A), BPD-PDT radiant exposures (B), and BPD-PDT + oxaliplatin combinations (C) in MP2+CAF6 microtumor cultures in presence of 10 mM Metf (dotted lines) and in absence of Metf (solid lines, similar data as depicted in Fig. 2C). (D–F) Dose-response effects of oxaliplatin (D), BPD-PDT radiant exposures (E), and BPD-PDT + oxaliplatin combinations (F) in MP2 microtumor cultures in presence of 10 mM Metf (dotted lines) and in absence of Metf (solid lines, similar data as depicted in Fig. 2C). (G–I) Comparison of oxaliplatin IC50 (G) and BPD-PDT radiant exposure IC50 (H) in microtumor cultures of MP2 only (black bars) and MP2+CAF6 microtumor cultures (red bars), as determined in the presence of 10 mM Metf (semi-opaque bars) or absence of metformin (solid bars). IC50 values were statistically compared using extra-sum-of-squares F-tests. (I) Box-whisker plots (median, average, and 90% CI, N = 12–36) representing the viability of MP2 (black) and MP2+CAF6 (red) microtumor cultures following treatment with a combination of 10 J/cm2 BPD-PDT and 500 μM oxaliplatin, performed in the absence and presence of 10 mM Metf. Data depicts the mean ± SEM of N = 12–32 from 3 technical repeats). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
This study aimed to investigate the influence of stromal cells on the resistance of PDAC tumors to oxaliplatin and BPD-PDT. To do so, we first developed a heterotypic 3D co-culture model in which CAFs were introduced to pre-formed PDAC microtumors. CAFs rapidly integrated in the PDAC microtumors and compressed multiple nodules into larger masses. Heterotypic PDAC microtumors containing CAFs were significantly more resistant to oxaliplatin chemotherapy and photodynamic therapy, although this was not related to the increased microtumor size. However, the CAFs influenced the metabolic state of the PDAC microtumors, which was characterized with increased oxidative states, and elevated expression of oxidative stress-induced survival proteins. The effects of CAFs on tumor compression, PDT resistance, and increased redox states that were observed in vitro were recapitulated in vivo. To overcome this resistance, we evaluated combination treatments with the OxPhos inhibitor metformin. Our results demonstrated that metformin impaired the CAF-induced treatment resistance cascade by partially reverting the increased oxidative states. As a result, the metformin-based combination treatments demonstrated superior treatment efficacies compared to oxaliplatin or BPD-PDT alone, without exerting toxicity as a single agent. These findings suggest that targeting PDAC metabolism represents a promising strategy to overcome stroma-induced survival signaling.
Various 2D and 3D (co)culture models have been used to explore the interplay between PDAC cells and stromal cells. However, given that the 3D microtumor architectures can influence cancer treatment outcomes due to heterogeneous drug penetration [76], oxygen gradients [77–79], and cell adhesion-mediated drug resistance [80], it has been well established that 3D culture models are more representative of clinical treatment responses compared to 2D monolayer cultures [18,81]. When it comes to co-culture models, a major challenge relates to differences in cell size and proliferation rates that can lead to competition between the cell types, with one cell type typically dominating over the other. To overcome this, the influence of fibroblasts on cancer cells can be studied in 2D using cell culture inserts or conditioned media experiments. For instance, Sherman et al. investigated the effects of conditioned media on MP2 cells. Cancer-associated pancreatic stellate cells (CAPSC) were grown to confluence, and exposed to the vitamin D analogue calcipotriol for 48 h. Conditioned media was then collected and transferred to MP2 monolayers. Transcriptomics revealed that CAPSC-conditioned media promoted proliferation, survival, epithelial-mesenchymal transition, and chemoresistance pathways. These stimulatory effects were largely inhibited by the treatment of the CAPSC with calcipotriol [35]. In another instance, Sousa et al. demonstrated that PSCs cells engage in autophagy upon stimulation by various PDAC cell lines, as demonstrated in direct co-culture experiments and conditioned media experiments [16]. A major limitation of conditioned media studies is that the CAFs and PDAC cells do not physically interact, and the scope of PDAC-CAF interactions are limited to paracrine signaling only. In comparison, the benefit of the microtumor culture model in this study is that these limitations are mostly overcome, allowing 3D migration and de-novo heterocellular microtumor formation. As such, our findings indicate a clear difference in the behavior of different fibroblast and CAF lines on PDAC microtumor formation that cannot be recapitulated in 2D co-culture models or conditioned medium experiments.
In this study, the 3D microtumor cultures were composed of commercially available PDAC lines. While patient-derived models certainly have immense value as they fully represent the clinical heterogeneity of human cancers, reproducibility and highly controlled mechanistic studies are difficult to perform due to the inter- and intra-patient heterogeneity in extracellular matrix, cell populations, and genetic backgrounds. Moreover, these models are idiosyncratic: They are relevant for a specific clinical situation for a given patient, making it hard to generalize findings obtained from such models [18]. Although models with PDAC cell lines do not comprehensively reproduce all clinical features of all PDAC, a major benefit of using well-characterized PDAC and CAF lines is that they provide a means to investigate the effects of tumor-stroma interactions on treatment resistance and tumor biology in a highly controlled manner. This degree of reproducibility is not likely achieved with patient-derived PDAC cells, as such biopsies can contain heterogenous and uncharacterized cell types such as distinct PDAC populations, CAF populations, immune- and endothelial cells. The current study introduces the heterotypic microtumor cultures as a highly modular cancer model: the cancer cells and fibroblasts are interchangeable, the extracellular matrix can be supplemented or replaced, and additional cell types can be added to increase the complexity of the models, for example with patient-derived cell populations. Because each component of the system is controllable, we propose that a combination of cell line-based organoid models are highly suited for investigating detailed mechanistic studies and screening for potentially effective treatments, whereas the more precious and heterogenous patient-derived models can subsequently be employed to validate the most promising findings. The presented heterotypic microtumor models and imaging tools presented here can facilitate such studies to provide a wealth of translationally relevant information on new treatment strategies.
A major point of interest in undertaking this study was to determine whether CAFs have a specific influence that cannot be recapitulated by non-CAFs. However, a comparison between healthy pancreatic fibroblasts and pancreatic CAFs is challenged by the relative scarcity of such cell lines. As a proof of concept, we therefore explored the use of dermal CAFs and HDFs, as described and characterized by Goruppi et al. These investigators demonstrated that in comparison to HDFs, CAFs express high levels of glioma-associated factor 2 (GLI2) and exhibit reduced expression of the Notch repressor CSL. The combined loss of CSL with overexpression of GLI2 in CAFs was found to be essential for the promotion of squamous cell carcinoma growth in SCID mice [26]. In our study, we included a high GLI2 expression CAF line (CAF1), as well as a CAF line expressing lower GLI2 levels (CAF6). Similarly to pancreatic CAFs or CAPSCs, which are typically characterized by the high expression of FAP and α-SMA [34,35], the CAF1 and CAF6 lines exhibited similar features. Our study convincingly demonstrates that treatment resistance is more efficiently facilitated by CAFs compared to HDFs, with low-GLI2 expressing CAF6 cells inducing the highest degree of resistance. These findings associated with increased redox states in the heterotypic microtumors. This was accompanied by higher HO-1 and COX-2 expression levels, two prominent survival-associated proteins downstream the NRF2 antioxidant survival response and NF-κB signaling pathways [6]. These findings suggest that the use of CAFs is critical to investigate the effects of stromal fibroblasts in 3D co-culture models, rather than using more readily available non-cancer associated fibroblast lines.
Moreover, the current study has discovered a previously unknown link between the influence of CAFs on treatment resistance and the influence of CAFs on cancer metabolism. It was recently discovered that PDAC organoids constitutively activate the NRF2-mediated antioxidant survival response, and that knockdown of NRF2 in murine premalignant pancreatic ductal organoids significantly increased susceptibility to the pan-AKT inhibitor MK2206 [51]. Another study separately demonstrated that CAFs may cause elevated oxidative states in PDAC by transforming tumor metabolism. Stromal cells were found to modulate cancer metabolism through an autophagy-dependent excretion of amino acids [14] that were subsequently metabolized by PDAC cells to form pyruvate. This fueled the tricarboxylic acid (TCA) cycle [14], and increased mitochondrial respiration through oxidative phosphorylation (OxPhos). Our findings connect these two phenomena, as the metabolic influence of CAFs on PDAC metabolism increased the redox state of the microtumors and corollary instigated treatment resistance.
We hypothesized that metformin could block the CAF-induced metabolic cascade that culminated in treatment resistance. Our findings demonstrate that metformin effectively impairs redox metabolism in MP2 microtumor cultures that contain CAFs, potentially reducing the oxidative stress-mediated survival signaling in these microtumors. In AsPC-1-based microtumors, neither CAFs nor metformin had an effect on the overall microtumor redox states. The adjuvant effects of metformin likely stem from impaired metabolic plasticity that enables cancer tissues to survive cancer therapies [13,50]. Survival of the microtumors following PDT may be more strongly linked to the pre-existing redox state, as the strongest adjuvant effect was observed in the MP2+CAF6 microtumors that similarly displayed the strongest reduction in redox state upon metformin pre-treatment. Therefore, metformin represents a promising adjuvant agent as it impairs the cyto-protective effects of the tumor stroma without ablating the potentially cancer-repressive effects of the PDAC stroma.
These findings warrant further investigation using patient-derived cancer cells and pancreatic stromal cells, in which stratification of individual cancer tissues based on their metabolic phenotypes may be necessary for treatment optimization. The presented modular microtumor platform combined with the high-content imaging assays can be further engineered to design and evaluate personalized treatment strategies using patient-derived microtumor cultures/organoids with varying clinical presentations of desmoplasia. The approach outlined in this study can be leveraged to obtain a wealth of translationally relevant information on new treatment strategies.
5. Conclusion
Cancer-associated fibroblasts and pancreatic stellate cells have recently been attributed a prominent role in providing metabolic support to PDAC cells. The increased number of metabolic pathways available to cancer cells, coupled with increased metabolic plasticity, is likely to be advantageous for the initiation of treatment escape mechanisms. Our findings indicate that restricting cancer metabolism is a promising strategy to overcome stroma-induced treatment resistance and augment clinical therapies such as oxaliplatin chemotherapy and BPD-PDT.
Supplementary Material
Acknowledgements
The work was supported by grants from the National Cancer Institute/National Institutes of Health: P01 CA084203, R01 CA158415, and R01 CA160998 (TH), K99CA175292 and R00CA175292 (IR). MB and AL acknowledge the European Society for Photobiology for support in the form a travel award. ALB received financial support from the Bullock-Wellman Fund, and the Bettencourt-Schueller and Philippe foundations. SA was a Howard Hughes Medical Institute Medical Research Fellow. The authors acknowledge Dr. Sandro Goruppi and Prof. Gian Paolo Dotto (Cutaneous Biology Research Center, Massachusetts General Hospital, Charlestown, MA) for providing the patient-derived fibroblasts.
Footnotes
Conflicts of interest
The authors have no conflict of interest to report.
Data availability
The data required to reproduce these findings, both raw and processed, is available upon request.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2019.119421.
References
- [1].Ryan DP, Hong TS, Bardeesy N, Pancreatic adenocarcinoma, N. Engl. J. Med. 371 (2014) 1039–1049, 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- [2].Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet J-B, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe tumeurs digestives of unicancer, PRODIGE intergroup, FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer, N. Engl. J. Med. 364 (2011) 1817–1825, 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- [3].Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF, Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine, N. Engl. J. Med. 369 (2013) 1691–1703, 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang-Gillam A, Li C-P, Bodoky G, Dean A, Shan Y-S, Jameson G, Macarulla T, Lee K-H, Cunningham D, Blanc JF, Hubner RA, Chiu C-F, Schwartsmann G, Siveke JT, Braiteh F, Moyo V, Belanger B, Dhindsa N, Bayever E, Von Hoff DD, Chen L-T, NAPOLI-1 Study Group, Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial, Lancet Lond. Engl. 387 (2016) 545–557, 10.1016/S0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- [5].Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, Kent E, Bown SG, Hasan T, Pogue BW, Pereira SP, Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer, Br. J. Canc. 110 (2014) 1698–1704, 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Broekgaarden M, Weijer R, van Gulik TM, Hamblin MR, Heger M, Tumor cell survival pathways activated by photodynamic therapy: a molecular basis for pharmacological inhibition strategies, Canc. Metastasis Rev. 34 (2015) 643–690, 10.1007/s10555-015-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T, Imaging and photodynamic therapy: mechanisms, monitoring, and optimization, Chem. Rev. 110 (2010) 2795–2838, 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Castano AP, Mroz P, Hamblin MR, Photodynamic therapy and anti-tumour immunity, Nat. Rev. Canc. 6 (2006) 535–545, 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang H-C, Mallidi S, Liu J, Chiang C-T, Mai Z, Goldschmidt R, Ebrahim-Zadeh N, Rizvi I, Hasan T, Photodynamic therapy synergizes with irinotecan to overcome compensatory mechanisms and improve treatment outcomes in pancreatic cancer, Canc. Res. 76 (2016) 1066–1077, 10.1158/0008-5472.CAN-15-0391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spring BQ, Bryan Sears R, Zheng LZ, Mai Z, Watanabe R, Sherwood ME, Schoenfeld DA, Pogue BW, Pereira SP, Villa E, Hasan T, A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways, Nat. Nanotechnol. 11 (2016) 378–387, 10.1038/nnano.2015.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Broekgaarden M, Rizvi I, Bulin A-L, Petrovic L, Goldschmidt R, Celli JP, Hasan T, Neoadjuvant photodynamic therapy augments immediate and prolonged oxaliplatin efficacy in metastatic pancreatic cancer organoids, Oncotarget 9 (2018) 13009–13022, 10.18632/oncotarget.24425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA, The pancreas cancer microenvironment, Clin. Canc. Res. Off. J. Am. Assoc. Canc. Res. 18 (2012) 4266–4276, 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Perera RM, Bardeesy N, Pancreatic cancer metabolism: breaking it down to build it back up, Canc. Discov. 5 (2015) 1247–1261, 10.1158/2159-8290.CD-15-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, Asara JM, Evans RM, Cantley LC, Lyssiotis CA, Kimmelman AC, Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion, Nature 536 (2016) 479–483, 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rosow DE, Liss AS, Strobel O, Fritz S, Bausch D, Valsangkar NP, Alsina J, Kulemann B, Park JK, Yamaguchi J, LaFemina J, Thayer SP, Sonic Hedgehog in pancreatic cancer: from bench to bedside, then back to the bench, Surgery 152 (2012) S19–S32, 10.1016/j.surg.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee JJ, Perera RM, Wang H, Wu D-C, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, Bardeesy N, Beachy PA, Stromal response to Hedgehog signaling restrains pancreatic cancer progression, Proc. Natl. Acad. Sci. U.S.A. 111 (2014) E3091–E3100, 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Öhlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FHM, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RGJ, Clevers H, Tuveson DA, Organoid models of human and mouse ductal pancreatic cancer, Cell 160 (2015) 324–338, 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baker LA, Tiriac H, Clevers H, Tuveson DA, Modeling pancreatic cancer with organoids, Trend. Canc. 2 (2016) 176–190, 10.1016/j.trecan.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville T, Froeling FEM, Burkhart RA, Denroche RE, Jang G-H, Miyabayashi K, Young CM, Patel H, Ma M, LaComb JF, Palmaira RLD, Javed AA, Huynh JA, Johnson M, Arora K, Robine N, Shah M, Sanghvi R, Goetz AB, Lowder CY, Martello L, Driehuis E, Lecomte N, Askan G, Iacobuzio-Donahue CA, Clevers H, Wood LD, Hruban RH, Thompson ED, Aguirre AJ, Wolpin BM, Sasson A, Kim J, Wu M, Bucobo JC, Allen PJ, Sejpal DV, Nealon W, Sullivan JD, Winter JM, Gimotty PA, Grem JL, DiMaio DJ, Buscaglia JM, Grandgenett PM, Brody JR, Hollingsworth MA, O’Kane GM, Notta F, Kim EJ, Crawford JM, Devoe CE, Ocean A, Wolfgang CL, Yu KH, Li E, Vakoc CR, Hubert B, Fischer SE, Wilson JM, Moffitt RA, Knox JJ, Krasnitz A, Gallinger S, Tuveson DA, Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer, Cancer Discov, 2018, 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lazzari G, Nicolas V, Matsusaki M, Akashi M, Couvreur P, Mura S, Multicellular spheroid based on a triple co-culture: a novel 3D model to mimic pancreatic tumor complexity, Acta Biomater. 78 (2018) 296–307, 10.1016/j.actbio.2018.08.008. [DOI] [PubMed] [Google Scholar]
- [21].Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza-Alvarez P, Cabeçadas J, Alves PM, Gualda EJ, Sommergruber W, Brito C, 3D-3-culture: a tool to unveil macrophage plasticity in the tumour microenvironment, Biomaterials 163 (2018) 185–197, 10.1016/j.biomaterials.2018.02.030. [DOI] [PubMed] [Google Scholar]
- [22].Bulin A-L, Broekgaarden M, Hasan T, Comprehensive high-throughput image analysis for therapeutic efficacy of architecturally complex heterotypic organoids, Sci. Rep. (2017) 16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL, Skala MC, Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer, Canc. Res. 73 (2013) 6164–6174, 10.1158/0008-5472.CAN-13-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Georgakoudi I, Quinn KP, Optical imaging using endogenous contrast to assess metabolic state, Annu. Rev. Biomed. Eng. 14 (2012) 351–367, 10.1146/annurev-bioeng-071811-150108. [DOI] [PubMed] [Google Scholar]
- [25].Procopio M-G, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo S-H, Goruppi S, Menietti E, Ostano P, Ala U, Provero P, Hoetzenecker W, Neel V, Kilarski WW, Swartz MA, Brisken C, Lefort K, Dotto GP, Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation, Nat. Cell Biol. 17 (2015) 1193–1204, 10.1038/ncb3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goruppi S, Procopio M-G, Jo S, Clocchiatti A, Neel V, Dotto GP, The ULK3 kinase is critical for convergent control of cancer-associated fibroblast activation by CSL and GLI, Cell Rep. 20 (2017) 2468–2479, 10.1016/j.celrep.2017.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bulin A-L, Broekgaarden M, Simeone D, Hasan T, Low dose photodynamic therapy harmonizes with radiation therapy to induce beneficial effects on pancreatic heterocellular spheroids, Oncotarget (2019) 2625–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Le A, Ng A, Kwan T, Cusmano-Ozog K, Cowan TM, A rapid, sensitive method for quantitative analysis of underivatized amino acids by liquid chromatography-tandem mass spectrometry (LC-MS/MS), J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 944 (2014) 166–174, 10.1016/j.jchromb.2013.11.017. [DOI] [PubMed] [Google Scholar]
- [29].Walsh AJ, Poole KM, Duvall CL, Skala MC, Ex vivo optical metabolic measurements from cultured tissue reflect in vivo tissue status, J. Biomed. Opt. 17 (2012) 116015, 10.1117/1.JBO.17.11.116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I, Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation, Sci. Rep. 3 (2013) 3432, 10.1038/srep03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu S, Huang Y, Tang Q, Li Z, Horng H, Li J, Wu Z, Chen Y, Li H, Quantitative evaluation of redox ratio and collagen characteristics during breast cancer chemotherapy using two-photon intrinsic imaging, Biomed. Opt. Exp. 9 (2018) 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim DE, Procopio M-G, Ghosh S, Jo S-H, Goruppi S, Magliozzi F, Bordignon P, Neel V, Angelino P, Dotto GP, Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation, J. Exp. Med. 214 (2017) 2349–2368, 10.1084/jem.20170724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kalluri R, The biology and function of fibroblasts in cancer, Nat. Rev. Canc. 16 (2016) 582–598, 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- [34].Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, Chio IIC, Hwang C-I, Tiriac H, Baker LA, Engle DD, Feig C, Kultti A, Egeblad M, Fearon DT, Crawford JM, Clevers H, Park Y, Tuveson DA, Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer, J. Exp. Med. 214 (2017) 579–596, 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM, Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy, Cell 159 (2014) 80–93, 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Celli JP, Rizvi I, Blanden AR, Massodi I, Glidden MD, Pogue BW, Hasan T , An imaging-based platform for high-content, quantitative evaluation of therapeutic response in 3D tumour models, Sci. Rep. 4 (2014) 3751, 10.1038/srep03751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Huelsken J, Hanahan D, A subset of cancer-associated fibroblasts determines therapy resistance, Cell 172 (2018) 643–644, 10.1016/j.cell.2018.01.028. [DOI] [PubMed] [Google Scholar]
- [38].Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan A-C, Sams SB, Pillai MM, Elias AD, Robinson WA, Sartorius CA, Kabos P , Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen, Clin. Canc. Res. Off. J. Am. Assoc. Canc. Res. 23 (2017) 1710–1721, 10.1158/1078-0432.CCR-15-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R, Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival, Canc. Cell 28 (2015) 831–833, 10.1016/j.ccell.2015.11.002. [DOI] [PubMed] [Google Scholar]
- [40].Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, Huang D, Zhao J, Yang L, Liao D, Su F, Li M, Liu Q, Song E, CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness, Cell 172 (2018) 841–856, 10.1016/j.cell.2018.01.009e16. [DOI] [PubMed] [Google Scholar]
- [41].Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW, Finkelstein D, Hasan T, Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer, Canc. Res. 70 (2010) 9319–9328, 10.1158/0008-5472.CAN-10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Celli JP, Rizvi I, Evans CL, Abu-Yousif AO, Hasan T, Quantitative imaging reveals heterogeneous growth dynamics and treatment-dependent residual tumor distributions in a three-dimensional ovarian cancer model, J. Biomed. Opt. 15 (2010) 051603, , 10.1117/1.3483903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kleinovink JW, van Driel PB, Snoeks TJ, Prokopi N, Fransen MF, Cruz LJ, Mezzanotte L, Chan A, Löwik CW, Ossendorp F, Combination of photodynamic therapy and specific immunotherapy efficiently eradicates established tumors, Clin. Can. Res. Off. J. Am. Assoc. Canc. Res. 22 (2016) 1459–1468, 10.1158/1078-0432.CCR-15-0515. [DOI] [PubMed] [Google Scholar]
- [44].Zuluaga M-F, Lange N, Combination of photodynamic therapy with anti-cancer agents, Curr. Med. Chem. 15 (2008) 1655–1673. [DOI] [PubMed] [Google Scholar]
- [45].Bosc C, Selak MA, Sarry J-E, Resistance is futile: targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment, Cell Metabol. 26 (2017) 705–707, 10.1016/j.cmet.2017.10.013. [DOI] [PubMed] [Google Scholar]
- [46].Luengo A, Gui DY, Vander Heiden MG, Targeting metabolism for cancer therapy, Cell Chem. Biol. 24 (2017) 1161–1180, 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Wirapati P, Becette V, André S, Piccart M, Campone M, Brain E, Macgrogan G, Petit T, Jassem J, Bibeau F, Blot E, Bogaerts J, Aguet M, Bergh J, Iggo R, Delorenzi M, A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer, Nat. Med. 15 (2009) 68–74, 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- [48].Meads MB, Gatenby RA, Dalton WS, Environment-mediated drug resistance: a major contributor to minimal residual disease, Nat. Rev. Canc. 9 (2009) 665–674, 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- [49].Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E, Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling, Canc. Cell 27 (2015) 574–588, 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vander Heiden MG, DeBerardinis RJ, Understanding the intersections between metabolism and cancer biology, Cell 168 (2017) 657–669, 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N, Tuveson DA, NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer, Cell 166 (2016) 963–976, 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Na H-K, Surh Y-J, Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1, Free Radic. Biol. Med. 67 (2014) 353–365, 10.1016/j.freeradbiomed.2013.10.819. [DOI] [PubMed] [Google Scholar]
- [53].Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, Chan KS, Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance, Nature 517 (2015) 209–213, 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, Jones RG, Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis, PLoS Biol. 13 (2015) e1002309, 10.1371/journal.pbio.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Anedda A, Rial E, González-Barroso MM, Metformin induces oxidative stress in white adipocytes and raises uncoupling protein 2 levels, J. Endocrinol. 199 (2008) 33–40, 10.1677/JOE-08-0278. [DOI] [PubMed] [Google Scholar]
- [56].Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC, Glutamine supports pancreatic cancer growth through a Kras-regulated metabolic pathway, Nature 496 (2013) 101–105, 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B, Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation, Br. J. Canc. 111 (2014) 85–93, 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB, Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 19345–19350, 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fendt S-M, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, Patnaik A, Blouin MJ, Cantley LC, Guarente L, Blenis J, Pollak MN, Olumi AF, Vander Heiden MG, Stephanopoulos G, Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism, Canc. Res. 73 (2013) 4429–4438, 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, Schmidt S, Metallo CM, Dranka BP, Schwartz B, DeBerardinis RJ, Reductive carboxylation supports redox homeostasis during anchorage-independent growth, Nature 532 (2016) 255–258, 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mullen AR, Wheaton WW, Jin ES, Chen P-H, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ, Reductive carboxylation supports growth in tumour cells with defective mitochondria, Nature 481 (2011) 385–388, 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, Boudreau A, Mroue R, Corson L, O’Brien T, Qing J, Sampath D, Merchant M, Yauch R, Manning G, Settleman J, Hatzivassiliou G, Evangelista M, Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors, Proc. Natl. Acad. Sci. U.S.A. 112 (2015) E4410–E4417, 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo SJ Mulvihill, Phenotype and genotype of pancreatic cancer cell lines, Pancreas 39 (2010) 425–435, 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN, Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma, Canc. Cell 22 (2012) 547–560, 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P, PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress, Canc. Cell 23 (2013) 287–301, 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lee K, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sánchez V, Sanders ME, Lee T, Gómez H, Lluch A, Pérez-Fidalgo JA, Wolf MM, Andrejeva G, Rathmell JC, Fesik SW, Arteaga CL, MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation, Cell Metabol. 26 (2017) 633–647, 10.1016/j.cmet.2017.09.009e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, Scotland S, Larrue C, Boutzen H, Féliu V, Nicolau-Travers M-L, Cassant-Sourdy S, Broin N, David M, Serhan N, Sarry A, Tavitian S, Kaoma T, Vallar L, Iacovoni J, Linares LK, Montersino C, Castellano R, Griessinger E, Collette Y, Duchamp O, Barreira Y, Hirsch P, Palama T, Gales L, Delhommeau F, Garmy-Susini BH, Portais J-C, Vergez F, Selak M, Danet-Desnoyers G, Carroll M, Récher C, Sarry J-E, Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism, Canc. Discov. 7 (2017) 716–735, 10.1158/2159-8290.CD-16-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xu HN, Nioka S, Li LZ, Imaging heterogeneity in the mitochondrial redox state of premalignant pancreas in the pancreas-specific PTEN-null transgenic mouse model, Biomark. Res. 1 (2013) 6, 10.1186/2050-7771-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD , Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 15 (1997) 2403–2413, 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- [70].Soranna D, Scotti L, Zambon A, Bosetti C, Grassi G, Catapano A, La Vecchia C, Mancia G, Corrao G, Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis, The Oncologist 17 (2012) 813–822, 10.1634/theoncologist.2011-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li X, Li T, Liu Z, Gou S, Wang C, The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis, Sci. Rep. 7 (2017) 5825, 10.1038/s41598-017-06207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Inzucchi SE, Lipska KJ, Mayo H, Bailey CJ, McGuire DK, Metformin in patients with type 2 diabetes and kidney disease: a systematic review, J. Am. Med. Assoc. 312 (2014) 2668–2675, 10.1001/jama.2014.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD, Metformin and reduced risk of cancer in diabetic patients, BMJ 330 (2005) 1304–1305, 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen C-H, Bosenberg MW, McMahon M, Cantley LC, Zheng B, Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma, Proc. Natl. Acad. Sci. U.S.A. 110 (2013) 18226–18231, 10.1073/pnas.1317577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lipner MB, Marayati R, Deng Y, Wang X, Raftery L, O’Neil BH, Yeh JJ, Metformin treatment does not inhibit growth of pancreatic cancer patient-derived xenografts, PLoS One 11 (2016) e0147113, 10.1371/journal.pone.0147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Alderden RA, Mellor HR, Modok S, Hall MD, Sutton SR, Newville MG, Callaghan R, Hambley TW, Elemental tomography of cancer-cell spheroids reveals incomplete uptake of both platinum(II) and platinum(IV) complexes, J. Am. Chem. Soc. 129 (2007) 13400–13401, 10.1021/ja076281t. [DOI] [PubMed] [Google Scholar]
- [77].Griffith LG, Swartz MA, Capturing complex 3D tissue physiology in vitro, Nat. Rev. Mol. Cell Biol. 7 (2006) 211–224, 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- [78].Doublier S, Belisario DC, Polimeni M, Annaratone L, Riganti C, Allia E, Ghigo D, Bosia A, Sapino A, HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: a potential model of the chemoresistance of invasive micropapillary carcinoma of the breast, BMC Canc. 12 (2012) 4, 10.1186/1471-2407-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Broekgaarden M, Weijer R, Krekorian M, IJssel B, Kos M, Alles LK, Wijk AC, Bikadi Z, Hazai E, Gulik TM, Heger M, Inhibition of hypoxia-inducible factor 1 with acriflavine sensitizes hypoxic tumor cells to photodynamic therapy with zinc phthalocyanine-encapsulating cationic liposomes, Nano Res. 6 (2016) 1639–1662, 10.1007/s12274-016-1059-0. [DOI] [Google Scholar]
- [80].Stein WD, Litman T, Fojo T, Bates SE, Differential expression of cell adhesion genes: implications for drug resistance, Int. J. Canc. 113 (2005) 861–865, 10.1002/ijc.20671. [DOI] [PubMed] [Google Scholar]
- [81].Longati P, Jia X, Eimer J, Wagman A, Witt M-R, Rehnmark S, Verbeke C, Toftgård R, Löhr M, Heuchel RL, 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing, BMC Canc. 13 (2013) 95, 10.1186/1471-2407-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.