

Abstract
Understanding why some groups of organisms are more diverse than others is a central goal in macroevolution. Evolvability, or the intrinsic capacity of lineages for evolutionary change, is thought to influence disparities in species diversity across taxa. Over macroevolutionary time scales, clades that exhibit high evolvability are expected to have higher speciation rates. Cone snails (family: Conidae,  900 spp.) provide a unique opportunity to test this prediction because their toxin genes can be used to characterize differences in evolvability between clades. Cone snails are carnivorous, use prey-specific venom (conotoxins) to capture prey, and the genes that encode venom are known and diversify through gene duplication. Theory predicts that higher gene diversity confers a greater potential to generate novel phenotypes for specialization and adaptation. Therefore, if conotoxin gene diversity gives rise to varying levels of evolvability, conotoxin gene diversity should be coupled with macroevolutionary speciation rates. We applied exon capture techniques to recover phylogenetic markers and conotoxin loci across 314 species, the largest venom discovery effort in a single study. We paired a reconstructed timetree using 12 fossil calibrations with species-specific estimates of conotoxin gene diversity and used trait-dependent diversification methods to test the impact of evolvability on diversification patterns. Surprisingly, we did not detect any signal for the relationship between conotoxin gene diversity and speciation rates, suggesting that venom evolution may not be the rate-limiting factor controlling diversification dynamics in Conidae. Comparative analyses showed some signal for the impact of diet and larval dispersal strategy on diversification patterns, though detection of a signal depended on the dataset and the method. If our results remain true with increased taxonomic sampling in future studies, they suggest that the rapid evolution of conid venom may cause other factors to become more critical to diversification, such as ecological opportunity or traits that promote isolation among lineages.
900 spp.) provide a unique opportunity to test this prediction because their toxin genes can be used to characterize differences in evolvability between clades. Cone snails are carnivorous, use prey-specific venom (conotoxins) to capture prey, and the genes that encode venom are known and diversify through gene duplication. Theory predicts that higher gene diversity confers a greater potential to generate novel phenotypes for specialization and adaptation. Therefore, if conotoxin gene diversity gives rise to varying levels of evolvability, conotoxin gene diversity should be coupled with macroevolutionary speciation rates. We applied exon capture techniques to recover phylogenetic markers and conotoxin loci across 314 species, the largest venom discovery effort in a single study. We paired a reconstructed timetree using 12 fossil calibrations with species-specific estimates of conotoxin gene diversity and used trait-dependent diversification methods to test the impact of evolvability on diversification patterns. Surprisingly, we did not detect any signal for the relationship between conotoxin gene diversity and speciation rates, suggesting that venom evolution may not be the rate-limiting factor controlling diversification dynamics in Conidae. Comparative analyses showed some signal for the impact of diet and larval dispersal strategy on diversification patterns, though detection of a signal depended on the dataset and the method. If our results remain true with increased taxonomic sampling in future studies, they suggest that the rapid evolution of conid venom may cause other factors to become more critical to diversification, such as ecological opportunity or traits that promote isolation among lineages.
Keywords: Macroevolution, phylogenetics, venom evolution
Why are some taxa more diverse than others? Species richness and phenotypic diversity are not distributed evenly across the tree of life (Rabosky et al. 2013). For example, there exist over 10,000 species of birds, but their closest relatives (crocodiles and alligators) comprise only 23 species. Differences in evolvability, or the intrinsic capacity to adapt and diversify, is one reason commonly used to explain these disparities (Wagner and Altenberg 1996; Yang 2001; Jones et al. 2007; Pigliucci 2008; Losos 2010). Evolvability is thought to be determined by the underlying genetic architecture of organisms; the genomes of some taxa have a greater propensity to generate variation that may be adaptive in the future (Wagner and Altenberg 1996; Jones et al. 2007; Pigliucci 2008). For example, gene duplication increases evolvability because the copied gene is free from the selective pressures on the original gene (Crow and Wagner 2006). Mutation, selection, and drift can then act on the copied gene, facilitating the possibility of new phenotypes to arise. This shapes the extent to which taxa can diversify and exploit resources (Crow and Wagner 2006). Over long evolutionary time scales, clades that exhibit higher evolvability are predicted to have increased species richness and diversification rates (Yang 2001).
Despite the ubiquity of this concept in macroevolutionary theory, few studies explicitly test these predictions; this is possibly due to the difficulty of identifying genes responsible for phenotype (Hoekstra and Coyne 2007). Past studies that have attempted to test the impact of evolvability on diversification have produced mixed results (Santini et al. 2009; Soltis et al. 2009; Mayrose et al. 2011; Rabosky et al. 2013; Zhan et al. 2014; Tank et al. 2015; Malmstrøm et al. 2016). For example, whole genome duplication events, which are hypothesized to increase the genomic potential of organisms, have been documented to increase (Santini et al. 2009; Soltis et al. 2009; Tank et al. 2015), decrease (Mayrose et al. 2011), and have no impact (Zhan et al. 2014) on the long-term evolutionary success of clades. In another case, a positive correlation between evolvability and speciation rates was found to exist when measuring evolvability through morphological proxies (Rabosky et al. 2013). One limitation of past research on this hypothesis is the inability to tie genomic changes with ecological factors driving diversification patterns (Robertson et al. 2017). Although gene duplication and whole genome duplication events can increase the evolutionary capacity of organisms, genes that are ecologically relevant for adaptation may not be readily available for selection to drive divergence.
Here, we investigate the relationship between evolvability and diversification in cone snails (family, Conidae), a diverse group ( 900 spp.) of predatory marine gastropods (WoRMS Editorial Board 2019). These snails feed on either worms, mollusc, or fish by paralyzing their prey with a cocktail of venomous neurotoxins (conotoxins, Duda and Palumbi 1999). Each species’ venom repertoire is estimated to contain 50–200 conotoxins which target neuroreceptors and ion channels within their prey (Olivera et al. 1990). Conotoxin precursor peptides typically consist of three regions: the signal region that directs the protein down the secretory pathway, the prepro region that is cleaved as the protein matures, and the mature region that ultimately becomes the injected peptide (Robinson and Norton 2014). For some peptides, there exists a “post” region that is cleaved on the C-terminus end of the mature peptide (Robinson and Norton 2014). Conotoxins are grouped and classified into over 50 gene superfamilies (e.g., O1 superfamily, T superfamily, etc.) based on similarities in the signal sequence, as these are conserved within a gene superfamily across species (Robinson and Norton 2014). A minority of conotoxin gene superfamilies are classified based on their similarity to venom proteins found in other venomous taxa such as conkunitzins and conopressins (Robinson and Norton 2014).
900 spp.) of predatory marine gastropods (WoRMS Editorial Board 2019). These snails feed on either worms, mollusc, or fish by paralyzing their prey with a cocktail of venomous neurotoxins (conotoxins, Duda and Palumbi 1999). Each species’ venom repertoire is estimated to contain 50–200 conotoxins which target neuroreceptors and ion channels within their prey (Olivera et al. 1990). Conotoxin precursor peptides typically consist of three regions: the signal region that directs the protein down the secretory pathway, the prepro region that is cleaved as the protein matures, and the mature region that ultimately becomes the injected peptide (Robinson and Norton 2014). For some peptides, there exists a “post” region that is cleaved on the C-terminus end of the mature peptide (Robinson and Norton 2014). Conotoxins are grouped and classified into over 50 gene superfamilies (e.g., O1 superfamily, T superfamily, etc.) based on similarities in the signal sequence, as these are conserved within a gene superfamily across species (Robinson and Norton 2014). A minority of conotoxin gene superfamilies are classified based on their similarity to venom proteins found in other venomous taxa such as conkunitzins and conopressins (Robinson and Norton 2014).
Cone snails provide a unique opportunity to test predictions of evolvability and diversification for several reasons. First, cone snail species share an ecologically relevant trait: venom. Conids are globally distributed in tropical and subtropical regions, where  30 species can co-occur within the same habitat (Kohn 2001). It has been hypothesized that co-occurrence happens in some habitats because each conid species becomes prey-specialized through the evolution of prey-specific conotoxins (Duda and Palumbi 1999). Second, the genes that encode conotoxins are well-defined and diversify through gene duplication (Duda and Palumbi 2000; Kaas et al. 2010, 2012; Chang and Duda 2012). Diet specialization is thought to be enabled by the rapid evolution of the genes that underlie conotoxins—estimated rates of gene duplication and nonsynonymous substitutions rates for conotoxin genes are the highest across metazoans (Duda and Palumbi 2000; Chang and Duda 2012). Therefore, conotoxin genes provide a natural way to characterize differences in evolvability between clades.
30 species can co-occur within the same habitat (Kohn 2001). It has been hypothesized that co-occurrence happens in some habitats because each conid species becomes prey-specialized through the evolution of prey-specific conotoxins (Duda and Palumbi 1999). Second, the genes that encode conotoxins are well-defined and diversify through gene duplication (Duda and Palumbi 2000; Kaas et al. 2010, 2012; Chang and Duda 2012). Diet specialization is thought to be enabled by the rapid evolution of the genes that underlie conotoxins—estimated rates of gene duplication and nonsynonymous substitutions rates for conotoxin genes are the highest across metazoans (Duda and Palumbi 2000; Chang and Duda 2012). Therefore, conotoxin genes provide a natural way to characterize differences in evolvability between clades.
We employ a sequence capture technique previously used in cone snails (Phuong and Mahardika 2018) to recover phylogenetic markers and conotoxin genes from 314 described species. We use the phylogenetic markers to reconstruct a time-calibrated phylogeny and perform trait-dependent diversification analyses to test the impact of evolvability on diversification patterns. We predict that clades with a greater number of conotoxin gene copies should have higher speciation rates. In addition, we test other traits that may have an impact on diversification patterns, including diet and larval dispersal strategy.
Methods
Bait Design
We used a targeted sequencing approach to recover markers for phylogenetic inference and obtain an estimate of conotoxin gene diversity from conid species. For the phylogenetic markers, we identified loci using a previous conid targeted sequencing dataset (Phuong and Mahardika 2018) and the conid transcriptome data from Phuong et al. (2016). In the conid targeted sequencing dataset, the authors generated a phylogeny using 5883 loci across 32 species (Phuong and Mahardika 2018). For our sequencing experiment, we only retained loci that were  180 bp and were present in at least 26 out of 32 taxa with at least 10
180 bp and were present in at least 26 out of 32 taxa with at least 10 coverage. We chose to only include longer loci to increase confidence in identifying orthologous fragments in other conid species. To identify additional phylogenetic markers from the transcriptome data (Phuong et al. 2016), which consisted of venom duct transcriptomes from 12 species, we first identified reciprocal best blast hits between the assembled transcriptome and the Lottia gigantea protein reference (Simakov et al. 2013) using BLAST
coverage. We chose to only include longer loci to increase confidence in identifying orthologous fragments in other conid species. To identify additional phylogenetic markers from the transcriptome data (Phuong et al. 2016), which consisted of venom duct transcriptomes from 12 species, we first identified reciprocal best blast hits between the assembled transcriptome and the Lottia gigantea protein reference (Simakov et al. 2013) using BLAST v2.2.31 (evalue
v2.2.31 (evalue  1e-10). We also considered fragments that had their best hit to the protein reference, but to a nonoverlapping portion (
1e-10). We also considered fragments that had their best hit to the protein reference, but to a nonoverlapping portion ( 20% overlapping). Then, we mapped reads to these candidate phylogenetic markers using bowtie2 v2.2.7 (Langmead and Salzberg 2012) and removed duplicate reads using picard-tools v.2.1.1 (http://broadinstitute.github.io/picard). To fix assembly errors, we called single nucleotide polymorphisms (SNPs) using samtools v1.3 and bcftools v1.3 (Li et al. 2009). Then, we created alignments per locus from sequences from each species using MAFFT v7.222 (Katoh et al. 2005). To account for spurious alignments either due to potential paralogy or misalignment, we calculated uncorrected pairwise distances within each locus for all possible pairwise comparisons and removed sequences if the uncorrected pairwise distance was greater than the 90th percentile of pairwise distances across all loci for those pair of species. To identify potential markers for exon capture, we denoted exon boundaries by comparing the transcriptome sequences to the L. gigantea genome reference (Simakov et al. 2013), retaining exons
20% overlapping). Then, we mapped reads to these candidate phylogenetic markers using bowtie2 v2.2.7 (Langmead and Salzberg 2012) and removed duplicate reads using picard-tools v.2.1.1 (http://broadinstitute.github.io/picard). To fix assembly errors, we called single nucleotide polymorphisms (SNPs) using samtools v1.3 and bcftools v1.3 (Li et al. 2009). Then, we created alignments per locus from sequences from each species using MAFFT v7.222 (Katoh et al. 2005). To account for spurious alignments either due to potential paralogy or misalignment, we calculated uncorrected pairwise distances within each locus for all possible pairwise comparisons and removed sequences if the uncorrected pairwise distance was greater than the 90th percentile of pairwise distances across all loci for those pair of species. To identify potential markers for exon capture, we denoted exon boundaries by comparing the transcriptome sequences to the L. gigantea genome reference (Simakov et al. 2013), retaining exons  180 bp.
180 bp.
For all retained phylogenetic markers, we first generated an ancestral sequence using FastML v3.1 (Ashkenazy et al. 2012) between a Californiconus californicus sequence and another conid sequence that had the highest amount of overlap with the C. californicus sequence (we generated these ancestral sequences to decrease the genetic distances between the target sequence and the orthologous sequence from any conid species). Then, we filtered potential markers that had a GC content 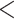 30% or
30% or 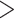 70% because extreme GC contents can reduce capture efficiency (Bi et al. 2012), removed loci that contained repeats identified through the RepeatMasker v4.0.6 web server (Smit et al. 2015), and performed a self-blast with the target sequences via blastn v2.2.31 (evalue
70% because extreme GC contents can reduce capture efficiency (Bi et al. 2012), removed loci that contained repeats identified through the RepeatMasker v4.0.6 web server (Smit et al. 2015), and performed a self-blast with the target sequences via blastn v2.2.31 (evalue  1e-10) and removed loci that did not blast to themselves with sequence identity
1e-10) and removed loci that did not blast to themselves with sequence identity 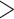 90%. The final set of target loci for phylogenetic inference included 1749 loci, with a total length of 470,435 bp.
90%. The final set of target loci for phylogenetic inference included 1749 loci, with a total length of 470,435 bp.
To recover conotoxin loci, we targeted sequences generated from both the previous targeted sequencing dataset (Phuong and Mahardika 2018) and the transcriptome dataset (Phuong et al. 2016). For conotoxin sequences discovered from the targeted sequencing dataset (Phuong and Mahardika 2018), we generated our target sequences by first trimming sequence to only retain the coding region and included 100 bp flanking the exon, merged sequences using cd-hit v4.6.4 (Li and Godzik 2006) at 95% sequence similarity to reduce redundancy among conotoxin loci, masked repeats using the RepeatMasker v4.0.6 web server (Smit et al. 2015), and retained loci 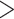 120 bp to ensure that the locus was longer than our desired bait sequence length. We concatenated all sequences below 120 bp to create a single, chimeric sequence for capture. The final set of target sequences from the previous targeted sequencing dataset consisted of 12,652 unique loci totaling 3,113,904 bp and a single concatenated sequence representing 351 merged loci with a total length of 37,936 bp. We also targeted conotoxin loci from the transcriptomes described in Phuong et al. (2016) to obtain conotoxin loci from gene superfamilies that were not targeted in Phuong and Mahardika (2018) or performed poorly. To finalize the set of conotoxin loci from the transcriptome data, we trimmed sequences from Phuong et al. (2016) to only include the coding region and 100 bp of the untranslated regions, merged sequences using cd-hit v4.6.4 (Li and Godzik 2006) at 97% sequence similarity to reduce redundancy among conotoxin loci, and masked repeats using the RepeatMasker v4.0.6 web server (Smit et al. 2015). We chose a 97% similarity threshold, which is 2% higher than the threshold for the phylogenetic markers, because we wanted to ensure that we recovered as many conotoxin sequences as possible. The higher similarity threshold would allow for a greater number of conotoxin sequences in the baits. This filtered dataset contained 395 conotoxin loci with a total length of 171,317 bp.
120 bp to ensure that the locus was longer than our desired bait sequence length. We concatenated all sequences below 120 bp to create a single, chimeric sequence for capture. The final set of target sequences from the previous targeted sequencing dataset consisted of 12,652 unique loci totaling 3,113,904 bp and a single concatenated sequence representing 351 merged loci with a total length of 37,936 bp. We also targeted conotoxin loci from the transcriptomes described in Phuong et al. (2016) to obtain conotoxin loci from gene superfamilies that were not targeted in Phuong and Mahardika (2018) or performed poorly. To finalize the set of conotoxin loci from the transcriptome data, we trimmed sequences from Phuong et al. (2016) to only include the coding region and 100 bp of the untranslated regions, merged sequences using cd-hit v4.6.4 (Li and Godzik 2006) at 97% sequence similarity to reduce redundancy among conotoxin loci, and masked repeats using the RepeatMasker v4.0.6 web server (Smit et al. 2015). We chose a 97% similarity threshold, which is 2% higher than the threshold for the phylogenetic markers, because we wanted to ensure that we recovered as many conotoxin sequences as possible. The higher similarity threshold would allow for a greater number of conotoxin sequences in the baits. This filtered dataset contained 395 conotoxin loci with a total length of 171,317 bp.
We submitted the following datasets to MYcroarray (Ann Arbor, Michigan, USA) for bait synthesis: (i) 1749 loci for phylogenetic inference, (ii) 12,652 conotoxin loci using data from Phuong and Mahardika (2018), (iii) a single concatenated sequence using data from Phuong and Mahardika (2018), and (iv) 395 additional conotoxin loci using transcriptome data from Phuong et al. (2016). We chose to synthesize a MYbaits-3 kit, which included 60,000 bait sequences to accommodate all the targeted loci. Because our aim was to recover sequences from species throughout Conidae, each bait sequence was 120 bp in length, which increases the efficiency of recovering divergent fragments. We used a 2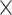 tiling density strategy (a new probe every 60 bp) across the sequences from datasets (1) and (2) and used a 4
tiling density strategy (a new probe every 60 bp) across the sequences from datasets (1) and (2) and used a 4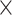 tiling density strategy (a new probe every 30 bp) across datasets (3) and (4). We chose to increase the tiling density for datasets (3) and (4) because the boundaries between exons were not denoted and we wanted to ensure effective capture of the conotoxin loci. The set of probe sequences are available on Dryad.
tiling density strategy (a new probe every 30 bp) across datasets (3) and (4). We chose to increase the tiling density for datasets (3) and (4) because the boundaries between exons were not denoted and we wanted to ensure effective capture of the conotoxin loci. The set of probe sequences are available on Dryad.
Genetic Samples, Library Preparation, Hybridization, and Sequencing
We performed the targeted sequencing experiment across 362 samples representing both described conid species and unique lineages/potential new species (Table S1 available on Dryad at http://dx.doi.org/10.5061/dryad.8d44d4q) identified during routine species verification using the mitochondrial locus CO1 (results not shown, Folmer et al. 1994). We focused our sampling efforts to represent as much of the extant diversity within Conidae as possible. Overall, we sampled all six known genera (Conus, Conasprella, Californiconus, Profundiconus, Lilliconus, and Pygmaeconus) and 53 out of 71 subgenera within Conidae (Table S1 available on Dryad). We also sequenced Bathyoma sp. as an outgroup based on a recent molecular phylogeny of the conoideans, a clade of gastropods that includes Conidae (Table S1 available on Dryad, Puillandre et al. 2011). We obtained these genetic samples from two field expeditions in Indonesia and Australia and from five museum collections (Table S1 available on Dryad).
We extracted DNA from tissue using the EZNA Mollusc DNA kit (Omega Bio-Tek, Doraville, GA, USA). There was slight variation in tissue preservation strategy among samples, with most tissues preserved directly in 95% ethanol (Table S1 available on Dryad). For 10 samples, tissue was not available but DNA was available from a previous extraction. For these samples, we ran the DNA through the EZNA Mollusc DNA kit to purify the DNA prior to library preparation. We extracted a minimum of 2000 ng per sample prior to library preparation, when possible. We sheared DNA using a Biorupter UCD-200 (Diagenode) when necessary and used a 1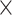 bead purification protocol to ensure that the DNA fragments per sample ranged from 250 to 600 bp, centered on
bead purification protocol to ensure that the DNA fragments per sample ranged from 250 to 600 bp, centered on 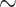 350 bp. We aimed to generate libraries with longer fragment sizes to ensure that we could recover exons containing the mature toxin region, which are often only recoverable because they are flanking conserved regions that are targeted by our bait design (Phuong and Mahardika 2018).
350 bp. We aimed to generate libraries with longer fragment sizes to ensure that we could recover exons containing the mature toxin region, which are often only recoverable because they are flanking conserved regions that are targeted by our bait design (Phuong and Mahardika 2018).
We prepared libraries following the Meyer and Kircher (2010) protocol with several modifications. We started library preparation with at least 2000 ng, rather than the 500 ng suggested by the protocol to increase downstream capture efficiency. Also, we performed 1 bead clean-up for all enzymatic reactions and generated dual-indexed libraries by incorporating adapters with unique 7 bp barcodes. We were able to reuse libraries for the 32 species sequenced in Phuong and Mahardika (2018) and incorporated new indexes for these samples.
bead clean-up for all enzymatic reactions and generated dual-indexed libraries by incorporating adapters with unique 7 bp barcodes. We were able to reuse libraries for the 32 species sequenced in Phuong and Mahardika (2018) and incorporated new indexes for these samples.
We generated equimolar pools of eight samples and hybridized probes with 2000ng of the pooled DNA for  24h. We substituted the adapter blocking oligonucleotides provided by MYcroarray with custom xGen blocking oligonucleotides (Integrated DNA technologies) because they have been shown to dramatically increase capture efficiency in targeted sequencing experiments (Portik et al. 2016). To increase library diversity, we performed three independent postcapture amplifications using 12 PCR cycles and pooled these products. We sequenced all samples across five Illumina HiSeq 4000 lanes with 100 bp paired-end reads. We multiplexed 80 samples per lane for the first four lanes and multiplexed the remaining 43 samples on the last lane. Sequencing was carried out at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley. We note that our third lane containing 80 samples was contaminated, with 65% of the reads belonging to corn DNA. We were able to resequence this entire lane, resulting in overall increased sequencing effort for samples belonging to our third lane.
24h. We substituted the adapter blocking oligonucleotides provided by MYcroarray with custom xGen blocking oligonucleotides (Integrated DNA technologies) because they have been shown to dramatically increase capture efficiency in targeted sequencing experiments (Portik et al. 2016). To increase library diversity, we performed three independent postcapture amplifications using 12 PCR cycles and pooled these products. We sequenced all samples across five Illumina HiSeq 4000 lanes with 100 bp paired-end reads. We multiplexed 80 samples per lane for the first four lanes and multiplexed the remaining 43 samples on the last lane. Sequencing was carried out at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley. We note that our third lane containing 80 samples was contaminated, with 65% of the reads belonging to corn DNA. We were able to resequence this entire lane, resulting in overall increased sequencing effort for samples belonging to our third lane.
Data Filtration and Initial Assembly
We filtered the raw data by first trimming reads using Trimmomatic v0.36 under several specific parameters. We used the ILLUMINACLIP option to trim adapters with a seed mismatch threshold of 2, a palindrome clip threshold of 40, and a simple clip threshold of 15. In addition, we performed quality trimming using the SLIDINGWINDOW option with a window size of 4 and a quality threshold of 20. Finally, we removed reads below 36 bp by setting the MINLEN option to 36, and we removed leading and trailing bases under a quality threshold of 15. We merged reads using FLASH v1.2.11 (Magoè and Salzberg 2011) with a min overlap parameter of 5, a max overlap parameter of 100, and a mismatch ratio of 0.05. We removed low-complexity reads using prinseq v0.20.4 (Schmieder and Edwards 2011) using the entropy method with a conservative threshold of 60. We assembled the filtered read data using SPAdes v3.8.1 using default parameters and reduced redundancy in the resultant assemblies with cap3 (Huang and Madan 1999) under default parameters and cd-hit v4.6 (Li and Godzik 2006, sequence identity threshold  99%).
99%).
Phylogenetic Data Processing and Filtering
To associate assembled contigs with the target sequences for phylogenetic inference, we used blastn v2.2.31 (word size  11, evalue
11, evalue  1e-10). For the set of target sequences that originated from the transcriptome dataset, we redefined exon/intron boundaries using EXONERATE v2.2.0 (Slater and Birney 2005) using the est2genome model because we found that several predicted exons actually consisted of several smaller exons. For each sample, we mapped reads using bowtie2 (very sensitive local and no discordant options enabled) to a reference that contained only sequences associated with the targeted phylogenetic markers. We marked duplicates using picard-tools v2.0.1 and masked all regions below 4
1e-10). For the set of target sequences that originated from the transcriptome dataset, we redefined exon/intron boundaries using EXONERATE v2.2.0 (Slater and Birney 2005) using the est2genome model because we found that several predicted exons actually consisted of several smaller exons. For each sample, we mapped reads using bowtie2 (very sensitive local and no discordant options enabled) to a reference that contained only sequences associated with the targeted phylogenetic markers. We marked duplicates using picard-tools v2.0.1 and masked all regions below 4 coverage and removed the entire sequence if more than 30% of the sequence was below 4
coverage and removed the entire sequence if more than 30% of the sequence was below 4 coverage. We called SNPs using samtools v1.3 and bcftools v1.3 and estimated average heterozygosity across all contigs within a sample. We removed sequences if a contig had a heterozygosity value greater than two standard deviations (SDs) away from the mean.
coverage. We called SNPs using samtools v1.3 and bcftools v1.3 and estimated average heterozygosity across all contigs within a sample. We removed sequences if a contig had a heterozygosity value greater than two standard deviations (SDs) away from the mean.
Conotoxin Assembly, Processing, and Filtering
Commonly used assembly programs are known to poorly reconstruct all copies of multilocus gene families (Lavergne et al. 2015; Phuong et al. 2016). To address this issue, we followed the conotoxin assembly workflow outlined in Phuong and Mahardika (2018). In brief, we first mapped reads back to our assembled contigs using the “very sensitive local” and no discordant’ options in bowtie2. Then, we identified conotoxins within our dataset by using blastn v2.2.31 (word size  11, evalue
11, evalue  1e-10) to associate our assembled contigs (from SPAdes) with conotoxins we targeted in the bait design. We generated a set of unique conotoxin “seed sequences” (a short stretch [
1e-10) to associate our assembled contigs (from SPAdes) with conotoxins we targeted in the bait design. We generated a set of unique conotoxin “seed sequences” (a short stretch [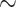 100 bp] of conotoxin-blasted sequence) using a combination of the pysam module (https://github.com/pysam-developers/pysam), cd-hit v4.6 (percent identity
100 bp] of conotoxin-blasted sequence) using a combination of the pysam module (https://github.com/pysam-developers/pysam), cd-hit v4.6 (percent identity  98%), cap3 (overlap percent identity cutoff
98%), cap3 (overlap percent identity cutoff  99%), blastn v2.2.31 (word size
99%), blastn v2.2.31 (word size  11, evalue
11, evalue 1e-10), and Tandem Repeats Finder v4.09 (Benson 1999, minscore
1e-10), and Tandem Repeats Finder v4.09 (Benson 1999, minscore  12, maxperiod
12, maxperiod  2). We mapped reads to these seed sequences using bowtie2 v2.2.6 (very sensitive local and no discordant options enabled) and built out the conotoxin sequences using the PRICE v1.2 algorithm, which uses an iterative mapping and extension strategy to build out contigs from initial seed sequences (Ruby et al. 2013). We ran PRICE on each seed sequence at five minimum percentage identity (MPI) values (90%, 92%, 94%, 96%, 98%) with a minimum overlap length value of 40 and a threshold value of 20 for scaling overlap for contig-edge assemblies. A reassembled sequence was retained if it shared 90% identity with the original seed sequence and we reduced redundancy by only retaining the longest sequence per seed sequence out of the five MPI assembly iterations. This approach is described in further detail in Phuong and Mahardika (2018). We note that the final conotoxin sequences per sample consisted of exon fragments, where each sequence represents a single conotoxin exon flanked by any adjacent noncoding region.
2). We mapped reads to these seed sequences using bowtie2 v2.2.6 (very sensitive local and no discordant options enabled) and built out the conotoxin sequences using the PRICE v1.2 algorithm, which uses an iterative mapping and extension strategy to build out contigs from initial seed sequences (Ruby et al. 2013). We ran PRICE on each seed sequence at five minimum percentage identity (MPI) values (90%, 92%, 94%, 96%, 98%) with a minimum overlap length value of 40 and a threshold value of 20 for scaling overlap for contig-edge assemblies. A reassembled sequence was retained if it shared 90% identity with the original seed sequence and we reduced redundancy by only retaining the longest sequence per seed sequence out of the five MPI assembly iterations. This approach is described in further detail in Phuong and Mahardika (2018). We note that the final conotoxin sequences per sample consisted of exon fragments, where each sequence represents a single conotoxin exon flanked by any adjacent noncoding region.
We updated our conotoxin reference database because we targeted additional conotoxin transcripts from Phuong et al. (2016). We used blastn v2.2.31 (word size  11, evalue
11, evalue  1e-10) and EXONERATE v2.2.0 to define exon/intron boundaries for these additional conotoxin transcripts and added them to our conotoxin reference database. The final conotoxin reference database consisted of conotoxin sequences with the coding regions denoted and gene superfamily annotated. We also annotated the conotoxin sequences for functional region (e.g., signal, pre, mature, post) using blastn v2.2.31 (word size
1e-10) and EXONERATE v2.2.0 to define exon/intron boundaries for these additional conotoxin transcripts and added them to our conotoxin reference database. The final conotoxin reference database consisted of conotoxin sequences with the coding regions denoted and gene superfamily annotated. We also annotated the conotoxin sequences for functional region (e.g., signal, pre, mature, post) using blastn v2.2.31 (word size  11, evalue
11, evalue  1e-10) with a conotoxin reference database that was previously categorized by functional region (Phuong and Mahardika 2018).
1e-10) with a conotoxin reference database that was previously categorized by functional region (Phuong and Mahardika 2018).
With the final conotoxin reference database, we performed blastn v2.2.31 (word size  11, evalue
11, evalue  1e-10) searches between the conotoxin reference and every sample’s re-assembled conotoxin sequences. We retained sequences if they could align across the entire coding region of the reference sequence. We estimated the coding region for each retained sequence by aligning the query sequence with the reference conotoxin using MAFFT v7.222 and denoting the coding region as the region of overlap with the exon in the reference conotoxin. We fixed misassemblies by mapping reads with bowtie2 (very sensitive local and no discordant options enabled, score min
1e-10) searches between the conotoxin reference and every sample’s re-assembled conotoxin sequences. We retained sequences if they could align across the entire coding region of the reference sequence. We estimated the coding region for each retained sequence by aligning the query sequence with the reference conotoxin using MAFFT v7.222 and denoting the coding region as the region of overlap with the exon in the reference conotoxin. We fixed misassemblies by mapping reads with bowtie2 (very sensitive local and no discordant options enabled, score min  L, 70, 1) back to each conotoxin assembly and marked duplicates using picard-tools v2.0.1. We masked regions below 5
L, 70, 1) back to each conotoxin assembly and marked duplicates using picard-tools v2.0.1. We masked regions below 5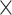 coverage and discarded sequences if coverage was below 5
coverage and discarded sequences if coverage was below 5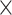 across the entire predicted coding region. To generate the final set of conotoxin sequences per sample, we merged sequences using cd-hit v4.6.4 (percent identity
across the entire predicted coding region. To generate the final set of conotoxin sequences per sample, we merged sequences using cd-hit v4.6.4 (percent identity  98%, use local sequence identity, alignment coverage of longer sequence
98%, use local sequence identity, alignment coverage of longer sequence  10%, alignment coverage of short sequence
10%, alignment coverage of short sequence  50%).
50%).
Targeted Sequencing Experiment Evaluation
We generated several statistics to evaluate the overall efficiency of the capture experiment. We calculated the % reads mapped to our targets by mapping reads to a reference containing all targets (both phylogenetic markers and conotoxin sequences) using bowtie2 v2.2.7 (very sensitive local and no discordant options enabled, score min  L, 70, 1). We quantified duplication by calculating % duplicates that were identified with picard-tools. Finally, we also assessed coverage by calculating average coverage across the phylogenetic markers and conotoxin sequences. We also evaluated the effect of tissue quality (measured by the maximum fragment length of the extracted DNA sample via gel electrophoresis) and genus (only on Conus, Profundiconus and Conasprella, the three genera with more than two samples included in this study) on these capture efficiency metrics using an analysis of variance (ANOVA). To assess the effectiveness of conotoxin sequence recovery, we compared our capture results with conotoxin diversity estimates from Phuong and Mahardika (2018) and calculated the average change in those estimates.
L, 70, 1). We quantified duplication by calculating % duplicates that were identified with picard-tools. Finally, we also assessed coverage by calculating average coverage across the phylogenetic markers and conotoxin sequences. We also evaluated the effect of tissue quality (measured by the maximum fragment length of the extracted DNA sample via gel electrophoresis) and genus (only on Conus, Profundiconus and Conasprella, the three genera with more than two samples included in this study) on these capture efficiency metrics using an analysis of variance (ANOVA). To assess the effectiveness of conotoxin sequence recovery, we compared our capture results with conotoxin diversity estimates from Phuong and Mahardika (2018) and calculated the average change in those estimates.
Phylogenetic Inference
In addition to the 362 cone snail samples that we sequenced in this study, we obtained sequences from 10 other species (Table S1 available on Dryad). For two of these species, we used data from another targeted sequencing study (Abdelkrim et al. 2018). We used blastn (word size  11, evalue
11, evalue  1e-10) to identify loci that were present in our phylogenetic marker reference. These sequences were filtered under conditions similar to the filtering strategy applied to the phylogenetic markers in this study. For the other eight species, we used data from venom duct transcriptomes provided by H. Safavi-Hemami et al. (Table S1 available on Dryad). With these transcriptomes, we trimmed data using trimmomatic v0.36 and merged reads using flash using parameters previously described above. We assembled each transcriptome using Trinity v2.1.1 (Grabherr et al. 2011), and reduced redundancy in these transcriptomes with cap3 and cd-hit (percent identity
1e-10) to identify loci that were present in our phylogenetic marker reference. These sequences were filtered under conditions similar to the filtering strategy applied to the phylogenetic markers in this study. For the other eight species, we used data from venom duct transcriptomes provided by H. Safavi-Hemami et al. (Table S1 available on Dryad). With these transcriptomes, we trimmed data using trimmomatic v0.36 and merged reads using flash using parameters previously described above. We assembled each transcriptome using Trinity v2.1.1 (Grabherr et al. 2011), and reduced redundancy in these transcriptomes with cap3 and cd-hit (percent identity  99%). We used blastn (word size
99%). We used blastn (word size  11, evalue
11, evalue 1e-10) to associate contigs with the phylogenetic markers present in our dataset. We used bowtie2 v2.2.7 (very sensitive local and no discordant enabled), samtools v1.3, and bcftools 1.3 to map reads and call SNPs. We removed sequences if they were below 4
1e-10) to associate contigs with the phylogenetic markers present in our dataset. We used bowtie2 v2.2.7 (very sensitive local and no discordant enabled), samtools v1.3, and bcftools 1.3 to map reads and call SNPs. We removed sequences if they were below 4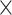 coverage for
coverage for  30% of the sequence and masked bases if they were below 4
30% of the sequence and masked bases if they were below 4 coverage. We also removed sequences if they had a heterozygosity value two SDs away from the mean heterozygosity within a sample. We used to MAFFT v7.222 to align loci across a total of 373 samples.
coverage. We also removed sequences if they had a heterozygosity value two SDs away from the mean heterozygosity within a sample. We used to MAFFT v7.222 to align loci across a total of 373 samples.
We inferred phylogenies under both maximum likelihood (Stamatakis 2006) and coalescent-based methods (Mirarab and Warnow 2015). We used RAxML v8.2.9 (Stamatakis 2006) to generate a maximum likelihood phylogeny using a concatenated alignment under a GTRGAMMA model of sequence evolution and estimated nodal support using 100 rapid bootstrap replicates of the data. We generated the coalescent-based phylogeny using ASTRAL-II v5.5.9 (Mirarab and Warnow 2015) with individual locus trees generated in RAxML v8.2.9 under a GTRGAMMA model of sequence evolution. We estimated local posterior probabilities as a measure of branch support (Sayyari and Mirarab 2017). Due to the underperformance of the capture experiment, we ran both phylogenetic analyses with loci that had 80% of the taxa, 50% of the taxa, and 20% of the taxa. For each iteration, we removed taxa that had 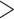 90% missing data.
90% missing data.
Time Calibration
We estimated divergence times using a Bayesian approach with MCMCTree implemented in PAML v4.9g (Yang 2007). Given the size of our alignments, we first estimated branch lengths using baseml and then estimated divergence times using Markov chain Monte Carlo (MCMC). We used a HKY85 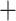
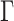 substitution model as this was the most complex model of sequence evolution available in MCMCTree and used an independent rates clock model. We left all other settings on default. We performed two independent runs of the analysis and checked for convergence among the runs. To account for uncertainty in branching order in our phylogeny, we executed dating analyses across all trees generated from RAxML.
substitution model as this was the most complex model of sequence evolution available in MCMCTree and used an independent rates clock model. We left all other settings on default. We performed two independent runs of the analysis and checked for convergence among the runs. To account for uncertainty in branching order in our phylogeny, we executed dating analyses across all trees generated from RAxML.
For time calibration, we applied a maximum constraint of 55 myr at the root of Conidae, which corresponds with the first confident appearance of Conidae in the fossil record (Kohn 1990). We assigned 12 additional fossils (Supplementary Table S2 and Fig. S1 available on Dryad, Hendricks 2009a, 2015, 2018) to nodes throughout the phylogeny as minimum age constraints. We note that all constraints within MCMCTree are treated as soft constraints, meaning that there is a probability that the bounds set in the analyses can be violated if the data supports alternative calibrations (Yang 2007). Further information on fossil placement on nodes can be found in the Supplementary Material. A recent paper showed that the number of species in Lautoconus may be overestimated (Abalde et al. 2017). To account for potential artificial inflation in the species richness of this clade, we artificially removed half the unique species in Lautoconus from our dataset and ran all dating analyses and downstream diversification analyses on this secondary dataset.
Characterizing Diversification Patterns
To visualize lineage accumulation patterns, we generated a log-lineage through time plot using the R package APE (Analyses of Phylogenetics and Evolution, Paradis et al. 2018). We estimated diversification rates and identified rate shifts using BAMM (Bayesian Analysis of Macroevolutionary Mixtures, Rabosky 2014), which uses reversible jump Markov chain Mone Carlo to explore potential lineage diversification models. To account for nonrandomness in species sampling across conid genera, we applied generic-specific sampling fractions. Using the number of valid conid names on WoRMS as estimates of total species diversity in each genus (WoRMS Editorial Board 2019), we applied a sampling fraction of 32.1% to Profundiconus, 50% to Lilliconus, 100% to Californiconus, 16.7% to Pygmaeconus, 28% to Conasprella, and 33.7% to Conus. We ran BAMM for 100,000,000 generations and assessed convergence by calculating ESS (Effective Sample Size) values. We analyzed and visualized results using the R package BAMMtools (Rabosky et al. 2014).
Trait Dependent Diversification
We tested for the impact of evolvability (measured as conotoxin gene diversity) on diversification patterns using two trait-dependent diversification methods, focusing on the genus Conus. We focused our hypothesis testing on Conus because conotoxin diversity is well-characterized in this group (Phuong et al. 2016) and the sequence capture approach used in this study likely represents uniform sampling in conotoxin gene diversity across the genus. This is in contrast to other genera in Conidae, such as Conasprella or Profundiconus, where low conotoxin diversity values are likely the result of poor knowledge of the venom repertoire of these genera (Supplementary Fig. S2 available on Dryad)
First, we used BiSSE (binary state speciation and extinction, Maddison et al. 2007) implemented in the R package diversitree (FitzJohn 2012), which employs a maximum likelihood approach to estimate the impact of a binary trait on speciation, extinction, and transition rates between character states. We coded the conotoxin gene diversity data as “low” or “high” across several thresholds (i.e., 250, 300, 350, 400, 500, 550, or 600 estimated conotoxin genes per species) and compared BiSSE models where speciation rates were allowed to vary or remain equal between traits. We applied a sampling fraction of 33.7%, taking the maximum number of Conus species to be the number of valid names on WoRMS (World Register of Marine Species, WoRMS Editorial Board 2019). We determined the best-fitting model using Akaike information criterion (AIC). Second, we used FiSSE (Fast, intuitive State-dependent Speciation-Extinction analysis), a nonparametric statistical test that assesses the effects of a binary character on lineage diversification rates (Rabosky and Goldberg 2017a). We followed the same coding strategy as in the BiSSE analyses to convert conotoxin gene diversity counts to binary character states. Finally, we used STRAPP (Structured Rate Permutations on Phylogenies, Rabosky and Huang 2016) implemented in the R package BAMMtools (Rabosky et al. 2014). STRAPP is a semiparametric approach that tests for trait-dependent diversification by comparing a test statistic with a null distribution generated by permutations of speciation rates across the tips of the phylogeny (Rabosky and Huang 2016). We generated the empirical correlation (method  Spearman’s rank correlation) between speciation rates and conotoxin gene diversity and compared this test statistic with the null distribution of correlations generated by permutations of evolutionary rates across the tree. We performed a two-tailed test with the alternative hypothesis that there is a correlation between speciation rates and total conotoxin gene diversity.
Spearman’s rank correlation) between speciation rates and conotoxin gene diversity and compared this test statistic with the null distribution of correlations generated by permutations of evolutionary rates across the tree. We performed a two-tailed test with the alternative hypothesis that there is a correlation between speciation rates and total conotoxin gene diversity.
We also tested the impact of diet and larval dispersal strategy on diversification patterns. Both piscivory and molluscivory is known to have evolved from the ancestral vermivory condition in cone snails (Duda et al. 2001; Puillandre et al. 2014a) and these diet transitions may be associated with increased diversification rates due to access to new dietary niches. In addition, differing larval dispersal strategies including long-lived larval stages (planktotrophy) and short-lived and/or direct developing larvae (lecithotrophy) are hypothesized to impact long-term diversification patterns (Jablonski 1986). We coded diet as either vermivory, molluscivory, and piscivory using natural history information from Tenorio and Tucker (2013). We tested the impact of speciation and extinction using MuSSE (multistate speciation and extinction, FitzJohn 2012) where speciation rates were allowed to vary or remained equal among traits. We excluded species that were documented to feed on multiple diet types from this analysis. For larval type, we used protoconch morphology described in Tenorio and Tucker (2013) to infer larval dispersal strategy, where multispiral protoconchs were assumed to be indicative of planktotrophic larvae (Shuto 1974; Jablonski and Lutz 1980; Kohn and Perron 1994; Röckel et al. 1995; Hendricks 2009b). We tested the impact of larval type on diversification patterns using BiSSE and FiSSE.
Results
Targeted Sequencing Data
We sequenced an average of 9,548,342 reads (range: 1,693,918–29,888,444) across the 363 samples (Table S1 available on Dryad). After redefining exon/intron boundaries in the phylogenetic marker reference, we ultimately targeted 2210 loci. On average, we recovered 1388 of these loci per sample (range: 30–1849, Table S1 available on Dryad) at an average coverage of 12.39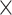 (range: 3.08
(range: 3.08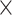 –27.87
–27.87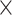 , Table S1 available on Dryad). For the conotoxin dataset, each sequence we reassembled contained a single conotoxin exon with any associated noncoding regions (referred to here as “conotoxin fragments”). We recovered on average 3416 conotoxin fragments per sample (range: 74–11,535 fragments, Table S1 available on Dryad) at an average coverage of 32.3
, Table S1 available on Dryad). For the conotoxin dataset, each sequence we reassembled contained a single conotoxin exon with any associated noncoding regions (referred to here as “conotoxin fragments”). We recovered on average 3416 conotoxin fragments per sample (range: 74–11,535 fragments, Table S1 available on Dryad) at an average coverage of 32.3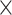 (range: 5.06
(range: 5.06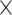 –65.77
–65.77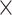 , Table S1 available on Dryad). When mapped to a reference containing both the phylogenetic markers and conotoxin genes, the % reads mapped to our targets was on average 14.86% (range: 0.7–38.07%, Table S1 available on Dryad) and the average level of duplication was 47.47% (range: 22.89–89.06%, Table S1 available on Dryad).
, Table S1 available on Dryad). When mapped to a reference containing both the phylogenetic markers and conotoxin genes, the % reads mapped to our targets was on average 14.86% (range: 0.7–38.07%, Table S1 available on Dryad) and the average level of duplication was 47.47% (range: 22.89–89.06%, Table S1 available on Dryad).
We found that the genus a sample belonged to had an impact on % mapped and % duplication, where non-Conus genera had lower % mapping and lower % duplication (Supplementary Fig. S2 available on Dryad). These differences likely occurred because conotoxin fragments were not easily recovered in these genera (ANOVA, p 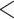 0.0001, Supplementary Fig. S2 available on Dryad). Genus did not have an impact on coverage or the number of phylogenetic markers recovered (ANOVA, p
0.0001, Supplementary Fig. S2 available on Dryad). Genus did not have an impact on coverage or the number of phylogenetic markers recovered (ANOVA, p  0.05, Supplementary Fig. S2 available on Dryad). We found that tissue quality, measured by the maximum fragment length visualized via gel electrophoresis, had a significant impact on the capture efficiency metrics (ANOVA, p
0.05, Supplementary Fig. S2 available on Dryad). We found that tissue quality, measured by the maximum fragment length visualized via gel electrophoresis, had a significant impact on the capture efficiency metrics (ANOVA, p  0.0001, Supplementary Fig. S3 available on Dryad). DNA samples with strong genomic bands at the top of the gel tended to have higher % mapping, less % duplication, higher coverage, and a greater number of targets recovered (Supplementary Fig. S3 available on Dryad).
0.0001, Supplementary Fig. S3 available on Dryad). DNA samples with strong genomic bands at the top of the gel tended to have higher % mapping, less % duplication, higher coverage, and a greater number of targets recovered (Supplementary Fig. S3 available on Dryad).
Our final conotoxin sequence dataset consists of exon fragments and we do not have information on exon coherence (which exons pair together on the same gene). We were unable to assemble full conotoxin genes because conotoxin introns are long (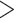 1 kilobases, Wu et al. 2013) and exceed the average insert size of our sequencing experiment (
1 kilobases, Wu et al. 2013) and exceed the average insert size of our sequencing experiment (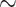 350 bp). We recovered fragments from all 58 gene superfamilies we targeted and obtained 159,670 sequences containing some or all of the mature toxin region (Table S3 available on Dryad). Total conotoxin gene diversity per species (estimated by summing across all signal region exon fragments and sequences containing the entire coding region) ranged from 5 to 1280 copies in Conus, 31 to 88 copies in Profundiconus, and 7 to 164 in Conasprella (Table S1 available on Dryad). Total conotoxin diversity was 311 copies for C. californicus, 12 copies for Pygmaeconus trailli, and 30 copies for the outgroup taxon, Bathyoma sp. (Table S1 available on Dryad). When compared with samples in Phuong and Mahardika (2018), the average change (increase or decrease) in total conotoxin gene diversity was
350 bp). We recovered fragments from all 58 gene superfamilies we targeted and obtained 159,670 sequences containing some or all of the mature toxin region (Table S3 available on Dryad). Total conotoxin gene diversity per species (estimated by summing across all signal region exon fragments and sequences containing the entire coding region) ranged from 5 to 1280 copies in Conus, 31 to 88 copies in Profundiconus, and 7 to 164 in Conasprella (Table S1 available on Dryad). Total conotoxin diversity was 311 copies for C. californicus, 12 copies for Pygmaeconus trailli, and 30 copies for the outgroup taxon, Bathyoma sp. (Table S1 available on Dryad). When compared with samples in Phuong and Mahardika (2018), the average change (increase or decrease) in total conotoxin gene diversity was 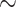 90 gene copies (Table S4 available on Dryad). If samples performed poorly in the number of phylogenetic markers recovered, conotoxin gene diversity estimates tended to be lower in this study than in Phuong and Mahardika (2018) and vice versa (Supplementary Fig. S4 available on Dryad). The average absolute change in the number of fragments recovered per gene superfamily by region was 3.7 for sequences containing the signal region, 12.2 for the prepro region, 9.6 for the mature region, 48.9 for the post region, and 3.4 for sequences containing the entire coding region (Supplementary Table S5 and Fig. S5 available on Dryad). We note several key outliers: the average absolute change in the number of fragments was 104.3 for the T gene superfamily containing the prepro region, 210.4 for the O1 gene superfamily prepro region, 57.4 for the O1 gene superfamily mature region, 219.9 for the O2 gene superfamily mature region, and 1417 for the T gene superfamily post region (Supplementary Table S5 and Fig. S5 available on Dryad).
90 gene copies (Table S4 available on Dryad). If samples performed poorly in the number of phylogenetic markers recovered, conotoxin gene diversity estimates tended to be lower in this study than in Phuong and Mahardika (2018) and vice versa (Supplementary Fig. S4 available on Dryad). The average absolute change in the number of fragments recovered per gene superfamily by region was 3.7 for sequences containing the signal region, 12.2 for the prepro region, 9.6 for the mature region, 48.9 for the post region, and 3.4 for sequences containing the entire coding region (Supplementary Table S5 and Fig. S5 available on Dryad). We note several key outliers: the average absolute change in the number of fragments was 104.3 for the T gene superfamily containing the prepro region, 210.4 for the O1 gene superfamily prepro region, 57.4 for the O1 gene superfamily mature region, 219.9 for the O2 gene superfamily mature region, and 1417 for the T gene superfamily post region (Supplementary Table S5 and Fig. S5 available on Dryad).
Phylogeny
The amount of missing data from the alignments was 15.4% when a minimum of 80% of the taxa were present in each locus, 26.8% when 50% of the taxa were present, and 38.6% when 20% of the taxa were present. The number of loci retained in the alignment was 387 (107,011 bp) when a minimum of 80% of the taxa were present in each locus, 976 (237,027 bp) when 50% of the taxa were present, and 1476 loci (336,557 bp) when 20% of the loci were present. Across all methods and datasets, we recovered phylogenies with a moderate level of resolution (average number of nodes resolved  71.1%, range
71.1%, range  61.4–79.2%, Table S6 available on Dryad). In general, as increased amounts of sequence data were given to the phylogenetic programs, more nodes became resolved (Table S6 available on Dryad). Though we recovered all six genera within Conidae with high confidence (bootstrap and PP
61.4–79.2%, Table S6 available on Dryad). In general, as increased amounts of sequence data were given to the phylogenetic programs, more nodes became resolved (Table S6 available on Dryad). Though we recovered all six genera within Conidae with high confidence (bootstrap and PP  100%, Supplementary Figs. S6–S8 available on Dryad), relationships among subgenera were less supported (Supplementary Figs. S6–S8 available on Dryad).
100%, Supplementary Figs. S6–S8 available on Dryad), relationships among subgenera were less supported (Supplementary Figs. S6–S8 available on Dryad).
Divergence Time Estimation
We found evidence for three major branching events during the Eocene: (i) a branching event leading to Profundiconus (56.5 mya, CI  46.3–65.3 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad), (ii) a branching event leading to Conus (54.7 mya, CI
46.3–65.3 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad), (ii) a branching event leading to Conus (54.7 mya, CI  42.5–63.6 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad), and (iii) a branching event separating Conasprella and Californiconus, Lilliconus, and Pygmaeconus (46.0 mya, CI
42.5–63.6 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad), and (iii) a branching event separating Conasprella and Californiconus, Lilliconus, and Pygmaeconus (46.0 mya, CI  36.5–53.2 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad). The branching event leading to Californiconus occurred during the Oligocene (26.1 mya, CI
36.5–53.2 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad). The branching event leading to Californiconus occurred during the Oligocene (26.1 mya, CI  13.8–36.5 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad) and the split between Lilliconus and Pygmaeconus occurred during the Miocene (17.8 mya, CI
13.8–36.5 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad) and the split between Lilliconus and Pygmaeconus occurred during the Miocene (17.8 mya, CI  9.25–25.1 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad).
9.25–25.1 mya, Fig. 1 and Supplementary Fig. S9 available on Dryad).
Figure 1.

Time calibrated maximum likelihood phylogeny of the cone snails. Phylogeny was estimated in RAxML using a concatenated alignment of loci and was calibrated using 13 fossils placed at nodes throughout the tree. Only loci with at least 20% of the taxa present were included in the alignment. Colors across the phylogeny show instantaneous diversification rates and are averaged across all rate models sampled from a BAMM analysis. Warmer colors indicate higher speciation rates. Log-lineage through time plot is shown below the phylogeny. First four columns shown next to tip represent the following from left to right: presence of vermivory (blue), presence of molluscivory (blue), presence of piscivory (blue), larval type (planktotrophy: light gray, lecithotrophy, blue), and missing data are represented as dark gray. Bars are shown at tips depicting variation in conotoxin gene diversity across the phylogeny. If bar is not shown, data are not available or were excluded from downstream diversification analyses. Histogram on the bottom right shows variation in conotoxin gene diversity. Plio., Pliocene; Pleis., Pleistocene.
Diversification Patterns
We found that most branching events within each genus began to occur in the Miocene and continued until the present (Fig. 1). When analyzing the entire dataset, we found support for diversification rate heterogeneity, where BAMM identified at least one rate shift across Conidae (Fig. 1 and Supplementary Fig. S10 available on Dryad). Across the 95% credible set of distinct shift configurations, BAMM detected an increase in diversification rates on the branch leading to Lautoconus, a clade consisting mainly of species endemic to the Cape Verde islands (Fig. 1 and Supplementary Fig. S10 available on Dryad). However, when examining an artificially reduced dataset consisting of half the species within Lautoconus, we detect no rate shift or a decrease in diversification rates leading to the Conus clade (Supplementary Fig. S11 available on Dryad).
Trait-Dependent Diversification
Across all thresholds for the BiSSE analysis, we found that diversification rates were not influenced by conotoxin gene diversity. In all cases, the null model was either preferred (delta AIC 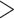 2, Table 1) or was indistinguishable from a model where speciation and extinction were allowed to vary (delta AIC
2, Table 1) or was indistinguishable from a model where speciation and extinction were allowed to vary (delta AIC 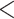 2, Table 1). Both the FiSSE and STRAPP analyses revealed that speciation rates were not correlated with conotoxin gene diversity (p
2, Table 1). Both the FiSSE and STRAPP analyses revealed that speciation rates were not correlated with conotoxin gene diversity (p 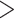 0.05). These results were consistent across both the full dataset and the reduced dataset.
0.05). These results were consistent across both the full dataset and the reduced dataset.
Table 1.
Conotoxin gene diversity BiSSE AIC results
| Dataset | Low/high gene diversity threshold | AIC—variable rates | AIC—equal rates (null) |
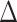 AIC AIC |
|---|---|---|---|---|
| Full dataset | 200 | 1667.74 | 1664.29 | 3.45 |
| Full dataset | 250 | 1645.03 | 1641.04 | 4.00 |
| Full dataset | 300 | 1607.43 | 1605.55 | 1.88 |
| Full dataset | 350 | 1571.78 | 1570.80 | 0.98 |
| Full dataset | 400 | 1556.94 | 1553.75 | 3.20 |
| Full dataset | 450 | 1512.22 | 1508.74 | 3.48 |
| Full dataset | 500 | 1493.84 | 1490.01 | 3.83 |
| Half of Lautoconus | 200 | 1475.91 | 1471.95 | 3.95 |
| Half of Lautoconus | 250 | 1466.18 | 1462.70 | 3.48 |
| Half of Lautoconus | 300 | 1428.96 | 1424.78 | 4.19 |
| Half of Lautoconus | 350 | 1398.14 | 1396.18 | 1.95 |
| Half of Lautoconus | 400 | 1383.51 | 1379.87 | 3.64 |
| Half of Lautoconus | 450 | 1338.68 | 1334.91 | 3.77 |
| Half of Lautoconus | 500 | 1320.32 | 1316.35 | 3.97 |
Notes: “Dataset” represents whether the full dataset was used or the reduced dataset. “Threshold” represents the conotoxin gene diversity value used to decide between “high” and “low” conotoxin diversity. Values above the threshold value were categorized as “high” and values below were categorized as “low.” “AIC—variable rates” shows AIC values for a model where speciation and extinction rates were allowed to vary depending on a trait. “AIC—equal rates” represents AIC values for the null model, where rates were not allowed to vary by trait.
We found that diversification rates were not dependent on diet when analyzing the full dataset (Table 2). However, in the reduced dataset, we found a signal for diet-dependent speciation rates (delta AIC 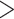 2, Table 2). We found that species with mollusc-feeding diets had the fastest speciation rates (0.33), followed by piscivory (0.24), and vermivory (0.16). For the larval dispersal trait, we found support for trait-dependent speciation rates in the full dataset (delta AIC
2, Table 2). We found that species with mollusc-feeding diets had the fastest speciation rates (0.33), followed by piscivory (0.24), and vermivory (0.16). For the larval dispersal trait, we found support for trait-dependent speciation rates in the full dataset (delta AIC 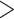 2, Table 3), where species with short-lived larvae had higher speciation rates (0.27 vs. 0.16). However, this result was not significant when examining the reduced dataset (Table 3).
2, Table 3), where species with short-lived larvae had higher speciation rates (0.27 vs. 0.16). However, this result was not significant when examining the reduced dataset (Table 3).
Table 2.
Diet BiSSE AIC results
| Dataset | Model | AIC |
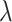 Molluscivory Molluscivory |
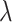 Piscivory Piscivory |
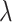 Vermivory Vermivory |
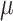
|
|---|---|---|---|---|---|---|
| Full dataset | Variable rates | 1819.407 | 0.24730 | 0.21886 | 0.18348 | 0.0000011 |
| Full dataset | Equal rates (null) | 1819.407 | 0.19083 | 0.19083 | 0.19083 | 0.00000 |
| Half of Lautoconus | Variable rates | 1638.943 | 0.33349 | 0.15917 | 0.24363 | 0.01275 |
| Half of Lautoconus | Equal rates (null) | 1643.52 | 0.23669 | 0.23669 | 0.23669 | 0.00686 |
Notes: Model values were generated under a variable rates model (where speciation was allowed to vary) or under an equal rates model (speciation rates across trait states were equal).
Table 3.
Larval dispersal type BiSSE AIC results
| Dataset | Model | AIC |
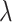 Planktotrophy Planktotrophy |
 Lecithotrophy Lecithotrophy |
|
|---|---|---|---|---|---|
| Full dataset | Variable rates | 2016.791 | 0.15978 | 0.27133 | 0.0000002 |
| Full dataset | Equal rates (null) | 2022.582 | 0.18881 | 0.18881 | 0.0000004 |
| Half of Lautoconus | Variable rates | 1836.08 | 0.27121 | 0.19455 | 0.03319 |
| Half of Lautoconus | Equal rates (null) | 1836.07 | 0.23735 | 0.23735 | 0.01279 |
Notes: Model values were generated under a variable rates model (where speciation was allowed to vary) or under an equal rates model (speciation rates across trait states were equal).
Discussion
Capture Results
Our targeted sequencing experiment underperformed initial testing of this sequencing method on cone snails (Phuong and Mahardika 2018). Although tissue quality impacted capture metrics (Supplementary Fig. S3 available on Dryad), the % of reads mapping to our targets for even our best samples was 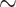 30% lower than expected (Phuong and Mahardika 2018). Whereas it is difficult to determine the exact cause of this depression in our capture statistics, we hypothesized that changes made in the bait design between this study and Phuong and Mahardika (2018) may have led to poorer capture results. For example, we recovered an overabundance of conotoxin sequences containing the postregion from the T gene superfamily that has no clear covariation pattern with phylogenetic relatedness (Supplementary Fig. S12 available on Dryad), which likely indicates a large amount of nonspecific binding due to conotoxin misclassification. In the future, we suggest redesigning the baits to only include sequences from only the most critical regions (signal region and mature region) to avoid nonspecific binding. Another source of the low mapping rate could be ascertainment bias, given that we were targeting a small fraction of genes per species. In general, if the target size (in bp) is small, the mapping rates will be low as well and this phenomenon may have contributed to the low mapping rates seen in this study. Although overall capture efficiency statistics were low, the absolute change in conotoxin diversity estimates per gene superfamily was generally minor (Table S5 available on Dryad). Therefore, we do not believe that total conotoxin diversity metrics were severely biased by the sequencing method.
30% lower than expected (Phuong and Mahardika 2018). Whereas it is difficult to determine the exact cause of this depression in our capture statistics, we hypothesized that changes made in the bait design between this study and Phuong and Mahardika (2018) may have led to poorer capture results. For example, we recovered an overabundance of conotoxin sequences containing the postregion from the T gene superfamily that has no clear covariation pattern with phylogenetic relatedness (Supplementary Fig. S12 available on Dryad), which likely indicates a large amount of nonspecific binding due to conotoxin misclassification. In the future, we suggest redesigning the baits to only include sequences from only the most critical regions (signal region and mature region) to avoid nonspecific binding. Another source of the low mapping rate could be ascertainment bias, given that we were targeting a small fraction of genes per species. In general, if the target size (in bp) is small, the mapping rates will be low as well and this phenomenon may have contributed to the low mapping rates seen in this study. Although overall capture efficiency statistics were low, the absolute change in conotoxin diversity estimates per gene superfamily was generally minor (Table S5 available on Dryad). Therefore, we do not believe that total conotoxin diversity metrics were severely biased by the sequencing method.
Phylogenetic Relationships
Below, we discuss the results of our phylogenetic analyses, how the phylogenetic relationships compare with past work, and their implications for conid taxonomy. Unless otherwise noted, the results we highlight below have at least 90% bootstrap support in the RAxML analyses and 90% posterior probabilities from the ASTRAL-II analyses (Supplementary Figs. S7 and S8 available on Dryad). When presenting results on subgeneric relationships within in genus, we move from top to bottom based on the tree shown in Supplementary Figure S6 available on Dryad.
We recovered all six major deep lineages representing genera in Conidae that were previously described in recent molecular phylogenetic studies using mtDNA (Fig. 1 and Supplementary Figs. S6–S8 available on Dryad, Puillandre et al. 2014a; Uribe et al. 2017). Specifically, we find strong support for Profundiconus, Californiconus, Lilliconus, Pygmaeconus, Conasprella, and Conus, as separate and distinct clades. We also confirm the branching order of these six genera that were recently described using mtDNA genomes (Uribe et al. 2017), with Profundiconus being sister to all other genera, Pygmaeconus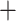 Lilliconus sister to Californiconus, Californiconus
Lilliconus sister to Californiconus, Californiconus Lilliconus
Lilliconus Pygmaeconus sister to Conasprella, and these four genera sister to Conus.
Pygmaeconus sister to Conasprella, and these four genera sister to Conus.
Based on the molecular phylogeny from three mtDNA genes, monophyletic groupings of species from Conasprella were classified into several subgenera (Puillandre et al. 2014a, 2014b). We note several differences between past results and our study in the relationships among these genera and their monophyly:
Ximeniconus is sister to all other Conasprella in some trees, or we reconstructed a polytomy at the base of Conasprella, which contrasts with Conasprella (Kohniconus) arcuata recovered at the base of Conasprella in previous work (Puillandre et al. 2014a).
Kohniconus is polyphyletic. In Puillandre et al. (2014a), only a single species from Kohniconus was included and we find evidence for the nonmonophyly of Kohniconus when we included an additional species, Conus centurio. Given these results, we propose that Conasprella emarginatus, Conusdelessertii, and C. centurio be placed in the subgenus Kohniconus and
 . arcuata placed in a new subgenus.
. arcuata placed in a new subgenus.Endemoconus is paraphyletic. When including an additional species (Conasprella somalica) not sequenced in Puillandre et al. (2014a), we find that Endemoconus is not monophyletic. Based on these results, C. somalica should be transferred to Conasprella.
Within Conus, our results largely confirm previous findings that Conus distans is sister to all other Conus species and the relationships among subgenera remain tenuous and difficult to resolve (Puillandre et al. 2014a). We note the following differences in subgenera relationships and classification between our results and past work:
We found support for a sister relationship between Turriconus and Stephanoconus, which has not been recovered in a previous study (Puillandre et al. 2014a).
We found support for the monophyly of Pyruconus across our RAxML analyses, but not our ASTRAL-II analyses. The monophyly of Pyruconus was not supported in Puillandre et al. (2014a).
Similar to Puillandre et al. (2014a), we found that Textila
 Afonsoconus is sister to Pionoconus. However, instead of the unsupported relationship of Asprella as sister to these three subgenera, we found support for Gastridium as the sister group.
Afonsoconus is sister to Pionoconus. However, instead of the unsupported relationship of Asprella as sister to these three subgenera, we found support for Gastridium as the sister group.We found support for the sister relationship between Asprella and Phasmoconus, which conflicts with the unsupported relationship shown in Puillandre et al. (2014a), where these subgenera branch in different parts of the phylogeny.
We find support for the following successional branch order: Tesselliconus, Plicaustraconus, Eugeniconus, and Conus. We found that Conus is sister to Leptoconus, Darioconus, and Cylinder, but the relationships among these three subgenera remained unresolved. This conflicts with Puillandre et al. (2014a) as Cylinder was paraphyletic, whereas in our results with increased sampling of Eugeniconus, Cylinder became monophyletic.
Conus sanderi was classified into its own subgenus (Sandericonus) based on morphological characters (Petuch and Sargent 2011; Tenorio and Tucker 2013; Puillandre et al. 2014b). Here, when sequence data were obtained, we found it nested within Dauciconus. Therefore, we synonymize Sandericonus with Dauciconus because C. sanderi is the type species for Sandericonus.
Conus granulatus was classified into its own subgenus (Atlanticonus) based on morphological characters (Petuch and Sargent 2011; Tenorio and Tucker 2013; Puillandre et al. 2014b). Here, we found that it was nested within Dauciconus. No other species within this subgenus have been sequenced up until this point. Therefore, we synonymize Atlanticonus with Dauciconus because C. granulatus is the type species for Atlanticonus.
Two species (Conus pergrandis and Conus moncuri) sequenced in this study were placed into the subgenus Embrikena (Puillandre et al. 2014b). Our results do not support the monophyly of Embrikena, as the sister relationship between C. moncuri and C. pergrandis was not supported in five out of six trees. Additional data are required to classify C. moncuri and C. pergrandis into the appropriate subgenus.
Conus cocceus was placed into Floraconus based on morphological characters in Puillandre et al. (2014b). With sequence data, we found that it was actually nested within Phasmoconus. Therefore, we transfer C. cocceus to the subgenus Phasmoconus.
Classification within Conidae is known to be highly unstable (Tenorio and Tucker 2013; Puillandre et al. 2014a, 2014b, Puillandre and Tenorio 2017). Although the phylogeny presented here improved understanding of subgeneric relationships and monophyly of subgenera, resolving relationships within Conidae still remains a significant challenge. Given the underperformance of our capture experiment (Table S1 available on Dryad), it is unclear if the reason for the moderate power in resolving relationships is due to insufficient data/incomplete data or due to short internal branches during the origination of conid subgenera that are extremely difficult to resolve. Overall, our results suggest that both additional data and increased sampling of conid species are reasonable pursuits to continue attempting to resolve the phylogeny and classification of this family of marine snails.
Timing of Diversification
The timing of splits between major lineages are largely congruent with past estimates from a study using mtDNA genomes (Uribe et al. 2017, Fig. 1 and Supplementary Fig. S9 available on Dryad). However, our age estimates for the branching events between Californiconus, Lilliconus, and Pygmaeconus are much younger (occurring across the Oligocene into the Miocene) than previous estimates (occurring across the Eocene into the Oligocene, Uribe et al. 2017, Fig. 1 and Supplementary Fig. S9 available on Dryad). This discrepancy may have been caused by differences in fossil calibration, as we included many more fossils in this study compared with previous studies. The conid fossil record and analyses of several molecular phylogenetic studies suggest a major radiation of Conus during the Miocene (Kohn 1990; Duda et al. 2001; Uribe et al. 2017). Although we noted that many branching events within Conus occurred during the Miocene into the present, we did not detect an increase in diversification on the branch leading to the origin of Conus (Fig. 1 and Supplementary Figs. S10 and S11 available on Dryad). This is congruent with diversification rates estimated from the fossil record (Kohn 1990), suggesting that the accumulation of species during the Miocene may have been a function of an increased number of lineages present rather than an increase in diversification rates. The number of species we included in the subgenus Lautoconus had an impact on the BAMM diversification analyses. With the full dataset, BAMM detected an increase in diversification rates leading to Lautoconus (Fig. 1 and Supplementary Fig. S10 available on Dryad), a known and documented radiation of cone snails (Cunha et al. 2005; Duda and Rolán 2005). However, when we remove half the species in response to recent work suggesting taxonomic inflation in this subgenus (Abalde et al. 2017), we do not detect the same shift. Rather, there is partial support for no shift across Conidae, or a slight decrease in diversification rates leading to Conus (Supplementary Fig. S11 available on Dryad). These results suggest that the original diversification analyses and identified radiation of Lauotoconus may have been due to taxonomic inaccuracies biasing the diversification analyses results, rather than a true radiation. What is even more striking about these results is that we found minimal diversification rate heterogeneity across Conidae, despite the expansive species richness across this group. It is unclear whether this signal is real, or due to other technical artifacts. For example, although we included over 300 species in this study, this only represents  30% of the total diversity in this group and may have hindered our ability to effectively estimate diversification rates. Similarly, new Conidae species are continually described, with over 100 species described over the last few years (WoRMS Editorial Board 2019). Therefore, our inability to estimate the number of living taxa may have weakened our ability to test the impact of diversification on this group.
30% of the total diversity in this group and may have hindered our ability to effectively estimate diversification rates. Similarly, new Conidae species are continually described, with over 100 species described over the last few years (WoRMS Editorial Board 2019). Therefore, our inability to estimate the number of living taxa may have weakened our ability to test the impact of diversification on this group.
Speciation Rates and Conotoxin Gene Diversity
Contrary to expectations based on macroevolutionary theory, we were unable to detect any relationship between speciation rates and conotoxin gene diversity across all trait-dependent diversification analyses (Fig. 1 and Supplementary Fig. S11 available on Dryad and Table 1). Our BAMM analyses found only minimal levels of diversification rate heterogeneity (at minimum, one shift, Fig. 1 and Supplementary Figs. S10 and S11 available on Dryad), leading to low power to correlate speciation rates with conotoxin gene diversity. Recent work suggests that BAMM tends to underestimate diversification rate heterogeneity in phylogenies, potentially masking true signals in our data (Meyer et al. 2018). However, even when performing the analyses with BiSSE, a method in recent years that has become the subject of criticism due to high false positive rates (Rabosky and Goldberg 2017a, 2017b), our analyses did not detect an impact of conotoxin gene diversity on diversification rates (Table 1).
One reason for the lack of a relationship between speciation rates and conotoxin gene diversity could be related to the nuances in using conotoxin gene diversity as a proxy for adaptive capacity. For example, we were not able to distinguish between pseudogenes and functional genes; therefore, we may not be accurately estimating “evolvability” across clades. In addition, proteomic studies have shown that not all conotoxin genes discovered through transcriptome sequencing are found in the actual venoms of cone snails (e.g., Safavi-Hemami et al. 2014), potentially confounding our results because conotoxin gene diversity may not be representative of the actual venoms conids use to capture prey. However, causes for discrepancies in venom composition between proteomic and transcriptomic studies are often difficult to rule out because there can be several nonbiological reasons for why peptides found through transcriptome sequencing are not found through proteomic methods such as rapid peptide degradation and strongly hydrophilic peptides remaining attached to commonly used chromatography columns. Finally, although we took great effort into only targeting well-studied conotoxin gene superfamilies, there may be still be errors in conotoxin gene classification, as documented in Safavi-Hemami et al. (2015) where the previously described Y2 gene superfamily was actually molluscan insulin. These potential classification errors may have also dampened the signal between conotoxin gene diversity and speciation rates.
As discussed previously, taxonomic instability in this group may have hindered our efforts to estimate past historical diversification patterns. However, we did find some signal for the impact of diet and larval dispersal strategy on diversification rates when using the BiSSe and MuSSE methods (Tables 2 and 3). Further work is needed to be fully confident in this signal given high false positive rates in these methods (Rabosky and Goldberg 2017a, 2017b) and given that our results depended on which dataset was used.
What is remarkable about these results is the lack of any signal on the impact of conotoxin gene diversity on diversification rates in cone snails, even as we found some signal for trait-dependent diversification in other conid characters. If this lack of signal is real, several biological factors may explain this decoupling between conotoxin gene diversity and speciation rates. A critical assumption in conid biology is that ecological diversification driven by diet specialization is a major factor governing diversification dynamics in cone snails (Duda and Palumbi 1999; Duda et al. 2001). Past studies have shown that cone snail venom repertoires track their dietary breadth, providing a link between diet and venom evolution (Phuong et al. 2016; Phuong and Mahardika 2018). However, it is unclear whether or not the relationship between diet and venom evolution leads to ecological speciation due to divergence in prey preference. Ecological speciation is often difficult to detect in marine ecosystems and long-term diversification patterns may be better explained by traits that limit dispersal and promote isolation (Bowen et al. 2013). Another possibility is that conotoxin phenotypic divergence may not be the rate-limiting factor in prey specialization and divergence (Duda et al. 2001). Conotoxin genes are under continuous positive selection and gene duplication, allowing venom components to change rapidly in response to the environment (Duda and Palumbi 1999; Duda et al. 2001; Puillandre et al. 2010; Chang and Duda 2012; Phuong and Mahardika 2018). This persistent evolutionary change in the venom cocktail suggests that perhaps venom evolution is not necessarily the factor limiting dietary shifts among species and ultimately, speciation among taxa. Ecological opportunity is hypothesized as a necessary component for diversification (Losos 2010) and may be a more critical factor limiting conid diversification. Indeed, evidence from the fossil record and past conid molecular phylogenetic studies indicate a concentration of lineage formation during the Miocene (Kohn 1990; Duda et al. 2001; Uribe et al. 2017), a period that is coincident with the formation of coral reefs in the Indo-Australian Archipelago (Cowman and Bellwood 2011). Our results also show a concentration of branching events during this period as well, though we do not detect a shift in diversification rates (Fig. 1 and Supplementary Figs. S10 and S11 available on Dryad). Overall, our results point to increased taxonomic sampling and a holistic approach to investigating factors shaping diversification in Conidae for future work.
Venom evolution is assumed to be a key innovation that led to the evolutionary success of venomous animal lineages (Pyron and Burbrink 2011; Sunagar et al. 2014) and a large body of work is devoted toward understanding how venom evolves and responds to the environment over time (Kordis and Gubensek 2000; Wong and Belov 2012; Casewell et al. 2013). However, the impact of venom evolution on higher-level diversification patterns is rarely tested. Here, we examined the effect of variation in the adaptive capacity of venom across conid species and found it had no influence on macroevolutionary diversification patterns. Although we do not detect a strong signal of conotoxin gene diversity shaping speciation rates in Conidae, it does not refute the importance of venom evolution in adaptation and prey specialization as venom may be necessary, but not sufficient, to promote speciation (Duda et al. 2009; Safavi-Hemami et al. 2015; Chang and Duda 2016; Phuong et al. 2016; Phuong and Mahardika 2018). Future work in other venomous animal systems may shed light on whether or not the ability to adapt to different prey through venom evolution translates to the long-term evolutionary success of taxa.
Data Availability
Raw read data is available at the National Center for Biotechnology Information Sequence Read Archive (BioProject ID# PRJNA526781). Bait sequences, conotoxin sequences, scripts, and final datasets used for analyses are uploaded onto Dryad (http://dx.doi.org/10.5061/dryad.8d44d4q).
Acknowledgments
A majority of the sampling material in this article originates from numerous shore-based expeditions and deep-sea cruises, conducted, respectively, by MNHN and Pro-Natura International (PNI) as part of the Our Planet Reviewed program (SANTO 2006, ATIMO VATAE, MAINBAZA, INHACA 2011, GUYANE 2014, PAPUA NIUGINI, KAVIENG 2014), by MNHN and AAMP (Pakaihi i Te Moana), and/or by MNHN and Institut de Recherche pour le Développement (IRD) as part of the Tropical Deep-Sea Benthos program (AURORA 2007, BIOPAPUA, EBISCO, EXBODI, MADEEP, MIRIKY, TAIWAN 2013, NANHAI 2014, BIOPAPUA, SALOMONBOA 3, CONCALIS, EXBODI, SALOMON BOA3, KARUBENTHOS 2015, NORFOLK 2, TERRASSES). Scientific partners included the University of Papua New Guinea (UPNG); National Fisheries College, Kavieng; Institut d’Halieutique et Sciences Marines (IH.SM), Université de Tuléar, Madagascar; Universidade Eduardo Mondlane, Maputo; the Madagascar bureau of the Wildlife Conservation Society (WCS); and Instituto Español de Oceanografia (IOE). Funders and sponsors included the Total Foundation, Prince Albert II of Monaco Foundation, Stavros Niarchos Foundation, Richard Lounsbery Foundation, Vinci Entrepose Contracting, Fondation EDF, European Regional Development Fund (ERDF), the Philippines Bureau of Fisheries and Aquatic Research (BFAR), the French Ministry of Foreign Affairs, Fonds Pacifique ,and the Government of New Caledonia. Additional field work included PANGLAO 2004 and PANGLAO 2005 (joint projects of MNHN and University of San Carlos, Cebu City, and the Philippines Bureau of Fisheries and Aquatic Research); KARUBENTHOS 2012 (a joint project of MNHN with Parc National de la Guadeloupe and Université des Antilles); sampling in Western Australia arranged by Hugh Morrison, with support of the Western Australian Museum. The Taiwan and South China Sea cruises were supported by bilateral cooperation research funding from the Taiwan Ministry of Science and Technology (MOST 102-2923-B-002-001-MY3, PI Wei-Jen Chen) and the French National Research Agency (ANR 12-ISV7-0005-01, PI Sarah Samadi). All expeditions operated under the regulations then in force in the countries in question and satisfy the conditions set by the Nagoya Protocol for access to genetic resources. We thank the Indonesian Ministry of State for Research and Technology (RISTEK) for providing permission to MAP and GNM to conduct fieldwork Bali in 2014 (permit number 277/SIP/FRP/SM/VIII/ 2013) and providing permission to PWHV TvR RMM and REP to conduct fieldwork across Indonesia in 2016 (permit number 414/SIP/FRP/E5/Dit.KI/X/2015). We thank DST Hariyanto, MBAP Putra, MKAA Putra, and the staff at the Indonesian Biodiversity Research Center in Denpasar, Bali for assistance during the 2014 field season; the staff at the Museum Zoologicum Bogoriense for assistance during the 2016 field season; F. Criscione, F. Köhler, A. Moussalli, A. Hogget, and L. Vail for logistical assistance for fieldwork at the Lizard Island Research Station in Australia; M. Reed, A. Hallan, and J. Waterhouse for access to specimens at the Australian Museum in Sydney, Australia; J. Finn, M. Mackenzie, and M. Winterhoff for access to specimens at the Museum Victoria in Melbourne, Australia; G. Pauley and A.M. Bemis for access to specimens at the Florida Museum of Natural History at the University of Florida; T.F. Duda Jr. and T. Lee for access to specimens at the University of Michigan Museum of Zoology; L. Kirkendale and C. Whisson for access to specimens at the Western Australian Museum in Perth, Australia; W.F. Gilly for access to the C. californicus specimen; H. Safavi-Hemami and Q. Li for access to 10 additional Conidae transcriptomes; K. Bi for advice on bait design; A. Devault and MYcroarray for great service and technical support for bait synthesis; L. Smith and the Evolutionary Genetics Laboratory at UC Berkeley for laboratory support; J. Chang, M.C.W. Lim and E.M. McCartney-Melstad for thoughtful advice and discussions throughout the entire process; E. Monnier for collating information on conid larval dispersal strategies and M.J. Tenorio for verifying conid diets and identifications. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. This work was supported by two Grants-in-Aid of research from Sigma Xi, a Grants-in-Aid of Research from the Society for Integrative and Comparative Biology, a research grant from the Society of Systematic Biologists, a Student Research Award from the American Society of Naturalists, a National Science Foundation Graduate Research Opportunities Worldwide to Australia, the Lerner Gray Fund for Marine Research from the American Museum of Natural History, research grants from the Department of Ecology and Evolutionary Biology at UCLA, the Melbourne R. Carriker Student Research Award from the American Malacological Society, an Academic Grant from the Conchologists of America, a Student Research Award from Unitas Malacologicas, a Lemelson Fellowship from the UCLA Indonesian Studies program, the Lewis and Clark Fund from the American Philosophical Society, a Young Explorer’s Grant from the National Geographic Society, a Travel Award from the UCLA Graduate Division, a Research Grant from the American Institute for Indonesian Studies and the Council of American Overseas Research Centers, a small award from the B Shaffer Lab, a National Science Foundation Graduate Research Fellowship, an Edwin W. Pauley fellowship, a Fulbright Fellowship to Indonesia, and a Chateaubriand fellowship awarded to M.A.P. This work was supported by the Service de Systématique Moléculaire (UMS 2700 CNRS-MNHN) and the CONOTAX project funded by the French National Research Agency (grant number ANR-13-JSV7-0013-01). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. The C. californicus specimen was collected under a California Department of Fish and Wildlife collecting permit granted to WF Gilly (SC-6426).
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.8d44d4q
Author Contributions
M.A.P. designed the study, conducted the field and laboratory work, carried out the bioinformatic analyses, and drafted the manuscript. G.N.M., R.M.M., R.E.P., T.v.R., and P.W.H.V. participated in field work, J.R.H. provided all of the fossil calibrations, and N.P. provided a majority of the specimens. All authors read, reviewed, and approved the manuscript.
References
- Abalde S., Tenorio M.J., Afonso C.M.L., Uribe J.E., Echeverry A.M., Zardoya R.. 2017. Phylogenetic relationships of cone snails endemic to Cabo Verde based on mitochondrial genomes. BMC Evol. Biol. 17:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkrim J., Aznar-Cormano L., Fedosov A.E., Kantor Y.I., Lozouet P., Phuong M.A., Zaharias P., Puillandre N.. 2018. Exon-capture-based phylogeny and diversification of the venomous gastropods (Neogastropoda, Conoidea). Mol. Biol. Evol. 35:2355–2374. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H., Penn O., Doron-faigenboim A., Cohen O., Cannarozzi G., Zomer O., Pupko T.. 2012. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 40:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyse DNA sequences. Nucleic Acids Res. 27:573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K., Vanderpool D., Singhal S., Linderoth T., Moritz C., Good J.M.. 2012. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics 13:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen B.W., Rocha L.A., Toonen R.J., Karl S.A.. 2013. The origins of tropical marine biodiversity. Trends Ecol. Evol. 28:359–366. [DOI] [PubMed] [Google Scholar]
- Casewell N.R., Wüster W., Vonk F.J., Harrison R.A., Fry B.G.. 2013. Complex cocktails: the evolutionary novelty of venoms. Trends Ecol. Evol. 28:219–229. [DOI] [PubMed] [Google Scholar]
- Chang D., Duda T.F.. 2012. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 29:2019–29. [DOI] [PubMed] [Google Scholar]
- Chang D., Duda T.F.. 2016. Age-related association of venom gene expression and diet of predatory gastropods. BMC Evol. Biol. 16:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman P.F., Bellwood D.R.. 2011. Coral reefs as drivers of cladogenesis: expanding coral reefs, cryptic extinction events, and the development of biodiversity hotspots. J. Evol. Biol. 24:2543–2562. [DOI] [PubMed] [Google Scholar]
- Crow K.D., Wagner G.P.. 2006. What is the role of genome duplication in the evolution of complexity and diversity? Mol. Biol. Evol. 23:887–892. [DOI] [PubMed] [Google Scholar]
- Cunha R.L., Castilho R., Rüber L., Zardoya R.. 2005. Patterns of cladogenesis in the venomous marine gastropod genus Conus from the Cape Verde islands. Syst. Biol. 54:634–650. [DOI] [PubMed] [Google Scholar]
- Duda T.F., Chang D., Lewis B.D., Lee T.. 2009. Geographic variation in venom allelic composition and diets of the widespread predatory marine gastropod Conus ebraeus. PLoS One 4:e6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda T.F. Jr., Kohn A.J., Palumbi S.R.. 2001. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc. 73:391–409. [Google Scholar]
- Duda T.F. Jr., Palumbi S.R.. 1999. Molecular genetics of ecological diversification: duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc. Natl. Acad. Sci. USA 96:6820–6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda T.F., Palumbi S.R.. 2000. Evolutionary diversification of multigene families: allelic selection of toxins in predatory cone snails. Mol. Biol. Evol. 17:1286–1293. [DOI] [PubMed] [Google Scholar]
- Duda T.F., Rolán E.. 2005. Explosive radiation of Cape Verde Conus, a marine species flock. Mol. Ecol. 14:267–272. [DOI] [PubMed] [Google Scholar]
- FitzJohn R.G. 2012. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 3:1084–1092. [Google Scholar]
- Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R.. 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3:294–299. [PubMed] [Google Scholar]
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B.W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A.. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks, J.R. 2009a. The genus Conus (Mollusca: Neogastropoda) in the Plio-Pleistocene of the Southeastern United States. Bull. Am. Paleo. 375:178. [Google Scholar]
- Hendricks J.R. 2009b. Sinistral snail shells in the sea: developmental causes and consequences. Lethaia 42:55–66. [Google Scholar]
- Hendricks J.R. 2015. Glowing seashells: diversity of fossilized coloration patterns on coral reef-associated cone snail (Gastropoda:Conidae) shells from the Neogene of the Dominican Republic. PLoS One 10:1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks J.R. 2018. Diversity and preserved shell coloration patterns of Miocene Conidae (Neogastropoda) from an exposure of the Gatun Formation, Colón Province, Panama. J. Paleontol. 92:804–837. [Google Scholar]
- Hoekstra H.E., Coyne J.A.. 2007. The locus of evolution: evo devo and the genetics of adaptation. Evolution 61:995–1016. [DOI] [PubMed] [Google Scholar]
- Huang X., Madan A.. 1999. CAP3: a DNA sequence assembly program. Genome Res. 9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski D. 1986. Larval ecology and macroevolution in marine invertebrates. Bull. Mar. Sci., 39:565–587. [Google Scholar]
- Jablonski, D. 1980. Molluscan larval shell morphology, ecological and paleontological applications. Skeletal Growth Aquat. Org. 323–377. [Google Scholar]
- Jones A.G., Arnold S.J., Bürger R.. 2007. The mutation matrix and the evolution of evolvability. Evolution 61:727–745. [DOI] [PubMed] [Google Scholar]
- Kaas Q., Westermann J.-C., Craik D.J.. 2010. Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon 55:1491–1509. [DOI] [PubMed] [Google Scholar]
- Kaas Q., Yu R., Jin A.-H., Dutertre S., Craik D.J.. 2012. ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40:D325–D330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Kuma K.-I., Toh H., Miyata T.. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn A.J. 1990. Tempo and mode of evolution in Conidae. Malacologia 32:55–67. [Google Scholar]
- Kohn A.J. 2001. Maximal species richness in Conus: diversity, diet and habitat on reefs of northeast Papua New Guinea. Coral Reefs 20:25–38. [Google Scholar]
- Kohn A.J., Perron F.E.. 1994. Life History and Biogeography: Patterns in Conus. Clarendon Press. [Google Scholar]
- Kordis D., Gubensek F.. 2000. Adaptive evolution of animal toxin multigene families. Gene 261:43–52. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavergne V., Harliwong I., Jones A., Miller D., Taft R.J., Alewood P.F.. 2015. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 112:E3782–E3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A.. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup.. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos J.B. 2010. Adaptive radiation, ecological opportunity, and evolutionary determinism. Am. Nat. 175:623–639. [DOI] [PubMed] [Google Scholar]
- Maddison W.P., Midford P.E., Otto S.P.. 2007. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 56:701–710. [DOI] [PubMed] [Google Scholar]
- Magoè T., Salzberg S.L.. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrøm M., Matschiner M., Tørresen O.K., Star B., Snipen L.G., Hansen T.F., Baalsrud H.T., Nederbragt A.J., Hanel R., Salzburger W., Stenseth N.C., Jakobsen K.S., Jentoft S.. 2016. Evolution of the immune system influences speciation rates in teleost fishes. Nat. Genet. 48:1204–1210. [DOI] [PubMed] [Google Scholar]
- Mayrose I., Zhan S.H., Rothfels C.J., Magnuson-ford K., Barker M.S., Rieseberg L.H., Otto S.P.. 2011. Recently formed polyploid plants diversify at lower rates. Science 333:1257. [DOI] [PubMed] [Google Scholar]
- Meyer M., Kircher M.. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protoc. 2010:1–10. [DOI] [PubMed] [Google Scholar]
- Meyer A.L.S., Román-Palacios C., Wiens J.J.. 2018. BAMM gives misleading rate estimates in simulated and empirical datasets. Evolution 72:2257–2266. [DOI] [PubMed] [Google Scholar]
- Mirarab S., Warnow T.. 2015. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 45:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera B.M., Rivier J., Clark C., Ramilo C.A., Corpuz G.P., Abogadie F.C., Mena E.E., Woodward S.R., Hillyard D.R., Cruz L.J.. 1990. Diversity of Conus neuropeptides. Science 249:257–263. [DOI] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K.. 2018. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Petuch E.J., Sargent D.M.. 2011. Rare and unusual shells of Southern Florida (Mainland, Florida Keys, and Dry Tortugas). Mount Dora, Florida: Conch Republic books; pp. 189. [Google Scholar]
- Phuong M.A., Mahardika G.N.. 2018. Targeted sequencing of venom genes from cone snail genomes improves understanding of conotoxin molecular evolution. Mol. Biol. Evol. 35:1210–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuong M.A., Mahardika G.N., Alfaro M.E.. 2016. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genomics 17:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigliucci M. 2008. Is evolvability evolvable? Nat. Rev Genet. 9:75–82. [DOI] [PubMed] [Google Scholar]
- Portik D.M., Smith L.L., Bi K.. 2016. An evaluation of transcriptome-based exon capture for frog phylogenomics across multiple scales of divergence (Class:Amphibia, Order:Anura). Mol. Ecol. Res. 16:1069–1083. [DOI] [PubMed] [Google Scholar]
- Puillandre N., Bouchet P., Duda T.F., Kauferstein S., Kohn A.J., Olivera B.M., Watkins M., Meyer C.. 2014a. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 78:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N., Duda T.F., Meyer C., Olivera B.M., Bouchet P.. 2014b. One, four or 100 genera? A new classification of the cone snails. J. Molluscan Stud. 81:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N., Kantor Y.I., Sysoev A., Couloux A., Meyer C., Rawlings T., Todd J.A., Bouchet P.. 2011. The dragon tamed? A molecular phylogeny of the Conoidea (Gastropoda). J. Molluscan Stud. 77:259–272. [Google Scholar]
- Puillandre N., Tenorio M.J.. 2017. A question of rank: DNA sequences and radula characters reveal a new genus of cone snails (Gastropoda: Conidae). J. Molluscan Stud. 83:200–210. [Google Scholar]
- Puillandre N., Watkins M., Olivera B.M.. 2010. Evolution of Conus peptide genes: duplication and positive selection in the A-superfamily. J. Mol. Evol. 70:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyron R.A., Burbrink F.T.. 2011. Extinction, ecological opportunity, and the origins of global snake diversity. Evolution 66:163–178. [DOI] [PubMed] [Google Scholar]
- Rabosky D.L. 2014. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS One 9:e89543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky D.L., Goldberg E.E.. 2017a. FiSSE: a simple nonparametric test for the effects of a binary character on lineage diversification rates. Evolution 71:1432–1442. [DOI] [PubMed] [Google Scholar]
- Rabosky D., Goldberg E.. 2017b. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst. Biol. 64:340–355. [DOI] [PubMed] [Google Scholar]
- Rabosky D.L., Grundler M., Anderson C., Title P., Shi J.J., Brown J.W., Huang H., Larson J.G.. 2014. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5:701–707. [Google Scholar]
- Rabosky D.L., Huang H.. 2016. A robust semi-parametric test for detecting trait-dependent diversification. Syst. Biol. 65:181–193. [DOI] [PubMed] [Google Scholar]
- Rabosky D.L., Santini F., Eastman J., Smith S.A., Sidlauskas B., Chang J., Alfaro M.E.. 2013. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nat. Commun. 4:1–8. [DOI] [PubMed] [Google Scholar]
- Robertson F.M., Gundappa M.K., Grammes F., Hvidsten T.R., Redmond A.K., Lien S., Martin S.A.M., Holland P.W.H., Sandve S.R., Macqueen D.J.. 2017. Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol. 18:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S., Norton R.. 2014. Conotoxin gene superfamilies. Mar. Drugs 12:6058–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röckel D., Korn W., Kohn A.J.. 1995. Manual of the living conidae. Verlag Christa Hemmen, Wiesbaden, Germany: Verlag Christa Hemmen; pp. 517. [Google Scholar]
- Ruby J.G., Bellare P., Derisi J.L.. 2013. PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3 (Bethesda) 3:865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H., Gajewiak J., Karanth S., Robinson S.D., Ueberheide B., Douglass A.D., Schlegel A., Imperial J.S., Watkins M., Bandyopadhyay P.K., Yandell M., Li Q., Purcell A.W., Norton R.S., Ellgaard L., Olivera B.M.. 2015. Specialized insulin is used for chemical warfare by fish-hunting cone snails. Proc. Natl. Acad. Sci. USA 112:1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H., Hu H., Gorasia D.G., Bandyopadhyay P.K., Veith P.D., Young N.D., Reynolds E.C., Yandell M., Olivera B.M., Purcell A.W.. 2014. Combined proteomic and transcriptomic interrogation of the venom gland of Conus geographus uncovers novel components and functional compartmentalization. Mol. Cell. Proteomics 13:938–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini F., Harmon L.J., Carnevale G., Alfaro M.E.. 2009. Did genome duplication drive the origin of teleosts? A comparative study of diversification in ray-finned fishes. BMC Evol. Biol. 9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E., Mirarab S.. 2017. Fast coalescent-based computation of local branch support from quartet frequencies article fast track. Mol. Biol. Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R., Edwards R.. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto T. 1974. Larval ecology of prosobranch gastropods and its bearing on biogeography and paleontology. Lethaia 7:239–256. [Google Scholar]
- Simakov O., Marletaz F., Cho S.-J., Edsinger-Gonzales E., Havlak P., Hellsten U., Kuo D.-H., Larsson T., Lv J., Arendt D., Savage R., Osoegawa K., de Jong P., Grimwood J., Chapman J.A., Shapiro H., Aerts A., Otillar R.P., Terry A.Y., Boore J.L., Grigoriev I. V, Lindberg D.R., Seaver E.C., Weisblat D.A., Putnam N.H., Rokhsar D.S.. 2013. Insights into bilaterian evolution from three spiralian genomes. Nature 493:526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S.C., Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinf. 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A.F.A., Hubley R., Green P.. 2013–2015. RepeatMasker Open-4.0. Avilable from http://www.repeatmasker.org. [Google Scholar]
- Soltis D.E., Albert V.A., Leebens-Mack J., Bell C.D., Paterson A.H., Zheng C., Sankoff D., Depamphilis C.W., Wall P.K., Soltis P.S.. 2009. Polyploidy and angiosperm diversification. Am. J. Bot. 96:336–348. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Sunagar K., Undheim E. a B., Scheib H., Gren E.C.K., Cochran C., Person C.E., Koludarov I., Kelln W., Hayes W.K., King G.F., Antunes A., Fry B.G.. 2014. Intraspecific venom variation in the medically significant Southern Pacific Rattlesnake (Crotalus oreganus helleri): biodiscovery, clinical and evolutionary implications. J. Proteomics 99:68–83. [DOI] [PubMed] [Google Scholar]
- Tank D.C., Eastman J.M., Pennell M.W., Soltis P.S., Soltis D.E., Hinchliff C.E., Brown J.W., Sessa E.B., Harmon L.J.. 2015. Nested radiations and the pulse of angiosperm diversification: increased diversification rates often follow whole genome duplications. New Phytol. 207:454–467. [DOI] [PubMed] [Google Scholar]
- Tenorio M.J., Tucker J.K.. 2013. Illustrated catalog of the living cone snails. Wellington (FL): MdM Publishing. [Google Scholar]
- Uribe J.E., Puillandre N., Zardoya R.. 2017. Phylogenetic relationships of Conidae based on complete mitochondrial genomes. Mol. Phylogenet. Evol. 107:142–151. [DOI] [PubMed] [Google Scholar]
- Wagner G.P., Altenberg L.. 1996. Perspective: complex adaptations and the evolution of evolvability. Evolution 50:967–976. [DOI] [PubMed] [Google Scholar]
- Wong E.S.W., Belov K.. 2012. Venom evolution through gene duplications. Gene 496:1–7. [DOI] [PubMed] [Google Scholar]
- WoRMS Editorial Board. 2019. World Register of Marine Species. Available from http://www.marinespecies.org at VLIZ. Accessed November 1, 2017.
- Wu Y., Wang L., Zhou M., You Y., Zhu X., Qiang Y., Qin M., Luo S., Ren Z., Xu A.. 2013. Molecular evolution and diversity of Conus peptide toxins, as revealed by gene structure and intron sequence analyses. PLoS One 8:e82495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A.S. 2001. Modularity, evolvability, and adaptive radiations: a comparison of the hemi- and holometabolous insects. Evol. Dev. 3:59–72. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhan S.H., Glick L., Tsigenopoulos C.S., Otto S.P., Mayrose I.. 2014. Comparative analysis reveals that polyploidy does not decelerate diversification in fish. J. Evol. Biol. 27:391–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw read data is available at the National Center for Biotechnology Information Sequence Read Archive (BioProject ID# PRJNA526781). Bait sequences, conotoxin sequences, scripts, and final datasets used for analyses are uploaded onto Dryad (http://dx.doi.org/10.5061/dryad.8d44d4q).