

Abstract
Families with breast and ovarian cancer are often tested for disease associated sequence variants in BRCA1 and BRCA2. Pathogenic sequence variants (PVs) in these two genes are known to increase breast and ovarian cancer risks in females. However, in most families no PVs are detected in these two genes. Currently, several studies have identified other genes involved in hereditary breast and ovarian cancer (HBOC). To identify genetic risk factors for breast and ovarian cancer in a Norwegian HBOC cohort, 101 breast and/or ovarian cancer patients negative for PVs and variants of unknown clinical significance (VUS) in BRCA1/2 were screened for PVs in 94 genes using next-generation sequencing. Sixteen genes were closely scrutinized. Nine different deleterious germline PVs/likely pathogenic variants (LPVs) were identified in seven genes in 12 patients: three in ATM, and one in CHEK2, ERCC5, FANCM, RAD51C, TP53 and WRN. Additionally, 32 different VUSs were identified and these require further characterization. For carriers of PV/LPV in many of these genes, there are no national clinical management programs in Norway. The diversity of genetic risk factors possibly involved in cancer development show the necessity for more knowledge to improve the clinical follow-up of this genetically diverse patient group.
Subject terms: Breast cancer, Cancer genetics, Genetics research
Introduction
A total of 3,589 new female breast cancer (BC) cases and 520 new ovarian cancer (OC) cases were reported in Norway in 20171, 5–10% are thought to be due to inherited pathogenic variants (PVs)2. Since 1994 when BRCA1 and BRCA2 were identified3,4, PVs in these two genes have been known to be the leading cause of hereditary breast and ovarian cancer (HBOC). Together, mutated BRCA1 and BRCA2 are responsible for about 15–25% of familial breast and ovarian cancer cases5,6. The risk estimates for PVs in these genes are 45–65% for BC and 11–44% for OC by age 707. Currently, approximately 3,000 BRCA1 variants and 3,400 BRCA2 variants are listed in ClinVar as PVs or likely pathogenic variants (LPVs) (https://www.ncbi.nlm.nih.gov/clinvar 8). Nevertheless, in a large proportion of HBOC families no PVs/LPVs in BRCA1/2 have been identified.
Next-generation sequencing (NGS) allows for rapid screening of several genes and with this technology several variants in other genes have been linked to increased risk of BC and/or OC. The largest study of its kind, so far, investigated 35,409 women with a single breast cancer diagnosis, where 93.2% met the National Comprehensive Cancer Network (NCCN) guidelines for HBOC genetic testing9. These patients were screened for PVs using a 25-gene panel, and identified PVs/LPVs in 9.3%. Nearly half (48.5%) of the identified PVs/LPVs were located in BRCA1 and BRCA2, meaning that more than half (51.5%) of all pathogenic findings were in other genes. Among the genes most frequently identified with PVs/LPVs were CHEK2, ATM and PALB29. Additional studies have identified these three genes as the most frequently mutated after BRCA1/210,11. In general, genes encoding proteins involved in homologous recombination repair, the same pathway in which BRCA1 and BRCA2 are involved, are frequently reported with pathogenic findings in HBOC cases. These genes include the previously mentioned CHEK2, ATM and PALB2, together with NBN, RAD50, RAD51C, RAD51D and BRIP19,10,12–14. In addition, PVs in genes from the overlapping Fanconi Anaemia (FA) pathway and mismatch repair (MMR) pathway have been identified in BC and OC patients7. Several NGS studies revealing PVs/LPVs in other genes than BRCA1/2 in HBOC cancer patients have been published over the last years9–19. However, no such study has been reported on HBOC patients in Norway. Identification of the population-specific mutation spectrum is critical, since accumulation of certain genetic aberrations may occur within a population. In the present study, we included Norwegian women diagnosed with BC and/or OC, for whom no BRCA1 or BRCA2 PV/LPV/variant of unknown clinical significance (VUS) have been identified.
Results
A total of 101 BC and/or OC patients with no BRCA1/2 PVs, LPVs or VUSs were included in the study (Supplementary Table S1). In Fig. 1, diagnosis and age at onset represented in 10-year intervals are displayed. The majority of the patients were diagnosed at age 50–59 in all three diagnosis groups (BC, bilateral BC and OC).
Figure 1.

Distribution of patient age and diagnosis. (a) Age distribution upon first BC/OC diagnosis, regardless of diagnosis. (b) Patients grouped according to BC/OC diagnosis. One patient presented both with BC and OC (P-18), another patient (P-68) had both bilateral BC and OC. Accordingly, these two patients were registered in both BC and OC or BC bilat and OC patient groups, respectively. (c) Combination of age and BC/OC diagnosis of patients. P-18 and P-68 are also here represented twice. BC = breast cancer. Bilat = bilateral. OC = ovarian cancer.
Samples from the 101 patients were investigated for the presence of nonsense or frameshift variants in 94 genes. In addition, 16 genes were scrutinized for missense, deletions, insertions, and possible splice-affecting variants (Table 1). The average coverage of the examined regions for samples from group 1 and 2 was 532.7 reads (S.D. 138.9). The average coverage information was not available for samples in group 3 where only Virtual Contact File (VCF) and Binary Alignment Map (BAM) files were studied. However, since all three sample groups were analyzed in an identical manner, similar coverage can be inferred.
Table 1.
Overview of the 18 genes (including BRCA1 and BRCA2) investigated more closely in this study (the 18 first genes in the table) together with four genes with additional findingsa.
| Gene symbol | Gene name | Reference sequence | Numbers of exons | Average coverage | Gaps |
|---|---|---|---|---|---|
| BRCA1 | BRCA1, DNA repair associated | NM_007294.3 | 23 | 514.6 | |
| BRCA2 | BRCA2, DNA repair associated | NM_000059.3 | 27 | 737.7 | |
| ATM | ATM serine/threonine kinase | NM_000051.3 | 63 | 795.6 | |
| BRIP1 | BRCA1 interacting protein C-terminal helicase 1 | NM_032043.2 | 20 | 714.6 | |
| CDH1 | Cadherin 1 | NM_004360.3 | 16 | 527.9 |
Ex2/in2: 63% 5′ in11: 16% |
| CHEK2 | Checkpoint kinase 2 | NM_007194 | 16 | 592.7 | |
| MLH1 | MutL homolog 1 | NM_000249.3 | 19 | 573.1 | |
| MSH2 | MutS homolog 2 | NM_000251.2 | 16 | 736.1 | |
| MSH6 | MutS homolog 6 | NM_000179.2 | 10 | 697.8 | Ex1/in1: 100% |
| NBN | Nibrin | NM_002485.4 | 16 | 764.3 | |
| NF1 | Neurofibromin 1 | NM_001042492.2 | 58 | 594.8 | |
| PALB2 | Partner and localizer of BRCA2 | NM_024675.3 | 13 | 578.8 | |
| PMS2 | PMS1 homolog 2, mismatch repair system component | NM_000535.6 | 15 | 735.3 | |
| PTEN | Phosphatase and tensin homolog | NM_00314.4 | 9 | 643.6 | |
| RAD51C | RAD51 paralog C | NM_058216.1 | 9 | 641.8 | |
| RAD51D | RAD51 paralog D | NM_002878.3 | 10 | 447.0 |
In4/ex5: 94% Start of ex1: 89% |
| STK11 | Serine/threonine kinase 11 | NM_000455.4 | 10 | 317.9 |
In3/ex4: 13% Ex7/in7: 54% Ex9/in9: 32% |
| TP53 | Tumor protein 53 | NM_000546.5 | 11 | 414.2 | 3′ in6: 5% |
| ERCC5a | ERCC excision repair 5, endonuclease | NM_000123.3 | 15 | 683.6 | |
| FANCFa | Fanconi anemia, complementation group F | NM_022725.3 | 1 | 744.0 | |
| FANCMa | Fanconi anemia, complementation group M | NM_020937.2 | 23 | 866.8 | |
| WRNa | Werner syndrome RecQ like helicase | NM_000553.4 | 35 | 679.8 |
Reported gaps (<30 reads) are listed in the last column, the percentage in the Gaps column represents the amount of samples with gaps in this region (gaps only present in one run or in other genes than the genes more closely scrutinized are not included). In = intron. Ex = exon.
aGenes only investigated for frame-shift and nonsense variants and with an identified pathogenic/likely pathogenic variant in this study.
Identified variants
For the analysed regions, on average 203.4 variants (range: 170–247) in all 94 genes were reported per patient. After filtration, on average 1.1 variant (range: 0–4) per patient remained, which resulted in a total of 77 unique variants. Of these 77 variants, nine were classified as PVs/LPVs (Table 2), 32 classified as VUSs (Table 3) and 36 were classified as benign/likely benign (Supplementary Table S2). The nine unique PVs/LPVs were found in seven genes (ATM, CHEK2, ERCC5, FANCM, TP53, RAD51C and WRN) in 12 patients (Table 2 and Fig. 2).
Table 2.
Pathogenic and likely pathogenic variants identified in a Norwegian breast and ovarian cancer cohort.
| Gene | Variant | Local. | Protein change | Databases | Pr. Ref. | Cl. | Patient | Diagn. & age | Additional VUS | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP | gnomAD | ClinVar | HGMDp | |||||||||
| ATM | c.3245_3247delinsTGAT | exon 22 | p.His1082Leufs*14 | — | — | RCV000159638.7; Pathogenic | CX983261; A-T: DM | 66 | 5 | P-31 | OC, 47y | BRIP1 c.2087C>T |
| ATM | c.3245_3247delinsTGAT | exon 22 | p.His1082Leufs*14 | — | — | RCV000159638.7; Pathogenic | CX983261; A-T: DM | 66 | 5 | P-2 | BC, 57y | NF1 c.5225A>G |
| ATM | c.5932G>T | exon 40 | p.Ser1974Ilefs*4 | rs587779852 | ALL: 0.00%; NFE: 0.01%; FIN: 0% | RCV000115219.8; Pathogenic | CM980147; A-T: DM | 37 | 5 | P-91 | BC, 54y | NF1 c.469A>G |
| ATM | c.8432delA | exon 58 | p.(Lys2811Serfs*46) | rs759472682 | ALL: 0.00%; NFE: 0.00%; FIN: 0% | RCV000131776.5; Pathogenic | CD000916; A-T: DM | 67 | 5 | P-62 | OC, 38y | — |
| CHEK2 | c.319+2T>A | intron 2 | p.(?) | rs587782401 | ALL: 0.01%; NFE: 0.01%; FIN: 0.06% | RCV000131434.11; Likely pathogenic | CS1617635; BC: DM | 16 | 4 | P-12 | OC, 27y | CDH1 c.136C>G |
| CHEK2 | c.319+2T>A | intron 2 | p.(?) | rs587782401 | ALL: 0.01%; NFE: 0.01%; FIN: 0.06% | RCV000131434.11; Likely pathogenic | CS1617635; BC: DM | 16 | 4 | P-16 | OC, 70y | — |
| ERCC5 | c.67G>T | exon 1 | p.(Glu23*) | — | — | — | — | — | 4 | P-44 | BC, 49y | NF1 c.378A>G |
| FANCM | c.5101C>T | exon 20 | p.(Gln1701*) | rs147021911 | ALL: 0.13%; NFE: 0.11%; FIN: 0,81% | RCV000115190.8; Risk factorb | CM147953; TF: DM | 68 | 4 | P-8 | BC, 56y | — |
| FANCM | c.5101C>T | exon 20 | p.(Gln1701*) | rs147021911 | ALL: 0.13%; NFE: 0.11%; FIN: 0,81% | RCV000115190.8; Risk factorb | CM147953; TF: DM | 68 | 4 | P-41 | BC, 69y | — |
| RAD51C | c.1026+5_1026+7delGTA | intron 8 | p.Arg322Serfs*22 | rs747311993 | ALL: 0.00%; NFE: 0.00%; FIN: 0% | RCV000116170.11; Likely pathogenic | CD1313340; BOC: DM | 69 | 4 | P-69 | OC, 52y | — |
| TP53 | c.818G>Aa | exon 8 | p.(Arg273His) | rs28934576 | ALL: 0.00%; NFE: 0.00%; FIN: 0% | RCV000115738.8; Pathogenic/Likely pathogenic | CM920677; LFS: DM | 70 | 5 | P-13 | BC, 36y | — |
| WRN | c.1105C>T | exon 9 | p.(Arg369*) | rs17847577 | ALL: 0.02%; NFE: 0.03%; FIN: 0.02% | RCV000005782.8; Pathogenicc | CM961463; WRN: DM | 71 | 5 | P-90 | BC, 57y | — |
Variants are named according to Human Genome Variation Society (HGVS) nomenclature. Local.: localization; Pr.Ref: Primary reference; cl.: class (4: likely pathogenic; 5: pathogenic); diagn.: diagnosis; NFE: Non-Finnish Europeans; FIN: Finnish Europeans; A-T: Ataxia telangiectasia; DM: Disease mutation; BC: Breast cancer; OC: Ovarian cancer; LFS: Li-Fraumeni Syndrome; DFP: Disease associated functional polymorphism; TF: Tetralogy of Fallot; WRN: Werner syndrome; y: years. The ClinVar references and the corresponding clinical significance are mainly linked to the condition “Hereditary cancer-predisposing syndrome”, exceptions are marked.
aVariant identified in 33% of reads.
bFanconi anemia.
cWerner syndrome.
Table 3.
Variants of unknown clinical significance identified in a Norwegian breast and ovarian cancer cohort.
| Gene | Variant | Local. | Protein change | Databases | Pr. Ref. | Patient | Diagn. & age | |||
|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP | gnomAD | ClinVar | HGMDp | |||||||
| ATM | c.1727T>C | Exon 11 | p.(Ile576Thr) | rs730881342 | ALL: 0.01%; NFE: 0.01%; FIN: 0.04% | RCV000159685.6; VUS | — | 72 | P-97 | BC, 50y |
| ATM | c.1986T>C | Exon 13 | p.(=) | rs1800055 | ALL: 0.05%; NFE: 0.08%; FIN: 0.08% | RCV000123724.5; Likely benign | — | 72 | P-99 | BC, 52y |
| ATM | c.2164T>C | Exon 14 | p.(=) | — | — | — | — | — | P-78 | BC, 49y |
| ATM | c.2220A>C | Exon 14 | p.(=) | — | — | — | — | — | P-56 | BC, 61y |
| ATM | c.2804C>T | Exon 19 | p.(Thr935Met) | rs3218708 | ALL: 0.01%; NFE: 0.01%; FIN: 0% | RCV000131651.8; Likely benign/VUS | CM177861; CRC: DM? | 73 | P-53 | BC, 38y |
| ATM | c.3549T>C | Exon 24 | p.(=) | rs767377764 | ALL: 0.00%, NFE: 0%; FIN: 0% | RCV000223274.2; Likely benign | — | — | P-88 | BC, 48y |
| ATM | c.3703C>T | Exon 25 | p.(Pro1235Ser) | rs779095853 | ALL: 0.00%, NFE: 0.00%; FIN: 0% | RCV000567940.1; VUS | — | — | P-80 | BC, 58y |
| ATM | c.4324T>C | Exon 30 | p.(Tyr1442His) | rs201666889 | ALL: 0.03%; NFE: 0.06%; FIN: 0.02% | RCV000115190.8; Benign/VUS | CM0910502; BC: DM? | 30 | P-75 | BC, 49y |
| ATM | c.5229A>G | Exon 36 | p.(=) | — | — | RCV000233826.2; Likely benigna | — | — | P-42 | BC, 63y |
| ATM | c.8734A>G | Exon 61 | p.(Arg2912Gly) | rs376676328 | ALL: 0.02%; NFE: 0.04%; FIN: 0.00% | RCV000131723.10; VUS | CM014034; BC: DM | 74 | P-14 | BC, 35y |
| BRIP1 | c.2087C>T | Exon 14 | p.(Pro696Leu) | rs147755155 | ALL: 0.00%; NFE: 0.01%; FIN: 0.00% | RCV000116135.9; VUS | — | — | P-31; P-32 | OC, 47y; OC, 52y |
| CDH1 | c.136C>G | Exon 2 | p.(Leu46Val) | — | — | — | — | — | P-12 | OC, 27y |
| CHEK2 | c.470T>C | exon 4 | p.(Ile157Thr) | rs17879961 | ALL: 0.49%; NFE: 0.53%; FIN: 2.50% | RCV000116018.12; Likely pathogenic /Pathogenic | CM993368; LFS, IR: DFP | 75 | P-59 | BC, 58y |
| CHEK2 | c.538C>T | Exon 4 | p.(Arg180Cys) | rs77130927 | ALL: 0.09%; NFE: 0.12%; FIN: 0.03% | RCV000116024.8; Benign/VUS | CM030417; PC: DM | 76 | P-66 | BC, 53y |
| CHEK2 | c.1205_1206delinsTC | Exon 11 | p.(Ala402Val) | — | — | RCV000537997.1; VUSe | — | — | P-74 | BC, 54y |
| FANCF | c.1087C>T | Exon 1 | p.(Gln363*) | rs201285915 | ALL: 0.01%; NFE: 0.01%; FIN: 0% | RCV000482395.1; VUSb | CM1824108; BOC: DM | 20 | P-48 | BC, 39y |
| MLH1 | c.1665T>C | Exon 14 | p.(=) | rs749204990 | ALL: 0.00%, NFE: 0.00%; FIN: 0% | RCV000167487.2; Likely benign | — | — | P-65 | BC bilateral, 51y |
| MSH2 | c.1667T>C | Exon 11 | p.(Leu556Ser) | rs587779101 | — | RCV000076234.2; VUSc | CM148293; LS: DM? | 77 | P-73 | BC, 50y |
| MSH2 | c.2503A>C | Exon 15 | p.(Asn835His) | rs41295296 | ALL: 0.00%; NFE: 0.01%; FIN: 0% | RCV000115519.10; Likely benign; VUS | — | 78 | P-82 | BC, 37 |
| MSH6 | c.3259C>T | Exon 5 | p.(Pro1087Ser) | rs63750998 | ALL: 0.01%; NFE: 0.02%; FIN: 0% | RCV000074827.4; VUSc | CM1210418; OC: DM | 79 | P-81 | BC, 64y |
| NBN | c.643C>T | Exon 6 | p.Arg215Trp | rs34767364 | ALL: 0.25%; NFE: 0.40%; FIN: 0.32% | RCV000115802.12; Benign/Likely benign/ VUS | CM044022; CRC: DM | 80 | P-75 | BC, 49y |
| NF1 | c.378A>G | Exon 4 | p.(=) | — | — | —— | — | — | P-44 | BC, 49y |
| NF1 | c.469A>G | Exon 4 | p.(Ile157Val) | — | — | RCV000566319.1; VUS | — | — | P-91 | BC, 54y |
| NF1 | c.587-6_587-5delTT | Intron 5 | p.(?) | — | — | — | — | — | P-15 | BC bilateral, 74/80y |
| NF1 | c.960T>A | Exon 9 | p.(=) | rs376447070 | ALL: 0.02%; NFE: 0.01%; FIN: 0.17% | RCV000167230.1; likely benign | — | — | P-64 | BC, 35y |
| NF1 | c.4926A>G | Exon 37 | p.(=) | — | — | — | — | — | P-88 | BC, 48y |
| NF1 | c.5225A>G | Exon 37 | p.(Asn1742Ser) | rs745407845 | ALL: 0.00; NFE: 0.00%; FIN: 0% | RCV000206576.4; VUSd | CM1512958; NF1: DM | 81 | P-2 | BC, 57y |
| NF1 | c.5793T>C | Exon 39 | p.(=) | rs779114598 | ALL: 0.00%; NFE: 0.01%; FIN: 0% | RCV000167490.1; likely benign | — | 82 | P-22 | BC, 86y |
| NF1 | c.7354C>T | Exon 50 | p.(Arg2452Cys) | rs377662483 | ALL: 0.00; NFE: 0%; FIN: 0% | RCV000203720.5; VUSd | — | — | P-1 | BC, 36 |
| NF1 | c.7595C>T | Exon 51 | p.(Ala2532Val) | rs148154172 | ALL: 0.07%; NFE: 0.05%; FIN: 0.00% | RCV000130730.3; Likely benign | — | — | p-89 | BC, 47y |
| PMS2 | c.1765G>C | Exon 11 | p.(Asp589His) | rs749727182 | ALL: 0.00%; NFE: 0.00%; FIN: 0% | RCV000483031.2; VUSb | — | — | P-49 | BC, 41y |
| PMS2 | c.1936A>C | Exon 11 | p.(=) | rs369582237 | ALL: 0.00%; NFE: 0.01%; FIN: 0% | RCV000163542.2; likely benign | — | — | P-67 | BC, 51y |
Variants are named according to Human Genome Variation Society (HGVS) nomenclature. Local.: localization; Pr.Ref: Primary reference; diagn.: diagnosis; NFE: Non-Finnish Europeans; FIN: Finnish Europeans; DM: Disease mutation; CRC: colorectal cancer BC: Breast cancer; BOC: Breast and ovarian cancer syndrome; PC: Prostate cancer; CRC: Colorectal cancer; LS: Lynch syndrome; OC: Ovarian cancer; NF1: Neurofibromatosis, type 1; LFS,IR: Li-Fraumeni Syndrome, increased risk; DFP: Disease associated functional polymorphism; y: years. P-75 has two VUS’s, one in ATM and one in NBN. The two MLH1 variants are listed together since they only have been identified in cis in Norway. The ClinVar references and the corresponding clinical significance are mainly linked to the condition “Hereditary cancer-predisposing syndrome”, exceptions are marked.
aAtaxia telangiectasia.
bAllHighlyPenetrant.
cLynch syndrome reviewed by expert panel (InSiGHT).
dNeurofibromatosis, type 1.
eFamilieal cancer of breast.
Figure 2.
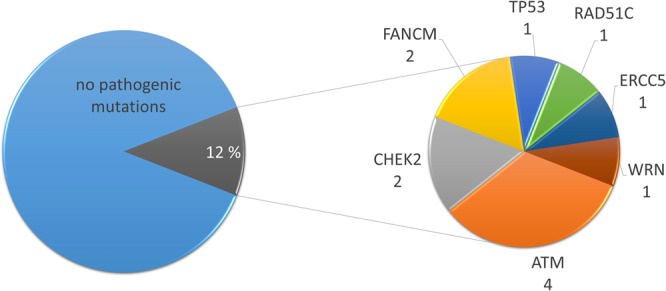
Distribution of patients with and without pathogenic/likely pathogenic variants. Twelve out of 101 patients were identified with pathogenic/likely pathogenic variants.
Variants according to diagnosis and age-groups
Four of the 12 patients with PVs/LPVs findings were diagnosed with BC between ages 50–59 years. Additional three patients with PVs/LPVs were diagnosed with BC between 30–39 years, 40–49 years and 60–69 years old. Five PVs/LPVs were identified in patients diagnosed with ovarian cancer. Interestingly, no PVs/LPVs/VUSs were identified in two patients diagnosed with both OC and BC (P-18 and P-68). In P-68 the only identified variant passed filtration was the likely benign MLH1 c.-7C>T, in cis (confirmed by manual investigation in the Integrative Genomics Viewer (IGV)) with MLH1 c.-28A>G (c.[-28A>G; -7C>T]) (Supplementary Table S2) and none were identified in P-18.
Variants in genes exclusively investigated for frameshift and stop variants
Five different variants were detected in the genes that were exclusively investigated for frameshift and stop variants. These five variants were located in ERCC5, FANCF, FANCM and WRN and were found in P-8, P-41, P-44, P-48 and P-90 (Tables 2 and 3). One of the patients (P-48) had two nonsense variants, in two different genes, FANCF c.1087C>T p.(Gln363*) and WRN c.4216C>T p.(Arg1406*). The FANCF variant was classified as VUS (Table 3) as this gene consists of only one exon and the variant therefor presumably results in the loss of the terminal 12 amino acids of a protein region, instead of undergoing nonsense mediated mRNA decay (NMD). However, the variant has a low population allele frequency in gnomAD (0.0071%) and has previously been reported in the literature as pathogenic by Quezada Urban et al.20. In ClinVar, it is reported as a VUS. The WRN c.4216C>T was classified as non-pathogenic due to its high allele frequency in the South Asian population (1.7% and 10 homozygotes in gnomAD). Additionally, this variant was classified as benign in ClinVar and listed as “DM?” (“Disease causing mutation?” = Variant reported as likely disease causing, but with questionable pathogenicity) in the Human Gene Mutation Database (HGMDp).
Two interesting cases
Five of the 12 patients with a PV/LPV also had a VUS in one of the 16 genes more closely scrutinized (Tables 1 and 2). One of these patients (P-31) was diagnosed with OC and was heterozygous for the pathogenic sequence variant c.3245_3247delinsTGAT in ATM. Her sister (P-32), who was equally diagnosed with OC and likewise included in this study, did not have this variant. However, both sisters were heterozygous for a VUS in BRIP1 (c.2087C>T p.(Pro696Leu)). Figure 3 depicts the pedigree of the two sisters (P-31 and P-32).
Figure 3.
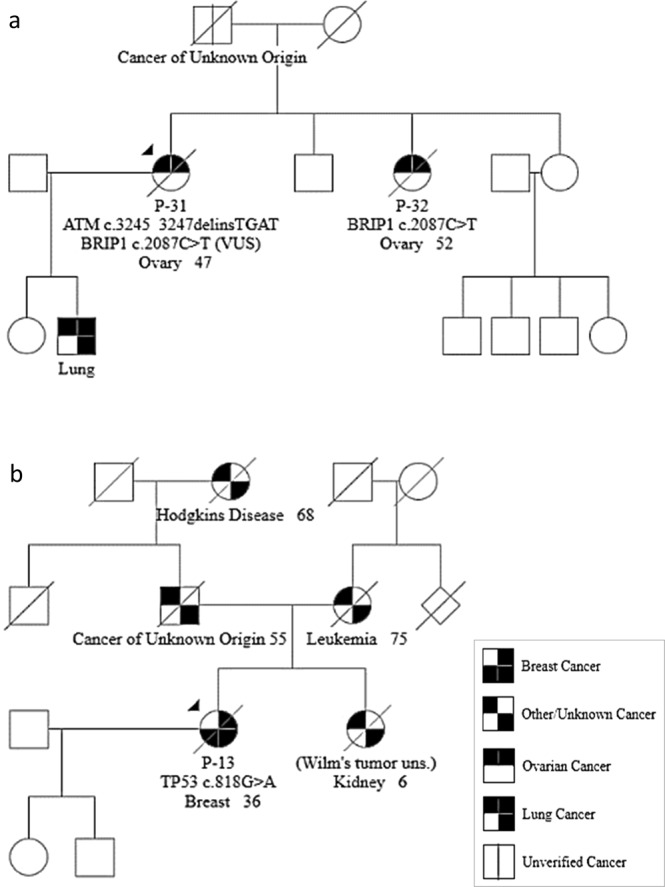
Illustration of two pedigrees. (a) Pedigree of P-31 and P-32 (ATM c.3245_3247delinsTGAT and BRIP1 c.2087C>T). (b) Pedigree of P-13 (TP53 c.818G>A).
The TP53 variant identified in this study was the pathogenic c.818G>A. This variant was identified in patient P-13, whose pedigree does not resemble a classic Li-Fraumeni family (Fig. 3).
Discussion
In this study, 101 patients were screened for the presence of deleterious sequence variants in 94 cancer associated genes. We identified PVs/LPVs in 12 patients, in seven different genes (Table 2 and Fig. 2). In total, 9 different PV/LPVs were identified, including one novel sequence variant in ERCC5 (c.67G>T). In addition, we detected 32 VUSs including six variants not previously described (Table 3).
These findings correspond to a total finding percentage of 12% in the investigated patient cohort (Fig. 2), which was in concordance with Pinto et al., Aloraifi et al. and Schubert et al.11,15,21. However, these finding percentages were higher than for several other studies (ranging from 4.7–9% in BRCA1/2-negative patients)9,14,16–19,22–24. In several of these studies, the majority of PVs/LPVs were identified in ATM, CHEK2 and PALB2. In our study, the majority of the PVs/LPVs were identified in ATM and CHEK2, in four and two patients, respectively (Fig. 2). However, whereas several studies identified PVs in PALB2, we did not detect any. Comparison of these studies was challenging, since the gene panels and/or the studied cohort differed between most of them.
The study included in total 83 patients with BC and 20 patients with OC (two overlapping, diagnosed with both BC and OC) (Fig. 1). Seven of the patients diagnosed with BC were found to carry a PV/LPV, corresponding to findings in 8.4%. Furthermore, five of the 20 patients (25%) diagnosed with OC were found to carry a PV/LPV. These percentages correspond well with the estimated disease burden of BC and OC cases due to inherited PV/LPVs, 5–10% and ~25%, respectively25,26. However, in the current study, a small number of patients were included and caution should be taken when comparing with other studies. Screening of a larger amount of patients might change the observed finding percentage.
One of the most frequently identified mutated genes in the studied cohort was ATM. Biallelic deleterious ATM variants cause Ataxia Telangiectasia (A-T)27. However, heterozygous carriers of deleterious variants have an increased risk of breast cancer28–32. Although we do not diagnostically test for ATM variants in HBOC families, patients with a family history of BC/OC and an identified PV/LPV in ATM (sequenced elsewhere) are offered additional follow-up, including mammography from 40 years of age. This has been established in concordance with the other Departments of Medical Genetics in Norway. However, the cancer risk of PVs in ATM may still be debatable. Patient P-2, diagnosed with BC at 57 years of age, was a carrier of the pathogenic ATM c.3245_3247delinsTGAT (Table 2). This variant has previously been identified as a pathogenic variant and is a Norwegian founder mutation33. The same variant was also identified in a patient diagnosed with OC (P-31). The latter patient had a sister (P-32) who was diagnosed with OC at age 52. However, the ATM c.3245_3247delinsTGAT variant was exclusively present in P-31 (Fig. 3). Furthermore, both sisters were carriers of a VUS in BRIP1 (c.2087C>T p.(Pro696Leu)) (Table 3). Pathogenic variants in BRIP1 have been associated with increased risk of OC34–36, but it remains to be investigated whether the germline variant in BRIP1 is the cause for the ovarian cancers in both sisters.
Another ATM variant identified in our cancer cohort was the pathogenic c.5932G>T variant. This variant was identified in a woman (P-91) diagnosed with BC at age 54. ATM c.5932G>T is predicted to be a nonsense variant, p.(Glu1978*), however, this variant has previously been shown to be a splice-affecting variant resulting in skipping of exon 40 and introducing a premature stop codon; p.Ser1974Ilefs*437. This variant has been shown to be associated with HBOC in several studies16,37,38.
A third ATM variant was the c.8432delA p.(Lys2811Serfs*46). This variant was identified in a patient diagnosed with OC at age 38.
The three variants in ATM found in the present study were frameshift variants leading to a premature stop-codon and have been previously identified as disease associated variants identified in the Scandinavian A-T cohort33. It has long been debated whether a monoallelic truncating ATM variant may increase cancer risks. Some studies indicate that truncating variants lead to increased cancer risk28,29,31, whereas others claim that missense variants exerting a dominant negative outcome are responsible for the associated increased cancer risk. In a meta-analysis of ATM variants, published by Tavtigian and colleagues (2009)30, they found marginal evidence that protein-truncating and splice-junction variants contribute to breast cancer risk, and stronger evidence that some evolutionary rare missense variants increase cancer risk.
The likely pathogenic CHEK2 c.319+2T>A variant identified in this study has previously been identified in another Norwegian patient diagnosed with thyroid cancer at age 31, BC at 43 and 48. Her family history included both BC and endometrial cancer39. Two of the patients in our cohort were carriers of this CHEK2 variant (P-12 and P-16; Table 2). P-12 was diagnosed with OC at age 27, while P-16 was diagnosed with OC at age 70. Interestingly, P-12 was also heterozygous for a VUS, the novel CDH1 c.136C>G p.(Leu46Val).
Another interesting CHEK2 variant is c.470T>C p.(Ile157Thr) in exon 4. This variant is well characterized and proposed as low-penetrant variant which is estimated to give a lifetime BC risk of 18.3%40. This variant was identified in P-59, diagnosed with BC at age 58 The variant is however categorized as a VUS, due to the high allele frequency in the Finnish population in gnomAD (2.50%), although this may be in concordance with the low increase in BC risk.
Deleterious variants in TP53 are the cause of Li-Fraumeni syndrome (LFS), a cancer predisposition syndrome associated with the development of various tumours: soft tissue sarcoma, osteosarcoma, pre-menopausal breast cancer, brain tumours, adrenocortical carcinoma and leukemias41. There is also an increased risk for Wilms’ tumour, skin, gastrointestinal, lung, endometrial, ovarian, prostate and gonadal germ cell cancers41,42. P-13 was a carrier of the known pathogenic sequence variant c.818G>A p.(Arg273His) in TP5343–46. However, the patient’s family does not meet the classic LFS criteria nor the revised Chompret criteria for LFS (Fig. 3)41. The patient was diagnosed with an early-onset BC (36 years), had a sister diagnosed with Wilms’ tumour at age 6 and a father with a cancer of unknown origin diagnosed at age 55, thereby fulfilling the Birch criteria for LFS-like47. Knowledge of her family history was sparse, which might explain why the Li-Fraumeni/Chompret criteria were not met for TP53 testing. Today, the family would have been offered testing for sequence variants in TP53, amongst others. The TP53 c.818G>A p.(Arg273His) variant, identified in this family, is located at a position in the TP53 gene which is characterized as a common hotspot for somatic mutations48. The variant was identified in 33% of the sequence reads from P-13. Somatic pathogenic sequence variants in TP53 have been shown to increase in blood of women who have endured chemotherapy treatment49. Accordingly, this patient might have a somatic sequence variant. However, a skewed amount of reads may also be due to a technical artefact. Further family studies are needed to determine the nature of this variant.
Although several NGS studies of patients with BC and/or OC have identified LPVs/PVs in the MMR genes9,12–14,17,23, we did not identify pathogenic variants in MLH1, MSH2, MSH6 or PMS2. We identified the MLH1 c.[-28A>G; -7C>T] in three patients. These variants are located in cis and have been shown to reduce the expression of MLH1 by 50% from this allele50. However, according to gnomAD these variants are identified with a minor allele frequency of 0.8% in the Finnish population. Furthermore, as there is still 50% MLH1 tumour suppressor function from the mutated allele50, it may provide a sufficient amount of MLH1 transcripts and accordingly not contribute to an increased cancer risk. Morak et al. investigated the promoter region of MLH1 in 480 patients with colorectal cancer (CRC) and 1150 controls. They identified the variant in an individual with MLH1-proficient CRC and two individuals with non-Lynch syndrome tumours, all part of one of the control groups in the study. Additionally, they found biallelic expression in cDNA from the three individuals with this variant.
RAD51C c.1026+5_1026+7delGTA was identified in P-69, diagnosed with OC at age 52 and the family history included BC, OC and prostate cancer. Janatova and colleagues (2015) identified this variant in a patient diagnosed with OC and later endometrial cancer. They classified this variant as likely pathogenic as it affects splicing by causing skipping of exon 8, resulting in a frameshift with an premature stop codon (p.Arg322Serfs*22)51. Only one other pathogenic RAD51C variant has been identified in the Norwegian population, as far as we know.
A novel ERCC5 c.67G>T p.(Glu23*) was identified in a woman diagnosed with BC at age 49 (P-44) (Table 2). The variant is predicted to introduce a stop codon, which will lead to transcripts that might be targeted for nonsense mediated mRNA decay (NMD). If ERCC5 is synthesized, it will lack most of the protein sequence. In addition, the variant is predicted to introduce a new cryptic 5′ splice site (ss) one nucleotide up-stream. The outcome of aberrant splicing using this cryptic splice site would lead to skipping of 23 nucleotides and a subsequent frameshift, introducing a premature stop codon (p.(Glu23Tyrfs*2)). Another possibility is that of an alternative translational start site down-stream of this variant, since it is located in the first exon of the gene. However, the next in-frame start-codon is Met169 in exon 5. Usage of this methionine as a start codon has not been reported.
The LPV FANCM c.5101C>T, p.(Gln1701*) was identified in two patients; P-8 and P-41 (Table 2). P-8 was diagnosed with BC at 56 years of age and P-41 was diagnosed with BC at age 69. Pathogenic variants in FANCM, including this variant, have previously been reported to confer an increased risk of BC21,52–55.
One of the patients in the study (P-90) carried the WRN variant c.1105C>T p.(Arg369*) and was diagnosed with BC at 57 years of age (Table 2). This variant introduces an early stop codon and has previously been reported in ClinVar and HGMDp as a pathogenic and disease mutation, respectively, in patients with Werner syndrome. Werner syndrome is an autosomal recessive disease characterized by the early appearance of features associated with normal aging and increased cancer risk56. Accordingly, heterozygous carriers might have an increased cancer risk57. This assumption is supported by another NGS study of breast cancer patients that identified a deleterious WRN sequence variant (c.4245dupT, p.(Asp1416*))13. In addition, Ding and colleagues (2007) have also reported association between WRN and breast cancer58.
For some of these variants, such as the variants in ERCC5 and WRN, the link between a heterozygous pathogenic variant and BC/OC is not well defined. For women carrying these variants, there is no clinical benefit from the discovery of these variant as there are currently no management plans or reliable risk data. However, the discovery of such variants in patients with BC/OC may in the future lead to better-documented associations, and subsequently to reliable risk data and management plans for these patients.
NGS gene panels generally has its limitations; variants in non-target regions cannot be detected, some regions have gaps due to insufficient probe coverage, pseudogenes can cause misalignments of reads, repetitive segments can create technical artefacts reported as deletions/insertions, deletions covering entire exons may not be detected, etc. Additional BC and OC cases might have been resolved if we had resequenced the gaps using Sanger sequencing, as well as investigated untranslated regions and regions further out in introns than +/−10 nucleotides. Furthermore, no copy number variation analysis using NGS-data or MLPA was used to investigate these genes; accordingly, large deletions or duplications could go undetected.
The challenge with pseudogenes is well illustrated with the PMS2 gene, which has several. Amongst these pseudogenes, one in particular confers problems during NGS, the PMS2CL. This pseudogene consists of exons almost identical to exon 9 and 11–15, including intronic sequences. Accordingly, the software has difficulties in aligning the sequences to the correct genomic position. Two of our samples, P-56 and P-57 (from the same family), initially seemed to have a deletion of a part of the PMS2 gene. However, secondary evaluations of reads using IGV revealed that most of the reads aligned with the PMS2CL reference sequence. This may be the result of gene conversion between PMS2 and PMS2CL59. Gene conversion might mask variants due to faulty alignment of reads to both PMS2 and PMS2CL. Consequently, both genes should therefore be manually investigated in IGV. Alternatively, to prevent overlooking PVs in PMS2, examination of PMS2 cDNA, as proposed by van der Klift and colleagues, could be included in the screening for PVs60.
Our current study is starting to reveal the diversity of genetic cancer risk factors in a Norwegian cancer cohort. However, a much larger patient study is warranted to assess the appropriate distribution of variants in Norway. Additionally, several sequence variants were identified, for which the clinical significance is currently unknown. Accordingly, there is a need for robust functional assays to study the biological consequences of these variants. The study demonstrates the necessity for more knowledge from similar studies and the investigation of families with these PVs/LPVs. Increased knowledge may contribute to the development of new and more specific clinical management programs.
Patients and Methods
Patients and samples
This study included samples from 101 (P-1–P-101) Norwegian patients from 93 unrelated families (referred to the Department of Medical Genetics at the University Hospital of North Norway) diagnosed with BC and/or OC) (Supplementary Table S1). All patients had previously been screened for PVs in BRCA1 and BRCA2, using Sanger sequencing/NGS and multiplex ligation-dependent probe amplification (MLPA), but no PVs, LPVs or VUSs were identified.
The cancer patients included in this study were divided in three groups, according to how they were recruited. Group 1 (n = 32) and 2 (n = 46) included patients previously tested for PVs in BRCA1/2 by Sanger sequencing. Samples from these patients were resequenced using the NGS technology. Group 1 represented samples from deceased patients and group 2 samples from surviving patients. Group 3 (n = 23) included patient samples previously sequenced using NGS technology, but where only BRCA1/2 had been analyzed. The sequence data for the additional 92 genes was available for group 3 patients and were further analyzed in this study.
For group 1 and 2, blood stored in the diagnostic biobank at the department was used. Genomic DNA was extracted using QIAsymphony (QIAGEN, Hilden, Germany) with the QIAsymphony DNA Mini Kit (QIAGEN), according to the manufacturer’s protocol.
Compliance with Ethical Standards
The project was approved by the Norwegian Regional Ethics Committee (ref. nr. 2016/980) and all experiments were performed in accordance with guidelines/regulations. The committee allowed inclusion of samples from deceased patients (group 1) without informed consent. The committee approved exemption from written informed consent from patients in group 2, where passive informed consent was obtained instead. Written informed consent was obtained from patients in group 3.
Analysed cancer genes
The TruSight cancer sequencing kit (Illumina, San Diego, CA, USA) containing probes to enrich 94 cancer related genes was used. All 94 genes were scrutinized for nonsense and frameshift variants, and 16 genes previously associated with BC or OC (Table 1) were investigated for all types of sequence variations. We also verified the normal results from the previous screening of BRCA1/2.
Library preparation and sequencing
Patients DNA samples were quantified using the Qubit dsDNA High Sensitivity (HS) assay kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) and measured on a Qubit 3.0 Fluorometer (Invitrogen, Thermo Fisher Scientific) according to manufacturer’s protocol. Quantification of DNA samples was performed prior to DNA tagmentation, before DNA libraries were pooled, and for end-library validation. The HS DNA kit and the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) were used according to manufacturer’s protocol for size-determination of tagmented fragments. Libraries were produced using the TruSight Rapid Capture kit (24 indexes) (Illumina) together with the TruSight Cancer sequencing panel. Sequencing was performed on a MiSeq Sequencer (Illumina).
Sequencing data analysis
Alignment and variant calling were performed using the MiSeq reporter software (version 2.6.2.3). The MiSeq reporter aligns the sequence reads against the reference genome hg19 using the Burrows-Wheeler Aligner (BWA) and calls variants using the Genome Analysis Toolkit (GATK).
Annotation and filtration of the sequenced variants were done using the Cartagenia Bench NGS software (Agilent). Variants were filtered based on call quality (≥30), genotype quality (≥20), R8 (deletion/insertion after eight mononucleotide or dinucleotide repeats), variant allele frequencies (≥0.2) and read depth (≥18). Variants which passed the quality filters were then filtered based on population allele frequencies (<1% in total populations of ESP6500, ExAC, 1000 genomes Phase 1 and 3, and dbSNP) and position in the gene (exons and up to +/−10 in introns).
The Integrative Genomics Viewer (IGV) (Broad Institute, Cambridge, MA, USA; https://www.broadinstitute.org/igv/) was used for manual inspection of certain regions. These regions included reported gaps (<30 reads) by the analysis software, inspection of variants passed filtering and the entire PMS2 gene, together with its pseudogene PMS2CL. In addition, one position was manually investigated for all samples, chr2:47641560 for variant MSH2 c.942+3A>T (intron 5). The position of the MSH2 variant needed manual investigation due to a poly A-stretch, inducing technical deletions/insertions artefacts that might mask this variant61.
Nomenclature
Variants were named following the guidelines proposed by the Human Genome Variation Society (HGVS) nomenclature62. Reference sequences used are listed in Table 1, and custom exon numbering was used for BRCA1 (missing exon 4).
Classification and Sanger sequencing confirmation
Primers were designed using the Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/) and evaluated using SNPCheck3 (www.snpcheck.net/). Primers were excluded if they aligned to sites that covered three or more single nucleotide polymorphism (SNP), if they included SNPs with a minor allele frequency above 0.5% or if SNPs occurred in the last five nucleotides of the primers63,64. Primers are listed in Supplementary Table S3. All primers included M13 forward and M13 reverse primer sequences, respectively, for sequencing purposes (M13.F: 5′-tgtaaaacgacggccagt-3′ and M13.R. 5′-caggaaacagctatgacc-3′).
In silico evaluation of the variants was done using Alamut® Visual v.2.11.0 (Interactive Biosoftware, Rouen, France), which includes the missense prediction programs Align GVGD, SIFT, MutationTaster and PolyPhen-2. Alamut also contains the splice prediction tools SpliceSiteFinder-like (SSF), MaxEntScan (MES), NNSPLICE, GeneSplicer (GS) and Human Splicing Finder (HSF). In addition, Alamut interactive software provides results and/or links to the following databases used in this study: the Exome Aggregation Consortium (ExAC)/the Genome Aggregation Database (gnomAD), the Exome Variant Server (EVS), the Database of Short Genetic Variation (dbSNP) and ClinVar. The Human Gene Mutation Database Professional (HGMDp) was queried independently.
Classification of variants was performed based on the ACMG guidelines65, with some modifications leading to stricter classification criteria.
Supplementary information
Acknowledgements
We would like to thank the members of the hereditary cancer research group at the Department of Medical Genetics at the University Hospital of North Norway. Additionally, we thank the patients for their consent to participate in this study, and thank Evy Johansen for administrative support. We would also like to thank Helse Nord, the Division of Child and Adolescent Health at the University Hospital of North Norway and Odd Fellow for financial support. The experiments reported here also feature in the doctoral thesis of E. J. This study was funded by Helse Nord (grant number SFP1161-14), the Division of Child and Adolescent Health at the University Hospital of North Norway and Odd Fellow - Medical Science Research Fund.
Author contributions
E.J. and S.S. carried out the experiments and analysed/interpreted the data. H.M.F.R.S., G.Å.M.H. and M.V.G. reviewed the interpretations. E.J. wrote the manuscript with the main support from H.M.F.R.S. and M.V.G., and additional support from G.Å.M.H., C.J., M.I. and N.S.. N.S. was responsible for inclusion of patients to the study and additional patient information, while M.I. and G.Å.M.H. contributed to the anonymization of samples. H.M.F.R.S. and M.V.G. supervised the project.
Data availability
The raw sequencing datasets generated during and/or analysed during the current study are not publicly available due to the privacy law/data protection law, which prohibit the disclosure or misuse of information about private individuals. However, screenshots from IGV of the reported sequence variants and surrounding regions can be obtained from the corresponding authors on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Elisabeth Jarhelle, Email: elisabeth.jarhelle@unn.no.
Marijke Van Ghelue, Email: marijke.van.ghelue@unn.no.
Supplementary information
is available for this paper at 10.1038/s41598-019-55515-x.
References
- 1.Cancer Registry of Norway. Cancer in Norway 2017 - Cancer incidence, mortality, survival and prevalence in Norway. (Oslo: Cancer Registry of Norway, 2018).
- 2.Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015;41:1–8. doi: 10.1016/j.ctrv.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 4.Wooster R, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 5.Frank TS, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J. Clin. Oncol. 2002;20:1480–1490. doi: 10.1200/JCO.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 6.Kast K, et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016;53:465–471. doi: 10.1136/jmedgenet-2015-103672. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat. Rev. Cancer. 2016;16:599–612. doi: 10.1038/nrc.2016.72. [DOI] [PubMed] [Google Scholar]
- 8.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buys SS, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017 doi: 10.1002/cncr.30498. [DOI] [PubMed] [Google Scholar]
- 10.Yadav S, Reeves A, Campian S, Paine A, Zakalik D. Outcomes of retesting BRCA negative patients using multigene panels. Fam. Cancer. 2017;16:319–328. doi: 10.1007/s10689-016-9956-7. [DOI] [PubMed] [Google Scholar]
- 11.Pinto P, et al. Implementation of next-generation sequencing for molecular diagnosis of hereditary breast and ovarian cancer highlights its genetic heterogeneity. Breast Cancer Res. Treat. 2016;159:245–256. doi: 10.1007/s10549-016-3948-z. [DOI] [PubMed] [Google Scholar]
- 12.LaDuca H, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet. Med. 2014;16:830–837. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang X, et al. Identification of a comprehensive spectrum of genetic factors for hereditary breast cancer in a Chinese population by next-generation sequencing. PLoS One. 2015;10:e0125571. doi: 10.1371/journal.pone.0125571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tedaldi, G. et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget, 10.18632/oncotarget.16791 (2017). [DOI] [PMC free article] [PubMed]
- 15.Aloraifi F, et al. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 2015;282:3424–3437. doi: 10.1111/febs.13352. [DOI] [PubMed] [Google Scholar]
- 16.Susswein Lisa R., Marshall Megan L., Nusbaum Rachel, Vogel Postula Kristen J., Weissman Scott M., Yackowski Lauren, Vaccari Erica M., Bissonnette Jeffrey, Booker Jessica K., Cremona M. Laura, Gibellini Federica, Murphy Patricia D., Pineda-Alvarez Daniel E., Pollevick Guido D., Xu Zhixiong, Richard Gabi, Bale Sherri, Klein Rachel T., Hruska Kathleen S., Chung Wendy K. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genetics in Medicine. 2015;18(8):823–832. doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tung N, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraus C, et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer. 2017;140:95–102. doi: 10.1002/ijc.30428. [DOI] [PubMed] [Google Scholar]
- 19.Singh Jaya, Thota Nishita, Singh Suhasini, Padhi Shila, Mohan Puja, Deshwal Shivani, Sur Soumit, Ghosh Mithua, Agarwal Amit, Sarin Ramesh, Ahmed Rosina, Almel Sachin, Chakraborti Basumita, Raina Vinod, DadiReddy Praveen K., Smruti B. K., Rajappa Senthil, Dodagoudar Chandragouda, Aggarwal Shyam, Singhal Manish, Joshi Ashish, Kumar Rajeev, Kumar Ajai, Mishra Deepak K., Arora Neeraj, Karaba Aarati, Sankaran Satish, Katragadda Shanmukh, Ghosh Arunabha, Veeramachaneni Vamsi, Hariharan Ramesh, Mannan Ashraf U. Screening of over 1000 Indian patients with breast and/or ovarian cancer with a multi-gene panel: prevalence of BRCA1/2 and non-BRCA mutations. Breast Cancer Research and Treatment. 2018;170(1):189–196. doi: 10.1007/s10549-018-4726-x. [DOI] [PubMed] [Google Scholar]
- 20.Quezada Urban Rosalía, Díaz Velásquez Clara, Gitler Rina, Rojo Castillo María, Sirota Toporek Max, Figueroa Morales Andrea, Moreno García Oscar, García Esquivel Lizbeth, Torres Mejía Gabriela, Dean Michael, Delgado Enciso Iván, Ochoa Díaz López Héctor, Rodríguez León Fernando, Jan Virginia, Garzón Barrientos Víctor, Ruiz Flores Pablo, Espino Silva Perla, Haro Santa Cruz Jorge, Martínez Gregorio Héctor, Rojas Jiménez Ernesto, Romero Cruz Luis, Méndez Catalá Claudia, Álvarez Gómez Rosa, Fragoso Ontiveros Verónica, Herrera Luis, Romieu Isabelle, Terrazas Luis, Chirino Yolanda, Frecha Cecilia, Oliver Javier, Perdomo Sandra, Vaca Paniagua Felipe. Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility. Cancers. 2018;10(10):361. doi: 10.3390/cancers10100361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schubert S, et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer. 2019;144:2683–2694. doi: 10.1002/ijc.31992. [DOI] [PubMed] [Google Scholar]
- 22.Slavin TP, et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer. 2017;3:22. doi: 10.1038/s41523-017-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Leary E, et al. Expanded Gene Panel Use for Women With Breast Cancer: Identification and Intervention Beyond Breast Cancer Risk. Ann. Surg. Oncol. 2017;24:3060–3066. doi: 10.1245/s10434-017-5963-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castera L, et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018;20:1677–1686. doi: 10.1038/s41436-018-0005-9. [DOI] [PubMed] [Google Scholar]
- 25.Caminsky NG, et al. Prioritizing Variants in Complete Hereditary Breast and Ovarian Cancer Genes in Patients Lacking Known BRCA Mutations. Hum. Mutat. 2016;37:640–652. doi: 10.1002/humu.22972. [DOI] [PubMed] [Google Scholar]
- 26.Walsh T, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 28.Goldgar DE, et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13:R73. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollestelle A, Wasielewski M, Martens JW, Schutte M. Discovering moderate-risk breast cancer susceptibility genes. Curr. Opin. Genet. Dev. 2010;20:268–276. doi: 10.1016/j.gde.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Tavtigian SV, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am. J. Hum. Genet. 2009;85:427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pylkas K, et al. Evaluation of the role of Finnish ataxia-telangiectasia mutations in hereditary predisposition to breast cancer. Carcinogenesis. 2007;28:1040–1045. doi: 10.1093/carcin/bgl237. [DOI] [PubMed] [Google Scholar]
- 32.Renwick A, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 33.Laake, K. et al. Characterization of ATM mutations in 41 Nordic families with ataxia telangiectasia. Hum. Mutat. 16, 232–246, https://doi.org/10.1002/1098-1004(200009)16:3<232::AID-HUMU6>3.0.CO;2-L (2000). [DOI] [PubMed]
- 34.Rafnar T, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 35.Ramus, S. J. et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 107, 10.1093/jnci/djv214 (2015). [DOI] [PMC free article] [PubMed]
- 36.Norquist BM, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA. Oncol. 2016;2:482–490. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Telatar M, et al. Ataxia-telangiectasia: identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am. J. Hum. Genet. 1998;62:86–97. doi: 10.1086/301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogdanova N, et al. A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer. Breast Cancer Res. Treat. 2009;118:207–211. doi: 10.1007/s10549-008-0189-9. [DOI] [PubMed] [Google Scholar]
- 39.Dominguez-Valentin M, et al. Potentially pathogenic germline CHEK2 c.319 + 2T > A among multiple early-onset cancer families. Fam. Cancer. 2018;17:141–153. doi: 10.1007/s10689-017-0011-0. [DOI] [PubMed] [Google Scholar]
- 40.Tung N, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat. Rev. Clin. Oncol. 2016;13:581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider, K., Zelley, K., Nichols, K. E. & Garber, J. Li-Fraumeni Syndrome, https://www.ncbi.nlm.nih.gov/books/NBK1311/ (Jan 19, 1999).
- 42.Dome, J. S. & Huff, V. Willms Tumor Predisposition, https://www.ncbi.nlm.nih.gov/books/NBK1294/ (Dec 19, 2003).
- 43.Moran O, et al. Revisiting breast cancer patients who previously tested negative for BRCA mutations using a 12-gene panel. Breast Cancer Res. Treat. 2017;161:135–142. doi: 10.1007/s10549-016-4038-y. [DOI] [PubMed] [Google Scholar]
- 44.Park KJ, Choi HJ, Suh SP, Ki CS, Kim JW. Germline TP53 Mutation and Clinical Characteristics of Korean Patients With Li-Fraumeni Syndrome. Ann. Lab. Med. 2016;36:463–468. doi: 10.3343/alm.2016.36.5.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zerdoumi Y, et al. Germline TP53 mutations result into a constitutive defect of p53 DNA binding and transcriptional response to DNA damage. Hum. Mol. Genet. 2017;26:2812. doi: 10.1093/hmg/ddx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, et al. Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum. Mutat. 2014;35:575–584. doi: 10.1002/humu.22528. [DOI] [PubMed] [Google Scholar]
- 47.Birch JM, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 48.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swisher EM, et al. Somatic Mosaic Mutations in PPM1D and TP53 in the Blood of Women With Ovarian Carcinoma. JAMA Oncol. 2016;2:370–372. doi: 10.1001/jamaoncol.2015.6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hesson LB, et al. Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression. Hum. Mutat. 2015;36:622–630. doi: 10.1002/humu.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janatova M, et al. Mutation Analysis of the RAD51C and RAD51D Genes in High-Risk Ovarian Cancer Patients and Families from the Czech Republic. PLoS One. 2015;10:e0127711. doi: 10.1371/journal.pone.0127711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katsuki Y, Takata M. Defects in homologous recombination repair behind the human diseases: FA and HBOC. Endocr. Relat. Cancer. 2016;23:T19–37. doi: 10.1530/ERC-16-0221. [DOI] [PubMed] [Google Scholar]
- 53.Kiiski JI, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA. 2014;111:15172–15177. doi: 10.1073/pnas.1407909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neidhardt G, et al. Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterlongo P, et al. FANCM c.5791C > T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum. Mol. Genet. 2015;24:5345–5355. doi: 10.1093/hmg/ddv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oshima, J., Martin, G. M. & Hisama, F. M. Werner Syndrome, https://www.ncbi.nlm.nih.gov/books/NBK1514/ (Dec 2, 2002).
- 57.Wang Z, et al. A polymorphism in Werner syndrome gene is associated with breast cancer susceptibility in Chinese women. Breast Cancer Res. Treat. 2009;118:169–175. doi: 10.1007/s10549-009-0327-z. [DOI] [PubMed] [Google Scholar]
- 58.Ding SL, Yu JC, Chen ST, Hsu GC, Shen CY. Genetic variation in the premature aging gene WRN: a case-control study on breast cancer susceptibility. Cancer Epidemiol. Biomarkers Prev. 2007;16:263–269. doi: 10.1158/1055-9965.EPI-06-0678. [DOI] [PubMed] [Google Scholar]
- 59.Hayward BE, et al. Extensive gene conversion at the PMS2 DNA mismatch repair locus. Hum. Mutat. 2007;28:424–430. doi: 10.1002/humu.20457. [DOI] [PubMed] [Google Scholar]
- 60.van der Klift HM, et al. Quantification of sequence exchange events between PMS2 and PMS2CL provides a basis for improved mutation scanning of Lynch syndrome patients. Hum. Mutat. 2010;31:578–587. doi: 10.1002/humu.21229. [DOI] [PubMed] [Google Scholar]
- 61.Mu W, Lu HM, Chen J, Li S, Elliott AM. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J. Mol. Diagn. 2016;18:923–932. doi: 10.1016/j.jmoldx.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 62.den Dunnen, J. T. & Antonarakis, S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12, https://doi.org/10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N (2000). [DOI] [PubMed]
- 63.Untergasser A, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 65.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vorechovsky I, et al. ATM mutations in cancer families. Cancer Res. 1996;56:4130–4133. [PubMed] [Google Scholar]
- 67.Li A, Swift M. Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. Am. J. Med. Genet. 2000;92:170–177. doi: 10.1002/(SICI)1096-8628(20000529)92:3<170::AID-AJMG3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 68.Grunert M, et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum. Mol. Genet. 2014;23:3115–3128. doi: 10.1093/hmg/ddu021. [DOI] [PubMed] [Google Scholar]
- 69.Golmard L, et al. Germline mutation in the RAD51B gene confers predisposition to breast cancer. BMC Cancer. 2013;13:484. doi: 10.1186/1471-2407-13-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malkin D, et al. Germline mutations of the p53 tumor-suppressor gene in children and young adults with second malignant neoplasms. N. Engl. J. Med. 1992;326:1309–1315. doi: 10.1056/NEJM199205143262002. [DOI] [PubMed] [Google Scholar]
- 71.Oshima J, et al. Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum. Mol. Genet. 1996;5:1909–1913. doi: 10.1093/hmg/5.12.1909. [DOI] [PubMed] [Google Scholar]
- 72.Skowronska A, et al. ATM germline heterozygosity does not play a role in chronic lymphocytic leukemia initiation but influences rapid disease progression through loss of the remaining ATM allele. Haematologica. 2012;97:142–146. doi: 10.3324/haematol.2011.048827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearlman R, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017;3:464–471. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teraoka SN, et al. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001;92:479–487. doi: 10.1002/1097-0142(20010801)92:3<479::AID-CNCR1346>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 75.Bell DW, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 76.Dong X, et al. Mutations in CHEK2 associated with prostate cancer risk. Am. J. Hum. Genet. 2003;72:270–280. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Loizidou MA, et al. The mutational spectrum of Lynch syndrome in cyprus. PLoS One. 2014;9:e105501. doi: 10.1371/journal.pone.0105501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.South SA, et al. Consideration of hereditary nonpolyposis colorectal cancer in BRCA mutation-negative familial ovarian cancers. Cancer. 2009;115:324–333. doi: 10.1002/cncr.24012. [DOI] [PubMed] [Google Scholar]
- 79.Pal T, et al. Frequency of mutations in mismatch repair genes in a population-based study of women with ovarian cancer. Br. J. Cancer. 2012;107:1783–1790. doi: 10.1038/bjc.2012.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steffen J, et al. Increased cancer risk of heterozygotes with NBS1 germline mutations in Poland. Int. J. Cancer. 2004;111:67–71. doi: 10.1002/ijc.20239. [DOI] [PubMed] [Google Scholar]
- 81.Bianchessi D, et al. 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol Genet Genomic Med. 2015;3:513–525. doi: 10.1002/mgg3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen-Dumont T, et al. Description and validation of high-throughput simultaneous genotyping and mutation scanning by high-resolution melting curve analysis. Hum. Mutat. 2009;30:884–890. doi: 10.1002/humu.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing datasets generated during and/or analysed during the current study are not publicly available due to the privacy law/data protection law, which prohibit the disclosure or misuse of information about private individuals. However, screenshots from IGV of the reported sequence variants and surrounding regions can be obtained from the corresponding authors on reasonable request.