

Abstract
In this article, we demonstrate an acoustofluidic device for cell lysis using the acoustic streaming effects induced by acoustically oscillating sharp-edged structures. The acoustic streaming locally generates high shear forces that can mechanically rupture cell membranes. With the acoustic-streaming-derived shear forces, our acoustofluidic device can perform cell lysis in a continuous, reagent-free manner, with a lysis efficiency of more than 90% over a range of sample flow rates. We demonstrate that our acoustofluidic lysis device works well on both adherent and non-adherent cells. We also validate it using clinically relevant samples such as red blood cells infected with malarial parasites. Additionally, the unique capability of our acoustofluidic device was demonstrated by performing downstream protein analysis and gene profiling without additional washing steps post-lysis. Our device is simple to fabricate and operate while consuming a relatively low volume of samples. These advantages and other features including the reagent-free nature and controllable lysis efficiency make our platform valuable for many biological and biomedical applications, particularly for the development of point-of-care platforms.
Graphical Abstract
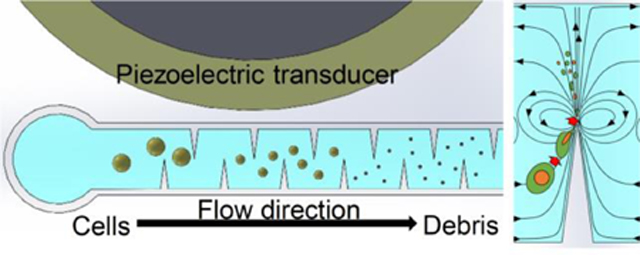
Shear stress induced by acoustic vibrating sharp edges lyse cells mechanically.
Introduction
Since many disease biomarkers cannot be detected outside of an individual cell’s membrane, cell lysis – the process of rupturing a cell’s membrane to release its contents – becomes a critical step in many diagnostic and therapeutic efforts. Lysing a sample provides access to protected factors such oncogenes and their expression products,1 multiple apoptosis regulators,2 and immune related regulators.3 From a broader perspective, an efficient cell lysis process, paired with downstream analysis, can help to combat the heterogeneity, and rapidly changing nature of biological systems and provide more accurate results. In this regard, a streamlined lysis procedure which can be used at the point-of-care (POC) could significantly improve clinical outcomes by allowing therapeutic intervention in a timely fashion. Therefore, developing cell lysis techniques that are easy to implement, require only small amounts of samples, and provide outputs that are conveniently analyzed downstream will greatly improve patient care by progressing the development of POC-based diagnostics and therapeutics.4–7
Conventional cell lysis is carried out using either chemical or physical rupture methods. Chemical-based lysis, which utilizes surfactants to solubilize lipid membranes, can efficiently rupture cell membranes to release intracellular contents; however, those surfactants can cause protein denaturation and inhibit downstream analysis.7, 8 To eliminate the adverse effects of the surfactants, additional washing and protein refolding steps are required, which can complicate the entire lysis process. Unlike chemical lysis, physical-based lysis methods often mechanically rupture cell membranes by exploiting heating effects and/or shear forces induced by sonication or other mechanical mechanisms. However, both of these conventional approaches are performed in a batch-wise fashion, which require multiple steps which are handled by trained personnel, thereby limiting their application at the POC.9, 10 A further concern is that with the increased sample consumption required by traditional methods, the heterogeneity of individual lesions and tumors cannot be differentiated within the bulk sample; that is, conventional cell lysis methods which consume large sample volumes lack the spatial and temporal resolution needed to detect heterogeneity.11, 12 This can lead to failure when attempting to identify disease-related mutations and biomarkers, which is especially important for cancer diagnosis and treatment where resistant mutations can induce non-ideal clinical outcomes.13, 14
With its streamlined operation, and ease of integration with downstream analysis methods, microfluidic-based lysis methods have shown strong potential as components in POC platforms. Many microfluidic-based lysis approaches have been developed based on various mechanisms including chemical,15–17 mechanical,18, 19 thermal,20–22 optical,23, 24 electrical,10, 25, 26 and acoustic based lysis.27–29 Among these methods, acoustic-based cell lysis methods offer certain advantages such as removing the need for mechanical or chemical pretreatment while maintaining cell-component integrity and the ability to be integrated into POC platforms.27–29 However, existing acoustic-based lysis approaches still require the fabrication of complex electrodes, which can generate excessive heat during the lysis process; previous acoustic methods are also operated in a batch-wise manner, limiting throughput. Overcoming these negative factors would yield an acoustic-based lysis method that could function in a continuous, reagent-free manner with minimal sample heating.
Here, we demonstrate an acoustofluidic (i.e., the fusion of acoustics and microfluidics)30–32 device that is capable of lysing cells in a continuous, reagent-free manner. Acoustofluidic technology introduces acoustic radiation force and acoustic derived streaming in microfluidic systems.33–36 Combined by acoustics and microfluidics, the systems have comprehensive functions and achieve multiple biological applications including cell and particle analysis,37–39 manipulation,40–42 sorting,43, 44 and separation.45–47 In our acoustofluidic lysis device, cells are primarily lysed in a mechanical manner by the shear forces that arise from fluid motion induced by acoustically oscillating sharp-edged structures;48–51 this fluid motion is termed acoustic streaming.52 By controlling the strength of the generated acoustic streaming, we can locally create shear forces that are controllable in magnitude and distribution. We first demonstrate the capability of our acoustofluidic lysis device by performing cell lysis on adherent cells (HeLa) and non-adherent cells (Jurkat). Increasing the strength of the acoustic streaming by raising the driving voltage of the acoustic transducer, we demonstrate significantly improved lysis efficiencies for both types of the cells. Our acoustofluidic device can achieve a lysis efficiency of more than 90% within a given range of sample flow rates. Furthermore, we demonstrate the capability of our acoustofluidic device to process clinically relevant and disease-related samples by conducting lysis on red blood cells infected with malaria parasites. With our reagent-free strategy, our device is able to yield high-quality lysis products that can be utilized directly for downstream analysis, without additional post-lysis washing steps.
Our acoustofluidic lysis device circumvents the need for complex transducer design, simplifying the fabrication and operation procedures of the device and alleviating the heat-generation problem. Relative to other existing microfluidic-based lysis devices, our device is simple in its design and operation, which allows for integration with other microfluidic components. It can also process small amounts of samples, which is well-suited for handling volume-limited samples and working at the POC. All of these features, when combined, make our acoustofluidic lysis device an important component for sample preparation towards the development of POC platforms.
Experimental section
Fabrication of sharp-edge-based acoustofluidic lysis device
The sharp-edge-based acoustofluidic lysis device is simply composed of a singly-layer polydimethylsiloxane (PDMS) channel constructed with 180 pairs of sharp-edged structures, an acoustic transducer, and a thin glass substrate. The PDMS channel was prepared using standard lithography, deep ion etching (DRIE), and PDMS replica molding. In short, a single-side polished silicon wafer was first lithographically patterned and chemically etched, creating a silicon master mold with patterns of microfluidic channels of 100 μm high for PDMS replica molding. Prior to PDMS casting, the silicon mold was exposed to silane vapor for 20 minutes using 1H,1H,2H,2H-perfluorooctyl-trichlorosilane (Sigma-Aldrich, MO, USA). Once coated with silane, the mold was caste with a 10:1 mixture of PDMS-Sylgard 184 silicone elastomer base and Sylgard 184 curing agent (Dow Corning, MI, USA) and placed under vacuum for 15 minutes to eliminate air bubbles, followed by baking at 65 °C for 1 hour to completely solidify the PDMS channels. The solidified PDMS channel was carefully peeled off from the mold and then punched with inlets and outlets. Then, the PDMS channel and one glass substrate (Cat. no. 48404–455, VWR, PA, USA) were treated with oxygen plasma (BD-10AS, Electro-Technic Products, IL, USA) and bonded together, followed by incubation at 65°C for 24 hours to stabilize the bonding. After a 24-hour incubation, an acoustic transducer (7BB-27–4L0, Murata Electronics, Japan) was bonded onto the stack consisting of the PDMS channel and the glass substrate using an epoxy (84101, Permatex, CT, USA).
Cell culture
HeLa cells (ATCC, VA, USA) and Jurkat cells (ATCC, VA, USA), which represent, respectively, adherent cells and non-adherent cells, were the two main cell lines chosen to validate the lysis performance of our acoustofluidic device. HeLa and Jurkat cells were grown in DMEM (Gibco, Life Technologies, MA, USA) and RPMI 1640 (Gibco, Life Technologies, MA, USA) culture media, respectively; 10% Fetal Bovine Serum (Gibco, Life Technologies, MA, USA) and Penicillin Streptomycin (Gibco, Life Technologies, MA, USA) were added to both cultures. Cells were cultured in an incubator (Nu-4750, NuAire, MN, USA) at 37 °C with a CO2 level of 5%. HeLa cells were harvested when growing to 90% confluency, while Jurkat cells were harvested when reaching a density of 8×105 cells/mL.
Cell lysis and staining
HeLa cells were harvested using trypsin–EDTA (Gibco, Life Technologies, NY, USA) and re-suspended in DMEM. Jurkat cells were directly centrifuged and re-suspended in RPMI 1640. For staining cytoplasm, Calcein-AM (Invitrogen, Life Technologies, MA, USA) was added to a final concentration of 5 μg/mL. After a 15-minute staining with Calcein-AM, cells were centrifuged and culture medium containing Calcein-AM was discarded. Cells were washed and re-suspended in phosphate buffered saline (Gibco, Life Technologies, MA, USA) at a final concentration ranging from 1 × 105 to 1 × 106 cells/mL. Before sample injection, the channel of the device was incubated with 5% Pluronic F127 (Sigma-Aldrich, MO, USA) for 5 minutes and washed by phosphate buffered saline (PBS) (Gibco, Life Technologies, MA, USA). This was done to prevent lysate attachment to the channel walls. Re-suspended cells were then injected into the acoustofluidic device using 1 mL syringes (BD Bioscience, NJ, USA); the injection was controlled by an automated syringe pump (neMESYS, Germany). The acoustic transducer of the acoustofluidic device was driven by a function generator (AFG3011C, Tektronix, OR, USA) and an amplifier (25A250A, Amplifier Research, PA, USA) at a working frequency of 5 kHz. Collected samples were stained with 1 μM SYTOX Orange (Invitrogen, Life Technologies, MA, USA) to identify cells that were damaged in their plasma membranes but maintained their cellular structures. The surface temperature of the transducer and the device’s temperature were monitored using a digital thermometer (HH804, Omega, CT, USA) and Thermal Camera (TG165, FLIR, MA, USA).
Characterization of lysis efficiency
Because the concentrations of cells varied among different experiments, an individual control group was established for each experiment to characterize the lysis efficiency. To avoid cross-contamination between the control and experimental groups, in each experiment we first collected the control groups (i.e., when the acoustic transducer was not activated) prior to lysis experiments. After collecting the control groups, the channel was rinsed with PBS and the experimental groups were then collected when the device was activated. In each experiment, the control and experimental groups used the same batch of cell samples with similar cell densities; as a result, the lysis efficiency for an individual experiment was calculated using the number of cells observed in the control or experiment groups that were derived from the same batch of cell samples.
Downstream analysis for cell lysis product
Lysed samples were collected at a flow rate of 5 μL/min while the acoustofluidic device was driven with a voltage of 50 VPP and a frequency of 5 kHz. Upon collection, the lysed samples were centrifuged at 800 rpm for 10 minutes to remove any un-lysed cells remaining in the samples, which was a critical step to completely eliminate the detection of target proteins and nuclear acids that were from un-lysed cells. After un-lysed cells were removed, the supernatant was collected for western-blot analysis. 5 μL of the supernatant was mixed with 10 μL Tricine Sample Buffer (Bio-Rad Laboratories, CA, USA) and heated to 95 °C for 5 minutes. The mixture was then processed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (Bio-Rad Laboratories, CA, USA) and transferred to a PVDF membrane (Bio-Rad Laboratories, CA, USA) through Trans-Blot Turbo Transfer System (Bio-Rad Laboratories, CA, USA). PVDF membranes were incubated with mouse anti-human beta-actin (Abcam, MA, USA) at 4 °C for 12 hours, followed by incubation with Goat anti-mouse HRP antibody (Abcam, MA, USA). Bands of target protein were imaged with the ChemiDoc Imaging System (Bio-Rad Laboratories, CA, USA). Polymerase chain reaction (PCR) reactions were processed using AmpliTaq Gold 360 Master Mix (Applied Biosystems, Life Technologies, NY, USA) with 26 PCR reaction circles. Primers are listed in Table 1. The amplicons were stained by SYBR green (Invitrogen, Life Technologies, MA, USA) and images were captured by ChemiDoc Imaging System (Bio-Rad Laboratories, CA, USA).
Table 1.
PCR primers for Human cells’ and malarial genome validation
| PCR primer for HeLa and Jurkat | Forward | ACCTGGGTCTTCAGTGAACC |
| Reverse | CAGTCAGATCCTAGCGTCGAG | |
| PCR primer for malaria | Forward | TTAAACTGGTTTGGGAAAACCAAATATATT |
| Reverse | ACACAATGAACTCAATCATGACTACCCGTC |
Lysis of malaria-infected RBCs and its downstream analysis
RBCs infected with malaria parasites were provided by the malaria lab at Duke University. Generally, infected RBCs were cultured in RPMI1640 culture medium at a concentration of 1 × 106 cells/mL, and 5% of RBCs were infected with 3D7 parasites. The infected RBCs were injected into the acoustofluidic device at a flow rate of 5 μL/min. The control group was first collected before the acoustofluidic device was activated; after the control group was collected, the experimental group was collected by activating the transducer with a driving voltage of 50 VPP and a frequency of 5 kHz (sinusoidal wave). A cell slide of collected samples was prepared following the instruction of Doublesytoslide system (Thermofisher, USA), and slide staining was performed by following the instructions of the Diff-Quik staining kit (Thermofisher, USA). For PCR analysis, the collected samples were centrifuged at 3000 g for 5 minutes and the supernatants were collected as templets. To perform PCR analysis on the lysis product using infected RBCs, we used PCR conditions and primers that were previously reported,53 as listed in Table 1.
Results and discussion
Device design and working mechanism
Fig. 1 shows the design and concept of our acoustofluidic device for cell lysis. Briefly, the device consists of a PDMS microchannel and an acoustic transducer, and they are bonded onto a thin glass substrate. The PDMS channel contains 360 sharp-edge structures (0.5 mm long and 1.0 mm apart) on both of its sidewalls (Fig. 1a), and features one inlet and one outlet for introducing cell samples and collecting lysis products, respectively (Fig. 1b). When activated at an optimal driving frequency, the acoustic transducer oscillates and causes the glass substrate to vibrate, which in turn oscillates the sharp-edge structures. The oscillating sharp-edge structures can induce acoustic streaming effects in the fluid surrounding their tips.48, 54 Once developed, the acoustic streaming effects can locally generate high shear forces that can mechanically lyse cells (Fig. 1a: lower-left). Because a large number of sharp-edged structures (180 pairs) are constructed and arranged evenly in the channel, intact cells loaded into the device will repeatedly be subjected to the acoustic-streaming-derived shear forces as they flow through the channel, thereby increasing the likelihood that the cells can be lysed and potentially improving the lysis efficiency. Thanks to this feature, our acoustofluidic device can perform cell lysis in a continuous manner, as opposed to a drop-wise (or batch-wise) manner reported elsewhere.34
Fig 1.

Design and concept of the sharp-edge-based acoustofluidic device for cell lysis. (a) Schematic elucidating the mechanism of our acoustofluidic lysis device. A number of sharp-edged structure are constructed on the sidewalls of the channel; when acoustically oscillated, they generate acoustic streaming effects (around the tip of each sharp-edged structure) that can catalyze the cell lysis process. (b) Photograph of the acoustofluidic lysis device. The device is composed of a serpentine channel with a large number of sharp-edged structures, an acoustic transducer, and a thin glass substrate. (c) Comparison of the cell density between upstream and downstream the channel. The cell density downstream the channel is visibly lower than that upstream the channel, which suggests that cells are being lysed as they flow through the channel. Microscopic images showing the lysis behavior observed. Device image (d) upstream and (e) downstream the channel. A number of floating cells are present as big particles upstream of the channel and are concentrated around the tip of each sharp-edged structure, while only negligible number of floating cells are present downstream the channel with significant amount of cellular debris observed. The difference in the number of floating cells indicates that the lysis process is taking place as the cells flow through the channel. Scale bars: 250 μm.
Our acoustofluidic device ruptures cells based primarily on the synergetic effects of two phenomena derived in the channel: the high shear force generated by the acoustic streaming, and the trapping effect induced by the acoustic radiation force and the drag force arising from the acoustic streaming.50, 54–56 The former is the main force to mechanically tear cells apart, while the latter will attract floating cells and force them to circulate along the streamline of the acoustic streaming. Moreover, the streamlines of the acoustic streaming effects generated by neighboring sharp-edged structures will partially merge and form a circulating streamline (Fig. 1a: lower-left); such a streamline will also allow cells to repeatedly experience the shear force, thereby increasing the chances of completely rupturing cells. Fig. 1c shows the Calcein-AM stained cells (fluorescent dots) flowing in the channel near the inlet (upstream) and outlet (downstream) in the channel in the presence of the acoustic streaming. The upstream channel contains a higher number of cells, while the downstream channel contains a much lower number of cells; this observation suggests that many cells are either being trapped by the acoustic microstreaming vortices around sharp-edge tips, or being lysed by shear stress generated by microstreaming as they flow through the channel. In the upstream channel, floating cells are concentrated around the tips of the sharp-edged structures (as indicated by the red-dashed circle) because of the acoustic streaming, and are being repeatedly ruptured via the high shear forces when circulating within the acoustic streaming (Fig. 1d). In the downstream channel, as shown in Fig. 1e, intact, floating cells are barely seen. Instead, a lot of lysis debris, which is smaller than the cells, is observed and not concentrated around the sharp-edged structures; this observation suggests that most cells are completely lysed after they have travelled through the beginning of the channel and arrived downstream. It is also worth mentioning that small sized cellular debris may avoid capture by the acoustic microstreaming vortices because they are subjected to smaller vortex derived drag forces and acoustic radiation forces than the larger intact cells. This phenomenon ensures that the lysis products can flow through the entire channel to be collected for subsequent analysis. We observe that the vast majority of cells are already lysed at the midpoint of the entire channel length (Fig. S1a). Acoustic microstreaming vortices trap large particles and whole cells (Fig. S1b), which allows the cells to remain in the high shear zone around the sharp-edge tip until they are lysed, after which their debris flows downstream (Fig. S1c, Video S1). This observation implies that the number of sharp-edged structures we have designed is more than sufficient for efficiently lysing cells in the current experimental setup. Additionally, increasing the number of sharp-edged structures may potentially improve the lysis efficiency and throughput of our acoustofluidic device.
In-channel acoustic streaming pattern and shear stress distribution
Our acoustofluidic device can perform mechanical lysis on cells by utilizing the shear forces derived from the acoustic streaming. To verify if the acoustic streaming induced by the oscillating sharp-edged structure does locally create high-shear zones, we first numerically investigated the generation of acoustic streaming and the resulting shear stress found in our acoustofluidic device. The simulation was performed based on the perturbation approach we adopted previously.49, 54, 57 Fig. 2a and 2b show, respectively, the simulated acoustic streaming patterns and the simulated distribution of shear forces. The value of the shear stress was calculated using Eqn. 1:
| (1) |
in which μ is the dynamic viscosity of fluid, and u, v are the x and y components of fluid velocity, respectively. The simulated acoustic streaming patterns are qualitatively in accordance with the experimentally observed acoustic streaming patterns (Fig. 2c). When the acoustic transducer is inactive (0 VPP), no acoustic streaming effects are observed and therefore, no high-shear zones are formed. Once the transducer is activated at 10 VPP, the acoustic streaming effect is generated, and as a result high-shear zones (though not strong in magnitude) are locally created around the tip of each sharp-edge structure. As the driving voltage of the transducer is further increased, the streaming velocity is significantly increased. Accordingly, with the increased streaming velocity, the high-shear zones become wider and the shear stress level is significantly increased (Fig. 2b). To validate the simulated streaming field with experimental results, digital particle image velocimetry (PIV) was recruited to analyze the streaming field by tracing 5 μm polystyrene beads (Fig. S2a and b).58 Streaming patterns and maximum streaming velocities between simulation results and PIV based experimental results are very similar, indicating that the simulation is accurate. Shear stress simulations at varying sharp-edge vibration amplitudes show a positive correlation to voltage increases (Fig. S2c) and shear stress also has a positive correlation with microstreaming velocity (Fig. S2d).
Fig 2.
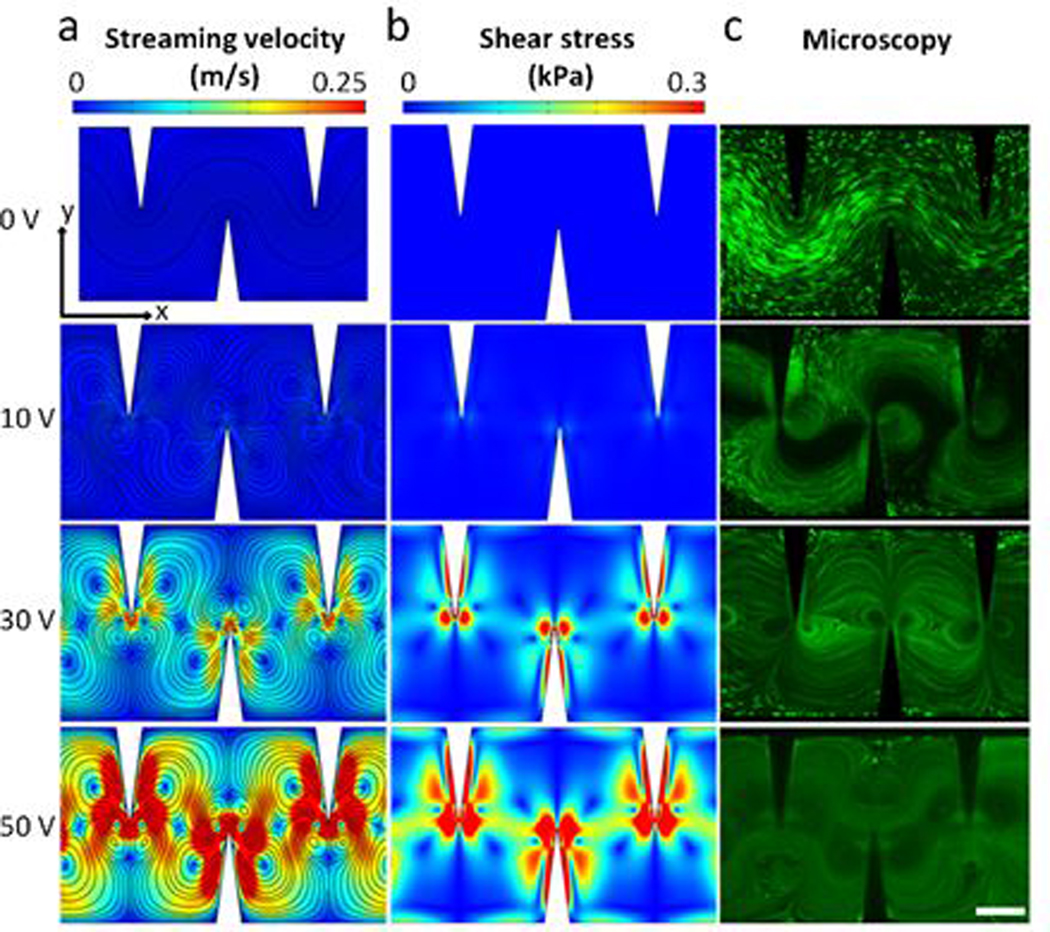
Acoustic streaming patterns and derived distribution of shear stress inside the channel of our acoustofluidic device. (a) Numerically simulated acoustic streaming patterns; maximum streaming velocities for 10, 30, and 50 V driving voltages of the acoustic transducer are 0.08, 0.36, and 0.58 m/s, respectively. (b) Simulated distribution of shear stress under varying driving voltages of the acoustic transducer; maximum shear stress for 10, 30, and 50 V excitation conditions are 0.575, 2.78, and 4.42 kPa, respectively. As the driving voltage is increased, the velocity (i.e., the strength) of the generated acoustic streaming increases, and high-shear zones are locally generated because of the increased streaming velocity. Similarly, with the increased streaming velocity, the resulting shear force increases in its magnitude and widens in its distribution. The simulation results reveal that by controlling the driving voltage of the transducer, the magnitude and distribution of the resulting shear forces can be adjusted. (c) Fluorescent images showing the trajectories of cells in the presence of acoustic streaming under varying driving voltages, which qualitatively match with the simulated streaming patterns. Scale bars: 250 μm.
These results demonstrate not only that the presence of acoustic streaming locally creates high-shear zones in the channel, but also that the shear stress level is directly proportional to the streaming velocity. Moreover, the results also reveal that by controlling the driving voltage of the transducer, we can control the streaming velocity and therefore the shear stress level. In addition to the shear stress level, the trapping effect can also be enhanced by increasing the driving voltage, as shown in Fig. 1c; the enhanced trapping effect would ensure that more cells can follow the streamline of the generated acoustic streaming and the lysis efficiency can therefore be improved. Dynamic processes showing the acoustic streaming patterns and the trapping effect on cells can be seen in Videos S2 and S3.
Validation of acoustofluidic-based cell lysis
Our acoustofluidic device mechanically lyses cells via the shear forces from the streaming induced by the acoustically oscillated sharp-edge structures. Upon numerically investigating the generation of shear stress in our device, we then experimentally validated whether the sharp-edged-based acoustic streaming does play a crucial role in our acoustofluidic lysis device. As a control experiment, we first perform cell lysis using acoustofluidic devices without any sharp-edge structure; besides the lack of sharp-edges, these devices were otherwise identical to the sharp-edge acoustofluidic lysis devices in terms of the device fabrication procedure and device dimensions. Fig. 3a and 3b show the representative lysis results obtained using the control devices and the sharp-edged acoustofluidic devices, respectively, for HeLa cells and Jurkat cells. Using the control devices, the vast majority of HeLa cells and Jurkat cells seem to remain intact and cell debris is barely observed, regardless of the activation of the transducer; this observation suggests that the control devices may be incapable of performing cell lysis because of the absence of acoustic streaming. With the sharp-edged acoustofluidic device, by contrast, intact HeLa cells and Jurkat cells are barely present with a considerable amount of cell debris observed in the channel when the transducer is activated; the relatively strong background of green fluorescence (Calcein-AM) present in the sharp-edged acoustofluidic device suggests that the fluorescent molecules that were originally stained into the cytoplasm of the cells have been released to the surrounding medium. These results demonstrate that by constructing the acoustofluidic device with sharp-edge structures, we can effectively lyse cells and thereby release molecules of interest from the cells. Most importantly, by comparing these results with those obtained using the control device, we believe that the sharp-edge structures and resulting acoustic streaming play a critical role in achieving cell lysis.
Fig 3.

Validation of the acoustic steaming effect on the lysis performance using (a) HeLa cells and (b) Jurkat cells. When using the control device, where no sharp-edged structure is constructed in the channel, most of the cells are intact and cellular debris is barely observed, regardless of the activation of the control device and the cell types. By contrast, when using the sharp-edged acoustofluidic device, where a number of sharp-edged structures are present in the channel, only negligible amount of cells are intact and cellular debris are clearly seen when the acoustofluidic device is activated, thus proving that the utilization of the sharp-edged structures and resulting acoustic streaming is essential for acoustofluidic cell lysis. Scale bars: 100 μm.
In acoustic-based cell lysis, heat generation arising from the application of acoustic energy, which can potentially become the primary force to lyse cells, are usually taken into consideration.7, 59, 60 The generated heat energy (regardless of lysis method) may degrade enzymes released from lysed cells.61, 62 In this work, to rule out heat as the main factor for cell lysis, we experimentally monitored the temperature distribution of our acoustofluidic device over a time interval of 20 minutes, which corresponds to the actual duration of each lysis experiment conducted. The temperature-monitoring experiment was carried out by activating the transducer mounted on our acoustofluidic device at a driving frequency of 5 kHz and a driving voltage of 50 VPP, which is the highest power used in this work. After our acoustofluidic device was continuously activated for 20 minutes, the highest surface temperature of the transducer as measured by a digital thermometer was 29.7 °C, and the temperature observed to come from the PDMS chip by thermal camera was only increased from 21.9 °C to 29 °C during operation (Fig. S3a and S3b). Since the transducer is the only heat source in the system, the temperature of the sample within the device cannot be higher than the transducer surface. Furthermore, the increased temperature is even lower than that of human body (Fig. S3c), and this level of heat generation is very unlikely to cause cell lysis according to previous studies.63 These results reveal that insignificant heat is generated using our acoustofluidic approach, and it rules out the thermal effect as the factor for cell lysis in our device.
Dependence of lysis efficiency on the driving voltage
The shear stress level can significantly influence the lysis efficiency of our acoustofluidic device and can be controlled by adjusting the driving voltage of the transducer. To evaluate the dependence of the lysis efficiency on the driving voltage, we conduct cell lysis at six different driving voltages including 0, 10, 20, 30, 40, and 50 VPP. Fig. 4a and 4b show the lysis results, respectively, for HeLa cells and Jurkat cells, using our acoustofluidic device under four representative driving voltages (0, 10, 30, and 50 VPP). When the device is inactive (0 VPP), most of the cells are intact and no cell debris is observed, because no shear forces (i.e., no acoustic streaming) are generated. Even when the device is activated at 10 VPP, no visible cell lysis phenomenon is observed because the generated shear stress and trapping effect may not be strong enough to initiate cell lysis. Further increasing the driving voltage to 30 VPP can significantly reduce the number of cells remaining in the channel, which suggests that the cells are being lysed due to the presence of shear forces. Once activated at 50 VPP, the acoustofluidic device can efficiently lyse the cells and intact cells are barely observed, indicating that driven at this acoustic power, our device can produce strong shear forces to rupture most of the cells.
Fig 4.

Dependence of the lysis efficiency on the driving voltage of the acoustic transducer. Fluorescent images of the representative lysis results for (a) HeLa cells and (b) Jurkat cells under the driving voltages of 0, 10, 30, and 50 VPP. The number of visually-observed cells reduces as the driving voltage is increased, which suggests improved lysis efficiency with the increment in the driving voltage. Also, the enhanced strong background of green fluorescence is observed as the driving voltage is increased, which implies that more cells are lysed into debris and more Calcein-AM molecules have been released to the surrounding medium upon the lysis. The percentage of remaining cells after lysis under different driving voltages for (c) HeLa cells and (d) Jurkat cells. Statistically, the percentage lowers as the driving voltage is increased, indicating that the lysis efficiency is improved by raising the driving voltage. The results presented in this figure are obtained at the same flow rate of 5 μL/min, and the number of remaining cells under the driving voltage of 0 VPP is regarded as 100 % of remaining cells. Scale bars: 100 μm. Error bars represents the standard deviation from three independent experiments (n=3).
Other than the observed number of cells, the effect of the driving voltage on the lysis efficiency can also be visually compared through the background of green fluorescence (Calcein-AM). As the driving voltage is increased, the intensity of the background noticeably increases, which suggests that the lysis efficiency is improved as we increase the driving voltage. We also note that the number of cells stained with SYTOX-orange (red fluorescence) changes insignificantly when our device is driven at higher voltages, presumably because the nuclei of lysed cells are also damaged. A complete set of images showing the visually observed lysis results under all the tested driving voltages can be found in Fig. S4.
To quantitatively and statistically characterize the lysis efficiency, we further calculate the percentage of un-lysed cells under different driving voltages from repeated experiments (n ⩾ 3). The percentage is calculated by dividing the number of un-lysed cells observed at each driving voltage by the number of un-lysed cells observed when the device is switched off (i.e., 0 VPP); a lower percentage represents a better lysis efficiency. As shown in Fig. 4c and 4d, the quantified lysis result reveals that the percentage of un-lysed cells is inversely proportional to the driving voltage applied. Additionally, the statistical analysis demonstrates that our acoustofluidic device can perform cell lysis in a reproducible manner, provided that the concentration/density of injected cell samples remains constant among different experiments. These results demonstrate that by increasing the driving voltage applied to our device, the lysis efficiency can be significantly improved; in other words, the lysis efficiency can be tuned by controlling the driving voltage. With such control over the lysis efficiency by simply adjusting the driving voltage, our acoustofluidic device may be well-suited for a wide range of applications where complete or partial cell lysis is needed.7, 64, 65
Dependence of lysis efficiency on the sample flow rate
The acoustic streaming generated by the oscillating sharp-edged structure is the key tool in our acoustofluidic-based cell lysis. However, based on our previously reported work,48, 49, 54, 66, 67 the generation of acoustic streaming can also be greatly influenced by the fluid velocity of the background flow in addition to the driving voltage of the transducer. In this work, since the device features only one inlet and one outlet, the fluid velocity of the injected cell sample is thus considered as the fluid velocity of the background flow. To investigate how the sample flow rate affects the lysis efficiency of our device, we conduct experiments on cell lysis at four different flow rates including 5, 7.5, 10, and 15 μL/min, under the same driving voltage (50 VPP). Fig. 5a and 5b show the lysis results obtained at these four different flow rates, respectively, for HeLa cells and Jurkat cells. When working with relatively low flow rates (i.e., 5 and 7.5 μL/min), our acoustofluidic device is able to efficiently lyse the cells with only negligible amounts of un-lysed cells observed. Once we raise the flow rate to 10 and 15 μL/min, the numbers of un-lysed cells significantly increases, compared to the numbers observed at lower flow rates. In addition, the reduced background of green fluorescence due to the raised flow rate also indicates that the lysis efficiency is compromised as the flow rate is increased.
Fig 5.

Dependence of the lysis efficiency on the sample flow rate. Fluorescent images showing the representative lysis results for (a) HeLa cells and (b) Jurkat cells at four different flow rates. Overall, as the flow rate is increased, the number of visually observed cells increases for both types of cells, suggesting that the lysis efficiency is degraded with the increase in the flow rate. The percentage of remaining cells after the lysis at different flow rates for (c) HeLa cells and (d) Jurkat cells. As the flow rate is raised, the percentage arises, which, once again, statistically indicates compromised efficiencies due to raised flow rates. Control groups presented in (c) and (d) display the results obtained at the flow rate of 15 μL/min with the acoustic transducer inactivated, and the number of remaining cells under these conditions is regarded as 100 % of remaining cells. Scale bars: 100 μm. Error bars represents the standard deviation from three independent experiments (n=3).
Likewise, we also quantitatively and statistically compared the percentage of un-lysed cells at the four flow rates, as shown in Fig. 5c and 5d. Overall, the percentage of un-lysed cells is directly proportional to the flow rate for both HeLa cells and Jurkat cells. When the flow rate is less than 7.5 μL/min, the percentage can be reduced to less than 10% and 3%, respectively, for HeLa cells and Jurkat cells. Once the flow rate is greater than 7.5 μL/min, there is a significant increase in the percentage (corresponding to a reduction in lysis efficiency) for both HeLa cells and Jurkat cells; nevertheless, the percentage of un-lysed cells at the highest flow rate tested (i.e., 15 μL/min) is below 50%, which might be still sufficient for downstream analysis of intracellular components.28, 62 The lysis efficiency at higher flow rates can be further improved by optimizing the design parameters and operating conditions of our acoustofluidic devices.
It is important to note that the degraded lysis efficiency with the increased flow rate is due in large part to the enhanced suppression of the generated acoustic streaming caused by the background flow (i.e., the sample flow). According to our previous findings,48, 54, 66 a background flow with a higher fluid velocity, compared to the one with lower fluid velocity, presents stronger resistance to the formation of acoustic streaming within our sharp-edge-based devices, assuming that the driving voltage remains constant. As a result, in this work, increasing the flow rate can adversely affect the formation of high-shear zones and in turn, compromise the lysis efficiency. Additionally, we note that the increased flow rate might also weaken the trapping effect — the other dominating phenomenon facilitating the cell lysis in our acoustofluidic device — and thereby degrade the lysis efficiency.
Analysis of cell lysate
Our acoustofluidic device relies primarily on mechanical forces (i.e., shear forces) to rupture cells, so it is a chemical-free approach to release intracellular components from cells for downstream analysis. Therefore, the key advantage of using our device is that the lysis products may be used directly for subsequent analysis without using post-lysis processes such as washing and purification, because the released intracellular components are present in their natural forms. To demonstrate this feature, we collect the cell lysate from our acoustofluidic device and use it directly for protein and genome analysis. Beta-actin, which is widely used as an internal reference for protein analysis, is chosen in this work as the target protein to carry out western-blot analysis on the lysates of HeLa and Jurkat cells.
Fig. 6a shows the western-blot analysis of the lysates obtained under different driving voltages, where the sample flow rate was held constant at 5 μL/min to ensure that the driving voltage is the only variable being changed to influence the lysis efficiency. For both Hela cells and Jurkat cells, the band corresponding to beta-actin is present for the samples lysed using more than 20 VPP. As the driving voltage is increased, the band of beta-actin becomes wider and darker. This western blot result suggests that the number of lysed cells (i.e., the number of target proteins present in the lysate) is increased as we elevate the driving voltage. To further analyze whether the lysates could be used for genetic profiling, we chose a 291 bp fragment on the exon 3 region of human P53 gene as the amplicon. By directly using the lysates of HeLa and Jurkat cells, the PCR reaction we designed can successfully amplify P53 fragments and present positive bands via agarose electrophoresis. Since the designed PCR reaction includes only 26 circles of amplification, the PCR result also suggests that the templates are abundant in the lysates and that genomic DNA of the lysed cells are not degraded during the lysis process. Additionally, western blot is based on antibody-specific capture of target molecules and PCR is based on the recognition between DNA single strands. We successfully use the lysates collected from our acoustofluidic device directly for these two analyses without additional washing procedures; this demonstrates that the biomarkers present in the lysates are bioactive and could be directly detected by both antibody-based and DNA-based methods. It should be noted that there may be lysates that remain in the channel, such as molecules of interest that stick to the channel surfaces or cellular debris that is trapped and circulates within the acoustic streaming vortices. Nevertheless, because our device performs cells lysis in a “continuous-flow” manner, we believe that the majority of the lysis products generated during device operation flow through the entire channel to be collected at the outlet. The amount of lysates remaining in the channel should therefore be minimal or negligible compared to the total amount of lysates that flow through the channel to be collected. These results reveal the potential of our acoustofluidic lysis device for the integration with existing microfluidic-based components towards the development of integrated, monolithic POC platforms.
Fig 6.

Downstream analysis on proteins and nucleic acids using the un-lysed and lysed samples for HeLa and Jurkat cells. (a) Western blot results of beta-actin under different driving voltages of the acoustic transducer. Improved lysis efficiencies by increasing the driving voltage lead to the increased expression (i.e., increased release of beta-actin from the lysed samples) of beta-actin for both types of the cells. (b) PCR results of targeted P53 gene. 291 bp fragments on one exon of P53 gene are observed only for the lysed cell samples, indicating that our acoustofluidic device can efficiently lyse the cells and then release nucleic acids of interest from the cells.
Lysis and analysis of malaria-infected RBCs
The ultimate goal for all of the existing lysis approaches is their utilization to process cell samples that are clinically relevant or disease-related. To demonstrate the capability of our acoustofluidic device towards this goal, we conduct the cell lysis on red blood cells infected with 3D7 malarial parasites (referred to as infected RBCs hereafter). Fig. 7a shows the morphological difference between infected RBCs that are lysed with our acoustofluidic device, and infected RBCs that flow through the device in the absence of acoustic streaming (i.e., un-lysed). In the un-lysed sample, the concentration of RBCs is higher, RBCs remain intact, and parasites (as indicated by white arrows) are enclosed within RBCs. In the acoustically lysed sample, by contrast, RBCs are present at a lower concentration, and there are partially-ruptured RBCs and many hollow, membrane-like structures observed. The membrane-like structures are believed to be RBCs that are ruptured and may have released their hemoglobin to the surrounding medium. As shown in Fig. 7b, the lysed sample contains less precipitation and its supernatant is pink, which proves that hemoglobin has been released from the ruptured RBCs. Most importantly, in the lysed sample, a few parasites and parasite debris are present as tiny black dots (as indicated by blue arrows), suggesting that parasites are released from ruptured RBCs and some of them may have been lysed. Based on the PCR result, a 205 bp amplicon on the SSUrRNA gene using designed primers can be detected using the lysis product of infected RBCs as templets, while no target band is present for the un-lysed sample. These results demonstrate that our acoustofluidic device can rupture parasite-infected RBCs to release malaria parasites, and that the lysis products can be directly used for subsequent analysis. Furthermore, the results also demonstrate the potential of our acoustofluidic devices for the lysis and detection of clinically relevant or disease-related samples such as bloodborne pathogens.
Fig 7.

Characterization of the lysis products using malaria-infected RBCs. (a) Morphological comparison between the un-lysed samples (left, OFF) and the lysed samples (right, ON). In the un-lysed samples, parasites are present and enclosed by normal RBCs, as indicated by white arrows; in the lysed samples, parasites are also observed but present as independent bio-particles because they are released from RBCs upon lysis. (b) Visual comparison between un-lysed and lysed samples. After centrifugation, the lysed sample (right, ON) exhibits less precipitation and a higher level of released hemoglobin than the un-lysed sample, which indicate that RBCs are lysed and hemoglobin is released to the surrounding medium. (c) PCR results using the un-lysed and lysed samples. The nucleic acid of malarial parasites could be detected from the lysed sample.
Conclusions
Utilizing the acoustic streaming induced by oscillating sharp-edged structures, we have demonstrated an acoustofluidic device that can mechanically lyse cells via the shear forces arising from the generated acoustic streaming, in a continuous, reagent-free, and controllable manner. The distribution and magnitude of the resulting shear forces can be controlled by adjusting the strength of the induced acoustic streaming effect, which can be achieved simply by controlling the driving voltage of the acoustic transducer. In other words, the lysis efficiency of our acoustofluidic device can be adjusted simply by altering the driving voltage. With the acoustofluidic device, we can perform cell lysis with both adherent and non-adherent cells. Currently, the cell lysis efficiency of our device is higher than 90%, and can even achieve 98% in some test runs. Compared with chemical microfluidic cell lysis (~86% efficiency),68 electrical cell lysis (74–99% efficiency),68 optical microfluidic cell lysis (78–80% efficiency),23 thermal lysis (~90% efficiency),21 and mechanical methods (65–99% efficiency),68 the lysis efficiency achieved by our sharp-edge acoustofluidic device is comparable. Because our acoustofluidic-based cell lysis approach is chemical-free, the lysis products can be used directly for downstream protein/DNA analysis without washing and purification steps. Additionally, we have demonstrated the ability of our acoustofluidic device to process clinically relevant or disease-related cell samples by conducting cell lysis with malaria-infected RBCs; the resulting lysate can also be used directly for the detection of representative malaria genes. Taken together, our acoustofluidic lysis device is simple in fabrication and operation, chemical-free in lysate quality, and controllable in lysis efficiency; all of these features render our acoustofluidic device a useful component for the development of automated, self-contained POC platforms.
Supplementary Material
Acknowledgements
We acknowledge support from the National Institutes of Health (R01GM132603, R01HD086325, and R01GM127714), United States Army Medical Research Acquisition Activity (W81XWH-18-1-0242), and National Science Foundation (ECCS-1807601).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
References
- 1.Rusling JF, Kumar CV, Gutkind JS and Patel V, Analyst, 2010, 135, 2496–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soper SA, Brown K, Ellington A, Frazier B, Garcia-Manero G, Gau V, Gutman SI, Hayes DF, Korte B. and Landers JL, Biosens. Bioelectron, 2006, 21, 1932–1942. [DOI] [PubMed] [Google Scholar]
- 3.Niemz A, Ferguson TM and Boyle DS, Trends Biotechnol., 2011, 29, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jung W, Han J, Choi J-W and Ahn CH, Microelectron. Eng, 2015, 132, 46–57. [Google Scholar]
- 5.Vashist SK, Luppa PB, Yeo LY, Ozcan A. and Luong JH, Trends Biotechnol., 2015, 33, 692–705. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh K, Ferguson BS, Eisenstein M, Plaxco KW and Soh HT, Acc. Chem. Res, 2015, 48, 911–920. [DOI] [PubMed] [Google Scholar]
- 7.Brown RB and Audet J, J. R. Soc., Interface, 2008, 5, S131–S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meacle F, Lander R, Ayazi Shamlou P. and Titchener‐Hooker N, Biotechnol. Bioeng, 2004, 87, 293–302. [DOI] [PubMed] [Google Scholar]
- 9.Gao J, Yin X-F and Fang Z-L, Lab Chip, 2004, 4, 47–52. [DOI] [PubMed] [Google Scholar]
- 10.Lu H, Schmidt MA and Jensen KF, Lab Chip, 2005, 5, 23–29. [DOI] [PubMed] [Google Scholar]
- 11.El-Ali J, Sorger PK and Jensen KF, Nature, 2006, 442, 403. [DOI] [PubMed] [Google Scholar]
- 12.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT and Martuza RL, Science, 2014, 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalerba P, Dylla SJ, Park I-K, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH and Simeone DM, PNAS, 2007, 104, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D. and Esposito D, Nature, 2011, 472, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahalanabis M, Al-Muayad H, Kulinski MD, Altman D. and Klapperich CM, Lab Chip, 2009, 9, 2811–2817. [DOI] [PubMed] [Google Scholar]
- 16.Santillo MF, Heien ML and Ewing AG, Lab Chip, 2009, 9, 2796–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sethu P, Anahtar M, Moldawer LL, Tompkins RG and Toner M, Anal. Chem, 2004, 76, 6247–6253. [DOI] [PubMed] [Google Scholar]
- 18.Di Carlo D, Jeong K-H and Lee LP, Lab Chip, 2003, 3, 287–291. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Jang SH, Jia G, Zoval JV, Da Silva NA and Madou MJ, Lab Chip, 2004, 4, 516–522. [DOI] [PubMed] [Google Scholar]
- 20.Liu RH, Yang J, Lenigk R, Bonanno J. and Grodzinski P, Anal. Chem, 2004, 76, 1824–1831. [DOI] [PubMed] [Google Scholar]
- 21.Privorotskaya N, Liu Y-S, Lee J, Zeng H, Carlisle JA, Radadia A, Millet L, Bashir R. and King WP, Lab Chip, 2010, 10, 1135–1141. [DOI] [PubMed] [Google Scholar]
- 22.Cheong KH, Yi DK, Lee J-G, Park J-M, Kim MJ, Edel JB and Ko C, Lab Chip, 2008, 8, 810–813. [DOI] [PubMed] [Google Scholar]
- 23.Huang S-H, Hung L-Y and Lee G-B, Lab Chip, 2016, 16, 1447–1456. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Liu A, Klaseboer E, Zhang J. and Ohl C, Lab Chip, 2013, 13, 1144–1150. [DOI] [PubMed] [Google Scholar]
- 25.Jokilaakso N, Salm E, Chen A, Millet L, Guevara CD, Dorvel B, Reddy B, Karlstrom AE, Chen Y. and Ji H, Lab Chip, 2013, 13, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H-Y and Lu C, Anal. Chem, 2006, 78, 5158–5164. [DOI] [PubMed] [Google Scholar]
- 27.Marentis TC, Kusler B, Yaralioglu GG, Liu S, Hæggström EO and Khuri-Yakub B, Ultrasound Med. Biol, 2005, 31, 1265–1277. [DOI] [PubMed] [Google Scholar]
- 28.Nan L, Jiang Z. and Wei X, Lab Chip, 2014, 14, 1060–1073. [DOI] [PubMed] [Google Scholar]
- 29.Taylor MT, Belgrader P, Furman BJ, Pourahmadi F, Kovacs GT and Northrup MA, Anal. Chem, 2001, 73, 492–496. [DOI] [PubMed] [Google Scholar]
- 30.Ozcelik A, Rufo J, Guo F, Gu Y, Li P, Lata J. and Huang TJ, Nat. Methods, 2018, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu M, Ouyang Y, Wang Z, Zhang R, Huang P-H, Chen C, Li H, Li P, Quinn D. and Dao M, PNAS, 2017, 114, 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruus H, Dual J, Hawkes J, Hill M, Laurell T, Nilsson J, Radel S, Sadhal S. and Wiklund M, Lab Chip, 2011, 11, 3579–3580. [DOI] [PubMed] [Google Scholar]
- 33.Bruus H, Lab Chip, 2012, 12, 1014–1021. [DOI] [PubMed] [Google Scholar]
- 34.Reboud J, Bourquin Y, Wilson R, Pall GS, Jiwaji M, Pitt AR, Graham A, Waters AP and Cooper JM, PNAS, 2012, 109, 15162–15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bourquin Y, Reboud J, Wilson R. and Cooper JM, Lab Chip, 2010, 10, 1898–1901. [DOI] [PubMed] [Google Scholar]
- 36.Wu M, Ozcelik A, Rufo J, Wang Z, Fang R. and Huang TJ, Microsystems & nanoengineering, 2019, 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ai Y, Sanders CK and Marrone BL, Anal. Chem, 2013, 85, 9126–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins DJ, Neild A, DeMello A, Liu A-Q and Ai Y, Lab Chip, 2015, 15, 3439–3459. [DOI] [PubMed] [Google Scholar]
- 39.Li P. and Huang TJ, Anal. Chem, 2018, 91, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Courtney CR, Demore CE, Wu H, Grinenko A, Wilcox PD, Cochran S. and Drinkwater BW, Applied Physics Letters, 2014, 104, 154103. [Google Scholar]
- 41.Melde K, Choi E, Wu Z, Palagi S, Qiu T. and Fischer P, Advanced Materials, 2018, 30, 1704507. [DOI] [PubMed] [Google Scholar]
- 42.Tian Z, Yang S, Huang P-H, Wang Z, Zhang P, Gu Y, Bachman H, Chen C, Wu M. and Xie Y, Science advances, 2019, 5, eaau6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faridi M, Ramachandraiah H, Iranmanesh I, Grishenkov D, Wiklund M. and Russom A, Biomedical microdevices, 2017, 19, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mutafopulos K, Spink P, Lofstrom C, Lu P, Lu H, Sharpe J, Franke T. and Weitz D, Lab Chip, 2019. [DOI] [PubMed] [Google Scholar]
- 45.Olm F, Urbansky A, Dykes JH, Laurell T. and Scheding S, Scientific Reports, 2019, 9, 8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeo LY, Chang HC, Chan PP and Friend JR, Small, 2011, 7, 12–48. [DOI] [PubMed] [Google Scholar]
- 47.Destgeer G, Lee KH, Jung JH, Alazzam A. and Sung HJ, Lab Chip, 2013, 13, 4210–4216. [DOI] [PubMed] [Google Scholar]
- 48.Huang P-H, Xie Y, Ahmed D, Rufo J, Nama N, Chen Y, Chan CY and Huang TJ, Lab Chip, 2013, 13, 3847–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nama N, Huang P-H, Huang TJ and Costanzo F, Biomicrofluidics, 2016, 10, 024124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ozcelik A, Nama N, Huang PH, Kaynak M, McReynolds MR, Hanna‐Rose W. and Huang TJ, Small, 2016, 12, 5120–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang PH, Zhao S, Bachman H, Nama N, Li Z, Chen C, Yang S, Wu M, Zhang SP and Huang TJ, Advanced Science, 2019, 6, 1900913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang SP, Lata J, Chen C, Mai J, Guo F, Tian Z, Ren L, Mao Z, Huang P-H and Li P, Nat. Commun, 2018, 9, 2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oyedeji SI, Awobode HO, Monday GC, Kendjo E, Kremsner PG and Kun JF, Malar. J, 2007, 6, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nama N, Huang P-H, Huang TJ and Costanzo F, Lab Chip, 2014, 14, 2824–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leibacher I, Hahn P. and Dual J, Microfluid. Nanofluid, 2015, 19, 923–933. [Google Scholar]
- 56.Shi C, Dubois M, Chen Y, Cheng L, Ramezani H, Wang Y. and Zhang X, Nat. Commun, 2016, 7, 11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi C, Dubois M, Wang Y. and Zhang X, PNAS, 2017, 114, 7250–7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thielicke W. and Stamhuis E, Journal of Open Research Software, 2014, 2. [Google Scholar]
- 59.Ciawi E, Rae J, Ashokkumar M. and Grieser F, J. Phys. Chem. B, 2006, 110, 13656–13660. [DOI] [PubMed] [Google Scholar]
- 60.Wu X, Joyce EM. and Mason TJ, Water Res., 2012, 46, 2851–2858. [DOI] [PubMed] [Google Scholar]
- 61.Ashokkumar M, Lee J, Zisu B, Bhaskarcharya R, Palmer M. and Kentish S, J. Dairy Sci, 2009, 92, 5353–5356. [DOI] [PubMed] [Google Scholar]
- 62.Shehadul Islam M, Aryasomayajula A. and Selvaganapathy P, Micromachines, 2017, 8, 83. [Google Scholar]
- 63.Lee C-Y, Lee G-B, Lin J-L, Huang F-C and Liao C-S, J. Micromech. Microeng, 2005, 15, 1215. [Google Scholar]
- 64.Lippi G, Montagnana M, Salvagno GL and Guidi GC, Arch. Pathol. Lab. Med, 2006, 130, 181–184. [DOI] [PubMed] [Google Scholar]
- 65.Coolbear T, Holland R. and Crow VL, Int. Dairy J, 1992, 2, 213–232. [Google Scholar]
- 66.Huang P-H, Chan CY, Li P, Nama N, Xie Y, Wei C-H, Chen Y, Ahmed D. and Huang TJ, Lab Chip, 2015, 15, 4166–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang P-H, Chan CY, Li P, Wang Y, Nama N, Bachman H. and Huang TJ, Lab Chip, 2018, 18, 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mun BP, Jung SM, Yoon SY, Kim SH, Lee JH and Yang S, Microfluid. Nanofluid, 2010, 8, 695–701. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.