

Abstract
Helicobacter (H.) pylori is the most important cause for peptic ulcer disease and a risk factor for gastric carcinoma. How colonization with H. pylori affects the intestinal microbiota composition in humans is unknown. We investigated the association of H. pylori infection with intestinal microbiota composition in the population-based cohort Study-of-Health-in-Pomerania (SHIP)-TREND. Anti-H. pylori serology and H. pylori stool antigen tests were used to determine the H. pylori infection status. The fecal microbiota composition of 212 H. pylori positive subjects and 212 matched negative control individuals was assessed using 16S rRNA gene sequencing. H. pylori infection was found to be significantly associated with fecal microbiota alterations and a general increase in fecal microbial diversity. In infected individuals, the H. pylori stool antigen load determined a larger portion of the microbial variation than age or sex. The highest H. pylori stool antigen loads were associated with a putatively harmful microbiota composition. This study demonstrates profound alterations in human fecal microbiota of H. pylori infected individuals. While the increased microbiota diversity associated with H. pylori infection as well as changes in abundance of specific genera could be considered to be beneficial, others may be associated with adverse health effects, reflecting the complex relationship between H. pylori and its human host.
Subject terms: Stomach diseases, Genetics research
Introduction
The Gram-negative proteobacterium Helicobacter (H.) pylori infects the gastric mucosa of approximately 50% of the global human adult population. It represents the major pathogen in the pathophysiology of diverse gastrointestinal conditions including gastritis, peptic ulcer disease, gastric adenocarcinoma, and mucosa-associated lymphoma1. In addition, several extra-gastrointestinal disorders such as iron deficiency anemia or idiopathic immunocytopenic thrombopenia have been associated with H. pylori infection2. However, despite chronic gastritis being histologically detectable in almost all cases3, the majority of affected individuals remain clinically asymptomatic during their lifetime.
In order to survive the hostile gastric environment, specifically gastric acid, H. pylori produces an alkaline ammonium cloud and moves towards the bicarbonate-rich mucous layer4. Although H. pylori does not invade the gastric epithelial layer its outer membrane proteins allow attachment to the epithelium5. Once infection is established, H. pylori persistently colonizes the gastric mucosa and dominates the gastric microbiome6. In Mongolian gerbils, H. pylori colonization has also been associated with changes in the large intestinal microbiota7. H. pylori infected transgenic insulin-gastrin mice were shown to have increased microbiota richness not only in the stomach, but also in cecal and colonic samples compared to non-infected controls8. However, no specific taxa with significantly altered abundance could be identified in their colon. In humans, only small studies analyzed changes in intestinal microbiota during and after eradication therapy of H. pylori9–11. Due to the dramatic effect of the antibiotics used for H. pylori eradication in those studies, changes in the microbiome cannot unambiguously be attributed to the absence of the pathogen. Older studies relied on culturing techniques12 which are inherently inappropriate to investigate the predominantly anaerobic gut milieu. In the present study we analyzed fecal microbiota profiles generated by 16S rRNA gene sequencing of 212 H. pylori infected and 212 phenotypically matched control individuals from the population-based Study-of-Health-in-Pomerania-TREND (SHIP-TREND)13.
Results
Phenotypic matching of the 212 H. pylori infected and 212 H. pylori negative subjects was performed to control for putative confounders known to influence intestinal microbiota such as age, sex, body mass index (BMI), alcohol consumption, smoking, proton pump inhibitor (PPI) intake, and diet14–18. A history of peptic ulcer disease was also considered for matching because affected individuals were more likely to have been subjected to eradication therapy which is assumed to influence gut microbiota10. After matching H. pylori cases and controls exhibited similar distribution patterns for all accounted phenotypic variables (Table 1 and Supplementary Table S1). None of the selected individuals were under antibiotic therapy at the time of sample collection.
Table 1.
Phenotype variables of H. pylori infected cases and matched H. pylori negative controls.
| Controls (n = 212) | H. pylori cases (n = 212) | p-value | |
|---|---|---|---|
| Age (years) | 53.0 (43.0–63.0) | 53.0 (44.0–62.0) | 0.826 |
| Male sex (%) | 42.5 | 42.9 | 1 |
| BMI (kg/m²) | 27.3 (24.5–30.3) | 27.3 (24.7–29.8) | 0.493 |
| Alcohol consumption (g/d) | 4.1 (1.1–10.4) | 3.9 (1.4–9.1) | 0.851 |
| Current smokers (%) | 17.0 | 18.4 | 0.799 |
| PPI users (%) | 1.9 | 2.4 | 1 |
| Individuals with history of PUD (%) | 4.2 | 3.3 | 0.800 |
Continuous variables are expressed as median (1st–3rd quartile). Binary variables are given as percentages. The statistical significance was assessed using the Mann-Whitney test for continuous and the Fisher’s exact test for binary variables. BMI: Body mass index; PPI: Proton pump inhibitor; PUD: Peptic ulcer disease; n: number of individuals.
Beta diversity analysis of H. pylori infected individuals as compared to non-infected controls
Beta diversity analysis estimates how samples differ from each other. We used the commonly applied Bray-Curtis dissimilarity which is calculated based on the minimal shared abundance of each taxon. Thus, dual absence of taxa is not treated as similarity. Figure 1 shows the result of a principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity including all 424 microbiota samples. H. pylori infection was associated with a clear shift mainly along the first principal coordinate axis. Permutational analysis of variance (PERMANOVA) based on Bray-Curtis dissimilarity confirmed a significant shift of H. pylori cases compared to controls (r² = 0.011, p < 0.001). Association of continuous H. pylori antigen levels with beta-diversity explained even more variation (r² = 0.023, p < 0.001). The association with continuous H. pylori antibody levels was much weaker (r² = 0.006, p = 0.014). In a next step, we investigated whether H. pylori antibody or H. pylori stool antigen levels showed a significant association with beta-diversity only within the group of controls or H. pylori cases, respectively. No significant association was found within the group of controls. Within the group of H. pylori cases, H. pylori antibody levels did not show an association with beta-diversity. In contrast, continuous H. pylori antigen levels were associated with distinct changes in beta-diversity (r² = 0.029, p < 0.001). This effect size was even larger than that of age (r² = 0.015, p = 0.005) or sex (r² = 0.018, p = 0.002). No significant associations with beta-diversity were found for BMI, alcohol, smoking, use of PPI, or history of peptic ulcer disease in the group of H. pylori cases.
Figure 1.

Principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity of H. pylori infected individuals and controls. (a) Shown are PCo1 and PCo2 of 424 gut microbiota samples. Orange triangles represent samples from H. pylori infected (n = 212) and blue squares samples from control individuals (n = 212), respectively. The centroids of both groups are displayed as diamonds and the respective samples are surrounded by a 95% data ellipse. H. pylori infected cases are shifted from controls. (b) Contribution of H. pylori stool antigen level and serology titer to beta-diversity. The association of H. pylori stool antigen level clearly exceeds that of serology titer.
Alpha diversity analysis of H. pylori infected individuals compared with non-infected controls
Alpha diversity estimators characterize the diversity of an ecological community within a sample. We determined several alpha diversity indices with different emphases. The ‘Shannon diversity index’ (H) and the ‘Simpson diversity number’ (N2) include information about the number of different operational taxonomic units (OTUs), as well as the abundance of each taxon in the respective samples. The ‘Phylogenetic diversity’ tries to predict the genetic relatedness of the taxa in each sample. We compared the alpha diversity scores between H. pylori cases and controls using the parametric two-tailed t-test in case of the normally distributed ‘Shannon diversity index’ (Supplementary Fig. S1). In case of the non-normal distributed ‘Simpson diversity number’ and ‘Phylogenetic diversity’ (Supplementary Fig. S1) the two-tailed Mann-Whitney test was applied. Alpha diversity estimations revealed significantly higher scores in H. pylori cases compared to controls for ‘Shannon diversity index’ and ‘Simpson diversity number’ (Table 2 and Fig. 2). Although the median and mean ‘Phylogenetic diversity’ scores of H. pylori cases were also higher compared to controls, this was not significant.
Table 2.
Alpha diversity estimations.
| Controls | H. pylori cases | p-value | |
|---|---|---|---|
| Shannon diversity index (H) |
4.03 (3.75–4.29) 4.01 ± 0.03 |
4.12 (3.87–4.31) 4.09 ± 0.02 |
0.032* (t-test) |
| Simpson diversity number (N2) |
25.23 (19.36–32.79) 26.92 ± 0.76 |
26.73 (21.83–34.01) 28.91 ± 0.74 |
0.026* (MW) |
| Phylogenetic diversity (PD) |
53.32 (44.07–64.70) 54.30 ± 0.97 |
55.84 (44.99–64.58) 54.91 ± 0.85 |
0.648 (MW) |
Alpha diversity comparison between H. pylori negative controls (n = 212) and infected cases (n = 212). Scores are expressed as median (1st–3rd quartile) and mean ± SEM. The statistical significance was assessed using the t-test or Mann-Whitney test (MW) for normally or non-normally distributed scores, respectively. *Indicates a significant test result.
Figure 2.
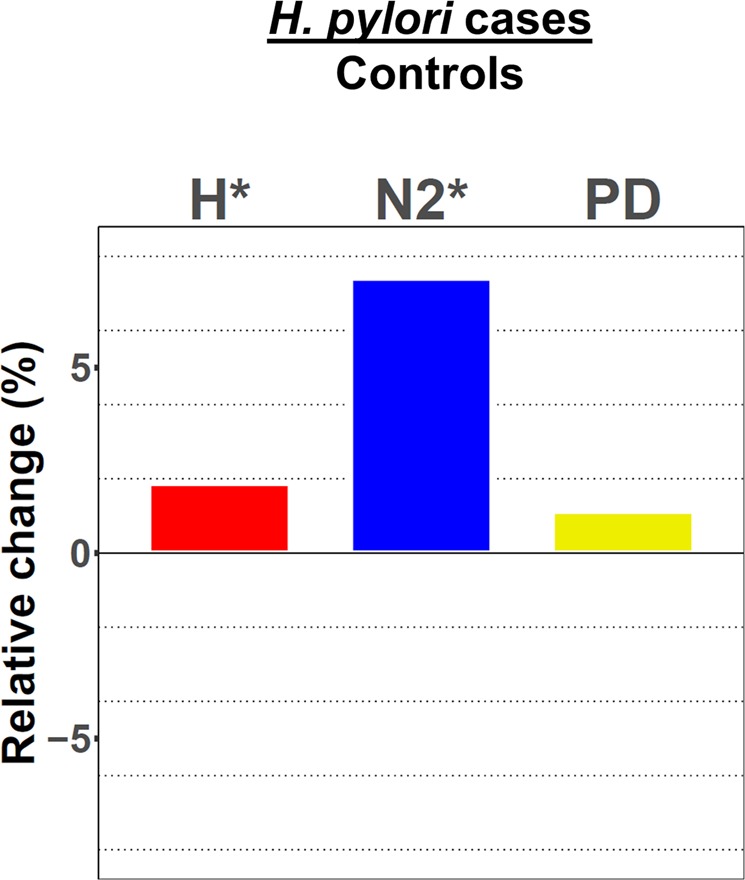
Alpha diversity analysis. Bars depict the relative change of the mean for Shannon diversity index (H, red), Simpson diversity number (N2, blue), and Phylogenetic diversity (PD, yellow) of H. pylori positive cases (n = 212) as compared to controls (n = 212). *Indicates a significant result (p < 0.05) according to t-test or Mann-Whitney test for H, N2, and PD, respectively.
Alterations of the intestinal microbiota in H. pylori infected individuals
In the complete study sample, the most abundant taxa at genus level were Bacteroides, unclassified members of the family Ruminococcacae, Faecalibacterium, and Prevotella comprising 41.4% of all microbiota. Supplementary Figs. S2 and S3 show the individual and the average composition of all participants at phylum and genus level, respectively.
Comparing genus abundance data between H. pylori cases and controls, we identified 13 taxa, namely Bacteroides, Prevotella, Parasutterella, unclassified Bacteroidales (order), unclassified Bacteroidetes (phylum), unclassified Burkholderiales (order), Holdemanella, unclassified Betaproteobacteria (class), unclassified Prevotellaceae (family), Pseudoflavonifractor, Haemophilus, Allisonella, and Howardella, that were differentially abundant in H. pylori cases. These taxa represented 32.6% of the total microbial abundance in the complete cohort. The majority of identified taxa including the highly abundant Prevotella were associated positively with H. pylori infection (Fig. 3a and Supplementary Table S2), whereas the most abundant genus Bacteroides was reduced by 16.6% in H. pylori cases compared to controls.
Figure 3.

Alterations of intestinal microbiota in H. pylori infected individuals. (a) Barplots (mean + SEM) are showing all taxa at genus level with significantly different (q < 0.05, Mann-Whitney test) abundance between controls (blue, n = 212) and H. pylori infected (orange, n = 212) individuals. Lower case letters in brackets indicate taxonomic rank of unclassified taxa at genus level: class (c), family (f), order (o), or phylum (p). (b) Pie charts showing the distribution of enterotype 1 (Bacteroides-dominated, brown) and enterotype 2 (Prevotella-dominated, yellow) in controls (left) and H. pylori cases (right).
Arumugam et al. described the existence of specific enterotypes that are either dominated by Bacteroides (enterotype 1), Prevotella (enterotype 2), or Ruminococcus (enterotype 3)19. To investigate whether the H. pylori carrier status affects the enterotype distribution, we performed enterotype clustering similar to the approach described by Arumugam et al. We found only two enterotypes in our dataset either dominated by Bacteroides or Prevotella. Ruminoccous or unclassified Ruminococcaceae were present in both groups and did not constitute a unique cluster. In controls, the enterotype 1 was present in 66.5% and enterotype 2 in 33.5% of cases. Analysis of H. pylori carriers revealed a shift from enterotype 1 (56.1%) towards enterotype 2 (43.9%) compared to controls (p = 0.036; Fisher’s exact test), (Fig. 3b).
Association of intestinal microbiota with H. pylori stool antigen load in H. pylori infected individuals
The results of the beta-diversity analysis revealed a significant microbiota variation even within the group of the 212 H. pylori cases with respect to the individual H. pylori load determined by stool antigen test. Hence, we performed linear regression analysis to identify the genera that are associated with the H. pylori stool antigen load. We analyzed all genera present in at least ten percent of all samples. Due to the reduced statistical power in this smaller data subset we focused on more prominent taxa by additionally excluding all genera with a mean abundance of less than or equal to 0.1% and all unclassified taxa at genus level. This analysis identified four genera that were all negatively associated with H. pylori stool antigen load, namely Bacteroides (q = 0.003), Barnesiella (q = 0.018), Alistipes (q = 0.035), and Fusicatenibacter (q = 0.046), (Fig. 4 and Supplementary Table S3). Performing a similar analysis for the individual H. pylori serology level instead of the stool antigen load did not yield a significant association.
Figure 4.

Genus association with H. pylori stool antigen load within H. pylori infected individuals. Shown are the age and sex adjusted abundance values (y-axis) of the four genera that were significantly associated with the H. pylori stool antigen level (x-axis) in H. pylori infected individuals. Only samples with presence of the respective genera were included. (a) Bacteroides, (b) Barnesiella, (c) Alistipes, and (d) Fusicatenibacter. r: Pearson correlation coefficient.
Discussion
We investigated changes in the intestinal microbial community structure of H. pylori infected individuals, finding significant alterations of the microbiota composition and diversity. Strikingly, in H. pylori cases, the alpha diversity estimators ‘Shannon diversity index’ and the ‘Simpson diversity number’ exhibited generally higher scores, indicating increased microbial diversity. High diversity is generally considered to be an indicator of a healthy gut microbiome, while a decrease is associated with poorer health or unhealthy habits. For example, a plant rich diet increases alpha diversity20, whereas a Western lifestyle or obesity are associated with reduced bacterial diversity21,22. In addition, conditions such as recurrent antibiotic-associated diarrhea23, Crohn’s disease24, or ulcerative colitis25, have all been reported to be associated with reduced intestinal diversity. Considering these findings, the observed positive correlation of H. pylori infection with increased diversity suggests a putative beneficial effect of H. pylori as it may strengthen the host’s resilience against microbiome perturbations or gastrointestinal infections. In more detail, a total of 13 taxa exhibited different abundance values between H. pylori infected cases and controls. We found Parasutterella to be decreased in case of H. pylori infection, a genus that has been reported to be increased in the ileal submucosa of Crohn’s disease patients26. H. pylori infected individuals exhibited increased levels of the facultative pathogen Haemophilus and decreased levels of Pseudoflavonifractor. The latter has been reported to be involved in the production of short-chain fatty acids (SCFA) such as butyrate27. SCFA represent an important energy source for colonic epithelia28 and have been shown to modulate the immune response by regulating colonic regulatory T-cell function and alleviate experimental colitis in rodents29. Thus, the decrease of the SCFA producer Pseudoflavonifractor may be disadvantageous for the human host as well.
In addition to the binary comparison of H. pylori infected individuals with controls we addressed putative differences within the group of H. pylori positive individuals and found that the H. pylori antigen load was associated with larger alterations in the fecal microbiota than age or sex. Of note, this association was not found for variations in the H. pylori antibody titer. The H. pylori antigen load was negatively associated with the four genera Bacteroides, Barnesiella, Fusicatenibacter, and Alistipes. Several of these taxa have previously been attributed health promoting features. The presence of Barnesiella was reported to be positively associated with higher eradication rates of antibiotic-resistant bacteria after fecal microbiota transplantation in mice30 and humans31, whereas higher rates of chemotherapy-related blood stream infections were observed in individuals with reduced Barnesiella counts32. Fusicatenibacter is known to be involved in SCFA production and to produce lactic acid33. It may therefore exert anti-inflammatory properties as shown for other lactic acid producing bacteria34. Finally, Alistipes is a supposed producer of the SCFA butyrate, which can alleviate intestinal inflammation35. Consequently, the observed alterations in individuals with a particularly high H. pylori antigen load may together cause adverse consequences for the human host. The putative health benefits mediated by the increased microbial diversity associated with H. pylori infection could be counteracted when H. pylori load is high.
Arumugam et al. first reported three different fecal microbiome clusters they designated as enterotypes19. It was proposed that each enterotype is characterized by a marked occurrence of Bacteroides (enterotype-1), Prevotella (enterotype-2), or Ruminococcus (enterotype-3). These authors also reported that enterotype-1 generates energy primarily by fermentation of carbohydrates and proteins, since genes encoding for galactosidases, hexosaminidases and proteases were more common. In contrast, the dominating genus of enterotype-2, Prevotella, is supposed to be a mucin degrader36. Furthermore, Arumugam et al. did not find a significant correlation of any of these enterotypes with BMI, sex, or age. In a later study, only two clusters dominated by Bacteroides or Prevotella were confirmed37. Ruminococcus which did not form an individual cluster was subsequently fused with that of Bacteroides. Both remaining clusters were primarily separated by dietary effects. The Bacteroides dominated enterotype was associated with the intake of animal protein and saturated fats, whereas the Prevotella dominated enterotype was associated with the intake of carbohydrates and a plant-based diet. In the present study, using a similar approach to that of Arumugam et al., two clusters either dominated by Bacteroides or Prevotella could be identified. The Prevotella dominated enterotype 2 was more common in H. pylori infected individuals. As our dataset was matched with respect to diet, this observation could not merely be explained by differences in intake of animal proteins or plant based products. There is currently an ongoing debate about the general concept of enterotypes. It was proposed that the clustering results which revealed separated enterotypes were rather guided by the dominant abundance of Bacteroides or Prevotella, respectively, and did not result from the presence of consistent microbial communities in each cluster38. Furthermore, it was emphasized that Bacteroides and Prevotella may form continuous gradients rather than being clearly segregated into two groups39,40. While contributing to the debate about the adequacy of the term ‘enterotype’ is beyond the scope of this study, we found H. pylori infection to be associated with a shift of the intestinal microbiota towards a Prevotella dominated microbiome, irrespective of the ‘enterotypes’ concept.
The underlying cause of the observed fecal microbiota changes associated with H. pylori infection is unknown. The altered gut microbiome in H. pylori-infected Mongolian gerbils has been explained by gastric hypochlorhydria7. It seems, indeed, plausible that a reduced production of gastric acid would promote the passage of acid-sensitive bacteria leading to an enriched diversity of the intestinal microbiome. During H. pylori infection there are generally two periods characterized by reduced gastric acid secretion: First, the initial infection phase can be followed by acute gastritis with temporarily impaired production of gastric acid4. Second, in a later stage of H. pylori infection many individuals develop pangastric, hypoacidic chronic gastritis caused by destruction of parietal cells41. However, this simple model would not be supported by the observation that gastric acid-suppressed PPI users were found to exhibit a reduced intestinal diversity14,16. Modulation of the distal gut microbiota diversity by H. pylori is therefore more complex. It has been proposed that the impact of H. pylori infection is not restricted to the gastrointestinal tract. In murine models, H. pylori demonstrated immunoregulatory features by preventing allergic asthma through the induction of regulatory T cells via IL-18 mediated tolerogenic reprogramming of dendritic cells, which then ensures the persistence of the pathogen42,43. In humans H. pylori could also regulate the intestinal microbial community composition in a similar fashion by modifying the host’s immune response.
In the complete dataset including H. pylori cases and controls, we did not find any stool microbiota profile with presence of the genus Helicobacter. Although PCR based approaches have managed to detect H. pylori in fecal samples before, these used distinct sample preparation (e. g. immunomagnetic separation) and/or specific primers for H. pylori genes for its detection44. Yet, other investigations failed to detect fecal H. pylori DNA even when using specific primers45. As the gastric pathogen H. pylori may mostly not survive under the anaerobic conditions of the distal intestine its fecal DNA concentrations are likely very low compared to the majority of anaerobic bacteria. It may therefore escape detection in fecal samples when investigating with an untargeted approach such as 16S rRNA gene sequencing.
The strength of the present study includes the large study population, the high specificity of the H. pylori diagnosis based on the assessment of both serology and stool antigen testing, and the thorough phenotypic matching of factors known to influence intestinal microbiota in order to avoid bias. Its main limitations result from the design as an association study. It may be possible that the H. pylori stool antigen levels were influenced by the gut microbiota composition, rather than the H. pylori load determining the gut microbiota. However, given the high sensitivity and specificity of the H. pylori stool antigen test in comparison to histology, culture, rapid urease test, or urea breath test46, it seems unlikely that the antigen levels were affected by gut microbiota to a great extent. A poorer performance of the stool antigen test could be predicted if defined gut bacteria would specifically degrade the target antigen.
It has been assumed that humans have been infected with H. pylori for at least ~100,000 years47 and distinctly more than half of the population in many developing and emerging countries are currently infected. In light of the sometimes life-threatening disorders that can arise in H. pylori infected individuals, this long lasting evolutionary relationship may be explainable by counterbalancing beneficial effects of the infection. Among them are the already mentioned protection against atopic diseases described in murine models42 and, according to our data, possible suppression of other gastrointestinal pathogens by increasing microbial diversity. Therefore, H. pylori eradication may trigger unwanted detrimental gut microbiota alterations, namely a reduced microbiota diversity, in individuals with low-grade infection. This underlines the need for careful decision making regarding H. pylori eradication in each individual. However, the complex relationship between H. pylori and its human host is once again demonstrated by the probably adverse microbial alterations found in individuals with high H. pylori antigen load. The large proportion of H. pylori infected individuals in the global population emphasizes the importance of this finding. As the applied method of 16S rRNA gene sequencing only allows taxon identification, future whole-genome sequencing approaches will have to define the functional potential of the enriched taxa in H. pylori cases.
Methods
Study participants
The longitudinal population-based Study-of-Health-in-Pomerania (SHIP) aims to determine the incidence and prevalence of common risk factors, subclinical disorders and clinical disease13. It comprises the two independent cohorts SHIP (recruitment 1997–2001) and SHIP-TREND (recruitment 2008–2012) with re-evaluations in 5-year intervals. All participants provided written informed consent. The study was approved by the ethics committee of the University Medicine Greifswald and carried out in accordance with its regulations.
H. pylori stool antigen test and serology
The H. pylori stool antigen test was performed using the H. pylori antigen ELISA Kit (Immundiagnostik AG, Bensheim, Germany). According to the manufacturer’s instructions, 100 mg stool were used per sample and all samples with an OD greater than or equal to 0.015 at 450 nm were considered positive for H. pylori. The H. pylori serology status was evaluated by anti-H. pylori serum IgG determination using the enzyme-linked immunoassay Pyloriset EIA-G III (Orion Diagnostica, Espoo, Finland). A positive test result was considered for antibody titers equal or greater to 20 U/ml48.
Definition of H. pylori infection
The initial dataset comprised 931 SHIP-TREND participants with available H. pylori stool antigen test as well as serology results. H. pylori infection was assumed when both, H. pylori stool antigen test and H. pylori serology, were positive. H. pylori negativity was assigned when both H. pylori stool antigen test and serology were negative. According to these criteria, 228 individuals were positive (24.5%) and 500 negative (53.7%) for H. pylori infection. A total of 183 (19.7%) participants demonstrated exclusively a positive H. pylori serology and 20 (2.1%) participants exclusively a positive stool antigen test. Of the total dataset of 931 individuals, we excluded 12 participants due to missing phenotype data and further 6 individuals because of an intake of antibiotics at the time of sample collection. This resulted in 221 H. pylori infected individuals of whom stool DNA for microbiota analysis by 16S rRNA sequencing was available for 212 individuals. As controls, 212 samples from individuals with both negative H. pylori stool antigen test as well as H. pylori serology were selected. All control samples were matched with respect to age, sex, BMI, alcohol consumption, smoking, PPI usage, history of peptic ulcer disease, and dietary habits using the ‘R’49 package ‘MatchIt’ (option ‘nearest’)50.
16S rRNA gene sequencing
Sequencing was performed as described before51. In brief, isolated DNA from fecal samples was used for amplification of the V1-V2 region of bacterial 16S rRNA genes on a MiSeq platform (Illumina, San Diego, USA). MiSeq Fast-Q files were created by CASAVA 1.8.2 (https://support.illumina.com/sequencing/sequencing_software/casava). After quality trimming of sequences with Sickle (https://github.com/najoshi/sickle), forward and reverse reads were merged and filtered using VSEARCH52. Subsequently all reads were quality filtered by FastX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit). At this step, only reads with a quality score of at least 30 (error probability 1 in 1,000) per base in 95% of sequenced nucleotides were included. To reduce redundancy among sequences de-replication was performed. OTUs were clustered using VSEARCH demanding a minimum sequence similarity of 97%. After chimera filtering by USEARCH53 each sample was normalized to 10,000 reads by random selection. Four of the 424 samples contained slightly less than 10,000 reads (9897, 9864, 9815, and 9408 reads). For assignment of taxonomy the SINTAX classifier was used54. A confidence of at least 80% for each taxonomic rank was ascertained. All taxa with a confidence below 80% were assigned to an arbitrary taxon as unclassified family, order, class, phylum, or bacteria, respectively.
Data analysis
All statistical analyses were performed using ‘R’. Bar plots were created with GraphPad Prism 5 (GraphPad Software, San Diego, USA). For calculation of ‘Shannon diversity index’ and ‘Simpson diversity number’ the R package ‘vegan’55 was used based on OTU counts. ‘Phylogenetic diversity’ was determined using the package ‘picante’56. Q-Q plots for normality assessment were generated using the qqplot function of the ‘stats’ package. The Bray-Curtis dissimilarity was calculated based on genus level data using the ‘vegan’ function vegdist. PCoA was performed with the cmdscale function from the ‘vegan’ package. Square root transformation of the Bray-Curtis dissimilarity was performed prior to the ordination to avoid negative eigenvalues. To determine the contribution of phenotypic variables to the Bray-Curtis dissimilarity, PERMANOVA was done using the ‘vegan’ function adonis and the significance assessed by 10,000 permutations. For regression of phenotypic variables on the two major principal coordinate axes the ‘vegan’ function envfit was used. The two-tailed Mann-Whitney test was performed for assessment of significance in case of continuous data. A two-tailed t-test was applied for significance assessment of the normally distributed ‘Shannon diversity index’. Fisher’s exact test was utilized for categorical data. Comparison of the relative abundance of all taxa that were present in at least ten percent of all samples at genus level between H. pylori cases and control individuals was performed using the two-tailed Mann-Whitney test. The resulting p-values were adjusted for multiple testing following the Benjamini-Hochberg procedure and called ‘q-values’. Association of the H. pylori antigen load with individual genera within the group of H. pylori infected individuals was examined as follows: All classified genera present in at least ten percent of all samples and with a mean abundance of greater than 0.1% were analyzed using a linear regression model (‘R’ function lm) based on log transformed abundance data. For this analysis zero-values were ignored, i.e. treated as missing values, to avoid biased linear regression estimates due to inflation of zeros. The putative confounder age and sex were included in the model and resulting p-values adjusted for multiple testing following the Benjamini-Hochberg procedure. P-values or q-values < 0.05 were considered significant. All p- and q- values were rounded to three significant digits.
Supplementary information
Acknowledgements
We thank Anja Wiechert, Jana Hoffmann, Kathrin Hollmann, Kathrin Rosa, Susanne Wiche, Doris Jordan, Stefan Bollmann, Ilona Urbach, Tonio Hauptmann, and Ines Spitzer for excellent technical assistance. This work was supported by a scholarship from the Gerhard Domagk program of the University Medicine Greifswald. Further support was received from the PePPP-project (ESF/14-BM-A55_0045/16), and the RESPONSE-project (BMBF grant number 03ZZ0921E). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 777111. SHIP is part of the Research Network Community Medicine of the University Medicine Greifswald, which is supported by the German Federal State of Mecklenburg-West Pomerania. We acknowledge support for the Article Processing Charge from the DFG (German Research Foundation) and the Open Access Publication Fund of the University of Greifswald.
Author contributions
Planning and concept of study: M.M.L., H.V., J.M., U.V. and A.F. Acquisition of data: F.F., T.K., M.R., C.B., F.U.W., K.Z., H.V. Statistical analysis: F.F., T.K., M.R. Data interpretation and manuscript revision: F.F., T.K., G.H., M.M.L., J.M., H.V., U.V., M.N., A.F., K.Z., C.S., R.B., M.R., C.B., F.U.W. Writing committee: F.F., G.H., M.M.L.
Data availability
All microbiome and phenotype data were obtained from the Study-of-Health-in-Pomerania (SHIP/SHIP-TREND) data management unit and can be applied for online through a data access application form (https://www.fvcm.med.uni-greifswald.de/dd_service/data_use_intro.php).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Georg Homuth and Markus M. Lerch.
Supplementary information
is available for this paper at 10.1038/s41598-019-56631-4.
References
- 1.Doorakkers E, Lagergren J, Engstrand L, Brusselaers N. Helicobacter pylori eradication treatment and the risk of gastric adenocarcinoma in a Western population. Gut. 2018;67:2092–2096. doi: 10.1136/gutjnl-2017-315363. [DOI] [PubMed] [Google Scholar]
- 2.Wong F, Rayner-Hartley E, Byrne MF. Extraintestinal manifestations of Helicobacter pylori: a concise review. World journal of gastroenterology. 2014;20:11950–11961. doi: 10.3748/wjg.v20.i34.11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dooley CP, et al. Prevalence of Helicobacter pylori Infection and Histologic Gastritis in Asymptomatic Persons. New England Journal of Medicine. 1989;321:1562–1566. doi: 10.1056/NEJM198912073212302. [DOI] [PubMed] [Google Scholar]
- 4.Waldum HL, Kleveland PM, Sørdal ØF. Helicobacter pylori and gastric acid: an intimate and reciprocal relationship. Therapeutic advances in gastroenterology. 2016;9:836–844. doi: 10.1177/1756283X16663395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Algood HMS, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clinical microbiology reviews. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz C, et al. The active bacterial assemblages of the upper GI tract in individuals with and without Helicobacter infection. Gut. 2018;67:216–225. doi: 10.1136/gutjnl-2016-312904. [DOI] [PubMed] [Google Scholar]
- 7.Heimesaat MM, et al. Helicobacter pylori induced gastric immunopathology is associated with distinct microbiota changes in the large intestines of long-term infected Mongolian gerbils. PloS one. 2014;9:e100362. doi: 10.1371/journal.pone.0100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lofgren JL, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh B, Kim JW, Kim B-S. Changes in the Functional Potential of the Gut Microbiome Following Probiotic Supplementation during Helicobacter Pylori Treatment. Helicobacter. 2016;21:493–503. doi: 10.1111/hel.12306. [DOI] [PubMed] [Google Scholar]
- 10.Yap TW-C, et al. Helicobacter pylori Eradication Causes Perturbation of the Human Gut Microbiome in Young Adults. PloS one. 2016;11:e0151893. doi: 10.1371/journal.pone.0151893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobsson HE, et al. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PloS one. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buhling A, Radun D, Muller WA, Malfertheiner P. Influence of anti-Helicobacter triple-therapy with metronidazole, omeprazole and clarithromycin on intestinal microflora. Alimentary pharmacology & therapeutics. 2001;15:1445–1452. doi: 10.1046/j.1365-2036.2001.01033.x. [DOI] [PubMed] [Google Scholar]
- 13.Völzke H, et al. Cohort profile. The study of health in Pomerania. International journal of epidemiology. 2011;40:294–307. doi: 10.1093/ije/dyp394. [DOI] [PubMed] [Google Scholar]
- 14.Imhann F, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65:740–748. doi: 10.1136/gutjnl-2015-310376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shanahan F, van Sinderen D, O’Toole PW, Stanton C. Feeding the microbiota. Transducer of nutrient signals for the host. Gut. 2017;66:1709–1717. doi: 10.1136/gutjnl-2017-313872. [DOI] [PubMed] [Google Scholar]
- 16.Jackson MA, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65:749–756. doi: 10.1136/gutjnl-2015-310861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hopkins MJ. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut. 2001;48:198–205. doi: 10.1136/gut.48.2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nature genetics. 2016;48:1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filippo Cde, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 23.Chang JY, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. The Journal of infectious diseases. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 24.Manichanh C, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boland K, et al. Microbiome Composition is Altered in Patients with IBD Independent of Endoscopic Activity. Gastroenterology. 2017;152:S991. doi: 10.1016/S0016-5085(17)33357-7. [DOI] [Google Scholar]
- 26.Chiodini RJ, et al. Microbial Population Differentials between Mucosal and Submucosal Intestinal Tissues in Advanced Crohn’s Disease of the Ileum. PloS one. 2015;10:e0134382. doi: 10.1371/journal.pone.0134382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polansky O, et al. Important Metabolic Pathways and Biological Processes Expressed by Chicken Cecal Microbiota. Applied and environmental microbiology. 2015;82:1569–1576. doi: 10.1128/AEM.03473-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function. Roles of resistant starch and nonstarch polysaccharides. Physiological reviews. 2001;81:1031–1064. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- 29.Smith PM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (New York, N.Y.) 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ubeda C, et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infection and immunity. 2013;81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bilinski J, et al. Fecal Microbiota Transplantation in Patients With Blood Disorders Inhibits Gut Colonization With Antibiotic-Resistant Bacteria. Results of a Prospective, Single-Center Study. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2017;65:364–370. doi: 10.1093/cid/cix252. [DOI] [PubMed] [Google Scholar]
- 32.Montassier E, et al. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome medicine. 2016;8:49. doi: 10.1186/s13073-016-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takada T, Kurakawa T, Tsuji H, Nomoto K. Fusicatenibacter saccharivorans gen. nov., sp. nov., isolated from human faeces. International journal of systematic and evolutionary microbiology. 2013;63:3691–3696. doi: 10.1099/ijs.0.045823-0. [DOI] [PubMed] [Google Scholar]
- 34.Menard S. Lactic acid bacteria secrete metabolites retaining anti-inflammatory properties after intestinal transport. Gut. 2004;53:821–828. doi: 10.1136/gut.2003.026252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borton MA, et al. Chemical and pathogen-induced inflammation disrupt the murine intestinal microbiome. Microbiome. 2017;5:47. doi: 10.1186/s40168-017-0264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wright DP, Rosendale DI, Robertson AM. Prevotella enzymes involved in mucin oligosaccharide degradation and evidence for a small operon of genes expressed during growth on mucin. FEMS microbiology letters. 2000;190:73–79. doi: 10.1111/j.1574-6968.2000.tb09265.x. [DOI] [PubMed] [Google Scholar]
- 37.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (New York, N.Y.) 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorvitovskaia A, Holmes SP, Huse SM. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4:15. doi: 10.1186/s40168-016-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeffery IB, Claesson MJ, O’Toole PW, Shanahan F. Categorization of the gut microbiota. Enterotypes or gradients? Nat Rev Micro. 2012;10:591–592. doi: 10.1038/nrmicro2859. [DOI] [PubMed] [Google Scholar]
- 40.Knights D, et al. Rethinking “enterotypes”. Cell host & microbe. 2014;16:433–437. doi: 10.1016/j.chom.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kusters JG, van Vliet AHM, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clinical microbiology reviews. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oertli M, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. The Journal of clinical investigation. 2012;122:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnold IC, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. The Journal of clinical investigation. 2011;121:3088–3093. doi: 10.1172/JCI45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabir S. Detection of Helicobacter pylori in faeces by culture, PCR and enzyme immunoassay. Journal of medical microbiology. 2001;50:1021–1029. doi: 10.1099/0022-1317-50-12-1021. [DOI] [PubMed] [Google Scholar]
- 45.van Zwet AA, Thijs JC, Kooistra-Smid AM, Schirm J, Snijder JA. Use of PCR with feces for detection of Helicobacter pylori infections in patients. Journal of clinical microbiology. 1994;32:1346–1348. doi: 10.1128/jcm.32.5.1346-1348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gisbert JP, La Morena Fde, Abraira V. Accuracy of monoclonal stool antigen test for the diagnosis of H. pylori infection. A systematic review and meta-analysis. The American journal of gastroenterology. 2006;101:1921–1930. doi: 10.1111/j.1572-0241.2006.00668.x. [DOI] [PubMed] [Google Scholar]
- 47.Moodley Y, et al. Age of the association between Helicobacter pylori and man. PLoS pathogens. 2012;8:e1002693. doi: 10.1371/journal.ppat.1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayerle J, et al. Identification of genetic loci associated with Helicobacter pylori serologic status. JAMA. 2013;309:1912–1920. doi: 10.1001/jama.2013.4350. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria., https://www.R-project.org/ (2017).
- 50.Ho, D. E., Imai, K., King, G. & Stuart, E. A. MatchIt. Nonparametric Preprocessing for Parametric Causal Inference. J. Stat. Soft. 42, 10.18637/jss.v042.i08 (2011).
- 51.Frost F, et al. Impaired Exocrine Pancreatic Function Associates With Changes in Intestinal Microbiota Composition and Diversity. Gastroenterology. 2019;156:1010–1015. doi: 10.1053/j.gastro.2018.10.047. [DOI] [PubMed] [Google Scholar]
- 52.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England) 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar, R. SINTAX. A simple non-Bayesian taxonomy classifier for 16S and ITS sequences (2016).
- 55.Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.4-2, https://CRAN.R-project.org/package=vegan (2017).
- 56.Kembel SW, et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics (Oxford, England) 2010;26:1463–1464. doi: 10.1093/bioinformatics/btq166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All microbiome and phenotype data were obtained from the Study-of-Health-in-Pomerania (SHIP/SHIP-TREND) data management unit and can be applied for online through a data access application form (https://www.fvcm.med.uni-greifswald.de/dd_service/data_use_intro.php).