

Abstract
This study aims to identify a miRNAs signature for predicting overall survival (OS) in esophageal squamous cell carcinoma (ESCC) patients. MiRNA expression profiles and corresponding clinical information of 119 ESCC patients were obtained from NCBI GEO and used as the training set. Differentially expressed miRNAs (DEmiRNAs) were screened between early-stage and late-stage samples. Cox regression analysis, recursive feature elimination (RFE)-support vector machine (SVM) algorithm, and LASSO Cox regression model were used to identify prognostic miRNAs and consequently build a prognostic scoring model. Moreover, promising target genes of these prognostic miRNAs were predicted followed by construction of miRNA-target gene networks. Functional relevance of predicted target genes of these prognostic miRNAs in ESCC was analyzed by performing function enrichment analyses. There were 46 DEmiRNAs between early-stage and late-stage samples in the training set. A risk score model based on five miRNAs was built. The five-miRNA risk score could classify the training set into a high-risk group and a low-risk group with significantly different OS time. Risk stratification ability of the five-miRNA risk score was successfully validated on an independent set from the Cancer Genome Atlas (TCGA). Various biological processes and pathways were identified to be related to these miRNAs, such as Wnt signaling pathway, inflammatory mediator regulation of TRP channels pathway, and estrogen signaling pathway. The present study suggests a pathological stage-related five-miRNA signature that may have clinical implications in predicting prognosis of ESCC patients.
Subject terms: Oncogenes, Diagnostic markers
Introduction
According to world statistics, esophageal cancer is among the ten most frequent cancers globally1. Its two main histological types are esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma. ESCC is the principal type and accounts for over 90% of esophageal cancer cases in China2. Approximately 398,000 ESCCs are reported globally in 20123. It has a poor prognosis, with overall five-year survival of less than 20%4.
MicroRNAs (miRNAs) are endogenous small noncoding RNAs, fine-tuning expression of genes in sequence-depending manner5. Growing studies have demonstrated that miRNAs play a role in initiation and progression of ESCC through regulating expression of oncogenes and tumor suppressors6,7. Prognostic application of miRNAs in ESCC has attracted recent interest. For instance, Chen et al. find a prognostic four-miRNA signature through analyzing miRNA expression profile of 119 ESCC samples by microarray8. There is evidence that miR-148 is associated with disease-free survival and overall survival (OS) in ESCC patients and could serve as a prognostic biomarker9. Additionally, circulating plasma miR-16 and miR-21 are of prognostic value for ESCC patients10. Furthermore, a recent study by Mao et al. demonstrates potential application of a six-miRNA signature for predicting survival of ESCC patients11. However, associations of miRNAs with prognosis of ESCC patients have not been fully elucidated.
Since pathological stage is an important prognostic indicator for ESCC, this study derived a prognostic five-miRNA signature from the differentially expressed miRNAs (DEmiRNAs) between early-stage and late-stage ESCC samples by using Cox regression analysis, recursive feature elimination (RFE)-support vector machine (SVM) algorithm, and LASSO Cox regression model. Moreover, potential target genes of the five miRNAs were predicted and miRNA-target gene networks were built. Function analysis was performed for these target genes to provide insights into the roles played by the five miRNAs in the molecular mechanisms of ESCC.
Methods
Retrieval of public data
This study included a training set and a validation set. GSE43732 was used as the training set of this study, including miRNA expression profiles of tumor tissue samples from 119 ESCC patients with clinical information downloaded from National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) based on Agilent-038166cbc_human_ miR18.0 platform (https://www.ncbi.nlm.nih.gov/geo/queryacc.cgi?Acc=GPL1654). The validation set consisted of miRNA expression data of 93 ESCC tissue samples with the corresponding clinical information downloaded from The Cancer Genome Atlas (TCGA) data portal (https://gdc-portal.nci.nih.gov/; IlluminaHiseq platform).
Clinical characteristics of all 119 ESCC patients in GSE43732 were shown in Table 1, and underwent uni-and multi-variate Cox regression analysis by using survival package12 of R. The clinical factors with log-rank p-value < 0.05 in uni-variate Cox regression analysis were further included in multi-variate Cox regression analysis. As a result, pathological stage was identified to be an independent prognostic factor (log-rank p < 0.05, Fig. 1 and Table 1) in multi-variate Cox regression analysis. According to pathologic stage, all patients of GSE43732 were separated into an early-stage group (stage I-II) and a late-stage group (stage III).
Table 1.
Clinicopathological characteristics of ESCC patients and identification of prognostic clinical factors.
| Clinical characteristics | GSE43732 (N = 119) | Uni-variate cox regression | Multi-univariate cox regression | ||||
|---|---|---|---|---|---|---|---|
| HR | 95%CI | P-value | HR | 95%CI | P-value | ||
| Age (mean ± SD) | 59.03 ± 8.93 | 1.027 | 0.999–1.057 | 0.06 | — | — | — |
| Gender (male/female) | 98/21 | 0.827 | 0.468–1.461 | 0.51 | — | — | — |
| Pathologic grade (poorly/moderately/well) | 32/64/23 | 0.819 | 0.575–1.169 | 0.27 | — | — | — |
| Pathologic stage (I/II/III) | 6/47/66 | 1.900 | 1.225–2.946 | <0.01 | 1.512 | 1.175–2.614 | 0.01 |
| Pathologic N (N1/N2/N3) | 8/20/62/29 | 1.126 | 0.838–1.514 | 0.43 | — | — | — |
| Pathologic T (T0/T1/T2/T3) | 54/42/13/10 | 1.443 | 1.143–1.821 | 0.02 | 1.243 | 0.915–1.688 | 0.16 |
| Tumor location (lower/middle/upper) | 36/69/14 | 1.104 | 0.755–1.614 | 0.61 | — | — | — |
| Alcohol (yes/no) | 74/45 | 1.053 | 0.656–1.689 | 0.83 | — | — | — |
| Tobacco (yes/no/reformed) | 80/39 | 0.859 | 0.532–1.388 | 0.54 | — | — | — |
| Death (dead/alive) | 46/73 | — | — | — | — | — | — |
| Overall survival days (months, mean ± SD) | 37.06 ± 24.25 | — | — | — | — | — | — |
SD, standard deviation.
Figure 1.
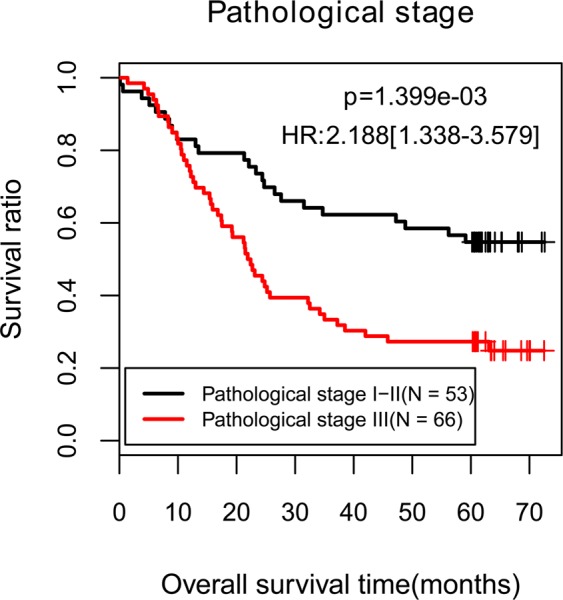
Kaplan-Meier curves for overall survival of patients in GSE43732 classified by pathological stage.
Screening DEmiRNAs between early-stage and late-stage samples
MiRNA-expression data of GSE43732 and the TCGA set was subject to unit scale normalization and median scale normalization as described previously13. Briefly, unit scale normalization was aimed to acquire samples with a norm scaled to 1 using the following formula:
For a given sample vector v = (v1, …, vn)
means the l2 norm of v, which is obtained using sqrt(sum(data2)) function of R.
Median scale normalization was conducted using the following two equations.
For a give feature vector x = (x1, …, xn):
The median absolute deviation (mad) is used to estimate variability of a uni-variate sample.
Following data normalization, differential expression analysis was performed between early-and late-stage samples in GSE43732 using limma package https://bioconductor.org/packages/release/bioc/html/limma.html) of R. The miRNAs with FDR < 0.05 and |log2FC| > 0.263 were identified as significant DEmiRNAs, followed by two-way hierarchical clustering analysis based on centered pearson correlation14 algorithm with pheatmap package of R.
SVM analysis
We employed survival package of R to conduct uni-variate Cox regression analysis to select the miRNAs significantly associated with OS in GSE43732 from the pre-selected DEmiRNAs, using log-rank p < 0.05 as the cutoff. Subsequently, we identified optimal feature miRNAs from these OS-related miRNAs within GSE43732 set by implementing RFE algorithm with caret package15 of R. Finally, SVM classifier (core: Sigmoid Kernel; cross: 100-fold cross validation) was built using the optimal feature miRNAs with SVM function16 of e1071 package of R. The classifier was used to distinguish early-stage samples from late-stage samples in GSE43732 and the TCGA set, respectively. Efficacy of the classifier was evaluated by concordance index (C-index)17, Brier score18, logRank p-value of cox-PH regression, and a number of receiver operating characteristic (ROC) curve-related metrics including area under ROC curve (AUROC), sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV).
Definition of a prognostic scoring model
In order to build a risk scoring model for survival prediction in ESCC patients, initially, we used the fore-mentioned optimal feature miRNAs to fit the LASSO Cox regression model for identification of optimal predictive miRNAs by employing penalized package of R. Optimal lambda value was calculated via a 1,000 cross-validations. Consequently, LASSO Cox regression coefficients and expression levels of the identified optimal predictive miRNAs were used to establish a prognostic scoring model as follows:
Using the prognostic model, risk scores were calculated for all patients within GSE43732. Using median risk score as the threshold, GSE43732 was separated into a high-risk group and a low-risk group. The two risk groups were compared for OS by using Kaplan-Meier curves19 and log-rank test. ROC curves were applied to estimate predictive value of the miRNAs-based prognostic model. To validate prognostic capability of the model in the TCGA set, similarly, the TCGA set was divided into a high-risk group and a low-risk group according to median risk score. Similarly, OS of the two risk groups was compared using Kaplan-Meier and log-rank methods.
Function analysis
As described above, the TCGA set was dichotomized by risk score into a high-risk group and a low-risk group. Using paired mRNA-seq data of miRNA-seq data of all 93 ESCC samples in the TCGA set, we screened differentially expressed genes (DEGs) between the two risk groups of the TCGA set. The cutoff for selection of DEGs was set at FDR < 0.05 and |log2FC| > 0.263. Besides, target genes of the identified prognostic miRNAs were predicted using starBase V3.020 (a http://starbase.sysu.edu.cn/). We reserved the target genes that were identified by at least one of five miRNA target prediction programs21 including targetScan (http://www.targetscan.org), picTar (http://pictar.mdc-berlin.de/), RNA22 (http://cbcsrv.watson.ibm.com/rna22.html), PITA (http://genie.weizmann.ac.il/pubs/mir07/) and miRanda (http://www.microrna.org/microrna/home.do). The overlapped genes between the identified target genes and the selected DEGs were chosen to construct a miRNA-target gene network with these prognostic miRNAs using Cytoscape software22. For genes in the network, gene ontology (GO)23 function and Kyoto Encyclopedia of Genes and Genomes24 pathway enrichment analyses were performed using database for annotation, visualization, and integrated discovery (DAVID)25 tool. When p-value <0.05, a GO term or a pathway was considered significant.
Results
Identification of DEmiRNAs
Overall study design was depicted in Fig. 2. GSE43732 (training set) comprised of 53 early-stage (stage I-II) samples and 66 late-stage (stage III) samples. MiRNAs expression profiles of these samples were analyzed. A total of 46 DEmiRNAs between the early-stage and late-stage samples were acquired, consisting of 6 down-regulated miRNAs and 40 up-regulated miRNAs in the late-stage samples compared to the early-stage samples (Fig. 3A). The two-way hierarchical- clustering heatmap showed that expression patterns of these DEmiRNAs were obviously different in the early-stage and late-stage samples (Fig. 3B).
Figure 2.

Graphic demonstration of DEmiRNAs. (A) Volcano plot of effect size (log2FC) and −log10(FDR) of miRNAs. Blue round spots represent DEmiRNAs, and black round spots represent non-DEmiRNAs. Horizontal dash line indicates FDR < 0.05, and two vertical dash lines indicate |logFC| > 0.263. (B) A heatmap for two-way hierarchical clustering analysis of DEmiRNAs.
Figure 3.

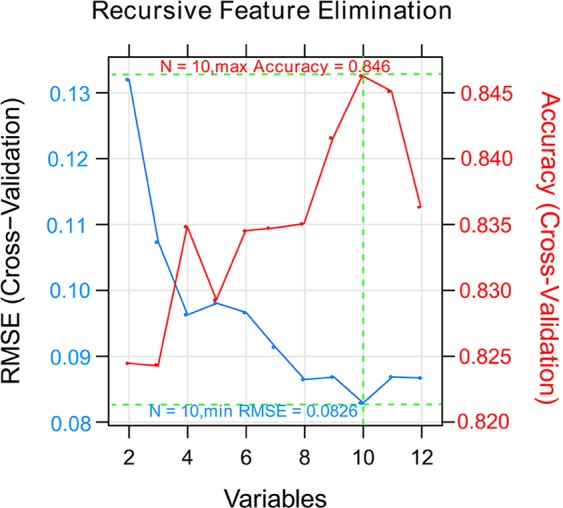
RFE algorithm optimization process. The horizontal axis is the number of selected miRNAs, and the vertical axis is cross-validation accuracy. When 10 feature miRNAs are selected, the model has the highest accuracy.
Definition of a SVM classifier of 10 miRNAs
Using data of GSE43732, we performed uni-variate Cox regression analysis to identify OS-related miRNAs from the identified 46 DEmiRNAs. As a result, 12 significant miRNAs (log-rank p < 0.05) were obtained. We then applied the RFE algorithm to filter the 12 OS-related miRNAs in order to identify the optimal combination of feature miRNAs in GSE43732. Finally, 10 miRNAs (maximal accuracy = 0.846, minimal RMSE = 0.0826) were identified as the optimal feature miRNAs to construct the classification model using an SVM (Fig. 4).
Figure 4.
Scatter plot for early-stage and late-stage samples predicted by the SVM classifier in the training set (A) and the validation set (B).
The constructed SVM classifier was applied on both GSE43732 and the TCGA set (validation set). As shown in Fig. 5A,B, the classifier could successfully differentiate early-stage samples from late-stage samples in both two datasets. Moreover, there was significant difference in OS time between predicted early-stage and late-stage samples in both two datasets (GSE43732, p-value = 1.05E−03; TCGA set, p-value = 1.47E−03, Fig. 6A,B). GSE43732 generated C-index of 0.867, Brier score of 0.0542 and AUROC of 0.948, while the TCGA set generated C-index of 0.819, Brier score of 0.0962 and AUROC of 0.902 (Table 2, Fig. 6A,B). These results illustrate that the classification model based on the 10 miRNAs could accurately discriminate between early-stage and late-stage ESCC samples.
Figure 5.

Kaplan-Meier survival curves (left) and ROC curves (right) to evaluate the SVM classification model based on ten miRNAs in the training set (A) and the validation set. (B) Patients are stratified into an early-stage group and a late-stage group by the classification model.
Figure 6.

Kaplan-Meier survival curves (left) and ROC curves (right) to evaluate the five-miRNA prognostic signature in the training set (A) and the validation set. (B) Patients are separated into a high-risk group and a low-risk group by risk score.
Table 2.
Effectiveness evaluation of the classifier of ten miRNAs on training set and validation set.
| Datasets | C index | Brier score | Log rank P | ROC | ||||
|---|---|---|---|---|---|---|---|---|
| AUROC | Sensitivity | Specificity | PPV | NPV | ||||
| Training set (GSE43732, N = 119) | 0.867 | 0.054 | 1.05 × 10−3 | 0.948 | 0.755 | 0.879 | 0.833 | 0.817 |
| Validation set (TCGA, N = 93) | 0.819 | 0.096 | 1.47 × 10−3 | 0.902 | 0.806 | 0.710 | 0.847 | 0.747 |
ROC, receiver operating characteristic curve; AUROC, area under the receiver operating characteristic curve; PPV, positive predictive value; NPV, negative predictive value.
Construction of a prognostic scoring model based on five miRNAs
To construct a risk score model for predicting survival in ESCC, we used the above-mentioned 10 feature miRNAs to fit the LASSO Cox regression model. With parameter lambda of 8.153 obtained by performing 1000 cross-validations, we identified a prognostic panel of five miRNAs (Table 3), including miR-181c-5p, miR-195-5p, miR-203, miR-212-3p and miR-28-5p. A prognostic prediction model was developed based on the five miRNAs as follows:
Table 3.
The five-miRNA signature for survival prediction.
| miRNA | coefficient | Hazard ratio | 95%CI | P-value |
|---|---|---|---|---|
| miR-181c-5p | 0.0368 | 1.129 | 1.085–1.510 | 0.041 |
| miR-195-5p | 0.0831 | 1.116 | 1.083–1.506 | 0.0471 |
| miR-203 | −0.1629 | 0.789 | 0.618–0.906 | 0.0156 |
| miR-212-3p | 0.0894 | 1.204 | 1.135–1.551 | 0.0151 |
| miR-28-5p | 0.0489 | 1.095 | 1.012–1.493 | 0.0454 |
With median risk score as threshold, GSE43732 was divided into a low-risk group and a high-risk group. OS time was significantly different between the two risk groups (p-value = 4.401E−04), with AUC of 0.952. These results suggest that the five-miRNA risk score could predict survival in ESCC patients. Moreover, the TCGA set was separated by the risk score model into two risk groups with significantly different OS time (p-value = 4.282E−02, AUC = 0.914, Fig. 7). These observations showed that prognostic value of the five-miRNA risk score was successfully validated in the TCGA set.
Figure 7.

A nomogram combining pathological stage and RS status for predicting prognosis of ESCC patients. (A) total points of pathological stage and RS status are used to decide probability of 5-year OS of each individual patient. (B) Calibration plots of nomogram for predicting 5-year OS.
Functional annotation for target genes of the five prognostic miRNAs
Using paired mRNA-seq data of miRNA-seq data of the TCGA set, 684 DEGs were found between the high-risk and low-risk samples of the TCGA set (FDR < 0.05 and log2FC| > 0.263), consisting of 526 up-regulated genes and 158 down-regulated genes in the high-risk samples relative to the low-risk samples. Out of these DEGs, the genes that were predicted to be target genes of the five prognostic miRNAs by using StarBase software were reserved. A total of 228 miRNA-target gene pairs were obtained and then used to construct a miRNA-target gene network (Fig. 8). The genes in the network were functionally related to 18 GO biological process (BP) terms, such as positive regulation of inflammatory response and Wnt signaling pathway (Table 4). Regarding KEGG pathways, cGMP-PKG signaling pathway, gastric acid secretion, vascular smooth muscle contraction, inflammatory mediator regulation of TRP channels, estrogen signaling pathway, pathways in cancer and aldosterone synthesis and secretion were important for these genes (Table 4).
Figure 8.

MiRNA-mRNA networks. Triangular nodes stand for miRNAs, and round nodes stand for mRNAs. Colors of nodes from green to red indicate values of log2FC.
Table 4.
Results of GO function and KEGG pathway enrichment analyses.
| Category | Term | Count of genes | P-value |
|---|---|---|---|
| GO biology process | Cytokine-mediated signaling pathway | 8 | 2.70 × 10−4 |
| Positive regulation of cell migration | 8 | 2.00 × 10−3 | |
| Negative regulation of angiogenesis | 5 | 2.95 × 10−3 | |
| Negative chemotaxis | 4 | 4.16 × 10−3 | |
| Establishment of skin barrier | 3 | 1.26 × 10−2 | |
| Melanocyte differentiation | 3 | 1.55 × 10−2 | |
| Signal transduction | 20 | 1.56 × 10−2 | |
| Thyroid gland development | 3 | 2.37 × 10−2 | |
| Mesenchymal-epithelial cell signaling | 2 | 2.85 × 10−2 | |
| Cornification | 2 | 2.85 × 10−2 | |
| Transcription, DNA-templated | 28 | 3.24 × 10−2 | |
| Positive regulation of inflammatory response | 4 | 3.30 × 10−2 | |
| Wnt signaling pathway | 6 | 3.43 × 10−2 | |
| Blood vessel remodeling | 3 | 3.76 × 10−2 | |
| Lung alveolus development | 3 | 4.20 × 10−2 | |
| Regulation of cell shape | 5 | 4.59 × 10−2 | |
| Positive regulation of glucose transport | 2 | 4.70 × 10−2 | |
| Negative regulation of cell cycle | 3 | 4.89 × 10−2 | |
| KEGG pathway | cGMP-PKG signaling pathway | 7 | 5.21 × 10−3 |
| Gastric acid secretion | 5 | 6.31 × 10−3 | |
| Vascular smooth muscle contraction | 6 | 6.56 × 10−3 | |
| Inflammatory mediator regulation of TRP channels | 5 | 1.73 × 10−2 | |
| Estrogen signaling pathway | 5 | 1.79 × 10−2 | |
| Pathways in cancer | 9 | 4.50 × 10−2 | |
| Aldosterone synthesis and secretion | 4 | 4.90 × 10−2 |
GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
EC is among the five most common causes of cancer-related death in China26. ESCC is the most common subtype of EC in China27. Accumulating evidence reveals that investigating esophageal carcinogenesis-related miRNAs is potentially useful for developing prognostic biomarkers28,29. The present study used miRNA expression profiles of ESCC samples from GEO to identify prognostic miRNAs. Total 46 DEmiRNAs were found between the early-stage and late-stage samples. A classification model based on 10 miRNAs for pathological stage was built by using RFE-SVM method. The SVM classifier performed well in classifying all ESCC samples into early-stage and late-stage groups on both GSE43732 set and the TCGA set, as evidenced by values of C-index, Brier score, AUROC, sensitivity, specificity, PPV and NPV. Moreover, by using LASSO Cox regression model, five prognostic miRNAs (miR-181c-5p, miR-195-5p, miR-203, miR-212-3p and miR-28-5p) were identified. The five-miRNA risk score could dichotomize the training set (GSE43732 set) into two risk groups with significantly different OS time. Moreover, prognostic capability of the five-miRNA risk score was successfully confirmed in an independent validation set (TCGA set).
Down-regulated miR-181c-5p is found in EC tissues compared to adjacent normal tissues30. MiR-181c-5p is observed to be dys-regulated in patients with pancreatic cancer relative to healthy controls and be significantly up-regulated in pancreatic cancer cases in comparison with chronic pancreatitis controls31. Up-regulation of miR-195-5p is reported in plasma of patients with laryngeal squamous cell carcinoma compared to healthy subjects32. Circulating miR-195-5p has been found to serve as a promising prognostic biomarker in head and neck cancer patients, with high expression indicative of poor prognosis33. There is evidence that miR-203 is associated with OS in esophageal adenocarcinoma patients9. Thomas et al. report that miR-203 has an oncogenic activity in pancreatic cancer and maybe a prognostic biomarker34. Down-regulated expression of miR-212-3p is related to radio-resistance in nasopharyngeal carcinoma35. Liu et al. suggest that miR-212-3p exerts an inhibitory effect on glioblastoma cell proliferation through targeting serum and glucocorticoid-inducible kinase 336. miR-28-5p is observed to be decreased in serum and tumor specimens of patients with renal cell carcinoma, and plays a tumor-suppressive role in this cancer37,38. To the best of our knowledge, this is the first time that the five-miRNA signature is suggested for survival prediction of ESCC patients. The prognostic model based on the five-miRNA signature enables differentiation of patients at high risk of mortality from patients at low risk of mortality, thus paving a way for development of personalized therapies.
To gain an understanding of functional roles of the five signature miRNAs in ESCC, we constructed a miRNA-target gene network using these miRNAs and their target genes, and analyzed possible biological processes and signaling pathways that involve these target genes. Results showed that the five miRNAs might be functionally related to Wnt signaling pathway, cGMP-PKG signaling pathway, inflammatory mediator regulation of TRP channels pathway, several inflammation- related biological processes, and estrogen signaling pathway. Abnormal activation of WNT signaling pathway contributes to esophageal tumorigenesis39. Recently, shi et al. uncover that up-regulated circRNAs are implicated in cGMP-PKG signaling pathway in ESCC40. TRP channels are associated with tumorigenesis and may represent promising therapeutic targets41. TRPC6 channel highly expressed in ESCC is critical for cell proliferation and cell cycle42. It has been established that inflammation plays an important role in cancer progression43. Several inflammatory biomarkers, such as neutrophil/lymphocyte ratio, platelet/lymphocyte ratio and lymphocyte/monocyte ratio have shown prognostic value in ESCC patients44. A recent study demonstrates that estrogen suppresses proliferation of human ESCC cells via estrogen-Ca2+ signaling pathway45. Nevertheless, further investigations are necessary to confirm these findings of our study.
Conclusion
In summary, our study identified a pathological stage-related five-miRNA signature as a promising predictor of OS for ESCC patients. Several biological processes and signaling pathways are unveiled to show that these miRNAs may participate in various molecular mechanisms of ESCC. Other independent cohorts of large sample size are needed to further validate prognostic value of the five-miRNA signature in ESCC.
Acknowledgements
The study was supported by Project of Jiangsu Natural Science Foundation (No. BK20161598), Project of Jiang-su Province Health and Family (No. H2017035) and Social Development Project of Jiangsu Provincial Science and Technology Department (No. BE2017759).
Author contributions
J.Y., M.Z., M.L. and X.L.W. participated in the design of this study, and they both performed the statistical analysis. X.M.Z. and Y.Y.Z. carried out the study and collected important background information. J.T.L. and Q.Z. drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jun Yu and Ming Zhu.
References
- 1.Mcguire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Advances in Nutrition. 2016;7:418. doi: 10.3945/an.116.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mei-Ling Z, et al. Potentially functional polymorphisms in the ERCC2 gene and risk of esophageal squamous cell carcinoma in Chinese populations. Scientific Reports. 2014;4:6281. doi: 10.1038/srep06281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melina A, Isabelle S, Jacques F, David F. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64:381–387. doi: 10.1136/gutjnl-2014-308124. [DOI] [PubMed] [Google Scholar]
- 4.Palladino-Davis AG, Mendez BM, Fisichella PM, Davis CS. Dietary habits and esophageal cancer. Diseases of the Esophagus. 2015;28:59–67. doi: 10.1111/dote.12097. [DOI] [PubMed] [Google Scholar]
- 5.Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends in Cell Biology. 2015;25:137–147. doi: 10.1016/j.tcb.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harada K, et al. The role of microRNA in esophageal squamous cell carcinoma. Journal of Gastroenterology. 2016;51:520–530. doi: 10.1007/s00535-016-1161-9. [DOI] [PubMed] [Google Scholar]
- 7.Chang Z-W, Zhong Y-L, Jia Y-X, Qin Y-R. The role of microRNAs in the occurrence and development of esophageal squamous cell carcinoma. Clinical Surgery Research Communications. 2017;1:1–9. [Google Scholar]
- 8.Chen Z, et al. MiRNA expression profile reveals a prognostic signature for esophageal squamous cell carcinoma. Cancer letters. 2014;350:34–42. doi: 10.1016/j.canlet.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 9.Hezova R, et al. Diagnostic and prognostic potential of miR-21, miR-29c, miR-148 and miR-203 in adenocarcinoma and squamous cell carcinoma of esophagus. Diagnostic pathology. 2015;10:42. doi: 10.1186/s13000-015-0280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li BX, Yu Q, Shi ZL, Li P, Fu S. Circulating microRNAs in esophageal squamous cell carcinoma: association with locoregional staging and survival. International journal of clinical and experimental medicine. 2015;8:7241–7250. [PMC free article] [PubMed] [Google Scholar]
- 11.Mao Yu, Fu Zhanzhao, Zhang Yunjie, Dong Lixin, Zhang Yanqiu, Zhang Qiang, Li Xin, Wang Chao. A six-microRNA risk score model predicts prognosis in esophageal squamous cell carcinoma. Journal of Cellular Physiology. 2018;234(5):6810–6819. doi: 10.1002/jcp.27429. [DOI] [PubMed] [Google Scholar]
- 12.Wang P, Wang Y, Hang B, Zou X, Mao JH. A novel gene expression-based prognostic scoring system to predict survival in gastric cancer. Oncotarget. 2016;7:55343–55351. doi: 10.18632/oncotarget.10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:1248–1259. doi: 10.1158/1078-0432.ccr-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, et al. RNA-seq analyses of multiple meristems of soybean: novel and alternative transcripts, evolutionary and functional implications. BMC plant biology. 2014;14:169. doi: 10.1186/1471-2229-14-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deist TM, et al. Machine learning algorithms for outcome prediction in (chemo)radiotherapy: An empirical comparison of classifiers. Medical physics. 2018;45:3449–3459. doi: 10.1002/mp.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Liu X. Screening of feature genes in distinguishing different types of breast cancer using support vector machine. Oncotargets & Therapy. 2015;8:2311. doi: 10.2147/OTT.S85271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayr A, Hofner B, Schmid M. Boosting the discriminatory power of sparse survival models via optimization of the concordance index and stability selection. Bmc Bioinformatics. 2016;17:288. doi: 10.1186/s12859-016-1149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rau, C. S. et al. Mortality prediction in patients with isolated moderate and severe traumatic brain injury using machine learning models. PLoS ONE. [DOI] [PMC free article] [PubMed]
- 19.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. International journal of Ayurveda research. 2010;1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids research. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, et al. Integrative analysis of mRNA and miRNA expression profiles in oral lichen planus: preliminary results. Oral Surgery Oral Medicine Oral Pathology & Oral Radiology. 2017;124:S2212440317308337. doi: 10.1016/j.oooo.2017.05.513. [DOI] [PubMed] [Google Scholar]
- 22.Kohl, M., Wiese, S. & Warscheid, B. In Data mining in proteomics 291–303 (Springer, 2011).
- 23.Gaudet, P. & Dessimoz, C. In The Gene Ontology Handbook 189–205 (Humana Press, New York, NY, 2017).
- 24.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dennis G, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology. 2003;4:1–11. doi: 10.1186/gb-2003-4-2-p1. [DOI] [PubMed] [Google Scholar]
- 26.Chen W, et al. National cancer incidence and mortality in China, 2012. Chinese Journal of Cancer Research. 2016;28:1. doi: 10.21147/j.issn.1000-9604.2016.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enzinger PC, Mayer RJ. Esophageal cancer. New England Journal of Medicine. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 28.Mathé EA, et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clinical Cancer Research An Official Journal of the American Association for Cancer Research. 2009;15:6192–6200. doi: 10.1158/1078-0432.CCR-09-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuxin H, et al. Prognostic significance of differentially expressed miRNAs in esophageal cancer. International Journal of Cancer Journal International Du Cancer. 2011;128:132–143. doi: 10.1002/ijc.25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenqiao Z, et al. Differential expression profiling of microRNAs and their potential involvement in esophageal squamous cell carcinoma. Tumour Biol. 2014;35:3295–3304. doi: 10.1007/s13277-013-1432-5. [DOI] [PubMed] [Google Scholar]
- 31.Kaur S, Krishn SR, Rachagani S, Batra SK. Significance of microRNA-based biomarkers for pancreatic cancer. Annals of Translational Medicine. 2015;3:277. doi: 10.3978/j.issn.2305-5839.2015.10.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayaz L, Yaroğlu HY, Özcan C, Tamer L. Differential expression of microRNAs in plasma of patients with laryngeal squamous cell carcinoma: potential early-detection markers for laryngeal squamous cell carcinoma. Journal of Cancer Research & Clinical Oncology. 2013;139:1499–1506. doi: 10.1007/s00432-013-1469-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summerer I, et al. Circulating microRNAs as prognostic therapy biomarkers in head and neck cancer patients. British Journal of Cancer. 2015;113:76–82. doi: 10.1038/bjc.2015.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas G, et al. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. International Journal of Cancer. 2010;126:73–80. doi: 10.1002/ijc.24687. [DOI] [PubMed] [Google Scholar]
- 35.Staff TPO. Correction: Genome-Wide Analyses of Radioresistance-Associated miRNA Expression Profile in Nasopharyngeal Carcinoma Using Next Generation Deep Sequencing. Plos One. 2013;8:e84486. doi: 10.1371/journal.pone.0084486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, et al. MiR-212-3p inhibits glioblastoma cell proliferation by targeting SGK3. Journal of neuro-oncology. 2015;122:431–439. doi: 10.1007/s11060-015-1736-y. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, et al. A panel of five serum miRNAs as a potential diagnostic tool for early-stage renal cell carcinoma. Scientific Reports. 2014;5:7610. doi: 10.1038/srep07610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, et al. miR-28-5p acts as a tumor suppressor in renal cell carcinoma for multiple antitumor effects by targeting RAP1B. Oncotarget. 2016;7:73888–73902. doi: 10.18632/oncotarget.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salahshor S, et al. Frequent accumulation of nuclear E-cadherin and alterations in the Wnt signaling pathway in esophageal squamous cell carcinomas. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2008;21:271–281. doi: 10.1038/modpathol.3800990. [DOI] [PubMed] [Google Scholar]
- 40.Shi P, et al. Profiles of differentially expressed circRNAs in esophageal and breast cancer. Cancer Management & Research. 2018;10:2207–2221. doi: 10.2147/CMAR.S167863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, et al. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Bioscience trends. 2014;8:1–10. doi: 10.5582/bst.8.1. [DOI] [PubMed] [Google Scholar]
- 42.Xia D, Zhuohao H, Yu S, Qun W, Yizheng W. Targeting TRPC6 channels in oesophageal carcinoma growth. Expert Opinion on Therapeutic Targets. 2010;14:513. doi: 10.1517/14728221003733602. [DOI] [PubMed] [Google Scholar]
- 43.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu JS, Huang Y, Yang X, Feng JF. A nomogram to predict prognostic values of various inflammatory biomarkers in patients with esophageal squamous cell carcinoma. American journal of cancer research. 2015;5:2180–2189. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang, Z., He, Q., Fu, S. & Zheng, Z. Estrogen Receptors in Regulating Cell Proliferation of Esophageal Squamous Cell Carcinoma: Involvement of Intracellular Ca 2+ Signaling. Pathology & Oncology Research, 1–6 (2017). [DOI] [PubMed]