

Abstract
The sudden infant death syndrome (SIDS) is the leading cause of postneonatal infant mortality in the United States today, with an overall rate of 0.39/1000 live births. It is defined as the sudden and unexpected death of an infant <12 months of age that remains unexplained after a complete autopsy, death scene investigation, and review of the clinical history. The serotonin brainstem hypothesis has been a leading hypothesis for SIDS over the last 2 decades. Our laboratory has studied this hypothesis over time with a variety of tissue techniques, including tissue receptor autoradiography, high performance liquid chromatography, Western blot analysis, immunocytochemistry, and proteomics. The purpose of this article is to review the progress in our laboratory toward supporting this hypothesis. We conclude that an important subset of SIDS infants has serotonergic abnormalities resulting from a “core lesion” in the medullary reticular formation comprised of nuclei that contain serotonin neurons. This lesion could lead to a failure of protective brainstem responses to homeostatic challenges during sleep in a critical developmental period which cause sleep-related sudden death.
Keywords: Autoresuscitation, Medulla oblongata, Proteomics, Raphe, Receptor, Rhombomeres, Tissue receptor autoradiography
INTRODUCTION
The phenomenon of sudden and unexpected infant death has long challenged health care providers, researchers and most of all, families. A full investigation after a sudden death may determine a cause of death, such as a metabolic disorder, infection, or cardiac arrhythmia. Many sudden deaths in infants, however, do not yield an identified cause, and are considered due to the “sudden infant death syndrome” (SIDS). In an attempt to create a relatively uniform category of infant deaths the National Institute of Child Health and Development (NICHD) in 1991 defined the term SIDS as infants for whom a cause of death remains unexplained after a complete autopsy, death scene investigation, and review of the clinical history (1). SIDS is the leading cause of postneonatal infant mortality in the United States today with an overall rate of 0.39/1000 live births (2). SIDS is characterized by the sudden death of a seemingly healthy infant typically associated with a sleep period—either during sleep itself or during one of the many spontaneous arousals that occur from sleep. Since the late 1980s, prone sleep has been recognized as a major risk factor for SIDS, leading to the national safe sleep campaigns in the 1990s and forward that advocated supine sleep. After the initiation of the campaigns there was a drop in the SIDS rate by 50% over the next decade (3). Over the last 2 decades the rate has plateaued (3), necessitating the need for continued research on the cause of SIDS and its prevention. Of note, several other improvements in perinatal care occurred alongside the Back-to-Sleep campaign, making it difficult to attribute unequivocally the decline in SIDS to supine sleep alone (3).
The first question confronting neural research in SIDS is whether or not a defined pathological lesion exists in the brain, and if so, where the lesion is located. Knowing the site of the pathologic lesion opens clues of discovery to the mechanism and cause of the underlying disorder(s). The site of the brain lesion in SIDS is evasive because there are no major anatomic markers of neural pathology—the standard autopsy is unrevealing and SIDS is called “mysterious.” Moreover, there is no naturally occurring, spontaneous animal model of SIDS, so human research in autopsy tissue is essential, with consideration of all caveats such as length of postmortem intervals. The strong epidemiological link of infant sleep to SIDS, however, suggests that dysfunction related to sleep is critical in the pathogenesis of these sudden deaths during a critical developmental time window. If SIDS is due to a brain lesion, then uncovering the lesion requires special research—quantitative tools at the cellular, neurochemical, and molecular levels, since standard histology shows few, if any, abnormal findings. The major focus of our research in SIDS over the last 2 decades has been to apply modern research tools to the analysis of the SIDS brainstem at autopsy, that is, directly in human tissue. We believe our major contribution has been to identify a serotonergic abnormality in the medullary reticular formation of the brainstem as a “core” lesion in SIDS. This review is focused on our research in serotonin in the SIDS brainstem and not a comprehensive review of the neuropathology of SIDS in general.
The purpose of this article is to review over 2 decades of our research studying SIDS autopsied infants and the serotonin (5-HT) brainstem hypothesis. We will first discuss the rationale for the brainstem hypothesis and the triple risk model for SIDS death. We begin with a brief review of the organization of the serotonergic system for background information, and then we provide evidence emphasizing 5-HT impairments in the ventral medulla, arcuate nucleus, and caudal raphe system as a segue into discovering the core lesion in the medullary reticular formation. This is followed by a discussion of initial work on selected neurotransmitter receptors in serially sectioned brainstems with tissue receptor autoradiography, searching for clues of abnormal neurotransmitters in nuclei related to cardiorespiratory control and homeostatic mechanisms. We discuss the interaction of the serotonergic system with the gamma-aminobutyric acid (GABA) system in the pathology of the brainstem in SIDS. We review the contribution of proteomics in attempting to identify the chain of events leading to 5-HT abnormalities. We propose a working animal model of the 5-HT brainstem abnormalities reported in SIDS with failure of autoresuscitation. We conclude with speculation about the pathogenesis of the brainstem lesion as originating prenatally. Direct human observations from autopsy analysis in SIDS cases in our laboratory is integrated with experimental observations in rodent models made in conjunction with collaborators at Harvard Medical School, Geisel School of Medicine at Dartmouth, and the University of Iowa.
Rationale for the Brainstem Hypothesis in SIDs
The brainstem is the primary anatomic site of sleep/waking regulation and homeostatic control in the brain (4), and impairments within it are known to result in sudden death (5–8). Broadly stated, the brainstem hypothesis in SIDS posits that there is a developmental abnormality in a brainstem region or regions that lead to a failure of protective mechanisms against life-threatening challenges during sleep, for example, hypoxia or asphyxia typically, but not solely, in the face down (prone) position, and sleep related sudden death. The rationale for the SIDS brainstem hypothesis is based upon several lines of evidence: (i) established human and animal data that demonstrate the brainstem’s critical role in respiratory and autonomic regulation, sleep, and arousal, that is, in the primary physiological processes considered abnormal in SIDS infants; (ii) analogy to human entities of sleep-related sudden death in children or adults in which isolated or primary pathology is found by neuroimaging and/or pathologic study in the brainstem (e.g. [8]); and (iii) reports of subclinical defects in cardiorespiratory control and/or arousal that are consistent with brainstem dysfunction in infants who are studied prospectively and subsequently die of SIDS (9–12).
In the 1980s, prone sleep was recognized as a major risk factor for SIDS, reinforcing the idea that death was related to the face down sleep position and positional asphyxia. In the late 1980s and early 1990s, national risk reduction campaigns advocated supine sleep position and by the mid-1990s, in the Back-to-Sleep era, exogenous impaired respiration due to chest-down (prone) breathing was considered an extrinsic stressor leading to a pocket of air in soft bedding and exposure to the rebreathing of exhaled gases. According to this model, the infant is normal, and develops progressive severe positional asphyxia for which no amount of physiological compensation works, and death results. Our work has examined whether infants dying under these circumstances are not “normal” but have abnormalities of brainstem homeostatic systems that would otherwise protect the infant against life threatening challenges during sleep. We propose that relatively modest asphyxia is all that is needed to trigger death in the face of the impaired brainstem system(s).
An additional line of evidence for the brainstem hypothesis in SIDS was the report in 1976 by Naeye et al (13) of gliosis in the medullary reticular formation of a subset of SIDS cases, highlighted by the astroglial fiber Holzer stain. He interpreted the subtle scarring as probable evidence of chronic alveolar hypoventilation and hypoxemia. The astroglial fibers correlated with the presence of so-called tissue markers of hypoxia that Naeye discovered in SIDS cases, for example, abnormal adrenal brown fat retention, retained extramedullary erythropoiesis, and greater than normal smooth muscle in the small pulmonary arteries (13). The presence of subtle gliosis in SIDS cases was confirmed by others using different astroglial markers and in additional brainstem regions, in sites directly related and not directly related to cardiopulmonary control (14–16). Kinney et al (14) reported in 1983 a subset of SIDS cases whose astrocytic counts exceeded that of the highest control; there were no other clinicopathologic features that distinguished this subgroup. This gliosis was postulated to be a marker of brainstem dysfunction secondary to hypoxia and chronic underventilation. Its presence, whether primary or secondary, supported a brainstem abnormality in SIDS and the validity of the brainstem hypothesis (15). Cerebral white matter gliosis and periventricular leukomalacia, a lesion of the perinatal brain linked to hypoxia-ischemia (17, 18) have also been reported by others and us, suggesting secondary hypoxic-ischemic injury in vulnerable areas in the young brain, that is, cerebral white matter and brainstem.
The Triple Risk Model in SIDs
Our laboratory work in the human brainstem in SIDS was based upon the triple risk model in SIDS, presented in an article in 1994, first authored by Dr. James J. Filiano and co-authored by Dr. Kinney (19). This model posits that SIDS results when 3 factors impinge upon the infant simultaneously: (i) a vulnerable infant with an underlying pathophysiologic process that puts him/her at risk for sudden death; (ii) a critical developmental window, that is, the first few months of life as the newborn makes the transition to independent extra-uterine life; and (iii) an exogenous homeostatic stressor that triggers sudden and unexpected death during a period of sleep (19) (Fig. 1). The exogenous risk factors are linked to unsafe sleep practices, including prone (face down) sleep position, over bundling, soft bedding and cosleeping. These stressors appear to have in common a shared potential to generate homeostatic/metabolic imbalances of hypoxia, asphyxia, hypercarbia, and overheating, all of which challenge the brainstem’s homeostatic response. An underlying abnormality in the brainstem of SIDS infants differentiates a SIDS infant from a healthy infant and explains why not all infants when placed prone die; healthy infants without the underlying brainstem abnormality do not die of SIDS because functioning protective brainstem responses come in to play in response to homeostatic challenges. Stemming from the triple risk model is the hypothesis that supine sleep removes one of the exogenous stressors allowing the vulnerable infant to pass through the critical period unharmed.
FIGURE 1.
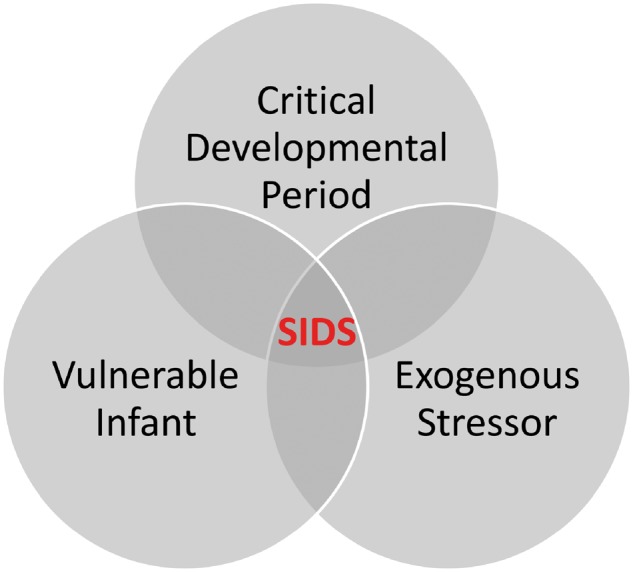
The Venn Diagram of the triple risk model. SIDS occurs when 3 factors impinge on the infant simultaneously: (i) the vulnerable infant; (ii) the critical developmental period; and (iii) the exogenous stressor(s). See text (19).
The Organization and Development of the Serotonergic System
Below we discuss the experimental evidence supporting the role of 5-HT in SIDS pathogenesis. The main finding is that there is a reproducible serotonergic lesion in 4 adjacent subnuclei in the reticular formation of the medulla: The raphe obscurus, paragigantocellularis lateralis, gigantocellularis nucleus, and the intermediate reticular zone (Table 1; Fig. 2). To better understand the implications of these data, an understanding of the serotonergic system, its organization and function, is critical. There are several comprehensive reviews of the organization of the serotonergic neuronal system in the human and rodent brain (20–27). Serotonergic neurons synthesize and release the neurotransmitter 5-HT. They are heterogeneous, collectively innervate most brain and spinal cord regions, and participate in a wide spectrum of neural functions (27) while differing from each other in cell body morphology, electrophysiology, neurochemistry, gene expression, development, projections and inputs (27).
TABLE 1.
Nuclei in Core Lesion in Reticular Formation in SIDS
| Nucleus | Difference in 5-HT1A Binding | Serotonin Neurons Present | Key Function |
|---|---|---|---|
| Raphe obscurus | + | + | Modulation of multiple brainstem reflexes (28) |
| Paragigantocellularis lateralis | + | + | Neurons expressing 5-HT1A autoreceptors modulate sleep state. Inhibition of 5-HT neurons in the LPGi leads to sleep fragmentation and decreases in REM sleep in piglets. The LPGi receives inputs from many sites in the caudal brainstem, and projects to the spinal cord and to the more rostral areas important for arousal and vigilance (29). |
| Gigantocellularis | + | + | Critically important for CNS arousal and behavioral responsiveness in postnatal mouse pups. Development of electrophysiological properties of gigantocellularis neurons correlate with increased CNS arousal (30). |
| Arcuate nucleus | + | + | Putative respiratory chemosensitive fields of ventral medulla in cat (31). A subpopulation of serotonergic neurons exist within the arcuate nucleus (32). |
| Intermediate reticular zone | + | + | 5-HT and catecholamine neurons are intermingled. The IRt brackets the line separating alar and basal visceral plates in embryo and is thought to be involved, via catecholamine activity, in control of sympathetic cardiovascular outflow, cardiorespiratory interactions, and reflex control of vasopressin (33) |
The nuclei of the core lesion in SIDS demonstrated in Figure 2.
Abbreviations: LPGi, paragigantocellularis lateralis; CNS, central nervous system; IRt, intermediate reticular zone.
FIGURE 2.

(A) Location of the core lesion in SIDS in the reticular formation of the medulla oblongata. (B) The nuclei that form the core lesion are paragigantocellularis lateralis (LPGi), the intermediate reticular zone (IRt), the gigantocellularis (Gi), the gigantocellularis pars alpha (GiA) (see text), raphe obscurus, (ROb), and arcuate nucleus (not shown). From the atlas of Paxinos et al (33) used with permission from Elsevier. (C) Computer-based plot of the topography of serotonergic neurons (as determined by the PH8 anybody) in a representative section of the rostral medulla in a postneonatal infant in the SIDS age range (37). Each circle represents a 5-HT neuron in the raphe midline (blue), extra raphe lateral to the midline (green), and ventral surface (red).
Initially Dahlstroem and Fuxe described 9 district clusters of nuclei, B1-9, extending from the caudal medulla through the pons and the midbrain to its rostral border in rats (34). In the caudal domain, the nuclei today in humans include the raphe obscurus, raphe pallidus, raphe magnus, gigantocellularis, gigantocellularis pars alpha, paragigantocellularis lateralis, and the intermediate reticular zone as delineated in the atlas of Paxinos and Huang (35) (Fig. 2). These are major nuclei that contain serotonergic and heterogeneous nonserotonergic neuronal cell bodies (21, 33, 35). Of note, the Paxinos and Huang atlas is 59 years newer than Olszewski and Baxter (1954) (36) and shows the medullary reticular formation parcellated into more subdivisions. Included in the caudal domain of Paxinos and Huang is the serotonergic nucleus, the intermediate reticular zone. In our work, this was a new subdivision that was not present in Olszewski and Baxter (Fig. 2; Table 1). Also, the original gigantocellularis of Olszewski and Baxter is now subdivided into the gigantocellularis and the gigantocellularis pars alpha (as per Paxinos and Huang [35]). The gigantocellularis pars alpha, according to this atlas, contains 5-HT-positive neurons that invade its ventral and lateral components (33). It should be highlighted that the serotonergic neurons form collections (nuclei) that flank the midline of the brainstem. A characteristic of primate species, including human, is a higher proportion of serotonergic neurons positioned laterally to the midline. The cytoarchitectonic boundaries of the serotonergic nuclei are poorly defined and vary among species and atlases. In preparation for analysis of the serotonergic medullary system, in our studies in SIDS (37), we mapped the distribution of 5-HT neurons with immunocytochemistry and the PH8 antibody from 7–8 gestational weeks to 6 postnatal months. Clusters of 5-HT neurons were very prominent off the midline, including in the gigantocellularis. They are also present in the far lateral reticular formation in the paragigantocellularis lateralis (Fig. 2). In our work, beginning over 2 decades ago, we used the label “gigantocellularis” to include serotonergic neurons lateral to the midline adjacent to the paragigantocellularis lateralis (Fig. 2). Going forward after the publication of this review article, we recommend the use of Paxinos and Huang as the atlas for this work, and the gigantocellularis be considered as a serotonergic nucleus in the caudal domain of the human brainstem based on our mapping studies (37). The dorsal raphe, median raphe, caudal linear nucleus, and B9 of the original Dahlstrom and Fuxe classification comprise the rostral 5-HT raphe domain in the pons and midbrain (34).
Acquiring generic 5-HT neuron identity from progenitor cells of the developing hindbrain involves turning on the expression of the transcription-factor-encoding genes, which include NKX2.2, Mash1/ASCL1, and Foxa2 (26, 38–40). This is then followed by expression of the transcription factor-encoding genes Pet1, Lmx1b, and Gata2/3 (26, 38, 39, 41–44). Sonic hedgehog, a signaling molecule secreted by nearby floor plate cells, initiates this cascade of cellular and molecular events which take the descendent postmitotic precursor cells to a state of 5-HT production, the hallmark of the 5-HT neuron (26, 45). Genetic tools, including intersectional genetic fate mapping and genome-wide RNA-Seq, now provide evidence for molecularly defined 5-HT subtypes with different molecular profiles, developmental origins, and migration pathways (20, 21, 26, 46). On the basis of these genetic approaches, evidence indicates that specialized 5-HT neuronal subtype identity may reflect, in part, different developmental origins within embryonic hindbrain segments or rhombomeres (26). Resident 5-HT progenitors and precursor cell subsets develop unique identities based on actions of rhombomere(r)-specific genetic programs (26). Rhombomeric lineage, in addition to anatomic domain and individual covariation of gene expression (including genes specific to other neurotransmitters such as glutamate, GABA, thyrotropin releasing hormone, and substance P) contribute to 5-HT subtypes that are molecularly and functional distinct, with different projection patterns (20, 47). As an example, in the mouse, the serotonergic neurons defined by their molecular identity as Tachykinin1-expressing Pet1 (Tac1-Pet1) neurons have been shown to blunt the ventilatory response in the respiratory CO2 chemoreflex, which normally increases ventilation in response to hypercapnic acidosis to pH and CO2 (47). Axons of this neuronal population project to respiratory brainstem nuclei involved in chemosensory motor processing (47). The Erg2-Pet1 subtype of 5-HT neurons, on the other hand, is specialized to modulate the respiratory CO2 chemoreflex via selective projections to respiratory chemosensory centers (48). Thus, there appear to be functionally specific neuron modules, defined by biophysical features, physiological outcome, and axonal projections. Moreover, these functional differences map onto developmental cell lineage—the originating rhombomere and gene expression code of the progenitor cell rather than the anatomical grouping of the mature neuron itself (B1-9) (48). A particular genetic defect or environmental insult may affect the functional capacity of one 5-HT neuron subtype but not others (48).
Outline of Autoradiographic Studies in SIDS
In the mid-1990s, we introduced the method of tissue receptor autoradiography into SIDS research (49) to investigate a likely brainstem neurochemical defect that required identification based upon a combination of neuroanatomy and neurochemistry. Receptor autoradiography involves the detection of receptor binding in anatomic tissue sections with the use of pharmacological methods, most notably the binding of radiolabeled ligands to neurotransmitter receptors (50). Using this method, the anatomic localization of receptors in a tissue structure is labeled and the binding of the ligand to the receptor is measured (Fig. 3). The binding measure reflects the receptor affinity for the radioligand and the number of receptors, which can be distinguished from each other by Scatchard analysis in tissue sections or homogenates (51). We considered tissue receptor autoradiography ideal for analyzing the SIDS problem because it detects a binding (neurochemical) abnormality in tissue sections that look histopathologically normal by light microscopy, or that demonstrate subtle, nonspecific gliosis, as in SIDS. The method also allows quantitative comparisons among cases and controls using radioactive standards for normalization of ligand binding across different cases and experiments. Altered levels of binding could reflect changes in receptor binding affinity and/or receptor number. An increase or decrease in binding can represent a compensatory response to too little or too much neurotransmitter at the synapse, respectively (52, 53). The abnormalities in binding affinity or number could be due, on the other hand, to a genetic deficiency in the receptor or an abnormality in its protein structure or expression. While further studies are needed to tell where the molecular defect lies—in receptor affinity or number—the defect highlights an abnormality in a specific neurotransmitter, an important first step in SIDS where microscopic tissue clues are lacking. In receptor-ligand autoradiography, the radiolabeled tissue section is used to generate an autoradiogram with receptor binding levels in fentomoles/mg. Using computer graphics, binding levels are quantitated in brainstem nuclei, as defined using conventional cell stains such as hematoxylin-and-eosin, and standard brainstem atlases (35, 36) (Fig. 3). We used Paxinos et al (35) for the identification of the intermediate reticular zone because it was not labeled separately in Olszewski and Baxter. To ensure we measured at standardized sites, we divided the brainstem into 15 levels with the use of the atlas of Olszewski and Baxter and reproducible landmarks (36). For example, the area postrema is used as a marker of the level of the caudal solitary nucleus. It is important to standardize the measurements to comparable levels of a nucleus because, as we showed in 3-D computer reconstructions, binding is typically not uniform through its rostrocaudal length, but rather heterogeneous at different levels, reflecting the three-dimensional topography of the different afferents and efferents entering and exiting at different levels (54, 55).
FIGURE 3.

Example of autoradiographic image in human postmortem tissue. (A) Autoradiogram of mid-medulla. The arcuate nucleus is located along the ventral surface. (B) Color-coded image of specific activity (fmol/mg tissue) in the same section as in A. The white arrows point to the arcuate nucleus. (C) Anatomic boundaries of nuclei. Arc, arcuate nucleus; CEN, nucleus centralis; CUL, nucleus cuneatus lateralis; DMX, dorsal motor nucleus of the vagus; DO, dorsal accessory olive; ICP, inferior cerebellar peduncle; MO, medial accessory olive; nXII, hypoglossal nucleus; SPvc, nucleus of the trigeminal nerve, pars caudalis; nTS, nucleus tractus solitarii; PO, principal inferior olive; PYR, pyramid, SUB, nucleus subtrigeminalis; VE, vestibular nucleus. Reprinted from (57) with permission from Oxford University Press.
In our analyses, SIDS cases were defined using the NICHD definition described above (1). Acute control cases were defined as infants less than one year whose clinical history, autopsy, and/or death scene investigation established a known cause of a death of acute duration, that is, less than one month (49). Chronic hypoxia controls were infants less than one year of age in which the disorder persisted longer than a month and was complicated by systemic hypoxia (49). This group was studied to control for the effects of hypoxia (and nonspecific metabolic factors of chronic illness) on receptor binding. We reasoned that if the medullary 5-HT abnormalities in SIDS were secondary to hypoxia-ischemia, neurochemical findings in SIDS cases would be identical to those in infants dying with clinically documented chronic hypoxia-ischemia. We found, however, that SIDS infants do not demonstrate the same 5-HT abnormalities (see below) as infants dying with known chronic oxygenation disorders, for example, cyanotic congenital heart disease (56), suggesting that the primary mechanisms underlying the 5-HT abnormalities are not necessarily directly related to chronic hypoxia. Intermittent hypoxia, however, cannot be excluded.
Initial Receptor Brainstem Tissue Autoradiographic Findings
We first applied radioligands to receptor subtypes in alternate sections of the same SIDS and control cases in one cohort of cases termed Dataset 1. These radioligands included the serotonergic, muscarinic cholinergic, α2-adrenergic, kainate, GABAA, and nicotinic cholinergic receptors (Table 2). Each of these receptors had comparable rationales for being involved in SIDS based on what was known at that time about their roles in homeostatic and cardiopulmonary function, and links to epidemiological risk factors in SIDS. Although the overall experiment in Dataset 1 seemed like a “fishing expedition,” we agree with the then NICHD program manager of SIDS research, the late Dr. Charlotte Catz, that we were fishing in “enriched waters” (Catz C personal communication) and the approach was thus valid as hypothesis generating with follow-up studies needed. The analysis was performed in a blinded fashion to the age, sex, and cause of death of each case and control.
TABLE 2.
Receptor Binding in Dataset 1
| Year/Reference | n = x (SIDS); n = y (Controls) | Finding | |
|---|---|---|---|
| Muscarinic; 3H-QNB | (49) | X = 44; y = 14 | Decreased binding in arcuate nucleus in SIDS |
| Kainate; 3H-kainate | (57) | X = 47; y = 15 | Decreased binding in arcuate nucleus in SIDS |
| Opioid: 3H-naloxone | (58) | X = 45; y = 14 | No difference |
| Nicotinic: 3H-nicotine | (59) | X = 42; y = 15 | Abnormal binding in selected mesopontine nuclei upon stratification of maternal smoking by history |
| Serotonergic receptors: 3H-LSD | (60) | X = 52; y = 15 | Core lesion in SIDS |
| Alpha adrenergic receptors; 3H-PAC | (61) | X = 10; y = 10 | No difference |
Abbreviations: 3H-QNB, 3H-quinuclidinyl benzilate; 3H-LSD, lysergic acid diethylamide; 3H-PAC, 3H-para-amino-clonidine.
In Dataset 1, there were no binding defects to mu opioid receptors using 3H-naloxone (58), or to α2-adrenerigic receptors using the radioligand [3H]para-aminoclonidine (61). There were, however, isolated binding deficits to the muscarinic cholinergic receptors using quinuclidinyl benzilate (3H-QNB) (49) and to the kainate receptors using 3H-kainate (57), both in the arcuate nucleus, a candidate region for the respiratory chemosensitive zones at the ventral medullary surface in experimental animals (31) responsive to hypercarbia and asphyxia. Given its role as a potential chemosensitive zone and emerging recognition of the risk for SIDS in the face-down sleep position with putative rebreathing of carbon dioxide and hypoxia, we recognized its potential significance. The studies that followed in the arcuate nucleus are discussed below. Of the neurotransmitters studied in Dataset 1 in SIDS (49, 57–61), 5-HT deficiencies proved to be the most extensive. In 2000 we published in Dataset 1 the first 5-HT receptor binding study in SIDS (60). This study showed deficiencies in serotonergic receptors in SIDS cases compared with acute and chronic controls, and in the medulla compared with the pons and midbrain (60). Subsequently we focused our attention on the serotonergic system and independently confirmed 5-HT abnormalities in 3 additional datasets (Datasets 2–4) with different nonoverlapping SIDS and controls (Tables 3 and 4).
TABLE 3.
Significant Serotonergic Abnormalities in SIDS Brainstem 1995–2001
| Dataset | Method | Findings |
|---|---|---|
| 1 (60) | Autoradiography | Abnormalities of 5-HT core lesion in rostral medulla |
| 2 (62) | Autoradiography | Abnormalities of 5-HT core lesion in rostral medulla |
| 3 (63) | Autoradiography | Abnormalities of 5-HT core lesion in rostral medulla |
| 3 (63) | Immunocytochemistry and quantitative cell counting | Increased number and density of immature 5-HT neurons in the raphe and extra-raphe 5-HT medullary system |
| 4 (56) | Autoradiography | Abnormalities of 5-HT core lesion in rostral medulla |
| 4 (56) | High pressure liquid chromatography | Decreased 5-HT in raphe obscurus and paragigantocellularis lateralis |
| 4 (56) | Western blotting | Decreased tryptophan hydroxylase 2 in raphe obscurus |
| 4 (64) | Autoradiography | GABAA abnormalities in 5-HT medullary system |
| 4 (65) | Proteomics | Deficiency of 14-3-3 involved in 5-HT metabolism |
TABLE 4.
The p Values for the Differences Between SIDS and Control Cases in the Nuclei of the Core Lesion in SIDS in the Reticular Formation of the Medulla Oblongata
| Data-set | SIDS/AC/CC (n) | Cohort | Arcuate n | Raphe Obs | LPGi | GC | IRt |
|---|---|---|---|---|---|---|---|
| 1(60) | 52/15/17 | Mixed | p = 0.0001* | p = 0.0001*,† | p = 0.003* | p = 0.022‡ | p = 0.048‡ |
| 2(62) | 23/6 | NP | p = 0.003* | NM | p = 0.043‡ | p = 0.01‡ | p = 0.026‡ |
| 3(63) | 16/7 | San Diego | p = 0.002* | p = 0.001* | p = 0.002* | p = 0.006* | p = 0.001* |
| 4(56) | 35/5 | San Diego | p = NS | p = 0.07‡ | p = 0.003‡ | p = 0.05‡ | p = 0.02‡ |
n, number of SIDS/acute controls (AC)/chronic controls (CC).
Decrease in age adjusted mean.
3 group p value.
Age versus diagnosis.
NM, not measured, cut in half; MIXED, cases from Massachusetts, Rhode lsland, San Diego Medical Examiner office; NP, Northern Plains; NS, nonsignificant; Arcuate n, arcuate nucleus; Raphe Obs, raphe obscurus; LPGi, paragigantocellularis lateralis; GC, gigantocellularis; IRt, intermediate reticular zone.
The Serotonergic System and the Ventral Medulla
When our work on the brainstem in SIDS began in the early 1990s, the anatomic site of the ventral chemoreceptive respiratory fields in the human was unknown, as was its relationship to the serotonergic system. Thus, we first undertook a comparative neuroanatomic study of the ventral surface of the cat and human infant (31). In 1990, we published homologous cell populations between the human infant and cat defined in the arcuate nucleus at the medullary surface (31). The homology was based upon cytoarchitecture and three-dimensional distribution in computer reconstructions (31). The human arcuate nucleus along the ventral surface had been considered historically as a precerebellar relay nucleus derived from downwardly displaced pontine neurons, and as rhombic lip derivatives via ventrolateral migrations (31). Yet, it contains neurons that are cytologically and spatially homologous to those in the respiratory chemosensitivity zones of the ventral medullary surface in the cat (31). The idea that the arcuate nucleus is involved in central chemosensitivity was strengthened by the independent case report of an infant with clinically recognized insensitivity to carbon dioxide, sudden death, and absence of the arcuate nucleus at autopsy (66). We also identified a SIDS infant with structural aplasia of the arcuate nucleus (67) in a computer reconstruction of the ventral medulla. Matturri et al (68) subsequently replicated arcuate nucleus hypoplasia in 56% of SIDS cases (n = 35) compared with controls.
DiI labeling studies in human midgestational fetal brains in our laboratory demonstrated connections between the arcuate nucleus and the caudal raphe in the midline of the medulla, suggesting a potential functional link between these 2 regions (69) (Fig. 4). Crystals of Dil, a lipophilic dye which labels cells and cell processes by lateral diffusion, were placed in the arcuate nucleus and labeled fibers and cell bodies in the medullary raphe and the external arcuate after plasma membrane diffusion over months (Fig 4). In the human infant medulla, scattered single fusiform 5-HT neurons extend ventrally from the raphe to line the median and ventral surface of the pyramids in the arcuate nucleus (Fig. 5) (37), forming a continuum of serotonergic raphe neurons ventrally. The arcuate nucleus is heterogeneous with predominately nonserotonergic neurons and a small subset of serotonergic neurons (31, 32). Past comparative studies by others suggest that the human arcuate nucleus is indeed homologous to the ventral raphe pallidus in nonhuman primates (37). The embryonic origin of the serotonergic arcuate neurons is presumably the same type as all other serotonergic neurons; that is, progenitor regions that flank the floor plate bilaterally. Utilizing the DiI track tracing method in the fetal human brainstem, we found that the connectivity of the arcuate nucleus is interrelated to the raphe obscurus and pallidus (69, 70), supporting the idea that the arcuate nucleus is a ventral extension of the caudal raphe system.
FIGURE 4.

Labeling of the caudal raphe from the arcuate nucleus. (A) Placement of DiI crystal in the arcuate nucleus (horizontal plane) with labeling of the caudal raphe, including the raphe pallidus (RPa) and the raphe obscurus (ROb). (B) Single DiI-labeled dorsoventral fiber in the ROb from the arcuate nucleus (×76). (C) Di-labeled fusiform cell body (arrow) (dorsal process and ventral process in the ROb) ×76. Reprinted from (69) with permission from Oxford University Press.
FIGURE 5.

Immunostained 5-HT neurons extend ventrally from the region of the raphe pallidus (midline) (A) along the ventral surface of the pyramids (B) in the human infant medulla (37). Abbreviations: pyr, pyramid; VMS, ventral medullary surface. A. 4×, scale bar = 200 μm. B. 10×, scale bar = 80 μm. Reprinted from (23) with permission from Elsevier.
On the basis of the findings of the DiI interconnections between the caudal raphe and arcuate nucleus, we hypothesized a priori that serotonergic receptor binding was decreased in the arcuate nucleus, as well in the interconnected raphe obscurus in SIDS cases compared with acute and chronic controls adjusted for postconceptional age (PCA). Using quantitative autoradiography, 3H-lysergic acid diethylamide (3H-LSD) binding to serotonergic receptors (5-HT1A-D and 5-HT2 subtypes) was measured blinded in 19 brainstem nuclei (60). At the time no radioligand was known to be specific for a single 5-HT receptor subtype. Cases were classified as SIDS (n = 52), acute controls (n = 15), and chronic cases with oxygenation disorders (n = 17) (60). Serotonergic binding was significantly lower in the SIDS cases compared with acute and chronic controls in the arcuate nucleus and the raphe obscurus, the latter located in both the rostral and caudal medullary reticular formation as postulated (Tables 3 and 4). Binding, however, was also significantly decreased in the inferior olive and in the paragigantocellularis lateralis, the latter nucleus in the rostral medullary reticular formation containing extra-raphe 5-HT neurons (60) (Tables 3 and 4). The gigantocellularis and intermediate reticular zone in Dataset 1 demonstrated the same pattern of binding, that is, an age versus diagnosis interaction showing that the binding decreased significantly in the SIDS cases but did not change with age in the controls (60) (Table 4).
The Delineation of a “Core Lesion” in the Rostral Reticular Formation of the Medulla in Datasets 1–4
After observing 5-HT binding deficits in Dataset 1 (see above), we subsequently examined 3 additional independent datasets of SIDS and control infants. In Dataset 2, we reproduced essentially the same serotonergic defects in site and pattern, and because of the robustness and reproducibility of the serotonergic binding defects, we decided to concentrate our time and resources on markers of the neurotransmission of 5-HT in the medulla (Tables 3 and 4). The analyses of the 3 subsequent datasets (Datasets 2–4) differed from the first and within themselves in the following ways: (i) Data set 2 was comprised of American Indians in the Northern Plains only. This study represented an analysis of this population alone because of its high rate of SIDS; the rate of SIDS among American Indian infants in the Northern Plains was almost 6 times higher than in U.S. white infants in the early 1990s, when tissue collection in Dataset 2 began (62); (ii) Dataset 2 did not have measurements in the raphe nuclei because the brainstem was cut in half for histological examination of one of the halves (62); (iii) Datasets 1 (60, 62) used the radioligand 3H-LSD to 5-HT1A-D and 5-HT2A receptors and Datasets 3 (63) and 4 (56) used the radioligand 3H-DPAT to 5-HT1A receptors (Tables 3 and 4). The recently discovered radioligand to the 5-HT1A receptor was used because it bound specifically to the 5-HT autoreceptor and in this way it labeled the nuclei with 5-HT neurons. (iv) Datasets 3 (63), and 4 (56) were nonoverlapping SIDS and controls cases from the San Diego medical examiner’s office only and were analyzed between 2003 and 2010; and (v) Sample size varied across all datasets (56, 60, 62, 63).
Despite the differences in SIDS and control cohorts, sample sizes, exact binding agents, and secular time frames, we continued to find statistically significant differences in either the binding pattern of age versus diagnosis interaction or absolute binding in the paragigantocellularis lateralis, gigantocellularis, and intermediate reticular zone in all datasets; significant differences in binding in the arcuate nucleus in 3 of the 4 datasets, and in binding differences in the raphe obscurus in 3 of the 3 datasets (Dataset 2 without raphe obscurus binding, see Tables 3 and 4). We would argue this group of nuclei with 5-HT binding abnormalities, all containing 5-HT cell bodies in the medullary rostral reticular formation, including the raphe obscurus, is the site of the core lesion in the SIDS group (Fig. 2). Most importantly, the nuclei in the core lesion are the major loci of the 5-HT neuronal cell bodies in the rostral medulla and comprise the reticular formation of the upper medulla, with each nucleus putatively interconnected to the other, though the connectivity is poorly understood. As noted, the serotonergic neurons in these 4 subnuclei are implicated in governing the vital functions of breathing, chemoreception, blood pressure, heart rate, temperature control, visceral reflexes, muscle tone, sleep, and arousal, all important for homeostasis (Table 1). We hypothesize that they form a neuronal network where pathology in the serotonergic neurons causes lethal dysfunction in arousal, cardiorespiratory control and autoresuscitation and represent the, or at least one of the, sites of vulnerability under stress in the triple risk model. It is unknown, however, which rhombomeres give origin to particular nuclei in the human brainstem, or to where the axons of the putative modules project. Thus, this speculation that the affected nuclei in the rostral reticular formation are rhombomere-specific forms the foundation for a hypothesis for future research. Other nuclei showing a difference between 5-HT1A binding between SIDS and control showed the difference in no greater than 2 datasets. The nuclei did not contain serotonergic neurons, for example, hypoglossal nucleus, nucleus of the solitary tract, and dorsal motor of the vagus.
Major knowledge to be gained is the detailed connectivity of these 5-HT neurons and the neural network they putatively form and integrate with one another in homeostatic modulation. What is known about the function and connectivity of these 5-HT neurons working together is based mainly upon extrapolations from animal models. The human homologue of the preBotzinger complex, a central rhythm generator, is apparently more caudal than the core nuclei in SIDS (71); the raphe obscurus of the core nuclei projects directly to the preBotzinger complex to assert 5-HT influence upon this key respiratory-related site via 5-HT receptors (72). The preBotzinger complex does not itself contain 5-HT cell bodies (71).
Dataset 4 did not confirm the lowered 5-HT binding in the arcuate nucleus seen in Datasets 1–3 (Table 4). It could be a false negative, or the arcuate nucleus might not be the most important of these 5-HT structures in the causation of SIDS. The consistent finding of significant interactions between age and diagnosis warrants mention. Although interpretation is impossible without longitudinal study, the reduced binding in older SIDS cases may reflect a progressive decrease with age in those infants with the “SIDS abnormality” (56). Alternatively, it may reflect the possibility that infants with a stronger abnormality take longer to outgrow the risk period for SIDS and may be at risk to die at older ages (56).
Parameters of 5-HT Neurotransmission Examined in Datasets 3 and 4 Other than by Receptor Autoradiography
In Datasets 3 and 4, we tested additional measures of 5-HT neurotransmission (Table 3). In Dataset 3, there was an ∼2-fold increase in 5-HT neuronal density in the raphe obscurus, with mainly morphologically immature serotonergic neurons contributing to the increase (63). This finding supports a developmental origin of the serotonergic defect, with a defect in cell number and cellular differentiation. In Dataset 4 a partial (26%) reduction of 5-HT (p < 0.05) as determined with high pressure liquid chromatography (HPLC) was found in the raphe obscurus and paragigantocellularis (Table 3) (56). Also, there was partial (33%) (p = 0.030) reduction of tryptophan hydroxylase 2 (TPH2), the key biosynthetic enzyme for 5-HT in the raphe obscurus by western blotting (56). We did not find abnormalities in levels of norepinephrine, dopamine, or their metabolites by HPLC in Dataset 4 (56), indicating that the brainstem pathology in SIDS does not affect all neurotransmitters or regions, but is neurotransmitter- and region-specific.
In support of our work described above, there have been additional studies using immunocytochemical methods that have confirmed abnormalities in the medullary 5-HT system in SIDS. Machaalani et al (73) examined the 5-HT1A receptor by quantitative immunocytochemistry in the medulla of 67 SIDS and 25 controls and found significant decreases in 5-HT1A immunoreactivity in 6 medullary nuclei, including nuclei reported previously to be deficient in 5-HT1A. In a study of 31 SIDS and 25 controls, Ozawa et al (74) used 5-HT1A immunocytochemistry to show a decrease in 5-HT1A immunoreactivity in the dorsal motor nucleus of the vagus and the nucleus of the solitary tract. In addition to 5-HT1A, Ozawa also used immunocytochemistry to examine 5-HT2A and found decreased 5-HT2A immunostaining in the same medullary nuclei (74). Interestingly, Ozawa found a significant increase in immunostaining for 5-HT1A and 5-HT2A in the periaqueductal gray matter of the midbrain in SIDS infants (74). In support of our 5-HT neuronal density data, Bright et al (75) reported in an Australian cohort of cases that there was a significantly higher 5-HT neuron number and density in SIDS cases (n = 41) compared with controls (n = 25) with significantly altered 5-HT neuronal morphology in the SIDS cases. These independent studies further support the hypothesis that complex abnormalities in the medulla contribute to the pathogenesis of a subset of SIDS.
GABA Related Abnormalities in the Medullary 5-HT System in SIDS Cases
GABA neurons in the medulla oblongata help regulate homeostasis, in part through interactions with the medullary serotonergic system (76). We tested the hypothesis that markers of GABAA receptors are altered in the medullary 5-HT system in SIDS cases compared with controls in Dataset 4 of the serotonergic binding studies (64). Using tissue receptor autoradiography with the radioligand 3H-GABA, we found 25%–52% reductions in GABAA receptor binding density in 7 of 10 key nuclei sampled of the medullary 5-HT system in the SIDS cases (PCA = 51.7 ± 8.3, n = 28) versus age-adjusted controls (PCA = 55.3 ± 13.5, n = 8) (p ≤ 0.04) (64). These nuclei included the raphe obscurus, paragigantocellularis lateralis, gigantocellularis, and intermediate reticular zone (but not the arcuate nucleus)—components of the core rostral medullary reticular formation. By Western blotting there was a 46.2% reduction in GABAAα3 subunit levels in the gigantocellularis of SIDS cases (PCA = 53.9 ± 8.4, n = 24) versus controls (PCA = 55.3 ± 8.3, n = 8) (56.8% of standard in SIDS cases vs 99.35% in controls; p = 0.026) (64). These data suggest that medullary GABAA receptors are abnormal in SIDS infants and that SIDS is a complex disorder of a 5-HT homeostatic network in the medulla that involves deficits of the GABAergic system (64). The question arises whether the GABAA receptor deficit in SIDS is secondary to the 5-HT deficits or vice-versa—or are the GABA and 5-HT defects separate and independent of each other, although still in the same SIDS cases? Substantial evidence suggests that 5-HT-related mechanisms help regulate GABA function, potentially involving G-coupled signal transduction pathways (76–79). The GABA and 5-HT defects in the SIDS medulla are likely to interact with each other to compound medullary dysfunction. The loss of GABAA receptor binding may lead to altered GABAergic modulation, including on 5-HT neurons which we have shown in the human infant medulla to express GABAAα3 receptor subunits (76), and highlights the point that combined 5-HT and GABAA receptor defects are present in the same SIDS cases in key medullary sites involved. These data underscore the likelihood that multiple neurotransmitters in >1 brainstem region alter homeostatic functions.
14-3-3 Deficiency in the Medulla in SIDS
The etiology of the TPH2 and 5-HT deficiency in SIDS is unknown. We tested the hypothesis that proteomics would uncover previously unrecognized abnormal levels of proteins related to TPH2 and 5-HT regulation in SIDS cases compared with controls, which could provide novel insight into the basis of their deficiency (65). We first performed a discovery proteomic analysis of the gigantocellularis in the same data set with deficiencies of TPH2 and 5-HT levels (64). Analysis in 6 pilot SIDS cases and 4 controls revealed a 42%–75% reduction in abundance in 5 of the 6 isoforms identified of the 14-3-3 signal transduction family (p < 0.07) (p value considered significant < 0.1 in the gigantocellularis, part of the core lesion site). 14-3-3 proteins are a family of homologous proteins that bind to target proteins and alter the conformation, stability, subcellular localization or activity of the binding partners (80). Within the brain, evidence suggests that 14-3-3 proteins regulate function within the synapse, including synaptic transmission and plasticity (80). Specific to 5-HT, the 14-3-3 protein affects 5-HT synthesis through direct binding to TPH2, increasing its stability and catalytic activity (81–83). To confirm these proteomic results in a larger dataset (38 SIDS, 11 controls), we applied Western blot analysis in the gigantocellularis and found 4/7 14-3-3 isoforms identified were significantly reduced in SIDS cases (p < 0.02), with a 43% reduction in all 14-3-3 isoforms combined (p < 0.001) (64). Given that the signal transduction 14-3-3 proteins are involved in the regulation of TPH2, we propose that the 14-3-3 deficits in the SIDS cases cause or contribute to the reduced TPH2 and 5-HT levels, potentially bringing us closer to the molecular defect in the medullary 5-HT system in sudden infant death (64). While less is known about the role of 14-3-3 proteins in GABA function and signaling, there is evidence suggesting that 14-3-3 plays a role in the phosphorylation of specific GABAA subunits (84). There is also evidence of a direct interaction of 14-3-3 proteins with GABA receptors, albeit GABAB receptors, and a role of this interaction in the maintenance of GABAB receptor complexes (85). Given the broad function of 14-3-3 proteins in signaling and neurotransmission, it is possible that the deficits in the family of 14-3-3 proteins in the medulla of SIDS infants provide a common basis for the 5-HT, GABA, and potentially other, yet undetermined, neurotransmitter abnormalities in SIDS infants.
The Serotonin Brainstem Hypothesis in SIDS Expanded Upon
The changes in 5-HT tissue parameters in Datasets 1-4, concentrated in the rostral medullary reticular formation (Table 3) led to the 5-HT brainstem hypothesis that we now hold. The 5-HT brainstem hypothesis states that a SIDS subset is due to 5-HT abnormalities in the rostral medullary 5-HT network (core lesion) that helps mediate protective respiratory and autonomic responses to homeostatic challenges during sleep, autoresuscitation, and/or transitions to arousal, leading to sleep-related sudden death when the infant meets an exogenous stressor. In considering what we call the core lesion in SIDS, the 5-HT cell bodies of the affected 5-HT nuclei are presumed to be interconnected to one another and form a neural network among the paragigantocellularis lateralis, gigantocellularis, intermediate reticular zone, caudal raphe, and arcuate nucleus. They are further postulated to be interconnected with the serotonergic ascending arousal system: The rostral reticular formation transmits its dysfunction to the median raphe and dorsal raphe of the ascending serotonergic arousal system and causes a failure of arousal to life threatening metabolic challenges such asphyxia or hypoxia, leading to death. Measurements of 5-HT1A receptor binding are needed in the median and dorsal raphe nuclei of the pons.
A Prospective Case Report of Cardiorespiratory Impairment and Serotonergic Abnormalities in a SIDS Infant
The medullary serotonergic system has a strong interface with the autonomic nervous system, and thus abnormalities within it could potentially lead to imbalances in sympathetic and parasympathetic tone (23). We reported the case of a full-term American Indian boy who died of SIDS at 2 postnatal weeks, and who had subtle respiratory and autonomic dysfunction measured prospectively on the second postnatal day (86). Cardiorespiratory assessment of heart rate variability suggested that the ratio of parasympathetic to sympathetic tone was higher than normal in active sleep and lower than normal in quiet sleep in this case (Fig. 6). At autopsy, arcuate nucleus hypoplasia and 5-HT receptor-binding abnormalities in the arcuate nucleus and other components of the medullary 5-HT system were found. This case suggests that medullary 5-HT system abnormalities may be detectable by such physiological tests before death. Replication of these findings in a large population may lead to the development of predictive cardiorespiratory assessment tools for future screening to identify infants with medullary 5-HT abnormalities and SIDS risk.
FIGURE 6.

Decreased 5-HT receptor binding with 3H-LSD in the arcuate nucleus (encircled in red oval) in a SIDS case who died at 2 postnatal weeks compared with 2 age-related controls (Control A and Control B). This infant was studied prospectively with different parameters of central cardiorespiratory control at 2 days of life. The bottom 3 panels show the histograms and fitted normal curves for respiratory rate in active sleep, difference of heart rate between active and quiet sleep, and sustained change in beat-to-beat heart rate variability. The y-axis in each of the 3 graphs is the proportion of the subjects with a particular parameter. This infant had values for each of these parameters (red dot) that were outside the 10th percentile of values. Abbreviations: NTS, nucleus of the solitary tract; ION, inferior olivary nucleus; Arc, arcuate nucleus; br/min, breaths per minute; AS, active sleep; HR, heart rate; QS, quiet sleep; HRV, heart rate variability; %, percent. Reprinted from (23) with permission from Elsevier.
A Working Model of the Postulated Pathophysiology of 5-HT Brainstem-Mediated Mechanisms of Sudden Death in SIDS; Evidence from Animal Studies
The method of tissue autoradiography helped us to identify a system of 5-HT neurons involved as a core lesion in SIDS, supplemented with the methods of HPLC, Western blotting, and immunocytochemistry with cell counting and computer graphics. There may well be other parts of the core lesion, particularly in the rostral domain of the brainstem (midbrain and pons). It is not clear yet how the serotonergic dysfunction leads to sudden death, but we present here a working model, based on animal experiments designed to provide 5-HT mechanisms in light of our findings in the human brainstem.
We posit that in SIDS several protective mechanisms fail simultaneously or in series (87). This model emphasizes the triggering event as one that generates asphyxia (combined hypoxia and hypercapnia) or hypoxia, and is based upon death scene observations (see above), and analyses of cardiopulmonary monitor tracings of SIDS infants (87–92). First, a life-threatening event causes severe asphyxia or hypoxia, brain hypoperfusion, or both. Such hypoxic or asphyxial events include rebreathing of exhaled gases in the facedown position, in soft bedding, or in the face-covered (supine) position, reflex apnea originating from the laryngeal chemoreflex (93, 94), obstructive apnea, or nasal obstruction in the facedown position, and apnea, bradycardia, and/or asystole from an unwitnessed seizure. Second, the vulnerable infant does not wake up (arouse) and turn his or her head in response to hypoxia or asphyxia, resulting in rebreathing or an inability to recover from apnea. The 5-HT system is involved in arousal (see above) and 5-HT defective parameters in the caudal medullary reticular formation and its influence on ascending arousal systems could contribute to this defect in arousal. Third, progressive asphyxia leads to a loss of consciousness, a hypoxic coma, a step that is hypothesized to occur on the basis of extrapolations from studies in animals that indicate the rapid development of coma when a critical level of the partial pressure of arterial oxygen is reached (∼10 mm Hg) or when hypoperfusion results in extreme brain tissue hypoxia (87). Fourth, extreme bradycardia and hypoxic gasping ensue, changes that are evident in the terminal-event recordings in infants who were being monitored at home at the time of death from SIDS (90–92). In severe hypoxia or ischemia, normal breathing fails and is rescued by the autoresuscitation response. Gasping is the process that increases the volume of air in the lungs after an apnea, followed by oxygen transport to the heart, increased cardiac output, and finally brain perfusion and re-oxygenation. Normally eupneic breathing follows. Each gasp is associated normally with increased sympathetic activity and increased heart rate. This response of autoresuscitation is thought to fail in SIDS.
One implicated component of recovery from apneas in response to severe hypoxia is the serotonergic raphe system of Pet1-expressing (largely serotonergic) neurons (95). Serotonergic transmission has been implicated in the autoresuscitation response in several animal models (95–100). Acute in vivo perturbation of Pet1-neuron activity with molecular biological techniques (via triggering cell-autonomously the synthetic inhibitory receptor hM4Di) results in altered postnatal age-dependent cardiorespiratory outcome and recovery from asphyxia-induced apneas (95), and greatly diminished apnea survival. Respiratory more than heart rate recovery is impaired, uncoupling their normal linear relationship (95). These rodent data build mechanistic plausibility for the SIDS-associated findings of postmortem brainstem raphe neuron 5-HT abnormalities, for example, partial deficiency of 5-HT (56), and partial deficiency of Tph2 (56) in SIDS infants and sudden death. The results suggest that Pet1-neuron activity is required neonatally for managing baseline heart rate and ventilation and normal survival rates in response to apneas resulting from severe hypoxia (95). Post hoc analyses identified specific respiratory features associated with autoresuscitation failure. The results of these experiments indicate that Pet1 neurons in the medullary raphe experimentally are critical for proper cardiorespiratory coupling in P8 mouse pups (95).
Potential Causes(s) of 5-HT Brainstem Abnormalities in SIDS
SIDS is likely not due to a single cause but rather multiple causes, leading to a common neurochemical phenotype and/or clinical presentation, that is, sleep-related sudden death in a critical developmental period. A role for maternal and pregnancy factors in the pathogenesis of brain abnormalities in epidemiological studies is supported by pathological studies of the placentas in newborns who subsequently die of SIDS. These placental abnormalities include maternal vascular underperfusion, particularly in the setting of maternal cigarette smoking, and acute abruption (101–103). Together, the association of maternal and placental factors with SIDS suggests that the vulnerability in at least some infants originates during gestation, and then is expressed as death in a critical postnatal period of development. Certain of these factors have been linked to fetal hypoxia, for example, maternal vascular underperfusion of the placenta, maternal anemia and cigarette smoking, and have thereby linked such fetal hypoxic-induced injury to impaired brain or other organ development in the pathogenesis of postnatal death. The association of placental pathology with SIDS suggests that a “suboptimal intrauterine environment” is involved (101–103). It is also postulated that prenatal exposure to alcohol and/or nicotine and/or other toxins (in cigarette smoke) may be directly toxic to the developing fetal brain particularly to the central 5-HT system, as demonstrated in animal models (104–106).
Conclusions
Our brainstem research answers a major fundamental question in SIDS, that is, where is the lesion? It indicates reproducible serotonergic defects in the rostral reticular formation of the medulla in SIDS infants compared with autopsied controls. Our point of entry into the serotonergic system was the ventral medulla and the interrelationship of the arcuate nucleus with the caudal raphe system (Fig. 4). By defining a core lesion in SIDS in specific serotonergic subnuclei, we hypothesize about the origin of the defective 5-HT cell bodies. Considering the rhombomeric origins of 5-HT subtypes (20, 24, 48), we must consider the possibility that the core lesion in SIDS develops embryonically in a rhombomere-specific fashion with abnormalities specific to rhombomeres that form the medullary 5-HT network. Here we speculate that the brainstem defect originates prenatally and not postnatally. In this way, the 5-HT defects in the brainstem in SIDS infant integrate with serotonergic experimental biology that support a model in which 5-HT neuron modules exist within gestation. To confirm this model in the SIDS brainstem, the human 5-HT cell progenitor/rhombomere origins of the caudal raphe, intermediate reticular zone, paragigantocellularis lateralis, and gigantocellularis and their projections must be discovered through extrapolation from animal models and human embryology.
The observation that multiple nonacute 5-HT abnormalities of the brainstem have been reported in SIDS infants answers another important question: Are SIDS infants “normal” infants who accidently die by suffocation? Rather, the brainstem research suggests that they have an underlying vulnerability in the serotonergic alarm system that makes them susceptible to sudden death, that is, the triple risk model. While ongoing research may uncover additional sites in the core lesion, for example, rostral raphe domain in the pons and/or midbrain, the research to date points to a starting site to build upon, and an approach to discovering that site with quantitative research tools.
Yet, the cause(s) of the serotonergic defects is unknown, and constitutes the major direction for future research. It is also unknown if certain infants “survive” the vulnerable period and subsequently develop disorders of 5-HT or related neurotransmitter systems in later life, or alternatively, suffer from the static effects of sublethal hypoxic-ischemic damage to vulnerable forebrain and brainstem regions. It is also unknown how the many risk factors, for example, infection, impinge upon the serotonergic system to increase risk. The scenario that SIDS is not hyper acute suggests that it perhaps could be prevented if we had the correct biomarkers to identify living infants at risk and the correct drug or behavioral means to intervene. Ultimately, the means to identify living infants at risk will likely involve neural biomarkers and brain-related treatment strategies to prevent sudden death in the critical period. We have provided emerging evidence that elevated serum 5-HT is a biomarker for SIDS in ∼30% of cases for unknown reasons, a finding that is being actively pursued by us now (107).
Many important questions continue to challenge SIDS investigators, for example, how does an infant who has survived the stressful passage from placental gas exchange to air breathing at birth succumb months later? Do 5-HT and GABA defects potentiate each other to cause neural dysfunction? What makes the rostral medullary reticular injury vulnerable to pathology in SIDS infants? How is the infant apparently “normal” during the waking hours, with no clue of imminent death? How does sleep unmask the dysfunction and lead to sudden death? Could the brainstem, the key regulator of vital functions during sleep, play the deciding role, or are interactions with forebrain regions critical as well?
Even though many questions persist, we believe that it is an overstatement today to refer to SIDS as a “mystery.” Noam Chomsky divided mankind’s ignorance into “problems and mysteries.” To quote him: “When we face a problem, we may not know its solution, but we have insight, increasing knowledge, and an inkling of what we are looking for. When we face a mystery, however, we can only stare in wonder and bewilderment, not knowing what an explanation would even look like” (108). The last 2 decades of SIDS brainstem research have given us some insight, we believe, as to “an inkling of what we are looking for,” and rational hypotheses to pursue with scientific foundations through the combined efforts of epidemiology, physiology, genetics, omics technology, neuropathology, and basic science.
ACKNOWLEDGMENTS
The authors thank all the committed and wonderful colleagues who worked in the laboratory on this research over the years beginning with the first collaborator, Dr. James J. Filiano. We also thank Dr. Henry Krous, Elisabeth Hass, MPH, and the medical examiners of the San Diego Medical Office, San Diego, CA, for dedicated and steadfast participation in case attainment and adjudication. We also thank Drs. Benjamin Okaty, Susan M. Dymecki, Richard D. Goldstein, Dawna D. Armstrong, and Eugene E. Nattie for critical reading of the manuscript in preparation. We dedicate this research to the autopsied infants of this study and to their families.
This work was funded by the National Institute of Child Health and Development (R37-HD20991, PO1-HD036379, and P30-HD18655 [Developmental Disabilities Research Center, Children’s Hospital Boston]), CJ Foundation for SIDS, Cooper Trewin Brighter Days Fund, River’s Gift, Evelyn Deborah Barrett Fellowship for SIDS Research, Marley J. Cherella Fellowship for SIDS Research, First Candle/SIDS Alliance, CJ Murphy Foundation for Solving the Puzzle of SIDS, Barrett Tallman Memorial Fund, Florida SIDS Alliance, Jacob Neil Boger Foundation for SIDS, Jason Lutz SIDS Foundation, Three Butterflies Foundation, Bennett C. Endres Fellowship, The Family of Lyla Heffernan, and Robert’s Program on Sudden Unexpected Death in Pediatrics.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Willinger M, James LS, Catz C.. Defining the sudden infant death syndrome (SIDS): Deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr Pathol 1991;11:677–84 [DOI] [PubMed] [Google Scholar]
- 2.Prevention CDCA. Sudden unexpected infant death and sudden Infant death syndrome: Data and statistics. https://www.cdc.gov/sids/data.htm2018
- 3. Goldstein RD, Trachtenberg FL, Sens MA, et al. Overall postneonatal mortality and rates of SIDS. Pediatrics 2016;137:1–10 [DOI] [PubMed] [Google Scholar]
- 4. Richerson GB, Aston-Jones G, Saper CB.. The modulatory functions of the brain stem In: Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ, eds. Principals of Neural Science. 5th ed New York, NY: The McGraw-Hill Companies, Inc; 2012:1038–55 [Google Scholar]
- 5. Brown RE, Basheer R, McKenna JT, et al. Control of sleep and wakefulness. Physiol Rev 2012;92:1087–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Talman WT, Lin LH.. Sudden death following selective neuronal lesions in the rat nucleus tractus solitarii. Auton Neurosci 2013;175:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaster JH. Unexpected sudden death after lateral medullary infarction. J Neurol Neurosurg Psychiatry 2001;70:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rhodes RH, Wightman HR.. Nucleus of the tractus solitarius metastasis: Relationship to respiratory arrest? Can J Neurol Sci 2000;27:328–32 [DOI] [PubMed] [Google Scholar]
- 9. Franco P, Szliwowski H, Dramaix M, et al. Decreased autonomic responses to obstructive sleep events in future victims of sudden infant death syndrome. Pediatr Res 1999;46:33–9 [DOI] [PubMed] [Google Scholar]
- 10. Schechtman VL, Lee MY, Wilson AJ, et al. Dynamics of respiratory patterning in normal infants and infants who subsequently died of the sudden infant death syndrome. Pediatr Res 1996;40:571–7 [DOI] [PubMed] [Google Scholar]
- 11. Kahn A, Groswasser J, Rebuffat E, et al. Sleep and cardiorespiratory characteristics of infant victims of sudden death: A prospective case-control study. Sleep 1992;15:287–92 [DOI] [PubMed] [Google Scholar]
- 12. Southall DP. Can we predict or prevent sudden unexpected deaths during infancy? Pediatrician 1988;15:183–90 [PubMed] [Google Scholar]
- 13. Naeye RL. Brain-stem and adrenal abnormalities in the sudden-infant-death syndrome. Am J Clin Pathol 1976;66:526–30 [DOI] [PubMed] [Google Scholar]
- 14. Kinney HC, Burger PC, Harrell FE Jr, et al. ‘Reactive gliosis' in the medulla oblongata of victims of the sudden infant death syndrome. Pediatrics 1983;72:181–7. [PubMed] [Google Scholar]
- 15. Becker LE, Takashima S.. Chronic hypoventilation and development of brain stem gliosis. Neuropediatrics 1985;16:19–23 [DOI] [PubMed] [Google Scholar]
- 16. Takashima S, Armstrong D, Becker L, et al. Cerebral hypoperfusion in the sudden infant death syndrome? Brainstem gliosis and vasculature. Ann Neurol 1978;4:257–62 [DOI] [PubMed] [Google Scholar]
- 17. Kinney HC, Brody BA, Finkelstein DM, et al. Delayed central nervous system myelination in the sudden infant death syndrome. J Neuropathol Exp Neurol 1991;50:29–48 [DOI] [PubMed] [Google Scholar]
- 18. Takashima S, Armstrong D, Becker LE, et al. Cerebral white matter lesions in sudden infant death syndrome. Pediatrics 1978;62:155–9 [PubMed] [Google Scholar]
- 19. Filiano JJ, Kinney HC.. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: The triple-risk model. Biol Neonate 1994;65:194–7 [DOI] [PubMed] [Google Scholar]
- 20. Okaty BW, Freret ME, Rood BD, et al. Multi-scale molecular deconstruction of the serotonin neuron system. Neuron 2015;88:774–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hornung JP. The human raphe nuclei and the serotonergic system. J Chem Neuroanat 2003;26:331–43 [DOI] [PubMed] [Google Scholar]
- 22. Azmitia EC. Serotonin neurons, neuroplasticity, and homeostasis of neural tissue. Neuropsychopharmacology 1999;21:33S–45S [DOI] [PubMed] [Google Scholar]
- 23. Kinney HC, Broadbelt KG, Haynes RL, et al. The serotonergic anatomy of the developing human medulla oblongata: Implications for pediatric disorders of homeostasis. J Chem Neuroanat 2011;41:182–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bang SJ, Jensen P, Dymecki SM, et al. Projections and interconnections of genetically defined serotonin neurons in mice. Eur J Neurosci 2012;35:85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alonso A, Merchan P, Sandoval JE, et al. Development of the serotonergic cells in murine raphe nuclei and their relations with rhombomeric domains. Brain Struct Funct 2013;218:1229–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jensen P, Farago AF, Awatramani RB, et al. Redefining the serotonergic system by genetic lineage. Nat Neurosci 2008;11:417–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okaty BW, Commons KG, Dymecki SM.. Embracing diversity in the serotonin neuronal system. Nat Rev Neurosci 2019;20:397–424 [DOI] [PubMed] [Google Scholar]
- 28. Kinney HC, Richerson GB, Dymecki SM, et al. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol Mech Dis 2009;4:517–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Darnall RA, Harris MB, Gill WH, et al. Inhibition of serotonergic neurons in the nucleus paragigantocellularis lateralis fragments sleep and decreases rapid eye movement sleep in the piglet: Implications for sudden infant death syndrome. J Neurosci 2005;25:8322–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X, Pfaff DW, Calderon DP, et al. Development of electrophysiological properties of nucleus gigantocellularis neurons correlated with increased CNS arousal. Dev Neurosci 2016;38:295–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Filiano JJ, Choi JC, Kinney HC.. Candidate cell populations for respiratory chemosensitive fields in the human infant medulla. J Comp Neurol 1990;293:448–65 [DOI] [PubMed] [Google Scholar]
- 32. Paterson DS, Thompson EG, Kinney HC.. Serotonergic and glutamatergic neurons at the ventral medullary surface of the human infant: Observations relevant to central chemosensitivity in early human life. Auton Neurosci 2006;124:112–24 [DOI] [PubMed] [Google Scholar]
- 33. Paxinos G, Haung X, Sengul G, et al. Organization of brainstem nuclei In: Mai J, Paxinos G, eds. The Human Nervous System. 3rd ed London, UK: Academic Press; 2012: 260–327 [Google Scholar]
- 34. Dahlstroem A, Fuxe K.. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand Suppl 1964;Suppl. 232:1–55 [PubMed] [Google Scholar]
- 35. Paxinos G, Huang X.. Atlas of the Human Brain Stem. Cambridge, MA: Academic Press; 1995 [Google Scholar]
- 36. Olszewski J, Baxter D.. Cytoarchitechture of the Human Brain Stem. 2nd ed Basel, Switzerland: S Karger; 1954 [Google Scholar]
- 37. Kinney HC, Belliveau RA, Trachtenberg FL, et al. The development of the medullary serotonergic system in early human life. Auton Neurosci 2007;132:81–102 [DOI] [PubMed] [Google Scholar]
- 38. Cheng L, Chen CL, Luo P, et al. Lmx1b, Pet-1, and Nkx2.2 coordinately specify serotonergic neurotransmitter phenotype. J Neurosci 2003;23:9961–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pattyn A, Simplicio N, van Doorninck JH, et al. Ascl1/Mash1 is required for the development of central serotonergic neurons. Nat Neurosci 2004;7:589–95 [DOI] [PubMed] [Google Scholar]
- 40. Nefzger CM, Haynes JM, Pouton CW.. Directed expression of Gata2, Mash1, and Foxa2 synergize to induce the serotonergic neuron phenotype during in vitro differentiation of embryonic stem cells. Stem Cells 2011;29:928–39 [DOI] [PubMed] [Google Scholar]
- 41. Hendricks T, Francis N, Fyodorov D, et al. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci 1999;19:10348–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding YQ, Marklund U, Yuan W, et al. Lmx1b is essential for the development of serotonergic neurons. Nat Neurosci 2003;6:933–8 [DOI] [PubMed] [Google Scholar]
- 43. Zhao ZQ, Scott M, Chiechio S, et al. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci 2006;26:12781–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Craven SE, Lim KC, Ye W, et al. Gata2 specifies serotonergic neurons downstream of sonic hedgehog. Development 2004;131:1165–73 [DOI] [PubMed] [Google Scholar]
- 45. Ye W, Shimamura K, Rubenstein JL, et al. FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell 1998;93:755–66 [DOI] [PubMed] [Google Scholar]
- 46. Wylie CJ, Hendricks TJ, Zhang B, et al. Distinct transcriptomes define rostral and caudal serotonin neurons. J Neurosci 2010;30:670–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hennessy ML, Corcoran AE, Brust RD, et al. Activity of tachykinin1-expressing Pet1 raphe neurons modulates the respiratory chemoreflex. J Neurosci 2017;37:1807–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brust RD, Corcoran AE, Richerson GB, et al. Functional and developmental identification of a molecular subtype of brain serotonergic neuron specialized to regulate breathing dynamics. Cell Rep 2014;9:2152–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kinney HC, Filiano JJ, Sleeper LA, et al. Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 1995;269:1446–50 [DOI] [PubMed] [Google Scholar]
- 50. Receptors in the Human Nervous System. Mendelsohn FAO, Paxinos G, eds. Cambridge, MA: Academic Press; 1991 [Google Scholar]
- 51. Smith RG, Sestili MA.. Methods for ligand-receptor assays in clinical chemistry. Clin Chem 1980;26:543–50 [PubMed] [Google Scholar]
- 52. Cahir M, Ardis T, Reynolds GP, et al. Acute and chronic tryptophan depletion differentially regulate central 5-HT1A and 5-HT 2A receptor binding in the rat. Psychopharmacology (Berl) 2007;190:497–506 [DOI] [PubMed] [Google Scholar]
- 53. Gutknecht L, Araragi N, Merker S, et al. Impacts of brain serotonin deficiency following Tph2 inactivation on development and raphe neuron serotonergic specification. PLoS One 2012;7:e43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kinney HC, Panigrahy A, Rava LA, et al. Three-dimensional distribution of [3H]quinuclidinyl benzilate binding to muscarinic cholinergic receptors in the developing human brainstem. J Comp Neurol 1995;362:350–67 [DOI] [PubMed] [Google Scholar]
- 55. Kinney HC, Ottoson CK, White WF.. Three-dimensional distribution of 3H-naloxone binding to opiate receptors in the human fetal and infant brainstem. J Comp Neurol 1990;291:55–78 [DOI] [PubMed] [Google Scholar]
- 56. Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA 2010;303:430–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Panigrahy A, Filiano JJ, Sleeper LA, et al. Decreased kainate receptor binding in the arcuate nucleus of the sudden infant death syndrome. J Neuropathol Exp Neur 1997;56:1253–61 [DOI] [PubMed] [Google Scholar]
- 58. Kinney HC, Filiano JJ, Assmann SF, et al. Tritiated-naloxone binding to brainstem opioid receptors in the sudden infant death syndrome. J Auton Nerv Syst 1998;69:156–63 [DOI] [PubMed] [Google Scholar]
- 59. Nachmanoff DB, Panigrahy A, Filiano JJ, et al. Brainstem 3H-nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol 1998;57:1018–25 [DOI] [PubMed] [Google Scholar]
- 60. Panigrahy A, Filiano J, Sleeper LA, et al. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol 2000;59:377–84 [DOI] [PubMed] [Google Scholar]
- 61. Mansouri J, Panigrahy A, Filiano JJ, et al. Alpha2 receptor binding in the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol 2001;60:141–6 [DOI] [PubMed] [Google Scholar]
- 62. Kinney HC, Randall LL, Sleeper LA, et al. Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol 2003;62:1178–91 [DOI] [PubMed] [Google Scholar]
- 63. Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA 2006;296:2124–32 [DOI] [PubMed] [Google Scholar]
- 64. Broadbelt KG, Paterson DS, Belliveau RA, et al. Decreased GABAA receptor binding in the medullary serotonergic system in the sudden infant death syndrome. J Neuropathol Exp Neurol 2011;70:799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Broadbelt KG, Rivera KD, Paterson DS, et al. Brainstem deficiency of the 14-3-3 regulator of serotonin synthesis: A proteomics analysis in the sudden infant death syndrome. Mole Cell Proteomics 2012;11:M111 009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Folgering H, Kuyper F, Kille JF.. Primary alveolar hypoventilation (Ondine's curse syndrome) in an infant without external arcuate nucleus. Case report . Bull Eur Physiopathol Respir 1979;15:659–65 [PubMed] [Google Scholar]
- 67. Filiano JJ, Kinney HC.. Arcuate nucleus hypoplasia in the sudden infant death syndrome. J Neuropathol Exp Neurol 1992;51:394–403 [DOI] [PubMed] [Google Scholar]
- 68. Matturri L, Biondo B, Suarez-Mier MP, et al. Brain stem lesions in the sudden infant death syndrome: Variability in the hypoplasia of the arcuate nucleus. Acta Neuropathol 2002;104:12–20 [DOI] [PubMed] [Google Scholar]
- 69. Zec N, Filiano JJ, Kinney HC.. Anatomic relationships of the human arcuate nucleus of the medulla: A DiI-labeling study. J Neuropathol Exp Neur 1997;56:509–22 [DOI] [PubMed] [Google Scholar]
- 70. Zec N, Kinney HC.. Anatomic relationships of the human nucleus paragigantocellularis lateralis: A DiI labeling study. Auton Neurosci 2001;89:110–24 [DOI] [PubMed] [Google Scholar]
- 71. Schwarzacher SW, Rub U, Deller T.. Neuroanatomical characteristics of the human pre-Botzinger complex and its involvement in neurodegenerative brainstem diseases. Brain 2011;134:24–35 [DOI] [PubMed] [Google Scholar]
- 72. Ptak K, Yamanishi T, Aungst J, et al. Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci 2009;29:3720–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Machaalani R, Say M, Waters KA.. Serotoninergic receptor 1A in the sudden infant death syndrome brainstem medulla and associations with clinical risk factors. Acta Neuropathol 2009;117:257–65 [DOI] [PubMed] [Google Scholar]
- 74. Ozawa Y, Okado N.. Alteration of serotonergic receptors in the brain stems of human patients with respiratory disorders. Neuropediatrics 2002;33:142–9 [DOI] [PubMed] [Google Scholar]
- 75. Bright FM, Byard RW, Vink R, et al. Medullary serotonin neuron abnormalities in an Australian cohort of sudden infant death syndrome. J Neuropathol Exp Neur 2017;76:864–73 [DOI] [PubMed] [Google Scholar]
- 76. Broadbelt KG, Paterson DS, Rivera KD, et al. Neuroanatomic relationships between the GABAergic and serotonergic systems in the developing human medulla. Auto Neurosci 2010;154:30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sibille E, Pavlides C, Benke D, et al. Genetic inactivation of the Serotonin(1A) receptor in mice results in downregulation of major GABA(A) receptor alpha subunits, reduction of GABA(A) receptor binding, and benzodiazepine-resistant anxiety. J Neurosci 2000;20:2758–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vinkers CH, Oosting RS, van Bogaert MJ, et al. Early-life blockade of 5-HT(1A) receptors alters adult anxiety behavior and benzodiazepine sensitivity. Biol Psychiatry 2010;67:309–16 [DOI] [PubMed] [Google Scholar]
- 79. Bruening S, Oh E, Hetzenauer A, et al. The anxiety-like phenotype of 5-HT receptor null mice is associated with genetic background-specific perturbations in the prefrontal cortex GABA-glutamate system. J Neurochem 2006;99:892–9 [DOI] [PubMed] [Google Scholar]
- 80. Zhang J, Zhou Y.. 14-3-3 proteins in glutamatergic synapses. Neural Plast 2018;2018:8407609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Winge I, McKinney JA, Ying M, et al. Activation and stabilization of human tryptophan hydroxylase 2 by phosphorylation and 14-3-3 binding. Biochem J 2008;410:195–204 [DOI] [PubMed] [Google Scholar]
- 82. Banik U, Wang GA, Wagner PD, et al. Interaction of phosphorylated tryptophan hydroxylase with 14-3-3 proteins. J Biol Chem 1997;272:26219–25 [DOI] [PubMed] [Google Scholar]
- 83. Gao K, Mason P.. Somatodendritic and axonal anatomy of intracellularly labeled serotonergic neurons in the rat medulla. J Comp Neurol 1997;389:309–28 [PubMed] [Google Scholar]
- 84. Qian Z, Micorescu M, Yakhnitsa V, et al. Climbing fiber activity reduces 14-3-3-theta regulated GABA(A) receptor phosphorylation in cerebellar Purkinje cells. Neuroscience 2012;201:34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Couve A, Kittler JT, Uren JM, et al. Association of GABA(B) receptors and members of the 14-3-3 family of signaling proteins. Mol Cell Neurosci 2001;17:317–28 [DOI] [PubMed] [Google Scholar]
- 86. Kinney HC, Myers MM, Belliveau RA, et al. Subtle autonomic and respiratory dysfunction in sudden infant death syndrome associated with serotonergic brainstem abnormalities: A case report. J Neuropathol Exp Neurol 2005;64:689–94 [DOI] [PubMed] [Google Scholar]
- 87. Kinney HC, Thach BT.. The sudden infant death syndrome. N Engl J Med 2009;361:795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pasquale-Styles MA, Tackitt PL, Schmidt CJ.. Infant death scene investigation and the assessment of potential risk factors for asphyxia: A review of 209 sudden unexpected infant deaths. J Forensic Sci 2007;52:924–9 [DOI] [PubMed] [Google Scholar]
- 89. Thach BT. Some aspects of clinical relevance in the maturation of respiratory control in infants. J Appl Physiol 2008;104:1828–34 [DOI] [PubMed] [Google Scholar]
- 90. Sridhar R, Thach BT, Kelly DH, et al. Characterization of successful and failed autoresuscitation in human infants, including those dying of SIDS. Pediatric Pulmonol 2003;36:113–22 [DOI] [PubMed] [Google Scholar]
- 91. Meny RG, Carroll JL, Carbone MT, et al. Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics 1994;93:44–9 [PubMed] [Google Scholar]
- 92. Poets CF. Apparent life-threatening events and sudden infant death on a monitor. Paediatr Respir Rev 2004;5(Suppl. A):S383–6 [DOI] [PubMed] [Google Scholar]
- 93. Van Der Velde L, Curran AK, Filiano JJ, et al. Prolongation of the laryngeal chemoreflex after inhibition of the rostral ventral medulla in piglets: A role in SIDS?. J Appl Physiol 2003;94:1883–95 [DOI] [PubMed] [Google Scholar]
- 94. Downing SE, Lee JC.. Laryngeal chemosensitivity: A possible mechanism for sudden infant death. Pediatrics 1975;55:640–9 [PubMed] [Google Scholar]
- 95. Dosumu-Johnson RT, Cocoran AE, Chang Y, et al. Acute perturbation of Pet1-neuron activity in neonatal mice impairs cardiorespiratory homeostatic recovery. eLife 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Barrett KT, Dosumu-Johnson RT, Daubenspeck JA, et al. Partial raphe dysfunction in neurotransmission is sufficient to increase mortality after anoxic exposures in mice at a critical period in postnatal development. J Neurosci 2016;36:3943–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Erickson JT, Sposato BC.. Autoresuscitation responses to hypoxia-induced apnea are delayed in newborn 5-HT-deficient Pet-1 homozygous mice. J Appl Physiol 2009;106:1785–92 [DOI] [PubMed] [Google Scholar]
- 98. Cummings KJ, Commons KG, Hewitt JC, et al. Failed heart rate recovery at a critical age in 5-HT-deficient mice exposed to episodic anoxia: Implications for SIDS. J Appl Physiol 2011;111:825–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cummings KJ, Hewitt JC, Li A, et al. Postnatal loss of brainstem serotonin neurones compromises the ability of neonatal rats to survive episodic severe hypoxia. J Physiol (Lond) 2011;589:5247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cummings KJ, Commons KG, Trachtenberg FL, et al. Caffeine improves the ability of serotonin-deficient (Pet-1-/-) mice to survive episodic asphyxia. Pediatr Res 2013;73:38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li DK, Wi S.. Maternal placental abnormality and the risk of sudden infant death syndrome. Am J Epidemiol 1999;149:608–11 [DOI] [PubMed] [Google Scholar]
- 102. Getahun D, Amre D, Rhoads GG, et al. Maternal and obstetric risk factors for sudden infant death syndrome in the United States. Obstet Gynecol 2004;103:646–52 [DOI] [PubMed] [Google Scholar]
- 103. Klonoff-Cohen HS, Srinivasan IP, Edelstein SL.. Prenatal and intrapartum events and sudden infant death syndrome. Paediatr Perinat Epidemiol 2002;16:82–9 [DOI] [PubMed] [Google Scholar]
- 104. Cerpa VJ, Aylwin MdL, Beltrán-Castillo S, et al. The Alteration of neonatal raphe neurons by prenatal-perinatal nicotine. meaning for sudden infant death syndrome. Am J Respir Cell Mol Biol 2015;53:489–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Duncan JR, Garland M, Myers MM, et al. Prenatal nicotine-exposure alters fetal autonomic activity and medullary neurotransmitter receptors: Implications for sudden infant death syndrome. J Appl Physiol 2009;107:1579–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nattie E, Kinney H.. Nicotine, serotonin, and sudden infant death syndrome. Am J Respir Crit Care Med 2002;166:1530–1 [DOI] [PubMed] [Google Scholar]
- 107. Haynes RL, Frelinger AL 3rd, Giles EK, et al. High serum serotonin in sudden infant death syndrome. Proc Natl Acad Sci USA 2017;114:7695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pinker S. How the Mind Works. New York: Norton & Company; 2009. [Google Scholar]