

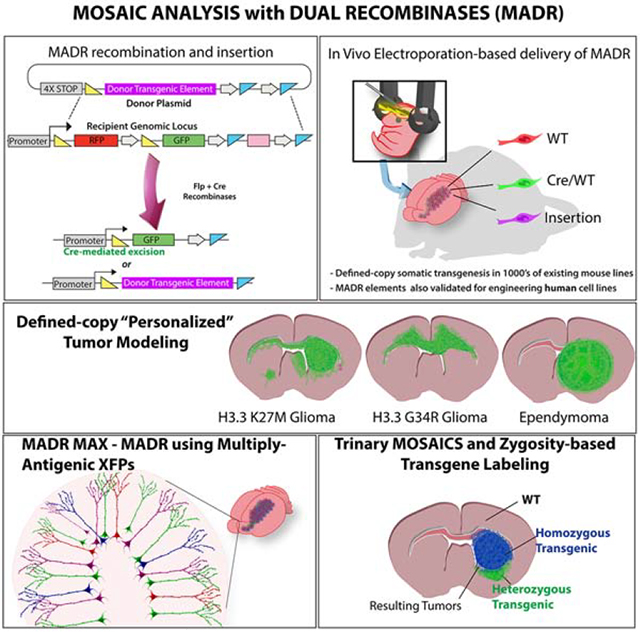
SUMMARY
In situ transgenesis methods such as virus and electroporation can rapidly create somatic transgenic mice, but lack control over copy number, zygosity and locus specificity. Here, we establish mosaic analysis by dual recombinase-mediated cassette exchange (MADR), which permits stable labeling of mutant cells expressing transgenic elements from precisely-defined chromosomal loci. We provide a toolkit of MADR elements for combination labeling, inducible/reversible transgene manipulation, VCre recombinase expression and transgenesis of human cells. Further, we demonstrate the versatility of MADR by creating glioma models with mixed, reporter-identified zygosity or with “personalized” driver mutations from pediatric glioma. MADR is extensible to thousands of existing mouse lines, providing a flexible platform to democratize the generation of somatic mosaic mice.
Keywords: RMCE, CRISPR/Cas9 base editors, ependymoma, epigenetics, gliogenesis, MADR MAX, AAVS1 locus, VCre, Brainbow, scRNA-seq, scATAC-seq
Graphical Abstract
In Brief
Mosaic analysis with dual recombinase-mediated cassette exchange (MADR) is a simple, fast, and generalizable method for the generation of stable, defined copy number somatic transgenic animals, which can accelerate research investigations of development and disease.
INTRODUCTION
Genetically engineered mouse models (GEMMs) are often used to analyze gene function in vivo in a temporal- and tissue-specific manner. As generating these mouse models is an expensive laborious process, alternative transgenic approaches such as gene delivery by electroporation (EP) and virus have been increasingly adapted as more rapid and efficient methods to create somatic mosaics (Breunig et al., 2015, Hambardzumyan et al., 2011). Both methods entail injecting specific tissues with virus or foreign DNA to transduce the surrounding cells and create somatic mosaics. EP can yield genomeinserted DNA using transposons (Breunig et al., 2015, Chen and LoTurco, 2012) or less efficiently with CRISPR/Cas9 and subsequent insertion of a donor template (Mikuni et al., 2016). Despite their speed, these methods have pitfalls that hinder more widespread use. Viruses have limited payloads, induce immune responses, and require special expertise. Both transposons and viruses have unpredictable genomic integration patterns, and exhibit epigenetic transgene silencing (Garrick et al., 1998, Woods et al., 2003), as well as transgene copy number variability and overexpression artifacts like cytotoxicity and transcriptional squelching (Akhtar et al., 2013, Gibson et al., 2013). These can lead to clonal genotypic/phenotypic variability as significant confounding factors.
With the identification of hundreds of recurrent, putative cancer driver mutations, many of which are gain-of-function (GOF) oncogenes, it is imperative to create a tractable in vivo platform that can model these potential oncogenes, especially in conjunction with loss-of-function (LOF) tumor suppressor mutations (Lawrence et al., 2014). Producing GEMMs for the myriad combinations of driver signatures is time-consuming, expensive, and prone to some methodological confounds. Alternatively, CRISPR/Cas9 systems can simultaneously induce multiple knockouts in vivo in mice (Chen et al., 2015), but can have significant variability and unintended off-target genome alterations (Kosicki et al., 2018).
Thus, to model the myriad driver mutations in cancer we sought a method that can inexpensively ensure defined gene copy number among transfected cells. To meet these needs we developed the mosaic analysis with dual recombinase-mediated cassette exchange (MADR) methodology. We demonstrate that MADR efficiently generates single-copy insertion in somatic cells in well-characterized mouse lines with definitive genetic labeling of recombined cells. Also, we show this system’s utility in generating mosaicism with a mixture of gain and loss of function mutations. Ultimately, our MADR tumor models reveal this method has a potential to become a higher-throughput pre-clinical platform for functionalizing various putative tumor driver mutations, and provides a rapid pipeline for preclinical drug discovery in a patient-specific manner.
RESULTS
MADR yields single-copy transgenesis
mTmG is a widely used mouse line that constitutively expresses membrane tdTomato and switches to EGFP expression upon Cre-mediated recombination (Muzumdar et al., 2007). To effect MADR in mTmG cells, we created a promoter-less donor plasmid with a transgene cassette flanked by loxP and FRT sites (Fig. 1A) The open reading frame (ORF) is preceded by PGK and trimerized SV40 polyadenylation signals (i.e. “4X PolyA” in Fig. 1A) to avoid transcription from un-integrated episomes and randomly integrated whole-plasmids. The ORF is followed by woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and PolyA element, (Fig. 1A). To create an in vitro MADR recipient, we generated a heterozygous mouse neural stem cell (mNSC) line from Rosa26WT/mTmGmice (mTmGHet). We then made two MADR lines by nucleofecting TagBFP2 or TagBFP2-HrasG12V donors (10 ng/μl) and Flp-Cre expression vector (Flp-Cre, S1A) (10 ng/μl) (Anderson et al., 2012) each of which yielded 3 distinct fluorescent colors: tdTomato+, EGFP+, or TagBFP2+ (Fig. 1B–C and S1B). Initially, about 5% of TagBFP2+ cells retained either tdTomato or EGFP (Fig. S1B) (Muzumdar et al., 2007). After another week of culturing the sorted cells, we confirmed the absence of residual EGFP or tdTomato and single-band HrasG12V by imaging and western blot, indicating that the recombined Rosa26 locus expressed a single correctly-sized polypeptide at the aggregate, polyclonal population level without antibiotic selection (Fig. S1C–D). To validate the single-copy insertion, we created donor plasmids carrying puromycin N-acetyl-transferase (PAC) and enriched the cells that correctly express the transgene via antibiotic selection (Fig. S1E). We confirmed the correct recombination and integration at Rosa26 locus by PCR and western blot in these cells (Fig. S1E–G). In order to assess protein production on a per-cell basis, we compared the TagBFP2 protein levels in mNSCs carrying piggybac-TagBFP2 and heterozygous TagBFP2+ MADR cells. The intensity of TagBFP2 in MADR cells had a tight distribution, whereas piggyBac cells had a broad dynamic expression range extending an order of magnitude (Figs. 1D–F). These findings demonstrate that MADR yields defined, single-copy insertion events in cell lineages leading to consistent transgene dosage.
Figure 1: MADR in mTmG mouse or human lines generates genetic reporter-defined populations in vitro.

A) Flp-Cre vector catalyzes either Cre-mediated excision or dRMCE on Rosa26mTmG allele in the presence of a MADR donor vector, resulting in two distinct recombinant products.
B) Nucleofection of heterozygous Rosa26WT/mTmG mNSCs result in three possible lineages: tdTomato+, EGFP+, and TagBFP2+.
C) Live imaging of representative cells with non-overlapping fluorescent colors. Scale bars, 100μm
D) Schematic of cell preparation for single-cell western blot.
E) Frequency of fluorescence intensities comparing MADR and PiggyBac transgenic cells.
F) Representative examples of single-cell western blots for PiggyBac and MADR groups. (Note that this is not a pure population and so some cells express the Histone H3 loading control protein but no TagBFP2. Also, many lanes are empty as is typical for this assay).
G) MADR-compatible TRE-smFP plasmids for MADR MAX.
H) Dox induces efficient smFP expression allowing for orthogonal imaging of 4 independent reporters in vitro. Scale bar, 100μm
I) High magnification confocal z-section demonstrates that each cell expresses a single smFP reporter. Scale bar, 10μm
J) Schematic of AAVS1 locus targeting for HUMAN MADR by TALEN or CRISPR/Cas9
K) HEK293T cells containing AAVS1-targeted MADR recipient site expressing tdTomato and TagBFP2-V5-nls Scale bar, 100μm
L) MADR-HEK293T cells transfected with MADR pDonor smFP-myc (Bright) or TagBFP-3XFlag showing GFP or BFP autofluorescence among non-inserted tdTomato+ cells. Scale bar, 100μm
M) High mag image of cells from L exhibiting tdTomato and smFP-myc in a mutually exclusive manner. Scale bar, 10μm
MADR-mediates “one shot” generation of multiple inducible in vitro cell lines
Assays for gene function are often performed using transduced or transfected cell lines in vitro, but the constitutive expression of some transgenes can hinder stable cell line generation if the mutations decrease fitness. To avoid this, inducible genetic systems, like TRE, can be employed to make the cell line first and later induce the gene(s) of interest (Akhtar et al., 2015). To showcase the utility of single-allele mTmGHet mNSCs, we established a pipeline for inducible cell line production by nucleofecting these cells with a MADR-compatible vector containing rtTA-V10 and TRE-Bi element (Figs. S1H) (Akhtar et al., 2015). This colorless TRE-Bi-EGFP cell line was enriched with puromycin selection and confirmed using standard in vitro Doxycycline (Dox) treatment (Figs. S1I–J).
This in vitro pipeline can be used to investigate the results of GOF mutations in various primary cell lines derived from any animal carrying loxP and FRT sites by generating more homogeneous, inducible stable lines. As proof-of-principle for this, and to determine whether the 3’ cistron of the TRE-Bi element was sporadically expressed because of distal promoter/enhancer regions, we generated a line that inducibly expresses the Notch ligand, Dll1, with a bicistronic TRE-Bi-Dll1/EGFP donor vector (Fig. S1K). This line showed small physiological levels of Dll1 without Dox, whereas both EGFP and Dll1 were expressed at similar levels by all cells with Dox treatment (Fig. S1L–M). Notch signaling is one of many gene-dosage sensitive molecular pathways where MADR can be used.
From the mTmGHet mNSCs, we also made distinct cell lines with 4 different “spaghetti monster” reporter fluorescent proteins (smFPs) in a single nucleofection (Viswanathan et al., 2015). We used this pipeline, which we name MADR with multiply-antigenic XFPs (MADR MAX) (Fig. 1G), to assess if more than one copy of each plasmid could be expressed per cell. smFPs were expressed in nearly all cells after antibiotic selection and Dox addition in proportionate ratios (Fig. 1H). Furthermore, we did not observe cells expressing more than one smFP, showing one-transgene-to-one-cell integration (Fig. 1I). This “one-shot” generation of stable, inducible cell lines can enable multiplex analysis of multiple transgenes in a common genetic background without causing differential genetic drift during antibiotic selection.
To test MADR in human cells, we engineered a MADR-compatible recipient site, and employed TALENs to create a HEK293T cell line with this cassette inserted at the AAVS1 locus. Here, the MADR reaction will replace a CAG-driven tdTomato flanked by loxP and FRT sites (Fig. 1J). To test MADR function, we transfected the cell line with a smFP(bright)-myc donor and an alternate TagBFP2-3XFlag donor. Immunofluorescent analysis confirmed the cell lines that lost tdTomato via excision expressed either the TagBFP2-3Flag or smFP(bright)-myc donor transgene (Figs. 1K–M). These results show that MADR can be used in human cells.
MADR efficiently yields single copy somatic transgenesis in vivo
To assess MADR in vivo, we electroporated (EPed) donor plasmids having fluorescent protein reporters (TagBFP2 or membrane-tagged smFP-myc) and Flp-Cre (0.5 μg/μl each) into the neural stem/progenitor cells lining the ventricular zone (VZ) of postnatal day 2 (P2) mTmGHet pups (Fig. 2A). Two days after, we noted the presence of TagBFP2+ cells along the VZ though some cells expressed detectable EGFP as well (Fig. S2A). At 7 days, many VZ radial glia and recently-migrated olfactory bulb neurons expressed the smFP-myc reporter (Movie S1). By two weeks, differentiated striatal glia and olfactory bulb neurons appeared (Fig. 2B and S2B). At this time point, we noticed some rare TagBFP2+ cells with persistent EGFP expression at the VZ with the morphological characteristics of ependymal-lineage cells (i.e. multi-ciliated with cuboidal morphology; Fig. S2B). We confirmed these double-positive cells are indeed Foxj1+ ependymal cells (Fig. S2C–G) and noted an inverse correlation between MADR reporter and EGFP. However, most TagBFP2+ cells lacked tdTomato and EGFP expression after the first week post-EP (Fig. S2B, S2H; Movie S2).
Figure 2: MADR in heterozygous mTmG allows for efficient tracing of lineages in vivo.

A) Standard postnatal EP protocol targeting the ventricular zone in P2 heterozygous Rosa26WT/mTmG pups with DNA mixture of a Flp-Cre vector and a donor plasmid
B) Postnatal EP recapitulates in vitro nucleofection experiment and yields TagBFP2+ MADR along with EGFP+ and tdTomato+ lineages at 2 weeks post-EP. Scale bar, 100μm
C) Different concentrations of recombinase and donor plasmids result in various efficiencies of both MADR and Cre-excision recombination reactions in vivo. All mixtures contained a nuclear TagBfp2 reporter plasmid. (See Supp. Fig. 2D for representative images from this quantitation). Error bars indicate standard error of the mean (SEM).
D) Schematic of plasmid delivery for combinatorial MADR MAX “brainbow” like multiplex labeling
E) Low mag image of olfactory bulb displaying multiplex smFP-based MADR MAX EPed cells and immunostaining for the smFP-linked epitope tags. Scale bar, 100μm
F) High mag image of cells from E exhibiting expression of a single smFP epitope tag per neuron. Scale bar, 10μm
G) Schematic of expansion microscopy and brightfield image example
H) MADR pDonor smFP-myc sh.Nf1 miR-E plasmid for simultaneous knockdown of Nf1 and smFP-myc labeling of transgenic cells
I) Image of EPed striatum showing two populations of reporter labeled cells—EGFP and smFP-myc (i.e. Nf1 knockdown cells).
J) Pre-expansion smFP-myc cell body
K) Post-expansion of cell in J
L) Post-expansion EGFP astrocyte displaying “super-resolution” detail.
M) Schematic of MADR pDonor-TagBFP2-P2A-VCre and FlEx VCre reporter plasmids for mosaic analysis with tertiary recombinase
N) EPed striatum with FlpO-2A-Cre, MADR pDonor-TagBFP2, HypBase and FlEx VCre reporter. Scale bar, 50μm
O) Striatum of littermate of mouse shown in N with FlpO-2A-Cre, MADR pDonor-TagBFP2–2A-VCre, HypBase and FlEx VCre reporter exhibiting VCre-dependent FlEx reporter (smFP-myc). Scale bar, 50μm
To test the effect of plasmid concentrations on the in vivo recombination efficiencies, we varied the concentrations of Flp-Cre plasmid and smFPY-myc for high-sensitivity detection of recombined cells (Viswanathan et al., 2015). We found that increasing recombinase dose or donor plasmid concentration led to more EGFP+ cells (Fig. 2C and Fig. S2I). However, since EGFP and the insertion donor were competing for the same locus, there is a zero-sum effect. Further, due to the perdurance of EGFP, at 2-days many cells expressed both transgenes. Notably, this was likely an inevitable consequence of the half-life of these fluorescent proteins and is similar to the overlap seen between tdTomato and EGFP cells at short survival time points after recombination in the mTmG where the reporter decay was estimated at over 9 days (Muzumdar et al., 2007).
To rule out the possibility that transgene expression was due to expression from randomly integrated or non-recombined episomes, we performed a series of control EPs (Fig. S2J). EP of highly concentrated TagBFP2-3XFlag multi-miRE or HrasG12Vdonors(~5 μg/μl) and piggyBac-EGFP reporter into wildtype pups (i.e. non-mTmG) resulted in no abnormal growth, hyperplasia, or tumorigenesis regardless of Flp or Cre presence (Fig. S2J; for examples of observed phenotypes after MADR of multi-miRE or HRasG12V phenotypes see below). Several independent EPs of the HrasG12V donor plasmid and Cre recombinase alone failed to produce tumor formation when examined at 2 weeks post-EP, indicating that Cre cannot induce marked stable integration of MADR donors without Flp-excision (Fig. S2J). In addition, we assessed Eped mTmG pups with HrasG12V harboring an inverted loxP and failed to detect blue recombined cells or hyperplasia by immunostaining, illustrating the specificity of MADR recombination reaction in vivo (Fig. S2K).
Although MADR is compatible with many existing mice, mTmG presented us with the drawback of being unable to use the red color channel (e.g. Fig. 2B) due to the native tdTomato. We solved this limitation two ways: by using a fifth laser channel with >750 nm wavelength fluorophores (Fig. S2L) or by bleaching and immunostaining the now available red channel (Fig. S2M–N). With bleaching, we tested for multiplex labeling of cell lineages in vivo by electroporating 4 smFP vectors simultaneously in mTmGHet pups (Fig. 2D). This resulted in four groups of distinctly colored olfactory neurons by 2 weeks, confirming one-transgene-to-one-cell stable integration (Figs. 2E–F; Movie S3) similar to the in vitro observations (Figs. 1H–I). These experiments suggest that MADR is a reliable method that depends on a well-known biochemical reaction specifically catalyzed at the target locus. Moreover, MADR is ideal for expansion microscopy approaches (Tillberg et al., 2016) that enable super resolution-like detail of the fine cellular features such as astrocytic processes due to the increased cell size combined with the excellent signal properties of the smFP-myc and EGFP reporters (Figs. 2G–L).
A potential limitation of MADR is its utilization of two commonly used recombinases, Flp and Cre. Thus, we tested overlaying conditional VCre-mediated activation of another transgene. To do this, we created a plasmid expressing VCre downstream of TagBFP2-P2A (Fig. 2M). Then we used an smFP-myc-based VCre FlEx reporter (Fig. 2M) to look for recombination with and without TagBFP2-P2A-VCre donor. Notably, smFP-myc was not detected when an alternate TagBFP2-3flag was inserted but was readily expressed when the VCre-containing donor was inserted (Fig. 2N–O). Taken together, MADR allows for precise and flexible, single-copy somatic transgenesis, which is further enabled by a suite of genetic tools for the investigation of cell lineages.
MADR loss of function phenotypes mimic glioma GEMMs
Given the stable genomic insertion and transgene expression that MADR provides, we sought to exploit MADR for generating single-copy in vivo tumor models. LOF tumor suppressor gene mutations such as Nf1, Pten, and Trp53 are some of the most prevalent driver genes in glioma patients (Mackay et al., 2017). Many mouse glioma models have shown that knockout of these tumor suppressors leads to high-grade gliomas. For example, it was elegantly demonstrated using MADM GEMMs that the dual Trp53/Nf1-knockouts promote the pre-malignancy hyperproliferation of oligodendrocyte progenitor cells (OPCs) (Liu et al., 2011). To test whether miR-E shRNAs against tumor suppressors are sufficient for tumorigenesis, we created a donor construct harboring TagBFP2 followed by 3 validated miR-Es targeting Nf1, Pten, and Trp53 (Fig. 3A–B). We observed the selective overgrowth of TagBFP2+/Pdgfra+ OPCs in vivo, aligning with similar observation in alternate LOF models based on Nf1 and Trp53 (Fig. 3C and S3A) (Liu et al., 2011, Breunig et al., 2015). Notably, the EGFP+ population with only Cre-excision yielded a smaller, mixed population of astrocytic cells and olfactory bulb neurons (Fig. 3C). These EGFP+ cells function as an EPed control cell population in MADR studies.
Figure 3: Loss-of-function manipulations using MADR transgenesis.

A) Donor construct for miR-E shRNAs against Nf1, Pten, and Trp53 tied to TagBFP2 reporter
B) Validation of knockdown efficacy of multi-miR-E function by qPCR.
C) 6-month-old mouse sagittal section showing a hyperplasia of TagBFP2+ cells but no tumor. Scale bar, 1mm
D) Plasmid for MADR of a TagBFP2-V5 reporter protein and SpCas9
E) Sequencing of TdTomato-/EGFP- glioma cells exhibit InDels in Nf1 and Trp53.
F) MADR insertion of TagBFP2-V5 reporter and Cas9 with co-EPed PCR-derived sgRNAs yields high grade glioma observable through labeling of 3 genetic reporter-defined populations in a coronal section of both hemispheres. Scale bar, 1000μm
G) Plasmid for MADR of an smFP-myc reporter protein and FNLS Cas9n base editor.
H) sgRNA-targeting sites (green letters) induce C->T base conversion (red lowercase ‘c’ are targeted) to produce premature stop codons in Nf1, Trp53, and Pten.
I) MADR insertion of myc reporter and FNLS Cas9n with co-EPed PCR-derived sgRNAs yields observable expansion of OPC progenitors at two months post-EP through labeling of three genetic reporter-defined populations in a coronal section. Scale bar, 1000μm
J) High magnification tdTomato (1), EGFP (2), and Myc tag (3) image showing myc+ populations. Scale bar, 100μm
To effect in vivo gene targeting with CRISPR/Cas9, we created a smBFP2-P2A-SpCas9 donor plasmid to simultaneously label and mutate cells, enabling faithful tracing of mutant cells in vivo (Fig. 3D). Co-delivery of this plasmid and sgRNAs to target Nf1 and Trp53 were enough to cause morbidity in EPed animals by 5 months, and pathological analysis diagnosed glioblastoma multiforme. Successful targeting in EPed cells was confirmed by genotyping (Fig. 3E). Confocal imaging demonstrated that the tumor was largely devoid of tdTomato-labeled populations, although the vasculature stayed red (Fig. 3F–F1, S3B).
To complement these Cas9-based LOF methods, we added the CRISPR/Cas base editor (FNLS) to MADR (Fig. 3G), which catalyzes C-to-T mutation near sgRNA-target site. We introduced smFP-myc reporter, FNLS, and sgRNAs designed such that they would create premature stop codons in Nf1, Trp53, and Pten (Fig. 3H) (Zafra et al., 2018). Amplicon sequencing of GFP-sorted MADR cells confirmed that the base editors could induce premature stops (Fig. S3C). Two months later, we noted a dramatic expansion of OPCs similar to the mir-E and Cas9 LOF studies (Fig. 3I–J). All of these knockdown vs knockout studies were done in the same mouse line (mTmG) and demonstrated MADR’s various means for multiplexing LOF analysis with combined lineage tracing. Moreover, we have generated MADR elements for CRISPR/Cas variants for gene knockdown/knockout (Fig. S3D) (Yeo et al., 2018, Konermann et al., 2018, Cox et al., 2017, Slaymaker et al., 2016). Taken as a whole, we have generated a diverse toolset for MADR LOF studies that recapitulate the phenotype observed in MADM mosaics.
MADR illuminates GOF oncogene dosage sensitivity
We made a HrasG12V-based MADR donor compatible with RCE reporter mouse and performed in utero EP (IU-EP) in E14 RCE-heterozygous embryos (Fig. 4A–B). PiggyBac-mediated HrasG12V-overexpression in mouse embryos has been shown to induce high-grade tumors within 15–20 days of birth (Glasgow et al., 2014). In contrast, we did not observe tumor growth when the MADR x RCE-het animals were examined at P15. However, we noted a marked cell-fate switch of TagBFP2-HrasG12V cells to the astroglial lineage (Fig. 4C, S4A). EGFP+ Cre-excised cells consisted of a mixed population of neurons and glia (Fig. 4C, S4A). This is an important case where MADR disagrees with multicopy-transgene based transposon models, highlighting the consequence of GOF oncogenes depending on gene dosage.
Figure 4: Generation of somatic glioma using in vivo MADR with HrasG12V indicates dosage effects of this oncogene and human oncofusion proteins generate ependymal tumors.

A-B) Schematic for in utero EP of MADR into E14.5 RCE +/− dams
C) In utero EP in RCE mice with HrasG12V oncogene produces mosaic patches of TagBFP+ astrocytes Rosa26HrasG12 but not evidence of invasive glioma
D) Schematic of possible outcomes after MADR in homozygous mt/mg recipient mice
E) P2 EP of homozygous mt/mg mice with TagBFP2-HrasG12V oncogene
F) Postnatal EP in homozygous Rosa26mTmG P2 pups with HrasG12V oncogene produces two different tumor types (Blue-only Rosa26HrasG12V×2 and blue-and-green Rosa26HrasG12V×1) Scale bars: 2mm
G) Representative tumor formation in homozygous mTmG 3 months post-EP. Blue-only Rosa26HrasG12V×2 cells occupy a larger section of the tumor than blue-and-green Rosa26HrasG12V×1, correlating with phosphor-Rb1 protein expression. Scale bars: 1mm
H) Zoom-in images of regions 1 and 2 from G show phosphorylated-Rb1 expression correlates largely with blue-only cells. Scale bars: 50μm
I) Plasmid schematics for expression of ependymoma-associated fusion proteins
J) Stitch of YAP1-MAML1D; p16/p19 Cas9 targeting induced ependymoma-like tumor.
K) Survival analysis of Ependymoma MADR model mice
L) Ependymoma-like tumor in a 3-month-old C11orf95-RELA; p16/p19 Cas9-targeted mouse
When the MADR TagBFP2-HrasG12V transgene was delivered postnatally to mTmGHet, HrasG12V+ cells similarly overproliferated when compared with EGFP+ populations (Fig. S4C–D). To definitively examine the effects of HrasG12V dosage, we EPed Hras in homozygous mTmG, in which we expected to be able to differentiate HrasG12V×1 or HrasG12V×2 cells (Fig. 4D–E). All mice rapidly developed glioma and reached morbidity within 3-4 months.
Interestingly, in homozygous mTmG mice, blue-only cells (HrasG12V×2) occupied a bigger patch of tumor cross-section than cells expressing both blue and green (HrasG12V×1) (Fig. 4F–G). Using piggyBac, we also observed that the patch of brighter EGFP-tagged HrasG12V cells expressed phosphorylated Rb1 (pRb1) more than the dimmer EGFP+ cells (Fig. S4D). In MADR, where the copy number is unambiguous, most of the Rosa26HrasG12V×2 cells seemed to express pRb1, whereas it was expressed in fewer hemizygous Rosa26HrasG12V×1 cells (Fig. 4G–H). Thus, MADR mosaics enable one to genetically distinguish the zygosity of two groups of cells on a single cell level and confirms that the copy number of oncogenes—which is uncontrollable in many somatic transgenic methods—can significantly alter the profile of resulting tumors.
Delivering ependymoma driver fusion with MADR yields phenotypically appropriate tumors
Many tumor drivers are fusion proteins, but it can be difficult to make a conditional GEMM mimicking chromosomal rearrangement. For example, the fusion protein drivers YAP1-MAML1D and C11orf95-RELA are recurrently seen in supratentorial ependymomas, and we made MADR vectors to express them (Pajtler et al., 2015, Parker et al., 2014) (Fig. 4I). Compared to MADR-KrasG12A tumor models - a genetic driver of glioma (Ceccarelli et al., 2016), YAP1-MAML1D and C11orf95-RELA MADR tumor cells showed remarkably different initiation patterns. Whereas KrasG12A cells rapidly invaded the striatum and proliferated (Fig. S4F), YAP1-MAML1D tumors delaminated into rosette-like structures and induced a non-cell autonomous reactive gliosis in the surrounding EGFP+ control cells (Fig. S4G–H). C11or95-RELA cells displayed a mixed phenotype, whereby they often stayed along the VZ or formed small clusters near the ventral VZ (Fig. S4I–J). To mimic the coincident loss of Cdkn2a that is frequently seen in ependymomas (Pajtler et al., 2015), we used Cas9 with sgRNAs against p16 and p19. YAP1-MAML1D × p16/19-knockout animals reached morbidity within roughly 1.5 months (Fig. 4J–K). However, the C11orf95-RELA × p16/19-knockout tumors showed a more protracted survival, reaching morbidity at approximately 3 months (Fig. 4K–L). Unlike the infiltrative margins of our glioma models and human glioma, the ependymomas exhibited defined margins with a lack of invading cells (Fig. S4J–K). Taken together, this data demonstrated MADR’s ability to model diverse tumor types, including those driven by fusion proteins.
Direct comparison of H3f3a G34R and K27M pediatric glioma drivers using MADR
Almost all human tumors present with a distinct set of somatic and germline mutations. With the ability to select mutations and to compare these sets of mutations, MADR can serve as a personalized tumor model platform tailored for studying genetic contributions to survival and drug resistance that are unique to each tumor. As a proof-of-principle, we chose to model pediatric glioblastoma multiforme where H3F3A K27M or G34R mutations are observed in more than 50% of patients, but co-occur with a variety of other mutations (Mackay et al., 2017). For example, H3F3A mutations are often coincident with recurrent dominant-active Pdgfra (D842V), and dominant-negative Trp53 (R270H) (Schwartzentruber et al., 2012, Mackay et al., 2017). Several K27M GEMMs have been introduced (Cordero et al., 2017, Funato et al., 2014, Larson et al., 2019, Pathania et al., 2017), but the ability to readily compare the functional effects of the K27M to G34R in vivo remains a challenge. To demonstrate MADR’s utility in this context, we made donor plasmids for modeling simultaneous H3f3a, Pdgfra, and Trp53 mutations—with variants differing only by missense mutations for G34R or K27M to study the differential effects of these driver genes (Fig. 5A).
Figure 5: MADR glioma models utilizing recurrent mutations observed in pediatric glioblastoma multiforme has phenotypes consistent with human subtypes.

A) Schematic of donor plasmid for MADR with multiple recurrent pediatric glioma driver mutations
B) Schematic of the plasmid delivery and electrode sweep employed to target striatal and cortical germinal niches simultaneously
C) Zoomed view from B showing the respective cortical (magenta) and striatal (orange) germinal niches that are targeted
D) Representative tumor formation in heterozygous mTmG 100 days post-EP. Nuclear EGFP+ Rosa26H3f3a-K27M/Pdgfra/Trp53 cells form a large striatal tumor. Inset D-1 shows a lack of significant cortical infiltration.
E) A littermate Rosa26H3f3aG34R/Pdgfra/Trp53 exhibits a glial hyperplasia in the striatum and cortex but no tumor is evident.
F) K27M tumor at 120 days post-EP is predominantly sub-cortical.
G) Cortically-infiltrating G34R tumor at 120 days post-EP.
H-I) Confocal pathology of K27M tumor at low mag (H), and high mag (I).
J) Low mag pathology of G34R tumor.
K) Comparison of survival across H3.3. groups (wildtype-blue, K27M-green, and G34R-red) all containing Pdgfra D842V and Trp53 R270H.
L) Chart of the site of K27M versus G34R tumors. *Because of the later onset of tumor growth in G34R groups and their inconsistent survival times, we were unable to collect 2 of 7 G34R samples before death to definitively ascertain initial tumor site.
M-N) Experimental schematic for co-EP of K27M and G34R plasmids
O-P) G34R and K27M immunostaining of co-EPed tumors in sequential sections. (smFP-myc shown in insets).
Q) Quantification of normalized cell counts from tumor
First, we checked for appropriate production of H3f3a, Pdgfra, and Trp53 by immunohistochemistry in vitro and in vivo and noted coincident expression of all proteins (Fig. S5A–B). Next, we introduced these plasmids by postnatal EP into sibling pups over several litters. To transfect the precursor cells in both cortical and striatal VZs, the electrodes were swept as shown (Fig. 5B–C). For the first 2–4 months, there was a diffuse expansion of EGFP+ cells in both G34R and K27M mice but no tumors were identifiable (Fig. S5C).
Patient tumors bearing either K27M or G34R/V mutations exhibit different transcriptomes as well as clinical features (Schwartzentruber et al., 2012, Sturm et al., 2012). Human K27M gliomas cluster along the midline, whereas G34R occur in the cerebral hemispheres (Sturm et al., 2012). K27M tumors manifest in younger patients than G34R/V (Sturm et al., 2012, Mackay et al., 2017). Seemingly in agreement with their earlier clinical presentation, some K27M+ mice exhibited midline gliomas by P100, at which time G34R+ displayed diffuse hyperplasias and very rare, small tumors (Fig. 5D–E). At P120, K27M tumors predominantly localized to the sub-cortical structures but cells could be observed in the white matter tracts (Fig. 5F). In contrast, G34R tumors localized to the corpus callosum and deeper cortical layers, often forming “butterfly” gliomas across the midline (Fig. 5G) in a pattern akin to patients. This happened despite the transduction of the striatal VZ, indicated by mG astrocytes (yellow arrow in Fig. 5G).
Pathological features included high cell density, microvascular proliferation, and necrosis at late stages (Figs. 5H–J). Both K27M, and G34R tumors were 100% penetrant and showed accelerated endpoints compared with H3f3a wildtype tumors containing Pdgfra and Trp53 mutations (Fig. 5K), but consistently exhibited a tumor “site-of-origin” (i.e. midline vs. cortical) matching to their patient counterparts (Fig. 5L). To ascertain the expression of the appropriate H3f3a mutation we employed monoclonal antibodies against the respective mutant residues (Fig. S5D–G).
To compare the cell autonomous properties of these cells we exploited unique properties of MADR whereby each allele can receive only one transgene insertion, and co-delivered K27M and G34R plasmids at a 1:1 ratio (Fig. 5M–N). The use of the aforementioned anti-K27M and anti-G34R antibodies in serial sections confirmed the co-expression of the respective transgenes (Fig. 5O–P) in individual tumors. We further confirmed that each smFP-myc+ cell expressed only one H3f3a mutant variant (Fig. S5H). Quantification of K27M and G34R cells demonstrated a highly significant increase in K27M, indicating their ability to out proliferate their G34R counterparts (Fig. 5Q). These findings indicate that the K27 and G34 residues given the same genetic background—or even animal—can alter the time and location of onset of these glioma subtypes similar to human phenotypes.
Several studies have shown that K27M mutations lead to hypomethylation at the H3K27 residue, and we confirmed the hypomethylation of K27M mutant cells by H3K27me3 antibody (Fig. S5I–J). The invasive tumor cells exhibited perineural satellitosis as has been described in human K27M tumors, and the juxtaposed EGFP+ K27M glia and neurons showed markedly different H3K27me3 levels at high resolution (Fig. S5K) (Venkatesh et al., 2015). Hypomethylation was not an artifact of tumor growth because in our CRISPR/Cas9-based Nf1/Trp53-knockout models, gliomas were normal or hypermethylated (Fig. S5L). These imaging findings demonstrate that MADR glioma models display many of the unique features of patient glioma.
MADR K27M recapitulates human tumor heterogeneity and developmental hierarchy
Immunohistological analysis demonstrated that tumor cells upregulated Bmi1 (Fig. S5M–O), which has recently been identified as being enriched in K27M glioma (Filbin et al., 2018) and as a population expressed markers of astrocytes and oligendrocytes (Fig. S5P–Q). This heterogeneity of glial markers was similar to recent findings in human K27M tumors (Filbin et al., 2018), which demonstrated a significant degree of intratumor heterogeneity by single-cell RNA sequencing (scRNA-seq). Given the availability of this analogous human K27M data we took the unique opportunity to credential the MADR model cells against their human counterparts and gain deeper insight into the MADR tumor heterogeneity through the use of scRNA-seq.
We subjected EGFP+ sorted tumor cells from 3 independent K27M tumors to droplet-based scRNA-seq (Fig. 6A, Table S1). Copy-number variation analysis demonstrated chromosomal abnormalities (Fig. S6A) as is observed in human K27M glioma (Filbin et al., 2018). Following sequencing, alignment, and quality control, we clustered the mouse K27M cells using Seurat (Fig. 6B–C, S6B–D) (Butler et al., 2018). For the choice of gene set for CCA-alignment, we used the four programs termed P1–4 that were identified in the human dataset, as this dataset and associated analysis represented a unique “ground truth” for comparison (Filbin et al., 2018).
Figure 6: Single-cell RNA-sequencing-based analysis of MADR glioma models.

A) Schematic of cell dissociation and scRNA-seq
B) UMAP depicting CCA alignment of 3 MADR mouse K27M scRNA-seq datasets from 3 distinct tumors, colored by cluster based on HVG programs P1-4 from (Filbin et al., 2018)
C) Heatmap depicting marker genes emerging from unbiased clustering of mouse K27M cells
D) Program and expression featureplots from CCA of mouse K27M tumors.
E) UMAP depicting CCA alignment of 6 human K27M datasets from 6 distinct tumors (Filbin et al., 2018), colored by cluster
F) Heatmap depicting markers genes emerging from unbiased clustering of human K27M cells
G) Program and expression featureplots from CCA of human K27M tumors.
H) UMAP depicting CCA alignment of 3 MADR mouse K27M datasets and 6 human K27M datasets (Filbin et al., 2018), colored by cluster
I) Program and expression featureplots from CCA of combined mouse and human K27M tumors.
J) UMAP depicting CCA alignment of 9 K27M datasets from the mouse and human brain colored by sample
K) Heatmap using gene list from (Filbin et al., 2018) demonstrates a high concordance of gene expression between murine and human K27M glioma cells.
L) scRNA-seq derived proliferation metrics are comparable across mouse and human sample
The “Cycle” cluster consisted of cells expressing markers of proliferation, including Top2a, mKi67, and Ccnb1 (Fig. 6B–C; Fig. S6E, Table S2). Astrocyte-like (AC) and oligodendrocyte-like (OC) clusters expressed genes associated with these more differentiated cell types (Fig. 6B–C; S6D, Table S2), while the largest cluster with features of oligodendrocyte progenitors and, thus, termed “OPC” based on the human P4 cluster (Filbin et al., 2018), expressed genes including Olig1, but didn’t seem to clearly fall into a differentiated cell lineage (Fig. 6B–C; S6D, Table S2). Scoring clusters based on gene lists derived from human K27M (Filbin et al., 2018) confirmed the enrichment of astroglial markers in AC and the enrichment of oligodendroglial markers in OC (Fig. 6B–D).
To conduct cross-species analysis of K27M gliomas, we repeated the Seurat clustering with all the cells from mouse and human K27M tumors (Fig. 6E–G; S6F–L) and saw that the 9 combined single-cell datasets continued to yield the four clusters seen in the individual mouse and human CCA alignments (Fig. 6H–J). By splitting the combined 9 sample UMAP into each respective sample, we noted relatively similar—though not uniform—contributions of cells from each sample to each individual cluster (Fig. 6J; S6M). Our specific combination of mutations closely matched patient MUV10, and this patient contained less AC cells than other patients, as our mouse K27M cells did (Fig. S6M).
As a “sanity check” for our credentialing approach of using the previously-identified P1-P4 programs (Filbin et al., 2018), we also performed clustering with the more common practice of employing highly-variable-genes for CCA, clustering, and UMAP analysis. This approach led to some almost identical clusters (e.g. cycling populations) but division of other populations into sub-clusters (e.g. OPC), which varied by the parameters chosen (Fig. S6N). This variability of clustering is an inherent issue in scRNA-seq due in part to batch effects, patient-specific transcriptome alterations, and in challenges associated with cross-species comparison (Butler et al., 2018, Stuart and Satija, 2019).
We also used the differentially expressed genes identified across human K27M, glioblastoma multiforme, IDH astrocytoma, IDH oligodendroglioma (Filbin et al., 2018) to plot a heatmap comparing our 3 mouse K27M tumors. The MADR K27M tumors exhibited gene expression comparable to human K27M tumors but not to other human glioma subtypes (Fig. 6K). Further, human K27M cells are characterized by a high proportion of cycling cells, as our mouse tumors did (Fig. 6L). Thus, MADR K27M tumors exhibit a high degree of correspondence with their human counterparts when compared by scRNA-seq.
MADR K27M regulatory network analysis uncovers unique features in cycling cells
We have shown a global correspondence between the MADR-based K27M mouse and the human K27M glioma transcriptomes, especially in that they show similar developmental hierarchies and over-representations of cycling cells. To our knowledge, our K27M scRNA-seq dataset is one of the first created to validate a mouse tumor model. Therefore, we subjected the datasets to further analysis to gain novel insights. The K27M mutation leads to widespread epigenetic perturbation, which led us to focus on whether similar transcription factor networks underlie human and mouse tumors. NANOG, OCT4, SOX2, MYC2, and embryonic stem cell (ESC)-expressed gene sets and the under-expression of PRC2, SUZ12, EED, and H3K27-bound gene sets have shown to indicate this poorly differentiated state (Fig. 7A–B) (Ben-Porath et al., 2008, Kim et al., 2010). In both human and mouse datasets, this embryonic stem cell-signature seemed to be strongest in the cycling cell types (Fig. 7A–B). As a further evidence, we performed Chip-seq on the three tumors, identified the genes that are specifically hypomethylated, and found that this subgroup of genes is highly expressed in the cycling cells (Fig. 7A–B; S7A–C). Further, SCENIC regulon-based analysis (Aibar et al., 2017a) on human and mouse K27M cells further indicated an enrichment for EZH2 and BRCA1 regulons—among others—specifically in the cycling populations despite their respective mRNA expression patterns being markedly less cycle-restricted (Fig. 7C–D, S7D–I). These findings point to a cell cycle-dependent enrichment of EZH2 and ESC—based transcriptional networks that might coordinate glioma self-renewal.
Figure 7: H3.3 K27M Transcriptional Network and snATAC-seq Analysis.

(A-B) t-SNE featureplots depicting cell type-specific upregulation NANOG, OCT4, SOX2, MYC target genes, and embryonic stem cell (ES)-associated gene sets and the underexpression of PRC2, SUZ12, EED, and H3K27-bound gene sets for human cells (A) and analogous genes/genesets in mouse (B).
C-D) Binary regulon activity and corresponding scRNA-seq featureplot for EZH2, BRCA1 (C; Filbin et al. dataset), Ezh2, and Brca1(D; K27M mouse dataset).
E) Schematic of snATAC-seq sample preparation
F) tSNE of sc- and snATAC datasets from P50, F) E18) and G) K27M mouse brains
I) K27M snATAC-identified MSigDB pathways
J) Genome browser alignments of snATAC-seq, scATAC-seq, and bulk ATAC-seq. *-Tumor MG is an overlaid(red/black) alignment of snATAC-seq microglial clusters captured with the K27M cells.
To examine the underlying epigenetic state through the examination of differentially accessible genome regions (DARs), we performed single-nucleus ATAC-seq of K27M mouse tumors and compared them to normal P50 and E18 mouse brains (Fig. 7E–H, Fig. S7J–R). While the P50 brain exhibited well-spaced, canonical marker gene defined clusters Fig. 7F, Fig. SJ-K); both the E18 brain (an alignment of 3 independent datasets; Fig. 7G; Fig. S7L–O) and tumor cells (but not the co-captured tumor microglia—which create distinct clusters) exhibited less well-defined DARs (Fig. 7H, Fig. S7P–R). Moreover, pathway analysis of K27M tumor clusters was notably altered when compared with the pure P50 astrocyte and OPC clusters (Fig. S7T), including a BRCA1-associated term consistent with SCENIC findings (Fig. 7C–D, I). Finally, alignment of DARs from these scATAC samples and a bulk K27M dataset further supported the tSNE findings that glial lineage-associated transcription factors like Olig2, Sox9, and Sox10 exhibit reduced relative accessibility when compared with P50 glial lineages and mutual exclusivity in terms of Sox9 and Sox10 (Fig. 7J). The K27M scRNA-seq data was consistent with this as Sox9 and Sox10 mRNA were co-expressed in each tumor cluster and often in individual cells, which is exceedingly rare in the normal adult brain (Zeisel et al., 2018). However, DARs found in the bulk samples were recapitulated in the scATAC datasets (i.e. Cacng8 in K27M tumors - 6.322 log2 ratio K27M bulk:mNSCs and Hes5 in mNSCs – 3.248 log2 ratio mNSCs:K27M tumors; Fig. 7J). Further, co-captured microglia retained robust DARs, arguing against dominant batch effects; Fig. 7J). Finally, the K27M tumor cells exhibited many immediate early gene motifs associated with cancer (Pylayeva-Gupta et al., 2011) and motifs for many of the embryonic stem cell-associated transcription factors (Fig. S7U) previously identified in aggressive tumors (Ben-Porath et al., 2008). Taken together, the K27M oncohistone leads to altered activity of a subset of transcription factors in the actively cycling subsets of these tumors by generating a primitive epigenetic state.
DISCUSSION
GEMMs still entail cumbersome mouse engineering and significant cross-breeding. Conversely, electroporation (EP) and viral transgenesis have enabled quick somatic transgenic investigations of development and disease but lack the precision of mouse models. MADR overcomes the intrinsic disadvantages associated with these methods, is a robust strategy for creating somatic mosaics with predefined insertion sites and copy numbers, and requires a negligible amount of colony maintenance. We demonstrated the versatility of MADR to generate combined modes (gain and loss of function) of mutations for multiple tumor drivers expeditiously and flexibly.
MADR compares favorably to CRISPR/Cas-based methodologies for in vivo mouse genetic manipulation and can be multiplexed with virtually all of them for mosaic generation. Further, there is an ever-increasing list of tissues that can be targeted by plasmid DNA EP, hydrodynamic infection, or lipofection, including retina, muscle, bone, thymus, pancreas, liver, skin, and bladder. Thus, MADR could be generalized for creating mosaics and tumors in a host of tissues. Additionally, non-integrating viral vectors could be employed to deliver MADR constituents to avoid insertional mutagenesis. It is important to note that besides the mTmG and RCE lines, MADR can be employed with any GEMM harboring dual recombinase sites, including thousands of Knockout Mouse Consortium lines where MADR can “trap” native cis-regulatory sequences (Data S1A) (International Mouse Knockout et al., 2007).
Next generation sequencing has exponentially increased the catalogue of recurrent somatic mutations seen in tumors. Further, it is now increasingly appreciated that histologically similar tumors can often have disparate genetic underpinnings with different phenotypes (e.g. K27M vs. G34R). We show proof of principle for using MADR as a platform for rapid ‘personalized’ modeling of diverse glioma types by combining gain and loss of function mutations. As an example, several K27M mouse models have been recently introduced (Cordero et al., 2017, Pathania et al., 2017, Funato et al., 2014, Larson et al., 2019). However, the midline-specific tumor emergence, a diagnostic feature in human K27M gliomas, was not exclusively observed in K27M piggyBac models (Pathania et al., 2017). A recent GEMM was able to generate midline K27M gliomas and uncovered key mechanisms of tumorigenesis, but suffered from a small but significant number of peripheral tumors and medulloblastomas (Larson et al., 2019). To our knowledge, our MADR-based model is the only one successful at recapitulating the spatiotemporal regulation of tumor growth by K27M vs G34R mutations. Further, by unambiguously comparing K27M and G34R mutant cells side-by-side in vivo in individual animals—a unique advantage of MADR— we have observed the increased ability of K27M to accelerate tumor growth compared to G34R. Thus, while our K27M and G34R models are both 100% penetrant, these distinct mutations at closely situated residues exert distinct and powerful influences over tumor growth dynamics and tumor sites of origin. We noted a similarly remarkable pattern in our novel side-by-side comparisons of YAP1-MAMLD1 and C11orf95-RELA ependymoma models, whereby synchronized MADR transgenesis in the same cell populations led to disparate survival times. This suggests that the clinical age of onset for tumor subtypes may not be reflective only of cell origin or time of mutation, but also is highly dependent on driver-mutation dictated growth dynamics. Very recent findings in the intestinal stem cell niche have uncovered that there is a “reverse chronology” in terms of enhancers that are activated after PRC2 complex inactivation (a manipulation which is presumably phenocopied by H3f3a K27M mutations) (Jadhav et al., 2019). Using our novel models combined with single-cell approaches, our observations that K27M tumor cells exhibit a protracted pre-tumor stage culminating in a primitive embryonic-like transcriptional and epigenetic state is consistent with the possibility that K27M mutation exhibits this same reverse chronology reactivation of developmental enhancers.
In summary, our findings establish MADR as a robust genetic methodology, one which promises to democratize the generation of high-resolution gain and loss of function mosaics, allowing a small lab to model a wide spectrum of genetic subtypes in vivo. Additionally, this genetic framework is adaptable to the thousands of mouse lines already engineered with dual recombinase recognition sites, and can easily be adapted to any cell, organoid or organism that can be engineered with a MADR recipient site. Given MADR’s ability to be combined with the existing arsenal of genetic approaches, its single-cell resolution, and its compatibility with sequencing technologies, these tools allow for efficient, higher throughput investigation of gene function in development and disease.
STAR★METHODS
EXPERIMENTAL PROCEDURES
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua Breunig (joshua.breunig@cshs.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were used in accordance with the Cedars-Sinai Institutional Animal Care and Use Committee. Embryonic day (E) 0.5 was established as the day of vaginal plug. Wild-type CD1 mice were provided by Charles River Laboratories. Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J and Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax mice (JAX Mice) were bred with wild-type CD1 mice (Charles River) or C57BL/6J mice to generate heterozygous mice. Male and female embryos between E12.5 and E15.5 were used for the in utero electroporations, and pups between postnatal day (P) 0 and P21 for the postnatal experiments. Pregnant dams were kept in single cages and pups were kept with their mothers until P21, in the institutional animal facility under standard 12: 12 h light / dark cycles.
Commercial cell lines
Human female embryonic kidney derived HEK293T and Mouse male Neuro-2a acquired from ATCC were used for MADR in vitro validation in human and mouse cells respectively. Both cell lines were maintained in DMEM high glucose (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS, GlutaMAX (Life Technologies 35050) and penicillin-streptomycin-amphotericin (Thermo Fisher Scientific, Waltham, MA).
De novo cell lines
Three heterozygous P0 mTmG pup brains were dissociated to establish the polyclonal mouse neural stem cell line used in the study. Sex was not noted due to the lack of reliable visual methods. The cell line was maintained as previously described (Breunig et al., 2015). Cells were grown in media containing Neurobasal®-A Medium (Life Technologies 10888–022) supplemented with B-27 without vitamin A (Life Technologies 12587-010), GlutaMAX (Life Technologies 35050), Antibiotic-Antimycotic (Life Technologies 15240), human epidermal growth factor (hEGF) (Sigma E9644), heparin (Sigma H3393), and basic fibroblast growth factor (bFGF) (Millipore GF003).
Additionally, 3 MADR-K27M tumor cell lines were generated by dissociation and sorting of GFP+ tumor cells from 3 different heterozygous mTmG mice (Table S1). K27M-1 cells were isolated from a female mouse, while K27M-2 and K27M-3 cell lines were generated from male mice. Finally, a non-tumor TdTomato+ cell line was isolated from the same region as K27M-1 by FACS of the non-transformed TdTomato+ peri-tumor populations. Newly isolated cell lines were cultured in Neurobasal media (Thermo Fisher Scientific, Waltham, MA) supplemented with penicillin-streptomycin-amphotericin (Thermo Fisher Scientific, Waltham, MA), B-27 supplement without Vitamin A (Thermo Fisher Scientific, Waltham, MA), Glutamax (Thermo Fisher Scientific, Waltham, MA), EGF (Shenandoah Biotechnology, Warwick, PA), FGF (Shenandoah Biotechnology, Warwick, PA), PDGF-AA (Shenandoah Biotechnology, Warwick, PA) and heparin (StemCell Technologies, Cambridge, MA); and cultured in a CELLstart CTS (Thermo Fisher Scientific, Waltham, MA) treated T25 Flask.
METHODS DETAILS
Plasmid cloning
The MADR pDonor plasmids were derived from PGKneotpAlox2, using In-Fusion cloning (Clontech) or NEBuilder HiFi DNA Assembly Master Mix (NEB) in combination with standard restriction digestion techniques (Breunig et al., 2015, Soriano, 1999) . Briefly, FRT site was created by annealing two oligos and infusing the insert into PGKneotpAlox2. Downstream generation of donor plasmids were done by removing the existing ORF and adding a new cassette using In-Fusion or ligation, as was done for the smFP-HA ORF (Addgene 59759). PiggyBac-CAG-plasmids were previously described and created using combination of In-Fusion, NEB assembly, and ligation strategies (Breunig et al., 2015, Breunig et al., 2012). Primer sequences used for In-Fusion or assembly reactions are available upon request. PCR was done using a standard protocol with KAPA HiFi PCR reagents. The original CMV Flp-2A-Cre and CMV Flp-IRES-Cre recombinase expression constructs were previously validated in the context of in vitro dRMCE (Anderson et al., 2012).
MADR + AAVS1 human cell line generation
AAVS1 targeting MADR vector was derived from AAVS1-targeting vector AAVS1_Puro_PGK1_3xFLAG_Twin_Strep (Addgene 68375). TagBFP2-V5-nls-P2A-puroR-Cag-LoxP-TdTomato-FRT was inserted into this AAVS1 vector, and a human cell line was transfected with it and selected in puromycin. MADR-smFP-myc (bright) and MADR-TagBFP2–3flag WPRE was transfected into the resulting stable cell line with Cag-Flpo-2A-Cre to induce the MADR reaction.
PCR analysis of MADR integration events
KAPA HiFi PCR reagents were used to PCR genomic DNA collected from mouse MADR lines. Amplicons were run on an E-Gel apparatus to assess size.
Mice and electroporation
Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J and Gt(ROSA)26Sortm1.1(CAG-EGFP)Fsh/Mmjax mice (JAX Mice) were bred with wild-type CD1 (Charles River) or C57BL/6J (JAX) mice to generate heterozygous mice. Postnatal lateral ventricle EPs were performed as previously described (Breunig et al., 2015). P1–3 pups were placed on ice for ~5 min. All DNA mixtures contained 0.5-1μg/μl of Flp-Cre expression vector, donor plasmid, hypBase, or CAG-reporter plasmids diluted in Tris-EDTA buffer, unless noted otherwise. Fast green dye was added (10%v/v) to the mixture, which was injected into the lateral ventricle. Platinum Tweezertrodes delivered 5 pulses of 120 V (50ms; separated by 950 ms) from the ECM 830 System (Harvard Apparatus). SignaGel was applied to increase conductance. Mice were warmed under a heat lamp and returned to their cages.
In utero electroporation.
In utero electroporation experiments were performed according to standard methods (McKenna et al., 2011). TagBFP2-HRasG12V and Flp-Cre plasmids were EPed into E14.5 RCE mice embryos. After electroporation, the embryos were allowed to survive to P15, at which time TagBFP2-HrasG12V (MADR mediated insertion), EGFP (non-MADR Cre-mediated recombination) and Sox2 expression was analyzed by immunostaining.
Note on MADR transduction
Mosaic analysis with dual recombinase-mediated cassette exchange (MADR) works with any approach that enables a minimum level of plasmid entry for subsequent FlpO and Cre transgene expression to facilitate donor plasmid insertion into the genome. In our experimentation, we have successfully employed in vivo electroporation, in vitro electroporation (i.e. nucleofection), and lipofection to effect MADR.
In vivo electroporation is believed to work by allowing plasmid DNA to permeate the plasma membrane and enter the nuclear space of cells undergoing mitosis (Stancik et al., 2010, Breunig et al., 2012, Ortiz-Alvarez et al., 2019). Thus, it is believed to be largely specific for the proliferating populations (Stancik et al., 2010, Breunig et al., 2012, Ortiz-Alvarez et al., 2019). However, postmitotic cells may be also targeted by mixing nuclear pore dilators with the DNA (De la Rossa et al., 2013, De la Rossa and Jabaudon, 2015).
As we have shown in our description of MADR, this approach facilitates stable expression of single-copy transgenes for studying development and disease. However, certain factors should be taken into account when designing experiments. The number of MADR transduced cells is largely dictated by the concentration of the MADR donor, the concentration of FlpO and Cre recombinases, and the proliferation rate of the targeted populations. Specifically, as we have shown, the number of MADR cells versus Cre recombined cells can be titrated in a defined population by varying the ratio of donor plasmid to recombinase plasmid.
However, as can be seen in our postnatal electroporations, we note that under the standard conditions that we have chosen (100 ng/ul of recombinase: 1000 ng/ul of donor plasmid), a pattern emerges whereby MADR transduction inversely correlates with the initial mitotic activity of the cells. Specifically, striatal glia are readily Cre recombined but are more rarely MADR transduced. Conversely, the radial glial populations, which are relatively more quiescent given their role as bona fide neural stem cells, make up a major population of MADR cells. Notably, ependymal cells, which have been recently reported to be the result of terminal asymmetric or symmetric divisions (Ortiz-Alvarez et al., 2019) tend to be readily targeted by MADR—presumably due to the fact that they don’t dilute the plasmids after the initial cell division targeted by electroporation. The cell cycle of the CNS lengthens over development, and postnatal cells are relatively more quiescent than their embryonic counterparts so smaller initial populations are typically transduced by postnatal electroporation. Thus, if large numbers of parenchymal glia or embryonically-generated neurons are desired, in utero electroporation should be performed targeting the local region (i.e. Fig. 4A–C of our manuscript).
We have noted that the perdurance of FlpO and Cre leads to a transient potential for “hopping” in and out of the genome by the donor cargo until the FlpO and Cre are diluted by subsequent cell divisions. Basically, MADR transgenes can theoretically play “musical chairs” with the recipient locus until the “music” stops (i.e. FlpO and Cre plasmid is diluted). (It should be noted that this or similar drawbacks are intrinsic to many of the somatic transgenic methods (Table S3). For example, retroviruses are readily silenced, and transposons suffer from an almost identical hopping in and out phenomenon with the additional complication that it is happening across the genome, causing insertional mutagenesis and may be similarly silenced in certain genomic regions.) Because the donor DNA copy numbers are presumably more substantial than the single-copy recipient transgene locus, insertion is typically favored. However, this necessitates that the titration of FlpO-2A-Cre and Donor plasmids and their relative ratios be determined empirically based on the desired ratio of starting populations (recombination only versus donor insertions).
In certain cases, where a quick but efficacious MADR insertion is needed without double labeled cells (e.g. multiplexing of transgenes to study developmental processes) we would advise using a self-excising FlpO-2A-Cre. In this case, FlpO and Cre are surrounded by FRT sites but the FlpO contains an intron to prevent bacterial self-recombination during DNA production (Fig. S1A).
In cases where only longer-term gene function is to be observed and where only one donor is used, the standard Cag FlpO-2A-Cre yields the highest level of MADR transduction. However, as we note in our results, transient reporter ambiguities can be seen in more quiescent populations such as ependymal cells. Specifically, double+ (Cre reporter and MADR reporter) cells are more frequent over the initial days and weeks post electroporation. We infer that this is potentially due to several factors including 1) ependymal cells are generated by terminal divisions, thus, trapping higher initial amounts of plasmids, and 2) the slower metabolism intrinsic to ependymal cells (Llorens-Bobadilla et al., 2015). Notably, though we have not empirically determined if this is the case, we observe a slow diminishment of mG EGFP over time (2 weeks) indicative of a much longer half-life compared with standard cytoplasmic EGFP. Again, such reporter ambiguities are often intrinsic to fluorescent proteins. For example, the original manuscript characterizing the mT/mG mouse candidly described the overlap of EGFP and TdTomato due to the presence of perdurant mT protein for up to 9.2 days after recombination (Muzumdar et al., 2007). Finally, random insertions are always theoretically possible and will theoretically increase with tranfection/transduction yields. However, spurious expression from such random integrants would presumably only result from donor plasmid insertion in active chromatin while simultaneously losing the 4XpolyA immediately upstream of the transgene coding sequence—theoretically a low likelihood event which we have not seen evidence of in our validation experiments. Although always important, when the experimental goal is to generate tumor models it is also critical to deliver DNA to the precise location and cell types, as more potent driver genes could yield tumors outside of the primary tumor site, confounding survival or interventional studies. In our experiments, fast green is used to confirm ventricular delivery of plasmid and the electrode orientation promotes VZ electroporation. However, cell type specific recombinases could also be employed as a safeguard.
Size considerations:
We have not observed significant differences in MADR efficiency based on donor plasmid size between the standard ranges of plasmid DNA (4Kb up to 18Kb). Empirically testing using time-lapse imaging of MADR donors into proxy cells in vitro at 3 days post lipofection is in agreement with in vivo observations (Data S2B). Plasmid mixes were based on identical molar ratios of individual donor variants. However, altering signaling pathways involved in cell fate, survival, proliferation, etc. will likely lead to changes in overall MADR cell numbers compared with using only genetic reporters.
Cis-regulatory elements:
We typically employ the strong CAG promoter due to its presence in the mouse lines that we utilize. However, there are several means of attenuating the strength of this promoter:
Any IKNM mouse allele (International Mouse Knockout et al., 2007) can be targeted with MADR so the transgenes could be regulated by the endogenous cis-regulatory elements.
We have demonstrated two orthogonal means for secondary induction of transgenes (Vcre, and Tet-On)—one of which is reversible and can be modulated by dosage of the induction agent (Tet-On). Moreover, other technologies (e.g. dimerization domains and destabilization domains) could also be employed to vary transgene function or expression.
Changes in the non-coding portion of the transcripts can have significant effects on transgene expression, including but not limited to WPRE removal, stuffer sequences, and miR-recognition sequences. WPRE has a potent effect on transcript perdurance and protein expression so removal will decrease expression of transgenes upstream. Also, one can specifically increase the number of elements in cistrons to create longer transcripts, which often leads to decreased overall expression. Finally, endogenous (or exogenous) miR-recognition sites can be used to tune expression in precise cell types (endogenous) or miR-hairpins with cognate or slightly mismatched targeting sequences can attenuate expression.
As is shown with our inducible plasmids (e.g. Fig. S1A), secondary cistrons with an attenuated or regulatable promoter can be inserted with MADR.
Injection site inflammation:
The pulled glass capillary tube has a very minute diameter—much smaller than a 30G syringe. We have performed serial sectioning of several animals and have been unable to identify any needle track. Also, there is rarely bleeding induced by the injection (see (Breunig et al., 2012) for a representative image). Thus, postnatal electroporation is considered a minimally invasive technique and a robust means of in vivo gene transduction.
One obvious concern is a possible microglial or astroglial reaction to the exogenous DNA at the injection site. However, we have not observed any significant inflammation compared to the control brain hemisphere (uninjected) in the days post-EP in the sections from our needle track analysis (Data S2C-E). However, going too ventral with the needle can lead to hydraulic trauma from the plasmid mixture, which can denude the surrounding ventricular walls.
For tumor-modeling purposes, there is a lengthy pre-tumor process (often spanning a few months), which gives substantial time for any tissue-injury-related inflammatory process to recede. This is still arguably better than viral-induced tumors or transplants into immunodeficient mice.
In utero electroporation (i.e. Fig. 4A–C) can be used as an alternate MADR delivery approach to additionally mitigate such issues by facilitating delivery into ventricles with a larger relative size and into embryos with a more immature immune system.
To facilitate the MADR workflow we have created a host of mouse N2a “proxy” lines of for in vitro prototyping of plasmids (Data S2F-G). In sum, MADR enable facile generation of in vitro lineages through precise transgene integration, including “one-shot” multiplexing of various donor plasmids.
To enable future non-invasive imaging and observation of tumor progenitor dynamics, we built in secondary constitutive cistrons for both non-invasive imaging (Data S2H-I) (Iwano et al., 2018), and cell cycle phase reporting with FUCCI (Data S2J) (Sakaue-Sawano et al., 2011, Grant et al., 2018). Notably, FUCCI elements are extremely sensitive to dosage due to the fact that high transgene copies can overwhelm the requisite proteasomal machinery (Grant et al., 2018). However, the consistent single-copy dosage of MADR is well-suited for proper FUCCI activity and accuracy of cell cycle indication (Data S2J).
It is important to note that MADR naturally lends itself to separating normal and tumor populations by the fluorescent markers (Data S2K). We used this feature to demonstrate that of two previously identified kinase inhibitors—Akt1/2 inhibitor and Vacquinol-1—that were found to be selectively toxic to K27M tumor cells (Pathania et al., 2017); the Akt1/2 inhibitor similarly inhibited NPC proliferation (Data S2L). Our confirmation that Vacquinol-1 does not alter NPC culture growth yet inhibits K27M growth provides evidence for continued investigation of this compound in the context of these tumors. Results are representative of 3 biological replicates.
Tissue preparation
After anesthesia, mouse brains were isolated and fixed in 4% paraformaldehyde on a rotator/shaker overnight at 4°C. Brains were embedded in 4% low-melting point agarose (Thermo Fisher) and sectioned at 70 μm on a vibratome (Leica).
Immunohistochemistry
Immunohistochemistry (IHC) was performed using standard methodology as previously described (Breunig et al., 2015)azide until use. Details on the primary antibodies can be found in the KEY RESOURCES TABLE. All primary antibodies were used in PBS-0.03% Triton with 5% normal donkey serum. All secondary antibodies (Jackson ImmunoResearch) were used at 1:1000. Care was taken when including fast green dye for ventricle targeting in shorter duration experiments. Though the dye rapidly diluted in longer survival experiments, it confounded early (0-2 day) single-copy reporter detection and was omitted in these cases because of fluorescence in the far red wavelengths. wavelengths.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken anti-EGFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| goat anti-V5 | Abcam | Cat# ab95038, RRID:AB_10676056 |
| rabbit anti-Sox9 | Abcam | Cat# ab185230, RRID:AB_2715497 |
| rabbit anti-ALDH1L1 | Abcam | Cat# ab56149, RRID:AB_879534 |
| human anti-C-Myc Epitope Tag | Absolute Antibody | Cat# Ab00100–10.0 |
| rabbit anti-H3.3S31ph | Active Motif | Cat# 39637 |
| chicken anti-C-Myc Epitope Tag | Aves | Cat# ET-MY100, RRID:AB_2313514 |
| rat anti-CD44 | BD Biosciences | Cat# 550538, RRID:AB_39373 |
| rat anti-PDGFRα | BD Pharmingen | Cat# 558774, RRID:AB_397117 |
| mouse anti-Foxj1 | Invitrogen | Cat# # 14-9965-82 RRID: AB_1548835 |
| rabbit anti-AU1 Epitope Tag | Biolegend | Cat# 903101, RRID:AB_256502 |
| sheep anti-p53 | Calbiochem | Cat# PC35, RRID: AB_2240806 |
| rabbit anti-H3K27Me3 | Cell Signaling | Cat# 9733, RRID:AB_2616029 |
| sheep anti-V5 | LSBio | Cat# LS-C136566, RRID: AB_10915392 |
| rat anti-GFAP | Invitrogen | Cat# 13-0300, RRID: AB_2532994 |
| rabbit anti-HA | Cell Signaling | Cat# 3724, RRID:AB_1549585 |
| rabbit anti-pRB 1 | Cell Signaling | Cat# 8516S, RRID:AB_11178658 |
| rabbit anti-Sox2 | Cell Signaling | Cat# 3579, RRID:AB_2195767 |
| rabbit anti-Bmi1 | Cell Signaling | Cat# 6964P, RRID:AB_10839408 |
| rabbit anti-H3K27Ac | Cell Signaling | Cat# 8173P, RRID:AB_10949887 |
| mouse anti-TetR | Clontech | Cat# 631132 |
| rabbit anti-Dsred | Clontech | Cat# 632496, RRID:AB_10013483 |
| mouse anti-V5 | Invitrogen | Cat# R960-25, RRID:AB_2556564 |
| rabbit anti-mCherry | Kerafast | Cat# EMU-106 |
| guinea pig anti-mKate2 | Kerafast | Cat# EMU108 |
| rat anti-Tdtomato | Kerafast | Cat# EST203, RRID:AB_2732803 |
| rabbit anti-H3F3A | Lifespan Biosciences | Cat# LS-C148509-100, RRID:AB_11135921 |
| rabbit anti-H3F3A K27M | Millipore | Cat# ABE419, RRID:AB_2728728 |
| rabbit anti-NG2 | Millipore | Cat# AB5320, RRID:AB_11213678 |
| sheep anti-Dll1 | R&D Systems | Cat# AF5026, RRID:AB_2092830 |
| goat anti-Olig2 | R&D Systems | Cat# AF2418, RRID:AB_2157554 |
| rabbit anti-H3.3G34R | Revmab | Cat# 31-1120-00, RRID:AB_2716433 |
| rabbit anti-Atrx | Sigma | Cat# HPA001906, RRID:AB_1078249 |
| mouse anti-Flag | Sigma Aldrich | Cat# F1804, RRID:AB_262044 |
| guinea pig anti-GFAP | Synaptic Systems | Cat# 173 004, RRID:AB_10641162 |
| Bacterial and Virus Strains | ||
| One Shot MAX Efficiency DH5-T1R cells | Invitrogen | Cat# 12297016 |
| Stellar chemically competent cells for cloning | Clontech | Cat# 636766 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tris-EDTA buffer | Sigma-Aldrich | Cat# E8008-100ML |
| Fast Green Dye | Sigma Aldrich | Cat# F7258-25g |
| SignaGel Electrode Gel | Medline Industries | Cat# PLI1525CSZ |
| Low-Melting Point Agarose | Fisher Bioreagents | Cat# bp1360-100 |
| Human Epidermal Growth Factor | Sigma-Aldrich | Cat# E9644 |
| Heparin | Sigma-Aldrich | Cat# H3393 |
| Basic Fibroblast Growth Factor (bFGF) | Millipore | Cat# GF003 |
| Doxycycline | Clontech | Cat# 631311 |
| Puromycin | Clontech | Cat# 631305 |
| Methanol | Sigma Aldrich | Cat# 179337 |
| Hydrogen peroxide solution | Sigma Aldrich | Cat# H1009 |
| Triton X-100 | Sigma Aldrich | Cat# X-100-500ML |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | Cat# D2650-5X10ML |
| Glycine | Sigma Aldrich | Cat# 410225-50g |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat# 017-000-121 |
| Dichloromethane | Sigma Aldrich | Cat# 270997 |
| Dibenzyl Ether | Sigma Aldrich | Cat# 108014 |
| Acryloyl-X, SE, 6-((acryloyl)amino)hexanoic Acid, Succinimidyl Ester | Thermo-Fisher | Cat# A20770 |
| NaCl | Sigma-Aldrich | Cat# S9888 |
| Sodium Acrylate | Sigma-Aldrich | Cat# 408220 |
| Tetramethylethylenediamine | Sigma-Aldrich | Cat# T9281 |
| Ammonium Persulfate | Sigma-Aldrich | Cat# A3678-25g |
| 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl | EMD Millipore | Cat# 840130 |
| Proteinase K | New England Biolabs | Cat# P8107S |
| Draq5 | Cell Signaling | Cat# 4084S |
| Eosin Y | Sigma-Aldrich | Cat# E4009 |
| Collagenase IV | Worthington Biochemical | Cat# LS004189 |
| DNAse I | Worthington Biochemical | Cat# LS002007 |
| ACK Lysis Buffer | Thermo Fisher Scientific | Cat# A1049201 |
| Neurobasal media | Thermo Fisher Scientific | Cat# 21103049 |
| DMEM, High Glucose | Thermo Fisher Scientific | Cat# 11965118 |
| Penicillin-Streptomycin-Amphotericin | Thermo Fisher Scientific | Cat# 15240096 |
| B-27 supplement without Vitamin A | Thermo Fisher Scientific | Cat# A3353501 |
| Glutamax | Thermo Fisher Scientific | Cat# 35050061 |
| Human EGF | Shenandoah Biotechnology | Cat# 100-26-500ug |
| Human FGF (Shenandoah Biotechnology, Warwick, PA), | Shenandoah Biotechnology | Cat# 100-146-100ug |
| PDGF-AA (Shenandoah Biotechnology, Warwick, PA) | Shenandoah Biotechnology | Cat# 100-16-100ug |
| Heparin Solution 0.2% | StemCell Technologies | Cat# 07980 |
| CELLstart | Thermo Fisher Scientific | Cat# A10142-01 |
| Akalumine-HCL | Sigma Aldrich | Cat# 808350 |
| Vacquinol-1 | Sigma-Aldrich | Cat# SML1187 |
| AKT 1/2 kinase inhibitor | Sigma-Aldrich | Cat# A6730 |
| Tween20 | Bio-Rad | Cat# 1610781 |
| Digitonin | Sigma-Aldrich | Cat# 300410 |
| Nonidet P40 substitute | Sigma-Aldrich | Cat# 74385 |
| Critical Commercial Assays | ||
| DNeasy | Qiagen | Cat# 69504 |
| Zero Blunt TOPO kit | Thermo Fisher | Cat# 450159 |
| Chromium™ Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | Cat# 120237 |
| SPRIselect Reagent Kit | Beckman Coulter | Cat# B23318 |
| Chromium Single-Cell 3’ Library Kit | 10X Genomics | Cat# PN-120237 |
| KAPA Library Quantification Kit | Roche | Cat# 07960140001 |
| KAPA HiFi PCR kit | Kapabiosystems | Cat# KR0368 |
| In-Fusion cloning | Clontech | Cat# 638920 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621L |
| Deposited Data | ||
| Mice raw and analyzed data | This paper | GEO: GSE117154, GSE131675, GSE131672 |
| Human data | GEO website | GEO: GSE70630, GSE89567, GSE102130 |
| P50 and E18 mouse scATAC data | 10X Genomics | https://www.10xgenomics.com/resources/datasets/ |
| Experimental Models: Cell Lines | ||
| Mouse MADR cell line: K27M-1 | This paper | N/A |
| Mouse MADR cell line: K27M-2 | This paper | N/A |
| Mouse MADR cell line: K27M-3 | This paper | N/A |
| Mouse mTmG cell line: TdTomato+ | This paper | N/A |
| Human: HEK293T | ATCC | Cat# CRL-3216 |
| Mouse: Neuro-2a | ATCC | Cat# CCL-131 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD1 | Charles River Laboratories | Strain Code 022 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX: 007676 |
| Mouse: Gt(ROSA)26Sortm1.1 (CAG-EGFP)Fsh/Mmjax | The Jackson Laboratory | JAX: 32037 |
| Oligonucleotides | ||
| sgRNA targeting sequence: Pten: gcCTCAGCCATTGCCTGTGTG | This paper | N/A |
| sgRNA targeting sequence: Trp53: GCCTCGAGCTCCCTCTGAGCC | This paper | N/A |
| sgRNA targeting sequence: Nf1: GCAGATGAGCCGCCACATCGA | This paper | N/A |
| sgRNA targeting sequence (BE): Pten: CCTcAGCCATTGCCTGTGTG | This paper | N/A |
| sgRNA targeting sequence (BE): Trp53: CTGAGCcAGGAGACATTTTC | This paper | N/A |
| sgRNA targeting sequence (BE): Nf1: TCCTcAGTCACACATGCCAG | This paper | N/A |
| Recombinant DNA | ||
| plasmid: MADR pDonor-TagBFP2-3XFlag (cyto) WPRE | This paper | Addgene Plasmid #129421 |
| plasmid: pCag TagBFP2-V5 Cyto PB | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-smFP-HA | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-smFP-Myc | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-smFP-Flag | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-SM TagBFP-V5 (weakly-fluorescent) | This paper | N/A |
| plasmid: pCag-FlpO-2A-Cre | This paper | Addgene Plasmid #129419 |
| plasmid: pCag-SE-FlpE-2A-Cre | This paper | Addgene Plasmid #130986 |
| plasmid: CMV FlpO-2a-Cre | This paper | N/A |
| plasmid: pAAV-Ef1a-flpo-2a-cre-wpre | This paper | N/A |
| plasmid: MADR pAAV-(inverted; promoterless) TagBFP2-3Flag cyto-wpre | This paper | N/A |
| plasmid: CMV Flp-Ires-Cre | This paper | N/A |
| plasmid: AAVS1_Tagbfp2-V5-nls-P2A-Puro_Cag LoxP myrTdtomato FRT | This paper | N/A |
| plasmid: MADR pDonor-smFP-myc (bright) WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-Flag (bright) WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-V5 (dark) WPRE | This paper | Addgene Plasmid #131006 |
| plasmid: MADR pDonor-smFP-Myc (dark) WPRE | This paper | Addgene Plasmid #130987 |
| plasmid: MADR pDonor-smFP-HA (dark) WPRE | This paper | Addgene Plasmid #131007 |
| plasmid: MADR pDonor-smFP-Flag (dark) WPRE | This paper | Addgene Plasmid #131005 |
| plasmid: MADR pDonor-mScarlet-3XSpot WPRE | This paper | N/A |
| plasmid: MADR pDonor-SM_TagBFP2-V5 (weakly-fluorescent) WPRE | This paper | N/A |
| plasmid: MADR pDonor-SM TagBFP2-V5-(cyto)-2A-Vcre WPRE | This paper | N/A |
| plasmid: pCag FlEx Vlox smFP-myc (dark) WPRE | This paper | N/A |
| plasmid: pCag TagBFP2-V5 Cypo PB triple miR-E shNf1.789:shTrp53.8914:shPten.1524 WPRE |
This paper | N/A |
| plasmid: MADR pDonor-SM-TagBFP2-V5-P2A-SpCas9 WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT-pTV1_FNLS-Cas9-BW WPRE | This paper | N/A |
| pC0043-SpCas9 BbsI (Empty) crRNA backbone (episomal) | This paper | N/A |
| pC0043-SpCas9 sg.Trp53 (episomal; for use with FNLS base editor) | This paper | N/A |
| pC0043-SpCas9 sg.Nf1 (episomal; for use with FNLS base editor) | This paper | N/A |
| pC0043-SpCas9 sg.Pten (episomal; for use with FNLS base editor) | This paper | N/A |
| plasmid: MADR pDonor-smFP-myc-P2A-EspCas9 WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-myc-P2A-Cas13b WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-myc-P2A-CasRX WPRE | This paper | N/A |
| pU6 BsmBi Empty SpCas9-crRNA Cag miRFP670-3X-HA WPRE PB | This paper | N/A |
| pU6 BsmBi Empty CasRX-crRNA Cag miRFP670-3X-HA WPRE PB | This paper | N/A |
| pU6 BsmBi Empty Cas13b-crRNA Cag miRFP670-3X-HA WPRE PB | This paper | N/A |
| plasmid: MADR pDonorRCE TagBFP2-Hras G12V Wpre (RCE donor compatible) | This paper | N/A |
| plasmid: MADR pDonor TagBFP2-Hras G12V Wpre (mtmg donor compatible) | This paper | Addgene Plasmid #129420 |
| plasmid: Ubi-EGFP-HRasG12V PB | Breunig et al. Cell Reports, 2015 | 10.1016/j.celrep.2015.06.012 |
| plasmid: MADR pDonor-smFP_Myc_p2a_YAP1-MAMl1D | This paper | N/A |
| plasmid: MADR pDonor-smFP_Myc_p2a_c11orf95-RELA | This paper | N/A |
| plasmid: MADR pDonor smFP_Myc_p2a_Kras G12A | This paper | N/A |
| plasmid: MADR pDonor-H3F3A-K27M-EGFP pTV1 Pdgfra D842V COTv1 Trp53-V5 WPRE | This paper | N/A |
| plasmid: MADR pDonor-H3F3A-G34R-EGFP pTV1 Pdgfra D842V COTv1 Trp53-V5 WPRE | This paper | N/A |
| plasmid: MADR pDonor-H3F3A-WT-EGFP pTV1 Pdgfra D842V COTv1 Trp53-V5 WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT-pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A K27M WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT-pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A G34R WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT-pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A WT WPRE | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT-pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A K27M WPRE::Ef1a-Akaluc(Inverted) | This paper | N/A |
| plasmid: MADR pDonor-PIP-NLS-Venus-P2A-mCherry-hGEM1/110 | This paper | N/A |
| plasmid: MADR pDonor-PIP-NLS-Venus-P2A-mIRFP670-hGEM1/110 | This paper | N/A |
| plasmid: MADR pDonor-PIP-NLS-mIRFP709-P2A-mIRFP670-hGEM1/110 (NIR-FUCCI) | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT- pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A K27M WPRE ::Ef1a-NIR-FUCCI (Inverted) | This paper | N/A |
| plasmid: MADR pDonor-smFP-mycBRIGHT- pTV1 Pdgfra D842V COTv1 Trp53 270h-P2ACO3-H3F3A K27M WPRE::Ef1a-NIR-FUCCI* (*-hGEM C-term NLS mutant; Inverted) | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-EGFP | This paper | N/A |
| plasmid: MADR pDonor rtTA-V10-AU1-P2a-puro-WPRE TRE-EGFP/mDll1 | This paper | N/A |
| Plasmid: pX330-dual U6-p16-p19-cdkn2a-Chimeric_BB-CBh-eSpCas9(1.1) | This paper | N/A |
| plasmid: pX330-U6-sg.ATRX-Chimeric_BB-CBh-eSpCas9(1.1) | This paper | N/A |
| plasmid: pX330-U6-sg.AAVS1-Chimeric_BB-CBh-eSpCas9(1.1) | This paper | N/A |
| plasmid: AAVS1-TALENs | Gift: Conklin and Mandegar (Mandegar et al., 2016) | N/A |
| plasmid: T7 FlpO-2A-Cre (mRNA generation) | This paper | N/A |
| plasmid: MC-FlpO-2A-Cre (parental) | This paper | N/A |
| minicircle: MC-FlpO-2A-Cre | This paper | N/A |
| plasmid: CMV Flp-2A-Cre | Gift: Y. Voziyanov (Anderson et al., 2012) | N/A |
| plasmid: mT/mG | (Muzumdar et al., 2007) | Addgene Plasmid #17787 |
| plasmid: CAG LF mTFP1 | Gift: I. Imayoshi (Imayoshi et al., 2012) | N/A |
| Software and Algorithms | ||
| Nikon’s Confocal NIS-Elements Package | Nikon | https://www.microscope.healthcare.nikon.com/products/software |
| Imaris 9.1 | Bitplane | https://imaris.oxinst.com/ |
| ImageJ software | NIH | https://imagej.nih.gov/ij/ |
| Syglass VR | IstoVisio | https://www.syglass.io/ |
| STAR/STARlong (version 2.5.1) | Dobin A et al. 2012 | https://github.com/alexdobin/STAR |
| Cell Ranger software version 2.0.0 (scRNA-seq) and 3.0.2 (snATAC-seq) | 10X Genomic | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/ |
| Seurat | Butler et al. 2018 | https://satijalab.org/seurat/ |
| Scanpy | Wolf FA et al. 2017 | https://scanpy.readthedocs.io/en/latest/ |
| SCENIC (1.0.0-02) | Aibar S et al. 2017 | https://github.com/aertslab/SCENIC |
| cisTOPIC | Bravo et al. 2019 | https://github.com/aertslab/cisTopic |
| SnapATAC | Fang et al. 2019 | https://github.com/r3fang/SnapATAC |
| Harmony | Korsunsky et al. 2019 | https://github.com/immunogenomics/harmony |
| ngs.plot v2.61 | Shen et al. 2014 | https://github.com/shenlab-sinai/ngsplot |
| IGV v.2.5.0 | Robinson et al. 2011 | https://software.broadinstitute.org/software/igv |
| bwa-mem | Li, H et al. 2009 | https://github.com/lh3/bwa |
| Other | ||
Immunohistochemistry with bleaching
For pre-bleached immunohistochemistry, 70 μm tissue sections were dehydrated with increasing concentrations of methanol (20%, 40%, 60%, 80%, 100%) for 15 minutes each in water at RT, and then treated overnight with 5% H2O2 in 100% methanol at 4°C. Tissue was then rehydrated using methanol (100%, 80%, 60%, 40%, 20%), 15 minutes each in water, and then washed with PBS before proceeding with normal immunostaining.
Cell nucleofection
Mouse NSC nucleofection was performed using the Nucleofector 2b device and Mouse Neural Stem Cell Kit according to manufacturer’s recommendations (Lonza AG). The nucleofection mixtures contained plasmids with equal concentrations of 10 ng/μl.
Live Cell Imaging
N2A proxy cells expressing PIP-Venus/mCherry-hGEM1/110 were plated in a 96-well format and imaged with at 20x objective lens under phase, red and green fluorescence using an Incucyte S3 System (Essen Bioscience, Ann Arbor, MI). Images were collected every 30 min using Incucyte S3 Software.
In high-throughput drug testing experiments, 10.000 cell from the cell lines generated from tumor dissociation and non-tumor control cells were plated in 96 well plates. 24 hours after the seeding appropriate concentration of each drug 1μM for Vacquinol-1(Sigma-Aldrich, SML1187) and 0.5μM for AKT 1/2 kinase inhibitor (Sigma-Aldrich, A6730)) was added to the media and cells were imaged for 92 hours in phase contrast using Incucyte S3 System. Images were collected every 2 hours using Incucyte S3 Software. Cell proliferation images analysis was done with Incucyte S3 software and normalized results presented and analyzed with Graphpad Prism 7.
Imaging and processing
All fixed images were collected on a Nikon A1R inverted laser confocal microscope. The live image of mNSCs was obtained on an EVOS digital fluorescence inverted microscope. For whole brain images, the automated stitching function of Nikon Elements was used. ND2 files were then imported into ImageJ to create Z-projection images, which were subsequently edited in Adobe Photoshop CS6. In several rotated images (e.g. Fig. 3F), rotation led to colorless space in the empty area completely outside of the tissue section and black fill was added. Adobe Illustrator CS6 was used for the final figure production.
Flow cytometry
Cells were collected as previously described (Breunig et al., 2015). Accutase (Millipore), pelleted at 250g for 3 min, and resuspended in the media. FACS was done on a Beckman Coulter MoFlo at the Cedars-Sinai Flow Cytometry Core. at the Cedars-Sinai Flow Cytometry Core.
Western blot
The cell pellets were resuspended in laemmli buffer and boiled for 5 min at 95°C. Protein concentrations were measured on a ThermoScientific NanoDrop 2000. After SDS-PAGE separation and transfer onto nitrocellulose membranes, proteins were detected using the antibodies listed in the KEY RESOURCES TABLE, diluted in 5% milk in 0.1% PBS-Tween. All secondary antibodies (Li-cor IRDye®) were used at 1:15000. Proteins were visualized by infrared detection using the Li-Cor Odyssey® CLX Imaging System.
Single-cell western blot
mTmG mNSCs were nucleofected (Lonza VPG-1004) with 6 μg of either piggybac or MADR TagBFP plasmid and 6 μg of FlpO 2A Cre in a T75 flask. After 4 days, cells were sorted through FACS, and 100,000-200,000 BFP+ cells were seeded onto Milo scWestern chips (ProteinSimple C300). Each chip was stained for guinea pig mKate (Kerafast EMU108) at 1:20 in Cy3 and rabbit histone H3 (Cell Signaling 4499) at 1:20 in 647. Imaging was performed using the Innoscan 710 microarray scanner.
Doxycycline and puromycin administration
Doxycycline (Clontech 631311) was added to culture media at the final concentration of 100ng/ml. Puromycin (Clontech 631305) was used at 1μg/ml.
Multi-miR-E knockdown efficiency quantification
We have previously used FlEx-based transgene expression, specifically Cre-mediated inversion and activation of EGFP cassette (FlEx-EGFP)(Breunig et al., 2015). To test our multi-miR-E targeting Nf1, Pten, and Trp53, we made a CAG-driven FlEx-based construct harboring the multiple miR-Es (FlEx-multi-miR-E). Postnatal mNSC line was established by dissociating CD1 pup brains, transfected with EGFP or FlEx-multi-miR-E and Cre-recombinase vector. Fluorescent cells were sorted and subjected to mRNA extraction and SYBR-based Fluidigm BioMark dynamic array using qPCR probes for Nf1, Pten, and Trp53.
Tissue clearing
For whole mount imaging, the iDisco tissue clearing method was used (Renier et al. 2014). Fixed samples were gradually dehydrated in 20%, 40%, 60%, 80%, 100%, 100% methanol/H2O, 1 hour each at RT, and then bleached overnight in 5% H2O2 in 100% methanol overnight at 4°C, followed by a gradual rehydration (80%, 60%, 40%, 20% methanol/H2O, then PBS with 0.2% Triton X-100, 1 hour each at RT). Samples were then incubated in PBS with 0.2% Triton X-100, 20% DMSO, and 0.3M glycine for 2 days at 37°C to permeabilize tissue, and then incubated in PBS with 0.2% Triton X-100, 10% DMSO, and 6% normal donkey serum for 2 days at 37°C to block the tissue for staining. Samples were then incubated with primary antibodies in PBS with 0.2% Triton and 10μg/ml heparin (PTwH), at 37°C for 5 days, followed by 5 washes of PTwH, 1 hour each at RT, plus 1 overnight wash at RT. Samples were then incubated in secondary antibodies in PTwH, at 37°C for 5 days, followed by 5 washes of PTwH, 1 hour each at RT, plus 1 overnight wash at RT.
Following staining, samples were again dehydrated gradually in 20%, 40%, 60%, 80%, 100%, 100% methanol/H2O, 1 hour each at RT, and then stored overnight in 100% methanol at 4°C. Samples were then incubated in a solution of 66% dichloromethane (DCM, Sigma 270997) in methanol for 3 hours at RT, followed by 2 washes with 100% DCM, 15 minutes each at RT, and then placed directly into dibenzyl ether (DBE, Sigma 108014) for clearing and imaging. Cleared samples were stored in DBE in glass containers at RT in the dark. Samples were imaged in DBE using a light sheet microscope (Ultramicroscope II, LaVision Biotec) equipped with an sCMOS camera (Andor NEO 5.5) and a 2x/0.5 objective lens with a 6mm WD dipping cap.
Light sheet datasets were imported into Imaris 9.1 (Bitplane) for 3D visualization. To digitally remove artifacts and fluorescent debris, the surface tool was used to create surface renderings of unwanted fluorescence, and the ‘mask all’ function in the surface menu was used to create fluorescence channels with debris removed. To create a digital surface of the whole sample, the volume-rendering tool was set to ‘normal shading’ and the color was set to gray. Movies of 3-D datasets were generated using the ‘animation’ tool.
Expansion microscopy
Samples were generated for expansion microscopy following the Pro-ExM protocol (Tillberg et al. 2015). Briefly, 100 μm sections were stained for EGFP and HA-tag. Before expansion, samples were imaged in water using a confocal microscope (Nikon A1R) for pre-expansion imaging.
Samples were anchored in 0.1 mg/ml Acryloyl-X, SE ((6-((acryloyl)amino)hexanoic acid, succinimidyl ester; Thermo-Fisher) in PBS with 10% DMSO, overnight at RT. After washing with PBS (3 x 10 minutes), samples were incubated for 30 minutes at 4°C in monomer solution (PBS, 2 M NaCl, 8.625% (w/w) sodium acrylate, 2.5% (w/w) acrylamide, 0.15% (w/w) N,N-methylenebisacrylamide), immediately after addition of 0.2% (w/w) tetramethylethylenediamine (TEMED), 0.2% (w/w) ammonium persulfate (APS), and 0.1% (w/w) 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-hydroxy-TEMPO). Slices were then incubated for 2 hours at 37°C for gelation. After incubation, samples were incubated overnight in a 6-well plate at RT with no shaking in a digestion solution containing Proteinase K (New England Biolabs) diluted to 8 units/ml in digestion buffer (50 mM Tris pH 8, 1 mM EDTA, 0.5% Triton X-100, 1 M NaCl). Following digestion, samples were washed with excess H2O 4 times, 1 hour per wash at RT, and then stabilized in 2% low melting agarose in H20 before imaging. Images were acquired using a confocal microscope (Nikon A1R) with a 40x long WD objective (Nikon CFI Apo 40xw NIR).
Pathology
After bleaching, immunohistochemistry was performed to stain for EGFP in the 405 channel. After incubation in secondary antibody, sections were incubated in 50μM Draq5 (Cell signaling 4084S) in PBS for 2 minutes at RT, followed by washes of PBS (3 x 5 minutes). Sections were then incubated in 2% w/w Eosin Y (Sigma E4009) in 80% ethanol for 2 minutes at RT, followed by washes with PBS (3 x 5 minutes). Finally, sections were incubated in another Draq5 solution (50μM in PBS) for 3 minutes, before washing with PBS, mounting, and imaging.
In vivo dRMCE efficiency titration
For each condition, pups were EPed with MADR pDonor-smFP-Myc and Flpo-2A-Cre. The brains were taken two days post-EP, and two non-adjacent sections from each brain were stained with Myc-Tag antibody and EGFP. For each section, cells were quantified for insertion (Myc expressed) and cre excision (only EGFP expressed) using Syglass VR with an Oculus Rift system. Quantifications were indicated as percentages of total cells counted per section. The proportions were averaged over two sections from different animals for each group. Fast green was omitted from these assays as the dye was found to fluoresce in the same wavelengths as Alexa647. Though the dye rapidly diluted in longer survival experiments, it confounded early (0-2 day) single-copy reporter detection.
PCR-generation of U6-sgRNA fragments
Reverse scaffold and forward primers (IDT DNA) were combined in a PCR reaction and subsequent purification to make concentrated sgRNAs (Ran et al., 2013). We used previously-validated target sites for tumor modeling (Xue et al., 2014, Heckl et al., 2014)(KEY RESOURCES TABLE)..
Sequencing InDel mutations in murine tumor cells
A pure population of tumor cells was obtained by FACS and genomic DNA was isolated (Qiagen DNeasy). Using primers flanking the gRNA target site, we PCR amplified the regions expected to contain InDel mutations for Nf1, Trp53, and Pten. The PCR amplified fragments were topo cloned using the Thermo Fisher Zero Blunt TOPO kit and transformed into One Shot MAX Efficiency DH5-T1R cells.
Confirmation of CRISPR base edits
For premature stop codon base conversions, EGFP+ cells were obtained by FACS, and genomic DNA was isolated (Qiagen DNeasy). Using primers flanking the sgRNA target site, we PCR-amplified the regions expected to contain base conversions for Nf1, Trp53, and Pten. The amplicons were normalized to 20ng/ul and sent for sequencing to the AMPLICON-EZ service (Genewiz).
Fastq files for each gene-primer pair were aligned to a custom genome file containing that gene locus using STARlong, and bwa-mem with default parameters, which all gave similar results. The BAM files were uploaded to IGV for visualization.
Tissue dissociation
Mice were euthanized in CO2 chamber and brains were collected in PBS. Immediately, EGFP+ tissue was microdissected under a Revolve Hybrid Microscope (Echo Labs, San Diego, CA). If allowed by the size of the tumor, some remains of the brain with residual tumor tissue was fixed in 4% PFA for tissue analysis. Microdissected tissue was mechanically dissociated into <1 mm pieces and further digested with Collagenase IV (Worthington Biochemical, Lakewood, NJ), and DNAse I (Worthington Biochemical, Lakewood, NJ). The resultant single cell suspension was filtered through 40mm cell strainer (Stellar Scientific, Baltimore, MD) and erythrocytes were lysed with ACK lysis buffer (Thermo Fisher Scientific, Waltham, MA). Single cell suspensions were split into 3 parts: First, for scRNAseq or sc-ATACseq experiments, GFP+ cells from single cell samples were FACS sorted (into 1.5ml tubes for 10X Chromium). A secondary fraction was used for in vitro cell line establishment. Specifically, cells were resuspended in Neurobasal media (Thermo Fisher Scientific, Waltham, MA) supplemented with penicillin-streptomycin-amphotericin (Thermo Fisher Scientific, Waltham, MA), B-27 supplement without Vitamin A (Thermo Fisher Scientific, Waltham, MA), Glutamax (Thermo Fisher Scientific, Waltham, MA), EGF (Shenandoah Biotechnology, Warwick, PA), FGF (Shenandoah Biotechnology, Warwick, PA), PDGF-AA (Shenandoah Biotechnology, Warwick, PA) and heparin (StemCell Technologies, Cambridge, MA); and cultured in a CELLstart CTS (Thermo Fisher Scientific, Waltham, MA) treated T25 Flask. Finally, the last third of the single cell suspensions were fixed in 80% methanol-PBS and stored at −80C.
ScRNA-seq library generation
Single-cell RNA-seq libraries were prepared per the Single Cell 3’ v2 Reagent Kits User Guide (10x Genomics, Pleasanton, California). Cellular suspensions were loaded on a Chromium Controller instrument (10X Genomics) to generate single-cell Gel Bead-In-EMulsions (GEMs). GEM-reverse transcription (RT) was performed in a Veriti 96-well thermal cycler (Thermo Fisher Scientific, Waltham, MA). After RT, GEMs were harvested and the cDNAs were amplified, cleaned up with SPRIselect Reagent Kit (Beckman Coulter, Pasadena, CA). Indexed sequencing libraries were constructed using Chromium Single-Cell 3’ Library Kit for enzymatic fragmentation, end-repair, A-tailing, adapter ligation, ligation cleanup, sample index PCR, and PCR cleanup. The barcoded sequencing libraries were quantified by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA). Sequencing libraries were loaded on a NovaSeq 6000 (Illumina, San Diego, CA) with a custom sequencing setting (26bp for Read 1 and 91bp for Read 2).
ScRNA-seq read alignment
The demultiplexed raw reads were aligned to the transcriptome using STAR (version 2.5.1) (Dobin et al., 2013) with default parameters, using a custom UCSC mouse reference with mm10 annotation, containing all protein coding and long non-coding RNA genes. Expression counts for each gene in all samples were collapsed and normalized to unique molecular identifier (UMI) counts using Cell Ranger software version 2.0.0 (10X Genomics). The result is a large digital expression matrix with cell barcodes as rows and gene identities as columns.
To obtain 2-D projections of the population’s dynamics, principal component analysis (PCA) was firstly run on the normalized gene-barcode matrix of the top 5,000 most variable genes to reduce the number of dimensions using Seurat package version 2.1-3 (Butler et al., 2018) in R v3.4.2-4.
Nuclei isolation for sc-ATACseq
GFP+ FACS sorted cells were processed following manufacture instruction for sc-ATACseq (10x Genomics, Pleasanton, California). Specifically, sorted cells were filtered through a 40 mm cell strainer, pelleted and resuspended in one volume of lysis buffer (Tris-HCl 10mM, NaCl 10mM, MgCl2 3mM, Tween-20 0.1% (Bio-Rad, 1610781), Nonidet P40 substitute 0.1% (Sigma-Aldrich, 74385), digitonin 0.01% (Sigma-Aldrich, 300410)and BSA 1% in Nuclease-fre water), cells were incubated on ice until optimal cell lysis. Then, lysis buffer was blocked by adding 10 volumes of Wash buffer (Tris-HCl 10mM, NaCl 10mM, MgCl2 3mM BSA 1%, Tween-20 0.1% in Nuclease-free water). Isolated nuclei were pelleted and resuspended in 1x nuclei buffer (10x Genomics, Pleasanton, California), Finally, nuclei concentration was calculated with an hematocytometer and proceeded immediately with sc-ATACseq library construction protocol.
scATAC-seq library construction
scATAC sequencing library was prepared on the 10X Genomics Chromium platform following the manufacturer’s protocol (10X Genomics 1000110). The isolated nuclei suspension was diluted and then incubated with transposition mix for a targeted nuclei recovery of 10,000 cells. GEMs were then captured on the Chromium Chip E (10X Genomics 1000082). Following GEM incubation, clean up was performed using Dynabeads My-One Silane beads (10X Genomics 2000048) and SPRIselect reagent (Beckman Coulter B23318). Finally, the library was amplified for a total of 10 SI PCR cycles.
Human single-cell RNA-seq Data processing
Three public processed Data (GSE70630, GSE89567, and GSE102130) were obtained from their respective GEO websites. GSE70630 and GSE89567 were back-converted to TPM values. GSE102130 was divided into K27M (GSE102130_ K27M) and glioblastoma multiforme (GSE102130_GBM) datasets (6 and 3 patients, respectively). To identify the non-malignant microglia and mOGs in the datasets, we used PCA-tSNE and Louvain clustering as implemented in Scanpy (Wolf et al., 2018). The clusters containing the markers of microglia (CSF1R, LAPTM5, CD74, TYROBP) or mOGs (MBP, MOG, PLP1), as double-checked by t-test and Wilcoxon, were removed. For each dataset, the number of malignant tumor cells matched closely with those determined by the original authors (GSE70630: 4044 vs 4050, GSE89567: 5157 vs 5097, GSE102130: 2270 vs 2259). GSE102130_GBM did not contain any microglia or mOGs. For processing in Seurat, GSE102130_ K27M was divided into 6 samples. All datasets, including the MADR mouse datasets, were normalized to have the library size of 10e5. For the comparative analysis across the tumor types, we used the relative expression as defined by (Filbin et al., 2018) to make the heatmap in Fig. 6K.
Mouse single-cell RNA-seq Data processing
The three 10X UMI count matrices (mK27M1, mK27M2, mK27M3) were normalized to have the library size of 10e5 for each cell. Then, we clustered in the same way as the public dataset to distinguish microglia and mOGs in Scanpy (Wolf et al., 2018). Cells that had more than 10% mitochondrial reads, less than 1000 unique reads, or more than 5000 unique reads were filtered out in Seurat (2.3.3) (Butler et al., 2018). After filtering, there were 2761, 562, and 3469 cells in mK27M1, mK27M2, and mK27M3, respectively.
Seurat processing
P1–4 genes were obtained from (Filbin et al., 2018) and used as the highly variable genes argument (genes.use) to identify the common substructures in each human and mouse dataset. The cells were clustered using CCA-UMAP (RunMultiCCA and DimPlot with ‘umap’), and the cluster-specific marker genes were identified using the Seurat function “find_all_markers” with the default arguments. To merge the mouse and human CCA-UMAPs, the mouse gene names were converted to their orthologous human counterparts using Ensembl BioMart (https://www.ensembl.org/biomart). For module scoring, the functions CellCycleScoring and AddModuleScore were used. The four gene lists (OC, AC, OPC, and Cycle) correspond to P1–4 genes. DoHeatmap function with at most top 50 genes for each cluster was used to make the heatmaps.
SCENIC on mouse and human dataset
SCENIC (1.0.0–02) was run with all default settings as described in (Aibar et al., 2017b). We used the two default databases for each species (500bp-upstream and tss-centered-10kb). The raw matrices with the library size of 10e5 for each cell and the metaData dataframe from Seurat processing were used as inputs for SCENIC. For the heatmap and tSNE plotting, we used the binary regulon output. The package component AUCell was used to select a threshold for each regulon and then score each regulon for their enrichment in each cell (Aibar et al., 2017b). The scores were then binarized (on vs off), and the outputs clustered according to this binary activity matrix (Aibar et al., 2017b).
Mouse single-cell ATAC-seq Data processing
CellRanger was used to identify and annotate open chromatin regions and perform aggregation of samples and initial clustering of cells and motif analysis. CellRanger outputs were used as inputs for cisTopic and SnapATAC and samples were processed according to recommended settings (Bravo Gonzalez-Blas et al., 2019, Fang et al., 2019) for annotating clusters, Topics, ontology, gene accessibility, and motifs. The Harmony package (Korsunsky et al., 2018) was used according default settings in conjunction with SnapATAC to align E18 datasets.
ChIP-seq preparation
H3K27me3 ChIP reactions were performed using 30 μg of mouse pediatric brain tumor chromatin and 4 μg of antibody (Active Motif, cat # 39155). The ChIP reactions also contained a drosophila chromatin spike in for the normalization of the sequencing data. A small fraction of the ChIP DNA was diluted and qPCR was performed using positive control primer pairs. For H3K27me3, the primer pair targeted to the promoter region of the active gene ACTB served as a negative control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Histological analyses
Nikon Elements and ImageJ software was used to analyze images. All results are shown as mean ± SEM, except when indicated otherwise. For statistical analyses, the following convention was used: *: p < 0.05, **: p < 0.01, ***: p < 0.001. “Student’s t-test” refers to the unpaired test.
Transcriptomic analyses
The three 10X UMI count matrices (mK27M1, mK27M2, mK27M3) were normalized to have the library size of 10e5 for each cell. Then, we clustered in the same way as the public dataset to distinguish microglia and mOGs in Scanpy [6]. Cells that had more than 10% mitochondrial reads, less than 1000 unique reads, or more than 5000 unique reads were filtered out in Seurat (2.3.3) [5]. After filtering, there were 2761, 562, and 3469 cells in mK27M1, mK27M2, and mK27M3, respectively. After filtering, there were 2761, 562, and 3469 cells in mK27M1, mK27M2, and mK27M3, respectively.
ChIP-seq analysis
ChIP-seq reads were aligned to the mouse reference genome mm10 using bwa. BigWig tracks were generated for each sample. H3K27me3 clustering was performed using ngs.plot (version 2.61) (Shen et al., 2014) for each sample with mm10 mouse genome build. The list of genes associated with 7 clusters (Fig. S7B) were imported to Seurat, and the expression for each cluster of genes was calculated using Seurat AddModuleScore.
Base editor genotyping
The cells expressing EDITOR were subject to PCR amplification. Fastq files for each gene-primer pair were aligned to a custom genome file containing that gene locus using STARlong and bwa-mem with default parameters, both of which gave similar results. The BAM files were uploaded to IGV for visualization.
Data AND SOFTWARE AVAILABILITY
The sequencing Data reported in this study has been deposited under GEO: Project GSE131675; Subseries - GSE117154, GSE131873, GSE131873, and GSE131940. P50 and E18 mouse brain scATAC-seq samples were downloaded from 10Xgenomics.com.
Supplementary Material
Supplementary Figure 2: Characterization of in vivo MADR and control experiments confirming specificity of integration, Related to Figure 2
A) At 2 days post-EP, cells start expressing TagBFP2. Scale bars: 50μm; Insets: 10μm
B) Gliogenesis and radial glia 2 weeks post-EP. Arrow indicates rare green-and-blue double positive cells at the VZ. Neurons with both markers can be observed in the OB at this time point. Scale bars: 100μm; Inset: 20μm
C) Projection of confocal z stacks showing EGFP (mG) and TagBFP (MADR) cells 1 week post EP.
D) Foxj1 immunostaining of same region depicted in C. Note the localized nuclear label along the VZ region. * -vascular staining due to “mouse on mouse” immunostaining.
E) MADR TagBFP single positive radial glial cell, displaying no EGFP (mG).
F) Three MADR TagBFP and EGFP (mG) double-positive cells—all expressing the Foxj1 transcription factor. Note that there seems to be an inverse correlation of TagBFP and EGFP expression.
G) Magnification of the white box from F showing that the cells with the brightest MADR labeling has the dimmest EGFP.
H) High-magnification confocal image of a pair of TagBFP2+ satellite glia, which are negative for tdTomato and EGFP. Scale bars: 10μm
I) Representative images of smFP-HA (donor), EGFP (mG), and TagBFP2-nls (blue) from VZ of the plasmid titration results depicted in Fig. 2D.
J) Lineage tracing of EP-ed cells in the VZ/SVZ with hyPBase-integrated EGFP reporter plus various donor vectors and recombinases do not show any integration by 2 weeks post-EP. Note that CD1 mice are an outbred mouse strain with no MADR recipient site. In the bottom panel, mTmG mice EPed with Cre but no Flp recombinase failed to show tumor formation or marked TagBFP2-Hras G12V insertion. Scale bars: 100μm
K) Donor vector with inverted loxP orientation fails to express HrasG12V and does not produce hyperplasia. (For comparison of integrated plasmid at same time point, see Supplementary Fig. 4A.) Scale bars: 100μm
L) Example of 5 color imaging for increasing spectral flexibility using Alexa 750 fluorophore.
M) Stitch of mT/mG brain immunostained with anti-EGFP in the 405 channel, anti-Olig2 in the 488 channel, and anti-Pdgfra in the 555 channel. H1) Note the intense tdTomato autofluorescence.
N) Stitch of same brain post-bleaching, showing significantly reduced mT tdTomato autofluorescence I1) Note the similar lack of detectable EGFP signal in the 488 wavelength due to bleaching.
{kind=link}
Supplementary Figure 3: Characterization of in vivo MADR loss of function lineages and comparison with CRISPR, Related to Figure 3
A) At 3 months post-EP, cells expressing multi-miR-E tied to TagBFP2 reporter are predominantly Pdgfra+ OPCs. Scale bars: 100μm
B) V5+ tumor-derived cell populations can be found juxtaposed to the Tdtomato+ vasculature in focal regions of the tumor.
C) Confirmation of base editing to induce a premature stop codon in Pten using genomic alignment of sequenced amplicon.
D) MADR CRISPR/Cas9 variants generated for knockdown by Crispri (dCas9-KRAB-MeCP2) or Cas13/RX, or for knockout/genome editing with HiFi EspCas9. U6/miRFP670 reporters plasmids for expressing appropriate sgRNA variants have been constructed with sites for the BsmBI type II restriction enzyme for seamless sgRNA cloning and expression. CS – dual BsmBI cloning site
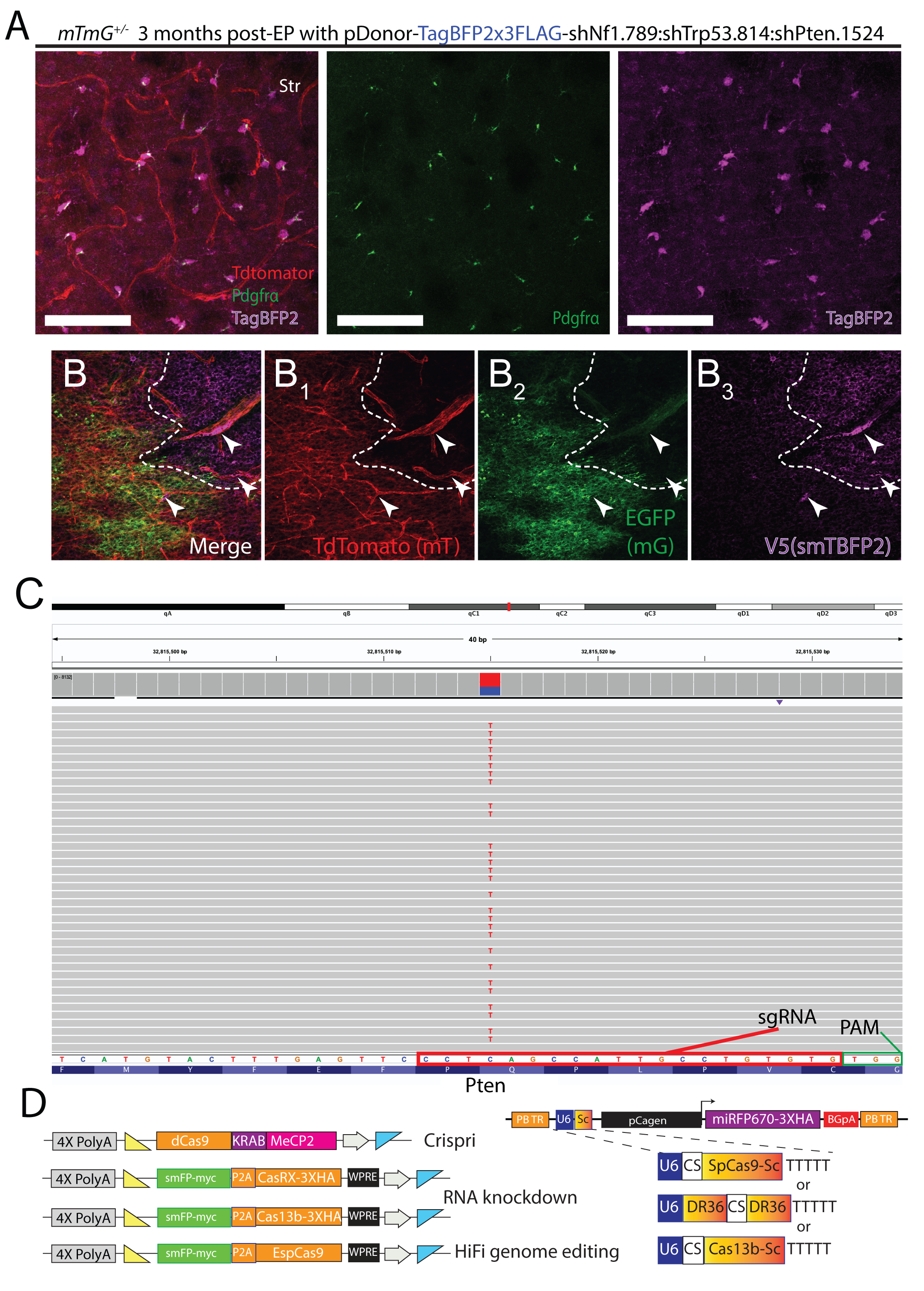{kind=link}
Supplementary Figure 4: Examination of MADR glioma and ependymoma cell fate changes and migratory dynamics, Related to Figure 4
A) RCE-based HrasG12V mosaic exhibiting Sox9/Gfap+ gliosis in TagBFP2+ regions.
B) Two weeks post-EP shows clear lineage divergence between EGFP+ cells that underwent Cre-mediated excision of tdTomato cassette and HrasG12V+ cells with successful MADR. Scale bars: 100μm
C) As low as 10 ng/μl recombinase-expression vector in EP mixture can catalyze MADR in vivo. Scale bars: 100μm
D) Brighter EGPF-HrasG12V cells after pBase-mediated integration express phosphorylated Rb1. Scale bars: 200μm
E-I) Striatal gliogenesis 1 month after electroporation of pDonor- (E) Kras G12A, (F-G) YAP1-MAML1D, or (H) C11orf95-RELA.
J-K) High magnification of ependymoma pushing margins displaying a lack of infiltration of these tumors.
{kind=link}
Supplementary Figure 5: Characterization of multi-cistronic tumors, secondary elements, and viability screens, Related to Figure 5
A) In vitro assessment of transgene expression after MADR in heterozygous mTmG mNSCs shows the co-expression of nuclear EGFP with Pdgfra, V5 (Trp53R270H), and P53. Note the presence of contaminating mG cells with membrane EGFP and no tdTomato or transgene expression. Scale bars: 50μm
B) Confirmation of Trp53 co-expression with nuclear EGFP (H3f3a). Scale bars: 50μm
C) Coronal section displaying pre-tumor phase of K27M-expressing lineages (nuclear EGFP; and mG EGFP – membrane EGFP).
D-G) Immunostaining of K27M (D,G) and G34R (E-F) tumors with anti-H3mutK27M and anti-H3mutG34R antibodies, confirming expression of the respective transgenes by specific immunolabeling with the appropriate antibodies.
H) Dual immunostaining of K27M and G34R in co-electroporated animals (K27M- and G34R-containing plasmids) confirms expression of only one H3f3a mutant variant per cell.
I) Rosa26H3f3aG34R/Pdgfra/Trp53 EGFP+ tumor cells are hypomethylated at H3K27.
J) High mag view of tumor margin.
K) Immunolabeling of K27M mutant tumor cells demonstrates perinuclear satellitosis and decreased H3K27Me labeling compared with neighboring neurons.
L) CRISPR/Cas9 targeting of Nf1/Trp53 to induce GBM does not yield reduced H3K27Me3.
M-O) Low mag (M-M2) and high mag images (N-O) of Bmi1 upregulation in K27M tumor cells. Arrows point to infiltrating cells and dotted line depicts tumor margin. P) Max projection of region in O-O1 showing that infiltrating K27M cells are juxtaposed to vessels.
P) A subset K27M and G34R mutant cells at the margins can be immunolabeling with the astrocyte marker Aldh1l1 and display hypertrophy
Q) Subpopulations of K27M and G34R mutant cells express the oligodendrocyte marker Cspg4.
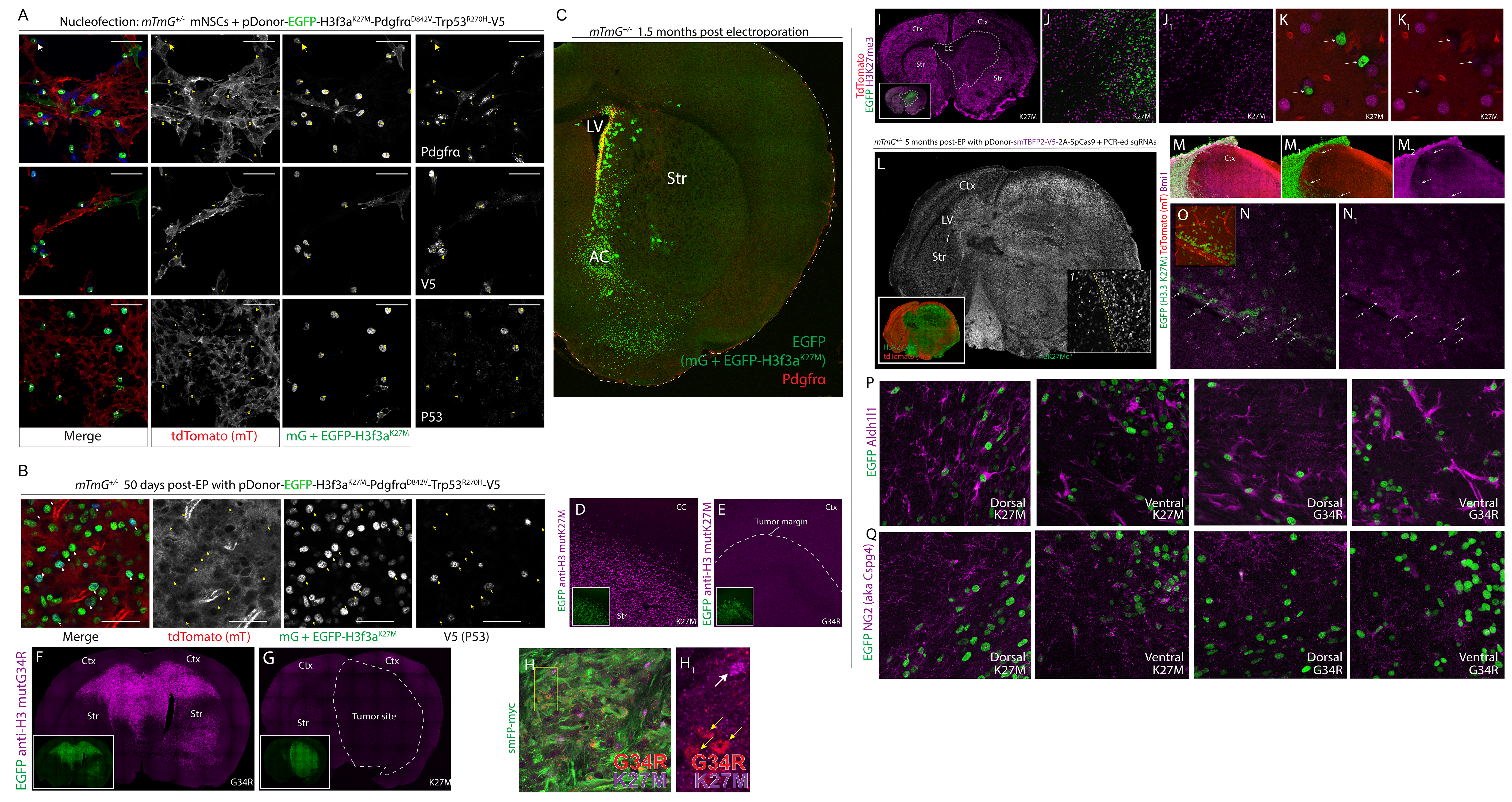{kind=link}
Supplementary Figure 6: Single-cell RNA-seq of MADR mutant models, Related to Figure 6
A) CNV analysis of 3 mouse K27M tumor scRNA-seq datasets.
B) CC1 and CC2 vector alignment of mouse K27M tumors.
C) Biweight midcorrelation plot of mouse K27M tumors across CCs.
D) UMAP depicting CCA alignment of 3 K27M datasets from the mouse brain colored by sample.
E) Featureplots depicting expression of genes in mouse K27M tumors.
F) Louvain clustering of human K27M scRNA-seq tumors from Filbin et al. (Science 2018).
G) CSF1R and H) MOG expression maps depicting clusters that are filtered before moving to CCA.
I) Clustering of Human K27M tumors post filtering.
J) Biweight midcorrelation plot of human K27M tumors across CCs.
K) UMAP of CCA alignment human K27M tumors colored by sample.
L) Featureplots depicting expression of genes in human K27M tumors.
M) Program featureplots split from CCA of all samples (i.e. Fig. 6H) and depicted by original sample.
A) Clustering by highly-variable genes rather than programs from Filbin et al. (Science 2018) leads to slightly altered clustering depending on the clustering parameters chosen.
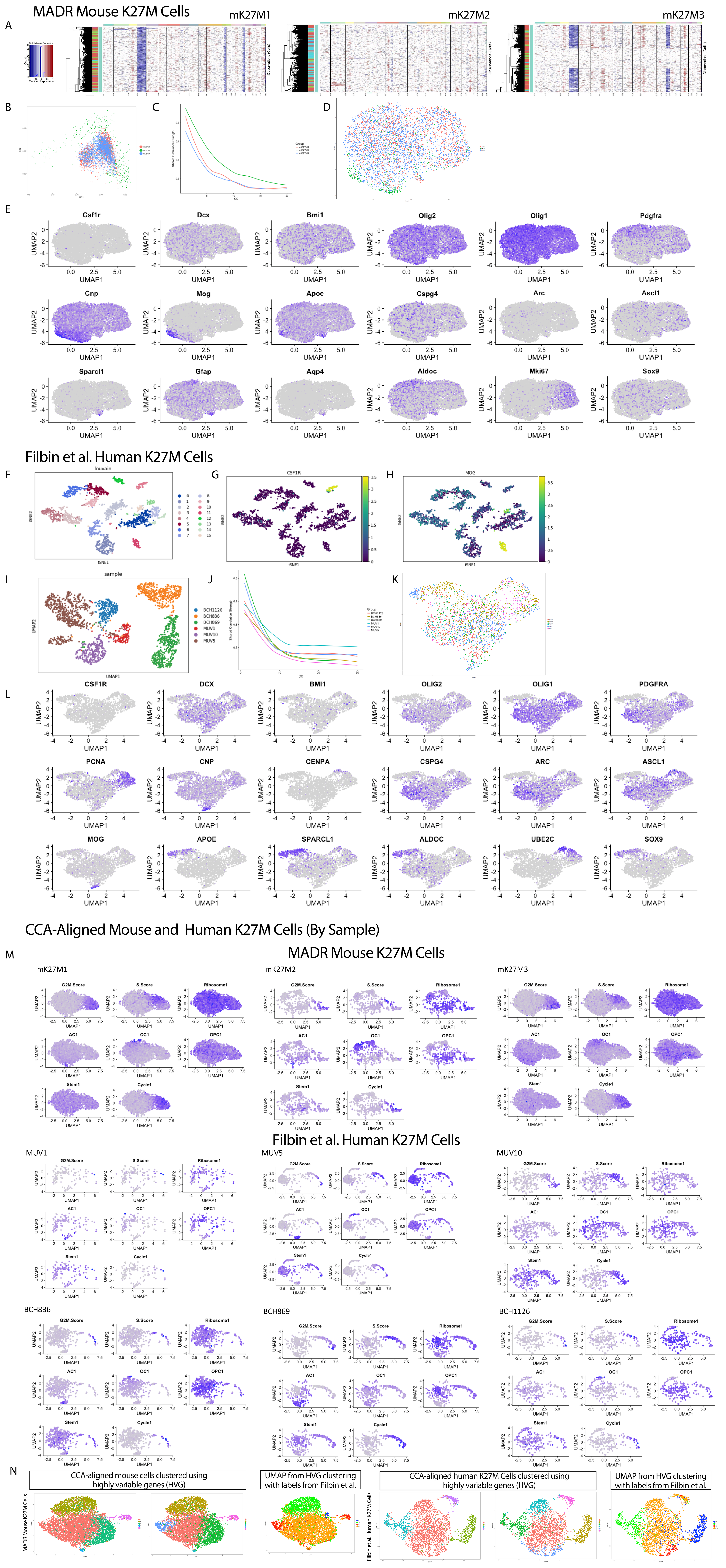{kind=link}
Supplementary Figure 7: SCENIC, H3K27me3 ChIP-seq, and snATAC-seq analysis of MADR mutant models, Related to Figure 7
A) General workflow for tumor dissociation and downstream analysis such as scRNA-seq, scATAC-seq, or ChIP-seq for H3K27Me3. Note same tumor source was used for scRNA-seq analysis and ChIP-seq but independent tumors were used for scATAC-seq samples.
B) Clustering of H3K27Me3 ChIP-seq Data from 3 mouse K27M tumors.
C) Scoring of scRNA-seq-derived UMAP for genes from clusters in B.
(D-I) t-Distributed Stochastic Neighbor Embeddings (t-SNEs) of SCENIC processed K27M human tumor cells (D,F,H) or SCENIC-derived t-SNEs of K27M mouse tumor cells (E,G,I). Samples are grouped by sample (D,E; i.e. patient or mouse of origin), cell type (F,G), and cell cycle phases (H,I).
J) tSNE of scATAC-seq dataset from P50 brain with numbered clusters.
K) Marker genes for oligodendrocyte (Mog), OPC (Pdgfra), astrocyte (Aqp4), microglia (C1qb), neuron (Snap25), and interneuron (Gad2, Pvalb, Sst) populations. Note the distinct signal to noise for each cluster.
L) tSNE of 3 combined scATAC-seq datasets from E18 brain after standard clustering.
M) tSNE of samples from L post-Harmony alignment.
N) tSNE of E18 datasets with clusters numbered.
O) Gene accessibility for Sox9 (astrocytes/stem cells), Olig2 (stem cells/oligodendrocyte lineage), Csf1r (microglia), and Gfap (astrocytes). Note the lack of clear population/cluster segregation for microglia or glial specificity of Sox9 and Olig2 compared with Gfap, which is more exclusive to discrete populations and readily associates with the glial clusters.
P) tSNE of scATAC-seq dataset derived from K27M tumor cells and co-capture innate immune populations
Q) Gene accessibility for Sox9, Olig2, Csf1r, and Gfap. Again, note the lack of clear population/cluster segregation of Sox9 and Olig2 compared with Gfap, which exhibits more accessibility. Csf1r is noticeably more accessible than Sox9 and Olig2 and is associated with innate immune clusters.
R) CisTopic and CellRanger-based clustering of K27M tumor populations leads to subtly different subclustering of tumor and immune populations.
S) Gene accessibility clearly defines microglial populations but Sox9 and Sox10 fail to co-segregate in tumor, unlike in P50 normal brain.
T) K27M scRNA-seq featureplots blending Sox10 (green) and Sox9 (red) demonstrate that Sox10 and Sox9 are highly expressed throughout the tumor clusters and even within individual cells in agreement with gene accessibility in S and genome browser Data (Fig. 7J).
U) mSigDB terms for P50 astrocyte and OPC clusters.
V) Motifs enriched in DARs from K27M tumor clusters. Note the enrichment of IEGs and ES-associated TFs.
{kind=link}
Supplementary Table 2: Labels for differentially-expressed markers on Heatmaps, Related to Figure 6.
Supplementary Table 1: Details on the mice used for scRNA-seq, Related to Figure 6.
Supplementary Table 3: Comparison of methods for conditional/somatic transgenesis, Related to STAR METHODS
Supplementary Movie 1: Light sheet image of cleared left brain hemisphere 7 days post-EP, Related to Figure 2. Hemisphere was immunostained for the Myc epitope tag.
Supplementary Movie 2: 3D rotation of confocal z-stack showing the non-overlapping genetically defined populations (red – non-EPed, Green – Cre recombined, Magenta – TagBFP2 MADR insertion), Related to Figure 2..
Supplementary Movie 3: Light sheet image of cleared olfactory bulbs 2 weeks post-EP. The bulbs were immunostained for the Myc, V5, HA, and Flag epitope tag, Related to Figure 2.
Supplementary Figure 1: Measurement of MADR efficiency in heterozygous mTmG mNSCs by FACS analysis, confirmation of correct protein translation at non-clonal population level, inducible MADR, and MADR “proxy” lines, Related to Figure 1
A) Schematic of recombinase-expressing plasmids (and minicircle) employed in this study.
B) FACS analysis indicates the approximate MADR efficiency in neural stem cells, and no obvious difference between Flp-2A-Cre and Flp-IRES-Cre in their catalytic efficiencies.
C) Sorted cells express HrasG12V but not tdTomato or EGFP. Scale bar, 50μm
D) Western blot indicating normal transgene production from non-clonal aggregate cells and lack thereof in FACSnegative population. Removal of tdTomato expression is also observed.
E) Schematics of plasmids and alleles subject to PCR analysis at denoted sites.
F) PCR screening analysis reveals that rtTA-V10-AU1 cassette is correctly integrated downstream of CAG-promoter in cells that are resistant to puromycin treatment.
G) Western blot analysis of the cell line from Fig. 2C showing the expression of rtTA-V10-AU1 and also EGFP upon doxycycline induction.
H) MADR-compatible TRE-EGFP plasmid.
I) Heterozygous mTmG mNSCs are nucleofected with plasmid in H, treated with puromycin, and turned into a colorless population. Scale bars: 10μm
J) Induction of EGFP expression in the cell line that constitutively express rtTA-V10-AU1. Scale bars: 50μm
K) TRE cell line with a bidirectional tet-response element that expresses EGFP and Dll1 upon doxycycline treatment.
L) Immunofluorescence of cells without and with Dox demonstrates the relative lack of leakiness and homogenous expression level of EGFP and mDll1. Scale bars: 20μm
M) qPCR measurement of mRNA abundance before and after Dox addition to medium. (Ctrl plasmid lacks mDll1 CDS but is otherwise identical to plasmid in K.)
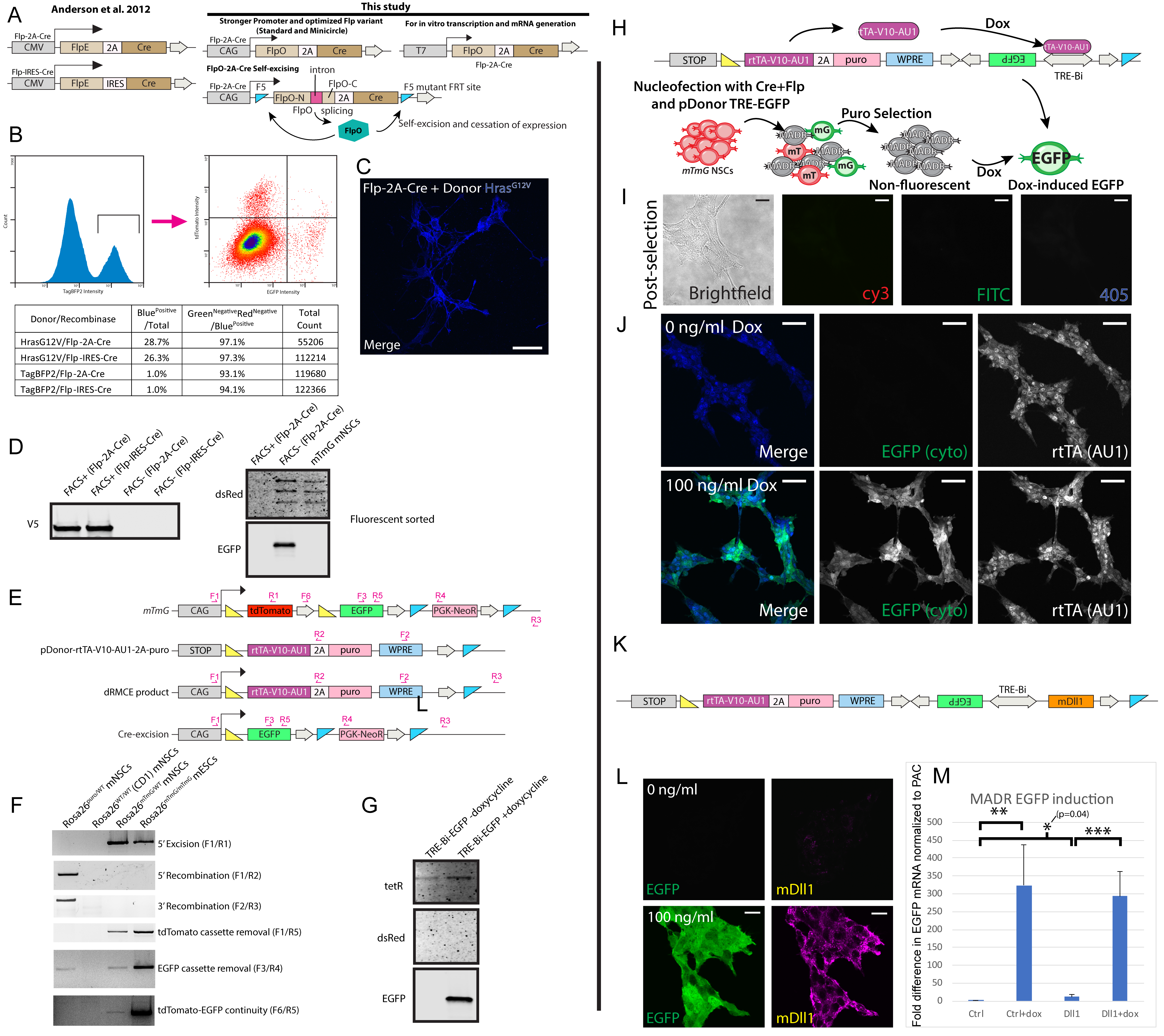{kind=link}
Highlights.
MADR enables single locus somatic genetic modification and lineage tracing in vivo
MADR can be used to interrogate the dosage response of transgenes in vivo
MADR allows “personalized” brain tumor modeling in vivo via CRISPR variants or transgenes
MADR is adaptable to hundreds of available mouse lines
ACKNOWLEDGEMENTS and FUNDING
We thank Y. Voziyanov for the gift of Flp-Cre expression vectors. We thank D. Cai for mKate2 and dsRed antibodies and L. Luo, H. Zeng, G. Miyoshi, G. Fishell, I. Imayoshi, and J. Huang for Cre reporter strain sequencing data. We thank S. Svendsen for editing. We acknowledge support from TJ’s Dream Team HEADing for a Cure, the Samuel Oschin Comprehensive Cancer Institute (SOCCI) Cancer Research Forum Award, the Board of Governors RMI of Cedars-Sinai (MD, JJB), the Smidt Family Foundation, and the Paul and Vera Guerin Family Foundation. JJB was supported by NIH grants R33CA202900, R33CA236687, R03NS101529, American Cancer Society grant RSG-16-217-01-TBG, and SOCCI Jack Mishkin Discovery, P&G, and Cancer Biology Awards.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Cedars-Sinai has filed for patent protection for in vivo dual recombinase-mediated cassette exchange and disease models thereof.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three tables, three movies, and a Data file and can be found with this article online
REFERENCES
- AIBAR S, GONZÁLEZ-BLAS CB, MOERMAN T, HUYNH-THU VA, IMRICHOVA H, HULSELMANS G, RAMBOW F, MARINE J-C, GEURTS P, AERTS J, VAN DEN OORD J, ATAK ZK, WOUTERS J & AERTS S 2017a. SCENIC: single-cell regulatory network inference and clustering. Nature Methods, 14, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AIBAR S, GONZALEZ-BLAS CB, MOERMAN T, HUYNH-THU VA, IMRICHOVA H, HULSELMANS G, RAMBOW F, MARINE JC, GEURTS P, AERTS J, VAN DEN OORD J, ATAK ZK, WOUTERS J & AERTS S 2017b. SCENIC: single-cell regulatory network inference and clustering. Nat Methods, 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKHTAR AA, MOLINA J, DUTRA-CLARKE M, KIM GB, LEVY R, SCHREIBER-STAINTHORP W, DANIELPOUR M & BREUNIG JJ 2015. A transposon-mediated system for flexible control of transgene expression in stem and progenitor-derived lineages. Stem Cell Reports, 4, 323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKHTAR W, DE JONG J, PINDYURIN AV, PAGIE L, MEULEMAN W, DE RIDDER J, BERNS A, WESSELS LF, VAN LOHUIZEN M & VAN STEENSEL B 2013. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell, 154, 914–27. [DOI] [PubMed] [Google Scholar]
- ANDERSON RP, VOZIYANOVA E & VOZIYANOV Y 2012. Flp and Cre expressed from Flp-2A-Cre and Flp-IRES-Cre transcription units mediate the highest level of dual recombinase-mediated cassette exchange. Nucleic Acids Res, 40, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEN-PORATH I, THOMSON MW, CAREY VJ, GE R, BELL GW, REGEV A & WEINBERG RA 2008. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics, 40, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRAVO GONZALEZ-BLAS C, MINNOYE L, PAPASOKRATI D, AIBAR S, HULSELMANS G, CHRISTIAENS V, DAVIE K, WOUTERS J & AERTS S 2019. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat Methods, 16, 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREUNIG JJ, GATE D, LEVY R, RODRIGUEZ J JR., KIM GB, DANIELPOUR M, SVENDSEN CN & TOWN T 2012. Rapid genetic targeting of pial surface neural progenitors and immature neurons by neonatal electroporation. Neural Dev, 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREUNIG JJ, LEVY R, ANTONUK CD, MOLINA J, DUTRA-CLARKE M, PARK H, AKHTAR AA, KIM GB, HU X, BANNYKH SI, VERHAAK RG & DANIELPOUR M 2015. Ets Factors Regulate Neural Stem Cell Depletion and Gliogenesis in Ras Pathway Glioma. Cell Rep, 12, 258–71. [DOI] [PubMed] [Google Scholar]
- BUTLER A, HOFFMAN P, SMIBERT P, PAPALEXI E & SATIJA R 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CECCARELLI M, BARTHEL FP, MALTA TM, SABEDOT TS, SALAMA SR, MURRAY BA, MOROZOVA O, NEWTON Y, RADENBAUGH A, PAGNOTTA SM, ANJUM S, WANG J, MANYAM G, ZOPPOLI P, LING S, RAO AA, GRIFFORD M, CHERNIACK AD, ZHANG H, POISSON L, CARLOTTI CG JR., TIRAPELLI DP, RAO A, MIKKELSEN T, LAU CC, YUNG WK, RABADAN R, HUSE J, BRAT DJ, LEHMAN NL, BARNHOLTZ-SLOAN JS, ZHENG S, HESS K, RAO G, MEYERSON M, BEROUKHIM R, COOPER L, AKBANI R, WRENSCH M, HAUSSLER D, ALDAPE KD, LAIRD PW, GUTMANN DH, NOUSHMEHR H, IAVARONE A & VERHAAK RG 2016. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell, 164, 550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN F & LOTURCO J 2012. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J Neurosci Methods, 207, 172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN F, ROSIENE J, CHE A, BECKER A & LOTURCO J 2015. Tracking and transforming neocortical progenitors by CRISPR/Cas9 gene targeting and piggyBac transposase lineage labeling. Development, 142, 3601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORDERO FJ, HUANG Z, GRENIER C, HE X, HU G, MCLENDON RE, MURPHY SK, HASHIZUME R & BECHER OJ 2017. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol Cancer Res, 15, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX DBT, GOOTENBERG JS, ABUDAYYEH OO, FRANKLIN B, KELLNER MJ, JOUNG J & ZHANG F 2017. RNA editing with CRISPR-Cas13. Science, 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA ROSSA A, BELLONE C, GOLDING B, VITALI I, MOSS J, TONI N, LϋSCHER C & JABAUDON D 2013. In vivo reprogramming of circuit connectivity in postmitotic neocortical neurons. Nature Neuroscience, 16, 193. [DOI] [PubMed] [Google Scholar]
- DE LA ROSSA A & JABAUDON D 2015. In vivo rapid gene delivery into postmitotic neocortical neurons using iontoporation. Nat Protoc, 10, 25–32. [DOI] [PubMed] [Google Scholar]
- FANG R, PREISSL S, HOU X, LUCERO J, WANG X, MOTAMEDI A, SHIAU AK, MUKAMEL EA, ZHANG Y, BEHRENS MM, ECKER J & REN B 2019. Fast and Accurate Clustering of Single Cell Epigenomes Reveals <em>Cis</em>-Regulatory Elements in Rare Cell Types. bioRxiv, 615179. [Google Scholar]
- FILBIN MG, TIROSH I, HOVESTADT V, SHAW ML, ESCALANTE LE, MATHEWSON ND, NEFTEL C, FRANK N, PELTON K, HEBERT CM, HABERLER C, YIZHAK K, GOJO J, EGERVARI K, MOUNT C, VAN GALEN P, BONAL DM, NGUYEN QD, BECK A, SINAI C, CZECH T, DORFER C, GOUMNEROVA L, LAVARINO C, CARCABOSO AM, MORA J, MYLVAGANAM R, LUO CC, PEYRL A, POPOVIC M, AZIZI A, BATCHELOR TT, FROSCH MP, MARTINEZ-LAGE M, KIERAN MW, BANDOPADHAYAY P, BEROUKHIM R, FRITSCH G, GETZ G, ROZENBLATT-ROSEN O, WUCHERPFENNIG KW, LOUIS DN, MONJE M, SLAVC I, LIGON KL, GOLUB TR, REGEV A, BERNSTEIN BE & SUVA ML 2018. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science, 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUNATO K, MAJOR T, LEWIS PW, ALLIS CD & TABAR V 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science, 346, 1529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARRICK D, FIERING S, MARTIN DI & WHITELAW E 1998. Repeat-induced gene silencing in mammals. Nat Genet, 18, 56–9. [DOI] [PubMed] [Google Scholar]
- GIBSON TJ, SEILER M & VEITIA RA 2013. The transience of transient overexpression. Nat Methods, 10, 715–21. [DOI] [PubMed] [Google Scholar]
- GLASGOW SM, ZHU W, STOLT CC, HUANG TW, CHEN F, LOTURCO JJ, NEUL JL, WEGNER M, MOHILA C & DENEEN B 2014. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat Neurosci, 17, 1322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANT GD, KEDZIORA KM, LIMAS JC, COOK JG & PURVIS JE 2018. Accurate delineation of cell cycle phase transitions in living cells with PIP-FUCCI. Cell Cycle, 17, 2496–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMBARDZUMYAN D, PARADA LF, HOLLAND EC & CHAREST A 2011. Genetic modeling of gliomas in mice: New tools to tackle old problems. Glia, 59, 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HECKL D, KOWALCZYK MS, YUDOVICH D, BELIZAIRE R, PURAM RV, MCCONKEY ME, THIELKE A, ASTER JC, REGEV A & EBERT BL 2014. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature Biotechnology, 32, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMAYOSHI I, HIRANO K, SAKAMOTO M, MIYOSHI G, IMURA T, KITANO S, MIYACHI H & KAGEYAMA R 2012. A multifunctional teal-fluorescent Rosa26 reporter mouse line for Cre- and Flp-mediated recombination. Neurosci Res, 73, 85–91. [DOI] [PubMed] [Google Scholar]
- INTERNATIONAL MOUSE KNOCKOUT C, COLLINS FS, ROSSANT J & WURST W 2007. A mouse for all reasons. Cell, 128, 9–13. [DOI] [PubMed] [Google Scholar]
- IWANO S, SUGIYAMA M, HAMA H, WATAKABE A, HASEGAWA N, KUCHIMARU T, TANAKA KZ, TAKAHASHI M, ISHIDA Y, HATA J, SHIMOZONO S, NAMIKI K, FUKANO T, KIYAMA M, OKANO H, KIZAKA-KONDOH S, MCHUGH TJ, YAMAMORI T, HIOKI H, MAKI S & MIYAWAKI A 2018. Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science, 359, 935–939. [DOI] [PubMed] [Google Scholar]
- JADHAV U, CAVAZZA A, BANERJEE KK, XIE H, O’NEILL NK, SAENZ-VASH V, HERBERT Z, MADHA S, ORKIN SH, ZHAI H & SHIVDASANI RA 2019. Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA. Mol Cell, 74, 542–554 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM J, WOO AJ, CHU J, SNOW JW, FUJIWARA Y, KIM CG, CANTOR AB & ORKIN SH 2010. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell, 143, 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONERMANN S, LOTFY P, BRIDEAU NJ, OKI J, SHOKHIREV MN & HSU PD 2018. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell, 173, 665–676. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORSUNSKY I, FAN J, SLOWIKOWSKI K, ZHANG F, WEI K, BAGLAENKO Y, BRENNER M, LOH P-R & RAYCHAUDHURI S 2018. Fast, sensitive, and accurate integration of single cell Data with Harmony. bioRxiv, 461954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSICKI M, TOMBERG K & BRADLEY A 2018. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology, 36, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARSON JD, KASPER LH, PAUGH BS, JIN H, WU G, KWON CH, FAN Y, SHAW TI, SILVEIRA AB, QU C, XU R, ZHU X, ZHANG J, RUSSELL HR, PETERS JL, FINKELSTEIN D, XU B, LIN T, TINKLE CL, PATAY Z, ONAR-THOMAS A, POUNDS SB, MCKINNON PJ, ELLISON DW, ZHANG J & BAKER SJ 2019. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell, 35, 140–155. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE MS, STOJANOV P, MERMEL CH, ROBINSON JT, GARRAWAY LA, GOLUB TR, MEYERSON M, GABRIEL SB, LANDER ES & GETZ G 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU C, SAGE JC, MILLER MR, VERHAAK RG, HIPPENMEYER S, VOGEL H, FOREMAN O, BRONSON RT, NISHIYAMA A, LUO L & ZONG H 2011. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell, 146, 209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLORENS-BOBADILLA E, ZHAO S, BASER A, SAIZ-CASTRO G, ZWADLO K & MARTIN-VILLALBA A 2015. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. [DOI] [PubMed] [Google Scholar]
- MACKAY A, BURFORD A, CARVALHO D, IZQUIERDO E, FAZAL-SALOM J, TAYLOR KR, BJERKE L, CLARKE M, VINCI M, NANDHABALAN M, TEMELSO S, POPOV S, MOLINARI V, RAMAN P, WAANDERS AJ, HAN HJ, GUPTA S, MARSHALL L, ZACHAROULIS S, VAIDYA S, MANDEVILLE HC, BRIDGES LR, MARTIN AJ, AL-SARRAJ S, CHANDLER C, NG HK, LI X, MU K, TRABELSI S, BRAHIM DH, KISLJAKOV AN, KONOVALOV DM, MOORE AS, CARCABOSO AM, SUNOL M, DE TORRES C, CRUZ O, MORA J, SHATS LI, STAVALE JN, BIDINOTTO LT, REIS RM, ENTZ-WERLE N, FARRELL M, CRYAN J, CRIMMINS D, CAIRD J, PEARS J, MONJE M, DEBILY MA, CASTEL D, GRILL J, HAWKINS C, NIKBAKHT H, JABADO N, BAKER SJ, PFISTER SM, JONES DTW, FOULADI M, VON BUEREN AO, BAUDIS M, RESNICK A & JONES C 2017. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell, 32, 520–537. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANDEGAR MA, HUEBSCH N, FROLOV EB, SHIN E, TRUONG A, OLVERA MP, CHAN AH, MIYAOKA Y, HOLMES K, SPENCER CI, JUDGE LM, GORDON DE, ESKILDSEN TV, VILLALTA JE, HORLBECK MA, GILBERT LA, KROGAN NJ, SHEIKH SP, WEISSMAN JS, QI LS, SO PL & CONKLIN BR 2016. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell, 18, 541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKENNA WL, BETANCOURT J, LARKIN KA, ABRAMS B, GUO C, RUBENSTEIN JLR & CHEN B 2011. Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. The Journal of Neuroscience, 31, 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIKUNI T, NISHIYAMA J, SUN Y, KAMASAWA N & YASUDA R 2016. High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell, 165, 1803–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUZUMDAR MD, TASIC B, MIYAMICHI K, LI L & LUO L 2007. A global double-fluorescent Cre reporter mouse. Genesis, 45, 593–605. [DOI] [PubMed] [Google Scholar]
- ORTIZ-ALVAREZ G, DACLIN M, SHIHAVUDDIN A, LANSADE P, FORTOUL A, FAUCOURT M, CLAVREUL S, LALIOTI ME, TARAVIRAS S, HIPPENMEYER S, LIVET J, MEUNIER A, GENOVESIO A & SPASSKY N 2019. Adult Neural Stem Cells and Multiciliated Ependymal Cells Share a Common Lineage Regulated by the Geminin Family Members. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAJTLER KW, WITT H, SILL M, JONES DT, HOVESTADT V, KRATOCHWIL F, WANI K, TATEVOSSIAN R, PUNCHIHEWA C, JOHANN P, REIMAND J, WARNATZ HJ, RYZHOVA M, MACK S, RAMASWAMY V, CAPPER D, SCHWEIZER L, SIEBER L, WITTMANN A, HUANG Z, VAN SLUIS P, VOLCKMANN R, KOSTER J, VERSTEEG R, FULTS D, TOLEDANO H, AVIGAD S, HOFFMAN LM, DONSON AM, FOREMAN N, HEWER E, ZITTERBART K, GILBERT M, ARMSTRONG TS, GUPTA N, ALLEN JC, KARAJANNIS MA, ZAGZAG D, HASSELBLATT M, KULOZIK AE, WITT O, COLLINS VP, VON HOFF K, RUTKOWSKI S, PIETSCH T, BADER G, YASPO ML, VON DEIMLING A, LICHTER P, TAYLOR MD, GILBERTSON R, ELLISON DW, ALDAPE K, KORSHUNOV A, KOOL M & PFISTER SM 2015. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell, 27, 728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKER M, MOHANKUMAR KM, PUNCHIHEWA C, WEINLICH R, DALTON JD, LI Y, LEE R, TATEVOSSIAN RG, PHOENIX TN, THIRUVENKATAM R, WHITE E, TANG B, ORISME W, GUPTA K, RUSCH M, CHEN X, LI Y, NAGAHAWHATTE P, HEDLUND E, FINKELSTEIN D, WU G, SHURTLEFF S, EASTON J, BOGGS K, YERGEAU D, VADODARIA B, MULDER HL, BECKSFORT J, GUPTA P, HUETHER R, MA J, SONG G, GAJJAR A, MERCHANT T, BOOP F, SMITH AA, DING L, LU C, OCHOA K, ZHAO D, FULTON RS, FULTON LL, MARDIS ER, WILSON RK, DOWNING JR, GREEN DR, ZHANG J, ELLISON DW & GILBERTSON RJ 2014. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature, 506, 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATHANIA M, DE JAY N, MAESTRO N, HARUTYUNYAN AS, NITARSKA J, PAHLAVAN P, HENDERSON S, MIKAEL LG, RICHARD-LONDT A, ZHANG Y, COSTA JR, HEBERT S, KHAZAEI S, IBRAHIM NS, HERRERO J, RICCIO A, ALBRECHT S, KETTELER R, BRANDNER S, KLEINMAN CL, JABADO N & SALOMONI P 2017. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell, 32, 684–700 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PYLAYEVA-GUPTA Y, GRABOCKA E & BAR-SAGI D 2011. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer, 11, 761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKAUE-SAWANO A, KOBAYASHI T, OHTAWA K & MIYAWAKI A 2011. Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biology, 12, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWARTZENTRUBER J, KORSHUNOV A, LIU XY, JONES DT, PFAFF E, JACOB K, STURM D, FONTEBASSO AM, QUANG DA, TONJES M, HOVESTADT V, ALBRECHT S, KOOL M, NANTEL A, KONERMANN C, LINDROTH A, JAGER N, RAUSCH T, RYZHOVA M, KORBEL JO, HIELSCHER T, HAUSER P, GARAMI M, KLEKNER A, BOGNAR L, EBINGER M, SCHUHMANN MU, SCHEURLEN W, PEKRUN A, FRUHWALD MC, ROGGENDORF W, KRAMM C, DURKEN M, ATKINSON J, LEPAGE P, MONTPETIT A, ZAKRZEWSKA M, ZAKRZEWSKI K, LIBERSKI PP, DONG Z, SIEGEL P, KULOZIK AE, ZAPATKA M, GUHA A, MALKIN D, FELSBERG J, REIFENBERGER G, VON DEIMLING A, ICHIMURA K, COLLINS VP, WITT H, MILDE T, WITT O, ZHANG C, CASTELO-BRANCO P, LICHTER P, FAURY D, TABORI U, PLASS C, MAJEWSKI J, PFISTER SM & JABADO N 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482, 226–31. [DOI] [PubMed] [Google Scholar]
- SHEN L, SHAO N, LIU X & NESTLER E 2014. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics, 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLAYMAKER IM, GAO L, ZETSCHE B, SCOTT DA, YAN WX & ZHANG F 2016. Rationally engineered Cas9 nucleases with improved specificity. Science, 351, 84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SORIANO P 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet, 21, 70–1. [DOI] [PubMed] [Google Scholar]
- STANCIK EK, NAVARRO-QUIROGA I, SELLKE R & HAYDAR TF 2010. Heterogeneity in ventricular zone neural precursors contributes to neuronal fate diversity in the postnatal neocortex. J Neurosci, 30, 7028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUART T & SATIJA R 2019. Integrative single-cell analysis. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- STURM D, WITT H, HOVESTADT V, KHUONG-QUANG DA, JONES DT, KONERMANN C, PFAFF E, TONJES M, SILL M, BENDER S, KOOL M, ZAPATKA M, BECKER N, ZUCKNICK M, HIELSCHER T, LIU XY, FONTEBASSO AM, RYZHOVA M, ALBRECHT S, JACOB K, WOLTER M, EBINGER M, SCHUHMANN MU, VAN METER T, FRUHWALD MC, HAUCH H, PEKRUN A, RADLWIMMER B, NIEHUES T, VON KOMOROWSKI G, DURKEN M, KULOZIK AE, MADDEN J, DONSON A, FOREMAN NK, DRISSI R, FOULADI M, SCHEURLEN W, VON DEIMLING A, MONORANU C, ROGGENDORF W, HEROLD-MENDE C, UNTERBERG A, KRAMM CM, FELSBERG J, HARTMANN C, WIESTLER B, WICK W, MILDE T, WITT O, LINDROTH AM, SCHWARTZENTRUBER J, FAURY D, FLEMING A, ZAKRZEWSKA M, LIBERSKI PP, ZAKRZEWSKI K, HAUSER P, GARAMI M, KLEKNER A, BOGNAR L, MORRISSY S, CAVALLI F, TAYLOR MD, VAN SLUIS P, KOSTER J, VERSTEEG R, VOLCKMANN R, MIKKELSEN T, ALDAPE K, REIFENBERGER G, COLLINS VP, MAJEWSKI J, KORSHUNOV A, LICHTER P, PLASS C, JABADO N & PFISTER SM 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell, 22, 425–37. [DOI] [PubMed] [Google Scholar]
- TILLBERG PW, CHEN F, PIATKEVICH KD, ZHAO Y, YU C-C, ENGLISH BP, GAO L, MARTORELL A, SUK H-J, YOSHIDA F, DEGENNARO EM, ROOSSIEN DH, GONG G, SENEVIRATNE U, TANNENBAUM SR, DESIMONE R, CAI D & BOYDEN ES 2016. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature Biotechnology, 34, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKATESH HUMSAS, JOHUNG TESSAB, CARETTI V, NOLL A, TANG Y, NAGARAJA S, GIBSON ERINM, MOUNT CHRISTOPHERW, POLEPALLI J, MITRA SIDDHARTHAS, WOO PAMELYNJ, MALENKA ROBERTC, VOGEL H, BREDEL M, MALLICK P & MONJE M 2015. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell, 161, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISWANATHAN S, WILLIAMS ME, BLOSS EB, STASEVICH TJ, SPEER CM, NERN A, PFEIFFER BD, HOOKS BM, LI WP, ENGLISH BP, TIAN T, HENRY GL, MACKLIN JJ, PATEL R, GERFEN CR, ZHUANG X, WANG Y, RUBIN GM & LOOGER LL 2015. High-performance probes for light and electron microscopy. Nat Methods, 12, 568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF FA, ANGERER P & THEIS FJ 2018. SCANPY: large-scale single-cell gene expression Data analysis. Genome Biology, 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOODS NB, MUESSIG A, SCHMIDT M, FLYGARE J, OLSSON K, SALMON P, TRONO D, VON KALLE C & KARLSSON S 2003. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood, 101, 1284–9. [DOI] [PubMed] [Google Scholar]
- XUE W, CHEN S, YIN H, TAMMELA T, PAPAGIANNAKOPOULOS T, JOSHI NS, CAI W, YANG G, BRONSON R, CROWLEY DG, ZHANG F, ANDERSON DG, SHARP PA & JACKS T 2014. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature, 514, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEO NC, CHAVEZ A, LANCE-BYRNE A, CHAN Y, MENN D, MILANOVA D, KUO CC, GUO X, SHARMA S, TUNG A, CECCHI RJ, TUTTLE M, PRADHAN S, LIM ET, DAVIDSOHN N, EBRAHIMKHANI MR, COLLINS JJ, LEWIS NE, KIANI S & CHURCH GM 2018. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods, 15, 611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAFRA MP, SCHATOFF EM, KATTI A, FORONDA M, BREINIG M, SCHWEITZER AY, SIMON A, HAN T, GOSWAMI S, MONTGOMERY E, THIBADO J, KASTENHUBER ER, SANCHEZ-RIVERA FJ, SHI J, VAKOC CR, LOWE SW, TSCHAHARGANEH DF & DOW LE 2018. Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol, 36, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEISEL A, HOCHGERNER H, LÖNNERBERG P, JOHNSSON A, MEMIC F, VAN DER ZWAN J, HÄRING M, BRAUN E, BORM LE, LA MANNO G, CODELUPPI S, FURLAN A, LEE K, SKENE N, HARRIS KD, HJERLING-LEFFLER J, ARENAS E, ERNFORS P, MARKLUND U & LINNARSSON S 2018. Molecular Architecture of the Mouse Nervous System. Cell, 174, 999–1014. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 2: Characterization of in vivo MADR and control experiments confirming specificity of integration, Related to Figure 2
A) At 2 days post-EP, cells start expressing TagBFP2. Scale bars: 50μm; Insets: 10μm
B) Gliogenesis and radial glia 2 weeks post-EP. Arrow indicates rare green-and-blue double positive cells at the VZ. Neurons with both markers can be observed in the OB at this time point. Scale bars: 100μm; Inset: 20μm
C) Projection of confocal z stacks showing EGFP (mG) and TagBFP (MADR) cells 1 week post EP.
D) Foxj1 immunostaining of same region depicted in C. Note the localized nuclear label along the VZ region. * -vascular staining due to “mouse on mouse” immunostaining.
E) MADR TagBFP single positive radial glial cell, displaying no EGFP (mG).
F) Three MADR TagBFP and EGFP (mG) double-positive cells—all expressing the Foxj1 transcription factor. Note that there seems to be an inverse correlation of TagBFP and EGFP expression.
G) Magnification of the white box from F showing that the cells with the brightest MADR labeling has the dimmest EGFP.
H) High-magnification confocal image of a pair of TagBFP2+ satellite glia, which are negative for tdTomato and EGFP. Scale bars: 10μm
I) Representative images of smFP-HA (donor), EGFP (mG), and TagBFP2-nls (blue) from VZ of the plasmid titration results depicted in Fig. 2D.
J) Lineage tracing of EP-ed cells in the VZ/SVZ with hyPBase-integrated EGFP reporter plus various donor vectors and recombinases do not show any integration by 2 weeks post-EP. Note that CD1 mice are an outbred mouse strain with no MADR recipient site. In the bottom panel, mTmG mice EPed with Cre but no Flp recombinase failed to show tumor formation or marked TagBFP2-Hras G12V insertion. Scale bars: 100μm
K) Donor vector with inverted loxP orientation fails to express HrasG12V and does not produce hyperplasia. (For comparison of integrated plasmid at same time point, see Supplementary Fig. 4A.) Scale bars: 100μm
L) Example of 5 color imaging for increasing spectral flexibility using Alexa 750 fluorophore.
M) Stitch of mT/mG brain immunostained with anti-EGFP in the 405 channel, anti-Olig2 in the 488 channel, and anti-Pdgfra in the 555 channel. H1) Note the intense tdTomato autofluorescence.
N) Stitch of same brain post-bleaching, showing significantly reduced mT tdTomato autofluorescence I1) Note the similar lack of detectable EGFP signal in the 488 wavelength due to bleaching.
Supplementary Figure 3: Characterization of in vivo MADR loss of function lineages and comparison with CRISPR, Related to Figure 3
A) At 3 months post-EP, cells expressing multi-miR-E tied to TagBFP2 reporter are predominantly Pdgfra+ OPCs. Scale bars: 100μm
B) V5+ tumor-derived cell populations can be found juxtaposed to the Tdtomato+ vasculature in focal regions of the tumor.
C) Confirmation of base editing to induce a premature stop codon in Pten using genomic alignment of sequenced amplicon.
D) MADR CRISPR/Cas9 variants generated for knockdown by Crispri (dCas9-KRAB-MeCP2) or Cas13/RX, or for knockout/genome editing with HiFi EspCas9. U6/miRFP670 reporters plasmids for expressing appropriate sgRNA variants have been constructed with sites for the BsmBI type II restriction enzyme for seamless sgRNA cloning and expression. CS – dual BsmBI cloning site
Supplementary Figure 4: Examination of MADR glioma and ependymoma cell fate changes and migratory dynamics, Related to Figure 4
A) RCE-based HrasG12V mosaic exhibiting Sox9/Gfap+ gliosis in TagBFP2+ regions.
B) Two weeks post-EP shows clear lineage divergence between EGFP+ cells that underwent Cre-mediated excision of tdTomato cassette and HrasG12V+ cells with successful MADR. Scale bars: 100μm
C) As low as 10 ng/μl recombinase-expression vector in EP mixture can catalyze MADR in vivo. Scale bars: 100μm
D) Brighter EGPF-HrasG12V cells after pBase-mediated integration express phosphorylated Rb1. Scale bars: 200μm
E-I) Striatal gliogenesis 1 month after electroporation of pDonor- (E) Kras G12A, (F-G) YAP1-MAML1D, or (H) C11orf95-RELA.
J-K) High magnification of ependymoma pushing margins displaying a lack of infiltration of these tumors.
Supplementary Figure 5: Characterization of multi-cistronic tumors, secondary elements, and viability screens, Related to Figure 5
A) In vitro assessment of transgene expression after MADR in heterozygous mTmG mNSCs shows the co-expression of nuclear EGFP with Pdgfra, V5 (Trp53R270H), and P53. Note the presence of contaminating mG cells with membrane EGFP and no tdTomato or transgene expression. Scale bars: 50μm
B) Confirmation of Trp53 co-expression with nuclear EGFP (H3f3a). Scale bars: 50μm
C) Coronal section displaying pre-tumor phase of K27M-expressing lineages (nuclear EGFP; and mG EGFP – membrane EGFP).
D-G) Immunostaining of K27M (D,G) and G34R (E-F) tumors with anti-H3mutK27M and anti-H3mutG34R antibodies, confirming expression of the respective transgenes by specific immunolabeling with the appropriate antibodies.
H) Dual immunostaining of K27M and G34R in co-electroporated animals (K27M- and G34R-containing plasmids) confirms expression of only one H3f3a mutant variant per cell.
I) Rosa26H3f3aG34R/Pdgfra/Trp53 EGFP+ tumor cells are hypomethylated at H3K27.
J) High mag view of tumor margin.
K) Immunolabeling of K27M mutant tumor cells demonstrates perinuclear satellitosis and decreased H3K27Me labeling compared with neighboring neurons.
L) CRISPR/Cas9 targeting of Nf1/Trp53 to induce GBM does not yield reduced H3K27Me3.
M-O) Low mag (M-M2) and high mag images (N-O) of Bmi1 upregulation in K27M tumor cells. Arrows point to infiltrating cells and dotted line depicts tumor margin. P) Max projection of region in O-O1 showing that infiltrating K27M cells are juxtaposed to vessels.
P) A subset K27M and G34R mutant cells at the margins can be immunolabeling with the astrocyte marker Aldh1l1 and display hypertrophy
Q) Subpopulations of K27M and G34R mutant cells express the oligodendrocyte marker Cspg4.
Supplementary Figure 6: Single-cell RNA-seq of MADR mutant models, Related to Figure 6
A) CNV analysis of 3 mouse K27M tumor scRNA-seq datasets.
B) CC1 and CC2 vector alignment of mouse K27M tumors.
C) Biweight midcorrelation plot of mouse K27M tumors across CCs.
D) UMAP depicting CCA alignment of 3 K27M datasets from the mouse brain colored by sample.
E) Featureplots depicting expression of genes in mouse K27M tumors.
F) Louvain clustering of human K27M scRNA-seq tumors from Filbin et al. (Science 2018).
G) CSF1R and H) MOG expression maps depicting clusters that are filtered before moving to CCA.
I) Clustering of Human K27M tumors post filtering.
J) Biweight midcorrelation plot of human K27M tumors across CCs.
K) UMAP of CCA alignment human K27M tumors colored by sample.
L) Featureplots depicting expression of genes in human K27M tumors.
M) Program featureplots split from CCA of all samples (i.e. Fig. 6H) and depicted by original sample.
A) Clustering by highly-variable genes rather than programs from Filbin et al. (Science 2018) leads to slightly altered clustering depending on the clustering parameters chosen.
Supplementary Figure 7: SCENIC, H3K27me3 ChIP-seq, and snATAC-seq analysis of MADR mutant models, Related to Figure 7
A) General workflow for tumor dissociation and downstream analysis such as scRNA-seq, scATAC-seq, or ChIP-seq for H3K27Me3. Note same tumor source was used for scRNA-seq analysis and ChIP-seq but independent tumors were used for scATAC-seq samples.
B) Clustering of H3K27Me3 ChIP-seq Data from 3 mouse K27M tumors.
C) Scoring of scRNA-seq-derived UMAP for genes from clusters in B.
(D-I) t-Distributed Stochastic Neighbor Embeddings (t-SNEs) of SCENIC processed K27M human tumor cells (D,F,H) or SCENIC-derived t-SNEs of K27M mouse tumor cells (E,G,I). Samples are grouped by sample (D,E; i.e. patient or mouse of origin), cell type (F,G), and cell cycle phases (H,I).
J) tSNE of scATAC-seq dataset from P50 brain with numbered clusters.
K) Marker genes for oligodendrocyte (Mog), OPC (Pdgfra), astrocyte (Aqp4), microglia (C1qb), neuron (Snap25), and interneuron (Gad2, Pvalb, Sst) populations. Note the distinct signal to noise for each cluster.
L) tSNE of 3 combined scATAC-seq datasets from E18 brain after standard clustering.
M) tSNE of samples from L post-Harmony alignment.
N) tSNE of E18 datasets with clusters numbered.
O) Gene accessibility for Sox9 (astrocytes/stem cells), Olig2 (stem cells/oligodendrocyte lineage), Csf1r (microglia), and Gfap (astrocytes). Note the lack of clear population/cluster segregation for microglia or glial specificity of Sox9 and Olig2 compared with Gfap, which is more exclusive to discrete populations and readily associates with the glial clusters.
P) tSNE of scATAC-seq dataset derived from K27M tumor cells and co-capture innate immune populations
Q) Gene accessibility for Sox9, Olig2, Csf1r, and Gfap. Again, note the lack of clear population/cluster segregation of Sox9 and Olig2 compared with Gfap, which exhibits more accessibility. Csf1r is noticeably more accessible than Sox9 and Olig2 and is associated with innate immune clusters.
R) CisTopic and CellRanger-based clustering of K27M tumor populations leads to subtly different subclustering of tumor and immune populations.
S) Gene accessibility clearly defines microglial populations but Sox9 and Sox10 fail to co-segregate in tumor, unlike in P50 normal brain.
T) K27M scRNA-seq featureplots blending Sox10 (green) and Sox9 (red) demonstrate that Sox10 and Sox9 are highly expressed throughout the tumor clusters and even within individual cells in agreement with gene accessibility in S and genome browser Data (Fig. 7J).
U) mSigDB terms for P50 astrocyte and OPC clusters.
V) Motifs enriched in DARs from K27M tumor clusters. Note the enrichment of IEGs and ES-associated TFs.
Supplementary Table 2: Labels for differentially-expressed markers on Heatmaps, Related to Figure 6.
Supplementary Table 1: Details on the mice used for scRNA-seq, Related to Figure 6.
Supplementary Table 3: Comparison of methods for conditional/somatic transgenesis, Related to STAR METHODS
Supplementary Movie 1: Light sheet image of cleared left brain hemisphere 7 days post-EP, Related to Figure 2. Hemisphere was immunostained for the Myc epitope tag.
Supplementary Movie 2: 3D rotation of confocal z-stack showing the non-overlapping genetically defined populations (red – non-EPed, Green – Cre recombined, Magenta – TagBFP2 MADR insertion), Related to Figure 2..
Supplementary Movie 3: Light sheet image of cleared olfactory bulbs 2 weeks post-EP. The bulbs were immunostained for the Myc, V5, HA, and Flag epitope tag, Related to Figure 2.
Supplementary Figure 1: Measurement of MADR efficiency in heterozygous mTmG mNSCs by FACS analysis, confirmation of correct protein translation at non-clonal population level, inducible MADR, and MADR “proxy” lines, Related to Figure 1
A) Schematic of recombinase-expressing plasmids (and minicircle) employed in this study.
B) FACS analysis indicates the approximate MADR efficiency in neural stem cells, and no obvious difference between Flp-2A-Cre and Flp-IRES-Cre in their catalytic efficiencies.
C) Sorted cells express HrasG12V but not tdTomato or EGFP. Scale bar, 50μm
D) Western blot indicating normal transgene production from non-clonal aggregate cells and lack thereof in FACSnegative population. Removal of tdTomato expression is also observed.
E) Schematics of plasmids and alleles subject to PCR analysis at denoted sites.
F) PCR screening analysis reveals that rtTA-V10-AU1 cassette is correctly integrated downstream of CAG-promoter in cells that are resistant to puromycin treatment.
G) Western blot analysis of the cell line from Fig. 2C showing the expression of rtTA-V10-AU1 and also EGFP upon doxycycline induction.
H) MADR-compatible TRE-EGFP plasmid.
I) Heterozygous mTmG mNSCs are nucleofected with plasmid in H, treated with puromycin, and turned into a colorless population. Scale bars: 10μm
J) Induction of EGFP expression in the cell line that constitutively express rtTA-V10-AU1. Scale bars: 50μm
K) TRE cell line with a bidirectional tet-response element that expresses EGFP and Dll1 upon doxycycline treatment.
L) Immunofluorescence of cells without and with Dox demonstrates the relative lack of leakiness and homogenous expression level of EGFP and mDll1. Scale bars: 20μm
M) qPCR measurement of mRNA abundance before and after Dox addition to medium. (Ctrl plasmid lacks mDll1 CDS but is otherwise identical to plasmid in K.)
Data Availability Statement
The sequencing Data reported in this study has been deposited under GEO: Project GSE131675; Subseries - GSE117154, GSE131873, GSE131873, and GSE131940. P50 and E18 mouse brain scATAC-seq samples were downloaded from 10Xgenomics.com.