

Abstract
Fusarium proliferatum causes diverse diseases of many economically important plants. The fungus produces several mycotoxins of which the fumonisins are the most toxic. Currently, deletion of key genes for mycotoxin biosynthesis is a laborious and time-consuming procedure. We developed a novel CRISPR/Cas9-based genome-editing tool for the direct delivery of preassembled Cas9 ribonucleoproteins into protoplasts of F. proliferatum. Our CRISPR–Cas9 system couples a site-specific double-strand DNA break mediated by two Cas9 ribonucleoproteins with microhomology recombination requiring only 50-bp regions flanking the target gene. This system reduces the risk of off-target mutations and minimizes the risk of altering any gene adjacent to the target region. We used this tool to delete a polyketide synthase gene (FUM1) required for fumonisin biosynthesis. The mutants generated are no longer able to produce fumonisins, confirming the key role of FUM1 in fumonisin biosynthesis. Our CRISPR-Cas9 system is an important new tool for genetic studies of Fusarium.
Subject terms: Fungi, Molecular biology
Introduction
Fusarium proliferatum is a globally distributed fungal pathogen that can attack diverse crops, including wheat, maize, rice, asparagus, date palm, garlic, onion, ornamental palms, and Chinese chive1. Fusarium proliferatum produces multiple mycotoxins, including fumonisins, moniliformin, beauvericin, fusaproliferin and fusaric acid2. The fumonisins are the most important of these toxins, not only for the mycotoxicoses with which they are associated, which includes esophageal cancer and neural tube defects in humans, cancer in rats, pulmonary edema in swine and leukoencephalomalacia in horses3, but also for their regulation in the international grain trade. There are four major classes of fumonisins – A, B, C and G – with the B-type the most toxic and the most commonly recovered. Fumonisin B1 (FB1) is categorized as a group 2B carcinogen by the International Agency for Research on Cancer4, and is potentially carcinogenic to humans.
The 16-gene fumonisin biosynthetic gene cluster (FUM) is known in multiple fumonisin-producing fungal species5–8, and includes genes that encode biosynthetic enzymes, regulatory factors and transport proteins9. Gene inactivation and/or heterologous expression studies10–14 have identified the functions of most of the genes in the FUM cluster. FUM1 encodes a polyketide synthase responsible for synthesis of the linear polyketide that forms the chemical backbone common to all fumonisins. The FUM cluster in F. proliferatum is collinear with the FUM clusters of Fusarium verticillioides and Fusarium oxysporum8,15,16. The F. proliferatum FUM1 sequence is 85% identical to the F. verticillioides homologue15. In F. verticillioides7,17, the FUM1 gene is required for fumonisin biosynthesis. The essential role of FUM1 in the biosynthesis of fumonisins by F. proliferatum has been demonstrated18.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system is a recently described genome-editing tool for gene knockout, gene insertion and gene replacement. The most popular type II CRISPR systems have two components: (i) a CRISPR-associated Cas9 endonuclease from Streptococcus pyrogenes, and (ii) a single-guide RNA (sgRNA), which is the fusion of a precursor CRISPR RNA (precrRNA) and a trans-activating CRISPR RNA (tracrRNA)19. The sgRNA recognizes the target DNA sequence and forms a DNA/RNA duplex at the recognition site. The Cas9endonuclease then cleaves this DNA/RNA duplex. The resulting double-strand DNA breaks are repaired by non-homologous end-joining, or by homology directed repair (HDR), if a donor DNA repair template is co-transformed into cell. The HDR repair system is particularly effective for complete gene knock-outs when coupled with dual in-vitro-assembled Cas9 ribonucleoproteins (RNPs) that target the 5ʹ and the 3ʹends of the gene coding region.
Type II CRISPR-Cas9 systems also have been used in other filamentous fungi including: Aspergillus nidulans and Aspergillus aculeatus20, Aspergillus oryzae21, Aspergillus fumigatus22–24, Aspergillus niger25, Aspergillus carbonarius26, Trichoderma reesei27, Neurospora crassa28, Alternaria alternata29, Penicillium chrysogenum30, F. oxysporum31, and Fusarium graminearum32. However, no application has been reported for F. proliferatum or other members of the Fusarium fujikuroi species complex.
In this study, we used a Type II CRISPR-Cas9 system to inactivate a key gene in the fumonisin biosynthetic cluster in F. proliferatum. Our working hypotheses were: (i) in-vitro-assembled RNPs could be used with a donor DNA repair template for direct co-transformation of F. proliferatum, and (ii) the FUM1 gene was essential for the synthesis of fumonisin. This work advances the field by providing a new, simple tool for making knockout mutants in members of the F. fujikuroi species complex, and by confirming the essential role of FUM1 in fumonisin biosynthesis in F. proliferatum.
Results
Designing sgRNA protospacers for deleting FUM1
Our goal was to develop a Cas9-mediated gene deletion system in F. proliferatum. We assembled in vitro dual Cas9 RNPs and a DNA repair template flanked by microhomology regions adjacent to the target gene.
To delete FUM1, we used two sgRNAs (Table 1) that directed Cas9 RNP DNA binding and cleavage to two specific locations. To reduce the possibility of deleting non-FUM1 coding sequences, sgRNAs nearest the start and stop codons were chosen. Based on predicted PAM sites and sgRNA scores, two sgRNAs were selected: (i) sgRNA1175, which was 15 bp downstream of the FUM1 start codon, and (ii) sgRNA9269 which was 11 bp upstream of the FUM1 stop codon (Fig. 1). This process leaves 26 (15 + 11) nucleotides of the FUM1 coding region flanking the HygB insertion. In silico analysis of off-target mutations by the selected sgRNAs confirmed the specificity of the two protospacers. Both of the selected sgRNAs had <15-bp off-target identity with other genomic regions in F. proliferatum.
Table 1.
sgRNAs used in this study
| ID | Protospacer sequence 5′→3′ | PAM site |
|---|---|---|
| sgRNA1175 | TCACCCCCGAGTACCGCTGT | AGG |
| sgRNA9269 | TGATGCGTATCTGGAAATGA | AGG |
Figure 1.

Schematic representation of the FUM1 gene deletion by in vitro-assembled dual Cas9 ribonucleoproteins coupled with homology directed repair (HDR). The cleavage sites of the in vitro-assembled Cas9 RNP1175 and RNP9269 (↓), the 50 bp microhomology regions for HDR (orange segment), the EcoRI cut sites, and the pksFUM1-specific probe (●●) are represented for the genomic locus of the wild-type ITEM 7595 and ΔFUM1 deletion strain.
Cas9-mediated FUM1 deletion in F. proliferatum
Cas9-mediated cleavage of the FUM1 gene and the HDR of the selectable marker (HygB) were coupled through microhomology recombination with dual RNPs directly into the cleavage sites. Microhomology recombination occurs in the 50-bp regions homologous to the nucleotide sequences adjacent to the sgRNA1175 and sgRNA9269 cleavage sites (Fig. 1).
The HDR-HygB repair template, which contains the HygB expression cassette, was successfully fused to the 50-bp flanking regions by PCR amplification and sequenced (GenBank accession no. MN226410). Following co-transformation of the dual RNPs and the HDR-HygB repair template, protoplasts were regenerated and mutants selected on PDA + hygromycin. Eleven putative ∆FUM1 transformants were obtained. Colony morphology and pigmentation of the F. proliferatum ∆FUM1 mutants were evaluated after 7 days of incubation at 25 °C on PDA. No significant differences were observed between the wild-type and mutant cultures in morphology or pigmentation under the tested growth conditions (Fig. 2).
Figure 2.

Colony morphology and pigmentation of F. proliferatum ∆FUM1 mutants obtained with the in-vitro-assembled RNPs coupled with HDR method. Front and reverse sides of the colonies are shown in the left and right panels, respectively. WT = wild-type strain ITEM 7595; M1-M5 = five arbitrarily selected F. proliferatum ∆FUM1 mutants. The phenotypes were evaluated after 7 days of incubation on PDA at 25 °C under white light (12 h light/dark).
PCR screening of F. proliferatum ∆FUM1 mutants
The putative ∆FUM1 mutants were screened by PCR amplification of a genomic sequence including both the upstream and the downstream cleavage sites in the RNPs. PCR amplification was performed on DNA from purified cultures of HygB resistant ∆FUM1 mutants. The resulting fragments were consistent with integration of the HygB cassette into the predicted cleavage sites in all 11 mutants (Fig. 3).
Figure 3.

PCR amplification of HDR-HygB repair template integration locus spanning the cleavage sites of the in-vitro-assembled RNP complexes. Expected amplicon size for the RNP1175 (A) and RNP9269 (B) complexes was 1734 bp and 1619 bp, respectively. M = 1 kb ladder; 1–11 = F. proliferatum ∆FUM1 mutants resistant to hygromycin B; 12 = wild-type strain ITEM 7595.
Southern blot analysis
To confirm the correct integration of the HDR-HygB repair template cassette by CRISPR-Cas9 mediated homologous recombination into the targeted gene locus, a Southern blot analysis was performed with five arbitrarily selected, purified ∆FUM1 transformants. Following EcoRI digestion of wild-type genomic DNA and hybridization with a FUM1-specific probe (Fig. 4), a 4035 bp genomic DNA fragment (located at position 64909–68942 on scaffold 50 of genome reference strain NRRL62905) was detected. This DNA fragment included portions of the FUM21 (810 bp) and FUM1 (2056 bp) genes. EcoRI digestion of DNA from all of the ∆FUM1 mutants resulted in a 6128 bp genomic segment. The DNA fragment from the ΔFUM1 mutants contains the HygB cassette (1619 bp) instead of most of the FUM1 gene and also includes a portion of the FUM6 gene (2160 bp) (Figs. 1 and 4).
Figure 4.
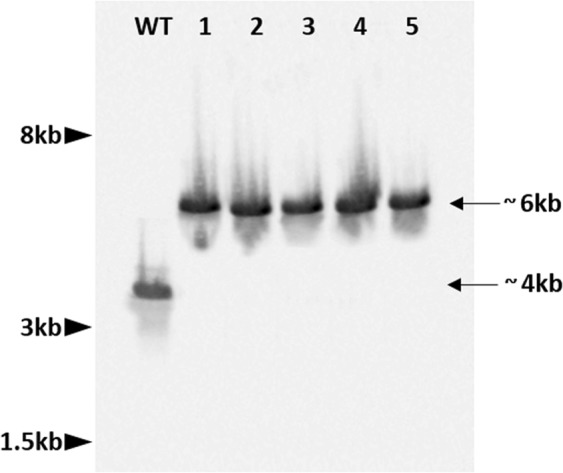
Southern blot of DNA from wild-type strain ITEM 7595 and five arbitrarily selected ∆FUM1 mutants following digestion of genomic DNA with EcoRI and hybridization with a FUM1-specific probe. In the wild type ITEM 7595 DNA (WT), a genomic fragment of ~4 kb was detected. Lanes 1–5 all contain an ~6 kb DNA fragment predicted from the FUM1 deletion pattern and HDR of the HygB cassette.
Fumonisin analysis
The polyketide synthase gene, FUM1, was the reporter target gene of this study. The encoded polyketide synthase catalyzes the synthesis of the linear polyketide backbone of the fumonisins. Deletion of FUM1 in F. proliferatum results in mutant strains that are resistant to hygromycin B and are unable to produce fumonisins, as expected.
We confirmed the role of FUM1 in fumonisin biosynthesis by analyzing cultures of wild-type strain ITEM 7595 and the ∆FUM1 mutants for the presence of FB1, fumonisin B2 (FB2) and fumonisin B3 (FB3) by HPLC/FLD. The amounts of FB1, FB2 and FB3 in cultures of ITEM 7595 after 19 days of growth were 272 µg/g, 78 µg/g and 88 µg/g, respectively. None of the ∆FUM1 mutants grown under the same conditions produced detectable FB1, FB2 or FB3 (Fig. 5).
Figure 5.

HPLC-FLD chromatograms of culture extracts of wild-type F. proliferatum strain ITEM 7595 (blue line) and one arbitrarily selected ∆FUM1 mutant strain (red line). Retention times: FB1, 16.250 min; FB2, 23.785 min; FB3, 24.968.
Discussion
The CRISPR/Cas9 system is an attractive, powerful tool that may replace the classical gene knockout approach based on homologous recombination in filamentous fungi. We describe the application of CRISPR/Cas9 tools to genome editing of F. proliferatum. This technique significantly expands the research tools available for F. proliferatum and related species in the F. fujikuroi species complex.
The tool we developed enables more comprehensive analysis and exploitation of gene function, especially of genes with roles in secondary metabolite biosynthesis. Genes coding for polyketide synthases (pks) have previously been used as reporter genes to assess the efficacy of the CRISPR-Cas9 genome editing system in other fungi, e.g., A. fumigatus33 and F. oxysporum31. Our study confirmed the key role of FUM1 in fumonisin biosynthesis by F. proliferatum. This report is the first of using a CRISPR-Cas9 system to delete a pks gene in a mycotoxin biosynthetic pathway. Given the length of the genomic region we successfully deleted (about 8000 bp), our study confirms that the CRISPR-Cas9 system can be used to target multiple genomic loci and/or large genomic regions at a single physical location with a single transformation event.
The method is a simple, near-universal system for gene deletion and has potentially widespread applicability in filamentous ascomycetes. In addition, the size of the genomic region being deleted is theoretically much larger with our system than with the traditional gene deletion approach.
Currently, deletion of genes involved in mycotoxin biosynthesis requires laborious and time-consuming construction of DNA fragments containing selectable markers with large (~750 to 1,500 bp) flanking homology regions17,34–36. The CRISPR–Cas9 system we used couples a dual site-specific DNA break, mediated by two in-vitro-assembled RNP complexes, with microhomology recombination dependent only on 50-bp regions flanking the targeted deletion site. A similar approach has been used in F. oxysporum31, but the HygB resistance cassette was flanked by ~600 bp homologous sequence both upstream to and downstream of the cleavage sites. In the current study, we demonstrated that 50 bp flanking regions are sufficient for specific recombination at a targeted genomic locus in F. proliferatum. The use of very short flanking regions for microhomology recombination offers two additional advantages: (i) it minimizes the risk of altering adjacent gene sequences; (ii) it allows targeting of a gene(s) that is closely spaced in a genomic region. The latter advantage makes this system particularly useful for studying genes located in a cluster that enables the biosynthesis of a secondary metabolite, where very tight targeting may be required to disrupt the function of just a single gene. Indeed, the possibility of deleting multiple clustered genes with a single transformation event could greatly improve the study of their interaction in the biosynthetic process.
The use of microhomology-site direct recombination coupled with Cas9-mediated cleavage was effective with a 60-bp flanking region in P. chrysogenum30, and with 50-bp or 35-bp flanking regions in A. fumigatus.45 The success of these approaches requires careful a priori studies of nucleotide sequences flanking the target genomic locus in order to avoid sequence homology with other genomic regions. The ability to target a specific genomic region by fusing only a 50-bp flank to the target is particularly important in fungi with significant amounts of DNA duplication, as it reduces the number of unwanted off-target recombination events and multiple site integrations by the HDR-repair template cassette.
The direct transformation of fungal protoplasts with in vitro-assembled RNPs also avoids the need to express the Cas9 protein in the fungal host cell and to assemble specific expression cassettes for each sgRNA. Usually both the Cas9 and the sgRNA expression cassettes are included in the same expression vector with a selectable marker that confers resistance to an antifungal agent. This vector is transformed into the fungal protoplasts for either transient expression20 or chromosomal integration32. Vector-based systems usually are laborious and time-consuming to assemble since they require the construction of multiple vectors, each with different sgRNAs for the different target genes. Cas9 expression usually is controlled by robust, constitutive promoters that guarantee high levels of active protein in the host cell. A significant drawback to high intracellular levels of Cas9 protein is off-target cleavage by the protein and unexpected, and often difficult to identify, genome alterations.
We used a commercially available Cas9 protein for in vitro assembly of RNP complexes with the appropriate sgRNAs. A similar approach has been used in other filamentous fungi, such as T. reesei27, P. chrysogenum30, A. fumigatus23, A. niger25, and F. oxysporum31. The efficiency of Cas9-mediated cleavage is linked to its nuclear localization, and to its binding to the targeted DNA regions. The nuclear import of the protein results from the fusion of a nuclear localization sequence (NLS) to the Cas9 protein. The commercial Cas9 protein used in this study was imported effectively into the F. proliferatum nucleus and cleaved its target site as expected. Indeed, the direct transformation of fungal protoplasts with in vitro-assembled RNPs has a much lower risk for off-target mutations than if Cas9 protein expression was mediated by vector transformation. The lowered risk of off-target mutations results from the limited time in which an active form of the Cas9 protein is present in the fungal nucleus. In this study, all eleven of the analyzed mutants had only a single integration event and the expected deletion profile. However, a genome-wide analysis of the ∆FUM1 mutants is needed to confirm our expectation that the genomic changes in the transformants are limited to the changes we report at FUM1. The lack of significant morphological differences between the wild-type strain and the ∆FUM1 mutants (Fig. 2) supports this expectation. These results differ from those reported by Sun et al.18, who found that deletion of FUM1 and FUM19 in their F. proliferatum strain altered growth rate and conidiation. Indeed, the potential influence of FUM1 and FUM19 on fungal growth and conidiation warrants further consideration with these characters evaluated in multiple mutants constructed in different genetic backgrounds that are grown on several media under a number of culture conditions.
Our process enables a strategy wherein RNPs are rapidly assembled in vitro with sgRNAs targeting multiple genomic loci. This capability is an important step forward for studies in F. proliferatum and other filamentous fungi, as multiple genes in the same metabolic pathway can potentially be deleted simultaneously.
Conclusion
We developed and applied for the first time in F. proliferatum a gene deletion process based on a CRISPR Cas9 Type II system that uses in vitro assembled RNPs and a donor DNA repair template. The use of in-vitro-assembled RNPs results in only transient exposure of the cells to Cas9, thereby minimizing off-target events due to rapid degradation of the RNPs. A protocol that combines sgRNAs synthesized in vitro with a commercially available Cas9 protein may be the most rapid and versatile option for deleting genomic regions in filamentous fungi, and could become the new standard for genetic studies with these organisms.
The ability to potentially target multiple genomic loci, both linked and unlinked, in a single transformation event is a powerful tool to study the relatedness of physically or functionally related genes. These genes might be clustered in a single chromosome region or dispersed throughout the genome. These properties are particularly relevant for studies of biosynthetic gene clusters involved in fungal secondary metabolism, and in the study of multi-copy genes that may have a role in plant pathogenicity. The phenotypes with these multi-locus genetic bases are of theoretical interest and practical importance and have been difficult to study with knock-out technology that permits only one-gene-at-a-time type studies.
Methods
Strains and preparation of conidia
We used a wild-type F. proliferatum strain, ITEM 7595, from the Agri-Food Toxigenic Fungi Culture Collection (Bari, Italy – http://server.ispa.cnr.it/ITEM/Collection/). The strain was routinely cultured on potato dextrose agar (PDA–Oxoid Ltd., Basingstoke, Hampshire, United Kingdom). The knock-out ∆pks FUM1 strains were grown on PDA supplemented with 100 mg/L hygromycin B (Thermo-Fisher Scientific, Waltham, MA, USA). Conidia suspensions were made from colonies grown on PDA for 7 days at 25 °C. The surface of the colony was scraped with a spatula, and mycelia and conidia were suspended in 10 mL of sterile distilled water. The suspension was filtered through Miracloth (Merck, Darmstadt, Germany). Spores in the resulting suspension were counted with a haemacytometer and the suspension diluted to a final concentration of 107 conidia/mL.
sgRNA design, synthesis and in vitro assembly of Cas9-gRNAs
The sgRNAs for FUM1 (GenBank: KU180047.1) deletion were designed with the Eukaryotic Pathogen CRISPR guide RNA/DNA Design Tool – EuPaGDT (grna.ctegd.uga.edu). The annotated sequence (NCBI accession number GCA_900029915.1) of reference strain NRRL62905 of F. proliferatum was used to design the sgRNAs. Parameters guiding design of the sgRNAs (Table 1) used for gene deletion were: (i) sgRNA cleavage sites were near the 5′ or the 3ʹ end of the target gene; (ii) the protospacer adjacent motif (PAM) sequence was (N)20NGG. The potential for off-target cleavage at other loci was checked by BLASTN of the F. proliferatum NRRL62905 genome sequence. For in vitro transcription, the sgRNA forward primer was fused to a T7 promoter. The sgRNA DNA templates were PCR-assembled and transcribed in vitro with the GeneArt Precision gRNA Synthesis kit (Thermo-Fisher Scientific), according to the manufacturer’s instructions. The sgRNA1175 and sgRNA9269DNA templates were assembled by using primers IVT-F-gRNA1175/IVT-R-gRNA1175 and IVT-F-gRNA9269/IVT-R-gRNA9269 (Table S1), respectively. Following in vitro transcription, the sgRNAs were purified and quantified with a Qubit RNA BR Assay kit (Thermo-Fisher Scientific). Purified sgRNAs were stored at −80 °C.
The RNP complexes were assembled in vitro by using TrueCut Cas9 Protein v2 (Thermo-Fisher Scientific). The RNP complexes were assembled in a final volume of 15 μl by combining in 1 × NEBuffer 3.1 (New England BioLabs, Ipswich, MA, USA), 500 nM of Cas9 enzyme and 500 nM of sgRNA1175 or sgRNA9269. The mixtures were incubated at 25 °C for 15 min to generate RNP complexes designatedRNP1175 or RNP9269, and used immediately to transform protoplasts of F. proliferatum (see below).
Construction of the HDR-HygB repair template
The HDR-HygB repair template, containing the hygromycin B resistance cassette (HygB) from plasmid pYTK079, was constructed by fusing the HygB cassette (1619 bp) to 50 bp sequences in NRRL 62905 that flank the upstream and downstream RNP cleavage sites. The HygB cassette was PCR amplified from pYTK079 with Platinum Super Fi PCR Master Mix (Invitrogen, Carlsbad, CA, USA) and 500 nM of primers HDR5ʹharmHygB_F and HDR3ʹharmHygB_R (Table S1). PCR amplification conditions were: 98 °C for 30 sec, 30 cycles of 98 °C for 10 sec, 63 °C for 15 sec, and 72 °C for 1 min, followed by a single 72 °C incubation for 5 min for final extension. PCR amplification products were checked following electrophoresis through a 1% agarose gel stained with 1 × GelRed (Biotium, Fremont, CA, USA). The resulting DNA fragment (1719 bp) was purified by using the GeneJET gel extraction kit (Thermo-Fisher Scientific) and cloned into the pCR-XL-2-TOPO vector with the TOPO XL-2 Complete PCR cloning kit (Invitrogen), according to the manufacturer’s instructions. Positive clones were sequenced and the resulting plasmid designated pHygR-FUM1. The HDR-HygB repair template for microhomology recombination was PCR amplified from plasmid pHygR-FUM1 with primers HR5ʹharmHygB_F and HR3ʹharmHygB_R. PCR amplification products were purified with the GeneJET PCR Purification Kit and checked following electrophoresis through a 1% agarose gel stained with 1 × GelRed.
Protoplast preparation and transformation
Protoplast preparation and transformation followed that of Coleman et al.37, with some modifications. Briefly, conidia of ITEM 7595 were inoculated into 100 mL of potato dextrose broth (PDB– Oxoid Ltd., Basingstoke, Hampshire, UK) at a final concentration of 104 conidia/mL and cultivated for 16–18 h at 28 °C with shaking at 250 rpm. Mycelia were harvested by filtration, washed with sterile 0.7 M NaCl, and resuspended in 1.2 M KCl protoplasting buffer (pH 6.5) containing 100 mg/mL of VinoTastePro (Novozyme, Bagsvaerd, Denmark) lytic enzyme mix. The mixture was incubated at 30 °C with gentle shaking (80 rpm) until protoplasts were released (3–4 hours). Protoplasts were separated from mycelial debris by filtering through Miracloth and then pelleted by centrifugation at 4 °C for 15 min at 2360 × g. Protoplasts were washed with SuTC (20% sucrose, 50 mM Tris-HCl, pH 7.0, 50 mM CaCl2), centrifuged at 4 °C for 15 min at 2360 × g, and finally resuspended in SuTC at a concentration of 107 protoplasts/mL. Protoplasts (200 μl, 2 × 106 protoplasts) were co-transformed with RNP1175 and RNP9269 (500 ng each) and 3 μg of purified HDR-HygB repair template, and then incubated without shaking at 25 °C for 20 min. Three transformation aliquots were prepared. One milliliter of PSuTC (20% sucrose, 50 mM Tris·HCl, pH 7.0, 50 mM CaCl2, 60% polyethylene glycol-3,350) was added to a transformation aliquot and the incubation continued at room temperature for another 20 minutes. The transformation aliquot was transferred to a 15 mL sterile tube containing 3 mL of TB3 medium (20% sucrose, 1% glucose, 0.3% yeast extract, 0.3% Cas-amino acids) and incubated on a rotary shaker (160 rpm) for 18–24 h at 25 °C. Regenerated fungal protoplasts were mixed with molten (45 °C) PDA containing 100 mg/L of hygromycin B, poured into sterile Petri dishes, and incubated at 25 °C for 5–8 days until hygromycin-resistant colonies appeared. Putative transformants were transferred to PDA containing 100 mg/L of hygromycin B. Purified mutant strains originated from colonies subcultured as single microconidia on selective Spezieller Nährstoffarmer agar (SNA) medium (0.2 g/L sucrose, 0.2 g/L glucose, 1.0 g/L KNO3, 1.0 g/L KH2PO4, 0.5 g/L MgSO4·7H2O, 0.5 g/L KCl, and 15 g/L agar) supplemented with 100 mg/L of hygromycin. Purified F. proliferatum ∆FUM1 mutants were maintained on selective PDA for further molecular analysis and stored at −80 °C as conidial suspensions in sterile 15% glycerol. Phenotypes of F. proliferatum ∆FUM1 mutants were evaluated on PDA plates inoculated with a drop (2 μL) of a conidial suspension (104 conidia/mL) in the center of the plate. Inoculated plates were incubated at 25 °C and inspected visually after 7 days.
PCR analysis of putative F. proliferatum ∆FUM1 mutants
Genomic DNA of wild-type ITEM 7595 and purified cultures of F. proliferatum ∆FUM1 mutants was extracted by using the GeneJET Plant Genomic DNA Purification Kit. Following quantification with a NanoDrop ND-1000 spectrophotometer (Thermo-Fisher Scientific) and an integrity check following electrophoresis through a 0.8% agarose gel, 10 ng of genomic DNA from each isolate was used for PCR amplification of the HDR recombination sites. A 1734-bp fragment beginning 115 bp upstream of the RNP1175 cleavage site (83 bp upstream of the FUM1 start codon), which includes the HygB cassette (1619 bp), was amplified with primers FUM1_F(−83up) and HygB_R (Table S1). Similarly, a 1744-bp fragment beginning 125 bp downstream of the RNP9269 cleavage site (114 bp downstream of the FUM1 stop codon), which includes the HygB cassette (1619 bp), was PCR amplified with primers HygB_F and FUM1_R(+114dw) (Table S1). PCR amplifications were made with Platinum SuperFi PCR Master Mix (Invitrogen) and 500 nM of each primer. PCR amplification conditions were: 98 °C for 30 sec, then 30 cycles of 98 °C for 10 sec, 63 °C for 15 sec, and 72 °C for 90 sec, and a final extension at 72 °C for 5 min. Amplification products were evaluated following electrophoresis through a 1% agarose gel stained with 1 × GelRed. Both fragments were purified with the GeneJET gel extraction kit. Purified DNA fragments were sequenced by using Sanger sequencing and primers FUM1_F(−83up) or FUM1_R(+114dw).
Southern blot analysis
A FUM1-specific probe was amplified from genomic DNA of ITEM 7595 by using Platinum SuperFi PCR Master Mix (Invitrogen) with 500 nM each of primers probe5ʹ_koFUM1_F and probe3ʹ_koFUM1_R (Table S1). PCR amplification conditions were: 98 °C for 30 sec, 30 cycles of 98 °C for 10 sec, 63 °C for 15 sec, and 72 °C for 1 min, followed by a final extension at 72 °C for 5 min. PCR amplification products were evaluated following electrophoresis on a 1% agarose gel stained with 1 × GelRed. The 591-bp PCR fragment was biotinylated with the PCR DIG Probe Synthesis kit (Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer’s instructions. Genomic DNA of five arbitrarily selected purified putative ∆FUM1 transformants was extracted by using the GeneJET Plant Genomic DNA Purification Kit. Following quantification with a NanoDrop ND-1000 spectrophotometer) and an integrity check following 0.8% agarose gel electrophoresis, genomic DNAs were digested with EcoRI overnight at 37 °C. Fragments separated on a 1% agarose gel and then transferred to a BrightStar-Plus Positively Charged Nylon Membrane (Thermo-Fisher Scientific). The membrane was hybridized with the FUM1-specific biotinylated probe and developed with the North2South chemiluminescent hybridization and detection kit (Thermo-Fisher Scientific). Chemiluminescent detection was performed with a ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA).
Fumonisin extraction and HPLC analysis
The wild type strain (ITEM 7595) and the ∆FUM1 mutants were inoculated on PDA in Petri dishes and incubated at 25 °C. After 19 days the contents of the Petri dishes were collected and the fumonisins extracted. Fumonisins were analyzed with a slightly modified version of the AOAC Official Method 2001.0438, which includes an immunoaffinity (IMA) column clean-up of extracts and toxin determination by HPLC/FLD of fumonisins derivatized with o-phtaldialdehyde (OPA). Briefly, ~10 grams of the colonized PDA substrate was extracted with 40 mL methanol/water (70:30, v/v) by shaking for 1 h at room temperature. The contents of the flask were filtered through Whatman no. 4 filter paper, and a 10 mL aliquot of the filtrate was mixed with 40 mL of distilled water (50 mL total volume). Ten milliliters of the diluted sample was passed through a FumoniTestTM Wide Bore IMA column (Vicam, Milford, MA, USA). The IMA column was washed with 10 mL PBS (10 mM phosphate buffer, 2.7 mM potassium chloride and 137 mM sodium chloride, pH 7.4). Fumonisins were eluted from the IMA columns with 2 mL of methanol followed by 2 mL of distilled water. Eluted extracts were evaporated under an air stream at 50 °C, dissolved in 1 mL acetonitrile/water (30:70, v/v), and analyzed by HPLC.
The HPLC/FLD determination and confirmation of fumonisins B1, B2 and B3 was performed as previously described39. Extracts were derivatized with OPA (1:1, v/v) and mixed for 50 sec in an Agilent 1100 HPLC auto sampler equipped with a binary pump, and the column thermostat set at 30 °C. Three minutes after adding the OPA reagent, 100 μL of the extract was injected by full loop. The analytical column was a Symmetry Shield RP18 150 × 4.6 mm, 5 μm (Waters) with a guard column inlet filter (0.5 μm × 3 mm diam., Rheodyne Europe GmbH Bensheim, Germany). The mobile phase was a binary gradient applied as: (i) 57% of solvent A (water/acetic acid, 99:1, v/v) and 43% of solvent B (acetonitrile/acetic acid, 99:1, v/v) for 5 min; (ii) solvent B was increased linearly to 54% at 21 min, and to 58% at 25 min, and (iii) 42% solvent A and 58% solvent B for 5 min. The flow rate of the mobile phase was 0.8 mL/min. The fluorometric detector was set at wavelengths, ex = 335 nm, em = 440 nm. The retention time for FB1, was about 17 min, about 24 min for FB2, and 25.5 min for FB3.
For fumonisin analyses, a 5 μg/mL FB1 and FB2 standard stock solution was prepared in acetonitrile/water (1:1, v/v) by dilution of a certified Biopure calibrant solution (Romer Labs Diagnostic, Tulln, Austria). FB1 and FB2 were measured by calculating peak areas and comparing the results with calibration curves prepared in acetonitrile/water (30:70, v/v) over the range 0.005–5.000 μg/mL. FB3 was measured by using the FB2 calibration curve. The limit of detection (LOD) of the analytical method was 30 μg/kg for each fumonisin.
Supplementary information
Acknowledgements
This work was supported in part by H2020-EU3.2-678781-MycoKey-“Integrated and Innovative Key Actions for Mycotoxin Management in the Food and Feed Chain” and USDA National Institute of Food and Agriculture Hatch Multi/state project KS1183A. Manuscript no. 19-320-J from Kansas Agricultural Experiment Station, Manhattan.
Author contributions
M.F. set up and performed the experiments, and wrote the first draft of the paper; M.H. conducted fumonisin analyses; J.F.L., A.F.L. and G.M. set up experiments and wrote the paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-56270-9.
References
- 1.Leslie, J. F. & Summerell, B. A. The Fusarium Laboratory Manual (Blackwell Professional Ames, IA, USA, 2006).
- 2.Desjardins, A. E. Fusarium Mycotoxins: Chemistry, Genetics and Biology (APS Press, St. Paul, MN, USA, 2006).
- 3.Howard PC, et al. Fumonisin B1 carcinogenicity in a two-year feeding study using F344 rats and B6C3F1 mice. Environ. Health Perspect. 2001;109(Suppl. 2):277–282. doi: 10.1289/ehp.01109s2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.IARC. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. IARC, Lyon, France (2002). [PMC free article] [PubMed]
- 5.Brown DW, Butchko RAE, Busman M, Proctor RH. The Fusarium verticillioides FUM gene cluster encodes a Zn(II)2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryotic Cell. 2007;6:1210–1218. doi: 10.1128/EC.00400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pel HJ, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nature Biotechnology. 2007;25:221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- 7.Proctor RH, Desjardins AE, Plattner RD, Hohn TM. A polyketide synthase gene required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating population A. Fung. Genet. Biol. 1999;27:100–112. doi: 10.1006/fgbi.1999.1141. [DOI] [PubMed] [Google Scholar]
- 8.Proctor RH, Busman M, Seo JA, Lee Y-W, Plattner RD. A fumonisin biosynthetic gene cluster in Fusarium oxysporum strain O-1890 and the genetic basis for B versus C fumonisin production. Fung. Genet. Biol. 2008;45:1016–1026. doi: 10.1016/j.fgb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Proctor RH, et al. Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol. Microbiol. 2013;90:290–306. doi: 10.1111/mmi.12362. [DOI] [PubMed] [Google Scholar]
- 10.Butchko RAE, Plattner RD, Proctor RH. FUM13 encodes a short chain dehydrogenase/reductase required for C-3 carbonyl reduction during fumonisin biosynthesis in Gibberella moniliformis. J. Agric. Food Chem. 2003;51:3000–3006. doi: 10.1021/jf0262007. [DOI] [PubMed] [Google Scholar]
- 11.Butchko RAE, Plattner RD, Proctor RH. Deletion analysis of FUM genes involved in tricarballylic ester formation during fumonisin biosynthesis. J. Agric. Food Chem. 2006;54:9398–9404. doi: 10.1021/jf0617869. [DOI] [PubMed] [Google Scholar]
- 12.Ding Y, Bojja RS, Du L. Fum3p, a 2-ketoglutarate-dependent dioxygenase required for C-5 hydroxylation of fumonisins in Fusarium verticillioides. Appl. Environ. Microbiol. 2004;70:1931–1934. doi: 10.1128/AEM.70.4.1931-1934.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi H, Bojja RS, Fu J, Du L. Direct evidence for the function of FUM13 in 3-ketoreduction of mycotoxin fumonisins in Fusarium verticillioides. J. Agric. Food Chem. 2005;53:5456–5460. doi: 10.1021/jf050062e. [DOI] [PubMed] [Google Scholar]
- 14.Zaleta-Rivera K, et al. A bi-domain non-ribosomal peptide synthetase encoded by FUM14 catalyzes the formation of tricarballylic esters in the biosynthesis of fumonisins. Biochemistry. 2006;45:2561–2569. doi: 10.1021/bi052085s. [DOI] [PubMed] [Google Scholar]
- 15.Waalwijk C, et al. Synteny in toxigenic Fusarium species: The fumonisin gene cluster and the mating type region as examples. Eur. J. Plant Pathol. 2004;110:533–544. doi: 10.1023/B:EJPP.0000032393.72921.5b. [DOI] [Google Scholar]
- 16.Proctor RH, Brown DW, Plattner RD, Desjardins AE. Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fung. Genet. Biol. 2003;38:237–249. doi: 10.1016/S1087-1845(02)00525-X. [DOI] [PubMed] [Google Scholar]
- 17.Yu F, Zhu X, Du L. Developing a genetic system for functional manipulations of FUM1, a polyketide synthase gene for the biosynthesis of fumonisins in Fusarium verticillioides. FEMS Microbiol. Lett. 2005;248:257–264. doi: 10.1016/j.femsle.2005.05.053. [DOI] [PubMed] [Google Scholar]
- 18.Sun L, et al. Effects of disruption of five FUM genes on fumonisin biosynthesis and pathogenicity in Fusarium proliferatum. Toxins. 2019;11:327. doi: 10.3390/toxins11060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nødvig Christina S., Nielsen Jakob B., Kogle Martin E., Mortensen Uffe H. A CRISPR-Cas9 System for Genetic Engineering of Filamentous Fungi. PLOS ONE. 2015;10(7):e0133085. doi: 10.1371/journal.pone.0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katayama T, et al. Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungus Aspergillus oryzae. Biotechnol. Lett. 2016;38:637–642. doi: 10.1007/s10529-015-2015-x. [DOI] [PubMed] [Google Scholar]
- 22.Fuller KK, Chen S, Loros JJ, Dunlap JC. Development of the CRISPR/Cas9 system for targeted gene disruption in Aspergillus fumigatus. Eukaryot. Cell. 2015;14:1073–1080. doi: 10.1128/EC.00107-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C, Meng X, Wei X, Lu L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet. Biol. 2016;86:47–57. doi: 10.1016/j.fgb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Weber J, et al. Functional reconstitution of a fungal natural product gene cluster by advanced genome editing. ACS Synth Biol. 2017;6:62–68. doi: 10.1021/acssynbio.6b00203. [DOI] [PubMed] [Google Scholar]
- 25.Kuivanen J, Jasmin Wang Y-M, Richard P. Engineering Aspergillus niger for galactaric acid production: Elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9. Microb. Cell Fact. 2016;15:210. doi: 10.1186/s12934-016-0613-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weyda I, et al. A comparison of Agrobacterium-mediated transformation and protoplast-mediated transformation with CRISPR-Cas9 and bipartite gene targeting substrates, as effective gene targeting tools for Aspergillus carbonarius. J. Microbiol. Meth. 2017;135:26–34. doi: 10.1016/j.mimet.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 27.Liu R, Chen L, Jiang Y, Zhou Z, Zou G. Efficient genome editing in the filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015;1:15007. doi: 10.1038/celldisc.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsura T, Baek M, Kwon J, Hong C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol. Biotechnol. 2015;2:4. doi: 10.1186/s40694-015-0015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenderoth M, Pinecker C, Voss B, Fischer R. Establishment of CRISPR/Cas9 in Alternaria alternata. Fung. Genet. Biol. 2017;101:55–60. doi: 10.1016/j.fgb.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Pohl C, Kiel JAKW, Driessen AJM, Bovenberg RAL. Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth. Biol. 2016;5:754–764. doi: 10.1021/acssynbio.6b00082. [DOI] [PubMed] [Google Scholar]
- 31.Wang Q, Cobine PA, Coleman JJ. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fung. Genet. Biol. 2018;117:21–29. doi: 10.1016/j.fgb.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardiner M, Kazan K. Selection is required for efficient Cas9-mediated genome editing in Fusarium graminearum. Fungal. Biology. 2018;122:131–137. doi: 10.1016/j.funbio.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Al Abdallah, Q., Ge, W. & Fortwendel, J. R. A simple and universal system for gene manipulation in Aspergillus fumigatus: In vitro assembled Cas9-guide RNA ribonucleoproteins coupled with microhomology repair templates. mSphere2:e00446-17, 10.1128/mSphere.00446.17 (2017). [DOI] [PMC free article] [PubMed]
- 34.Gallo A, et al. New insight into the ochratoxin A biosynthetic pathway through deletion of a non-ribosomal peptide synthetase gene in Aspergillus carbonarius. Appl. Environ. Microbiol. 2012;78:8208–8218. doi: 10.1128/AEM.02508-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallo A, et al. Identification and characterization of the polyketide synthase involved in ochratoxin A biosynthesis in Aspergillus carbonarius. Int. J. Food Microbiol. 2014;179:10–11. doi: 10.1016/j.ijfoodmicro.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Ferrara M, et al. Identification of a halogenase involved in the biosynthesis of ochratoxin A in Aspergillus carbonarius. Appl. Environ. Microbiol. 2016;82:5631–5641. doi: 10.1128/AEM.01209-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coleman JJ, White GJ, Rodriguez-Carres M, VanEtten HD. An ABC transporter and a cytochrome P450 of Nectria haematococca MPVI are virulence factors on pea and are the major tolerance mechanisms to the phytoalexin pisatin. Mol. Plant-Microbe Interact. 2011;24:368–376. doi: 10.1094/MPMI-09-10-0198. [DOI] [PubMed] [Google Scholar]
- 38.Visconti A, Solfrizzo M, De Girolamo A. Determination of fumonisins B1 and B2 in corn and cornflakes by liquid chromatography with immunoaffinity column clean-up: Collaborative study. J AOAC Int. 2001;84:1828–1837. [PubMed] [Google Scholar]
- 39.De Girolamo A, Pascale M, Visconti A. Comparison of methods and optimisation of the analysis of fumonisins B1 and B2 in masa flour, an alkaline cooked corn product. Food Addit. &Contam.: Part A. 2011;28:667–675. doi: 10.1080/19440049.2011.555846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.