

Abstract
[Pyr1]apelin-13 is the predominant apelin peptide isoform in the human cardiovascular system and plasma. To date, few studies have investigated [Pyr1]apelin-13 metabolism in vivo in rats with no studies examining its stability in humans. We therefore aimed to develop an LC-MS/MS method for detection and quantification of intact [Pyr1]apelin-13 and have used this method to identify the metabolites generated in vivo in humans. [Pyr1]apelin-13 (135 nmol/min) was infused into six healthy human volunteers for 120 minutes and blood collected at time 0 and 120 minutes after infusion. Plasma was extracted in the presence of guanidine hydrochloride and analysed by LC-MS/MS. Here we report a highly sensitive, robust and reproducible method for quantification of intact [Pyr1]apelin-13 and its metabolites in human plasma. Using this method, we showed that the circulating concentration of intact peptide was 58.3 ± 10.5 ng/ml after 120 minutes infusion. We demonstrated for the first time that in humans, [Pyr1]apelin-13 was cleaved from both termini but the C-terminal was more susceptible to cleavage. Consequently, of the metabolites identified, [Pyr1]apelin-13(1–12), [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6) were the most abundant. These data suggest that apelin peptides designed for use as cardiovascular therapeutics, should include modifications that minimise C-terminal cleavage.
Subject terms: Mass spectrometry, Translational research
Introduction
Apelin is an endogenous ligand of the apelin receptor, initially characterised from bovine stomach extracts as a 77-amino acid preproprotein1. The prepro-apelin is further cleaved into shorter but functional fragments including apelin-36, apelin-17, apelin-13 and [Pyr1]apelin-13 that contain an evolutionary conserved 12-amino acid C-terminal1–3. [Pyr1]apelin-13 was subsequently identified as the most predominant isoform of the apelin family of peptides in the cardiovascular system4,5, and the major circulating form of the peptide6.
In the cardiovascular system, apelin is the most potent endogenous inotropic agent yet identified4. Apelin modulates vascular tone in vivo, decreasing blood pressure when infused into rats and dilating resistance vessels when infused into human forearm7–9. In vitro, apelin causes nitric oxide-dependent vasodilation of human splanchnic artery10, although a nitric oxide-independent, prostanoid dependent vasodilation in humans has been reported4. Apelin acted as a vasoconstrictor in endothelium denuded vessels via a direct action on vascular smooth muscle cells whilst also acting as a potent angiogenic factor and mitogen of endothelial cells4,11. Based on these beneficial effects, apelin was proposed as a potential therapeutic target in cardiovascular diseases. For example, apelin administration showed cardioprotective effects in heart failure12, and ameliorated the development of pulmonary arterial hypertension in rats13,14 and humans15. In addition, the protective effects of apelin has been reported in metabolic diseases where it decreased adiposity, serum insulin and increased insulin sensitivity16,17; and in renal diseases where it decreased acute renal injury and fibrosis18. It has recently emerged that apelin has pro-tumorigenic effects in various cancer models possibly by promoting angiogenesis and that inhibition of the apelin pathway was protective against tumour growth14,19. However, these beneficial effects of apelin peptides are limited by the rapid in vivo metabolism.
Previous studies investigating the metabolism of apelin peptides were largely conducted in plasma in vitro or in rodent models neither of which may represent metabolism in humans. These studies demonstrated that apelin peptides are very labile in plasma with a half-life of less than 1–5 minutes in vitro20–24. This plasma instability has to date been attributed the enzymatic activity of neprilysin25 and angiotensin converting enzyme II (ACE2)22–24, and more recently plasma kallikrein26,27. Similarly, another recent study reported more rapid degradation of [Pyr1]apelin-13 in rat and mouse plasma when compared to dog, monkey and human plasma in vitro28. The authors also confirmed their findings in vivo in rat and mouse, and identified N-terminal metabolites of the peptide, particularly apelin-7 that was most abundant28. This study therefore highlighted species differences in the repertoire of proteases circulating and present in rodent and higher mammalian systems. However, to date no studies have investigated the metabolism of apelin peptides in vivo in humans. The aim of this study was to develop a highly sensitive mass spectrometry based method for detection and quantification of apelin peptides in plasma. We used this method to measure intact [Pyr1]apelin-13 and its metabolites generated in humans, following a constant 120 minutes infusion of the peptide. We found that [Pyr1]apelin-13 was cleaved into smaller fragments from both termini but that the C-terminal was more susceptible. We identified the biologically active C-terminal cleavage product, [Pyr1]apelin-13(1–12), as the most abundant, as well as identifying novel metabolites including [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6).
Results
Precision and accuracy of the extraction and quantification method
An 8-point calibration line was generated for [Pyr1]apelin-13 in human plasma (r2 = 0.99, data not shown), with a lower limit of quantification (LLOQ) of 1 ng/ml. The relative errors (% RE) for all calibration standards were less than 20% at the LLOQ and below 15% at other levels, conforming with typical bioanalytical method validation guidelines29. The precision and accuracy of the QC samples showed that the method was robust and accurate. The LLOQ samples returned a coefficient of variation (%CV) of 8.0 and %RE of 15.5, whilst the other QC levels had %CV’s below 6.1 and %RE’s below 8.4. Representative chromatograms obtained from calibration standards 1 and 8 are shown in Fig. 1.
Figure 1.

Representative chromatogram of calibration standards. LLOQ standard shows peaks corresponding to [Pyr1]apelin-13 at 1 ng/ml (A) and [Pyr1]apelin-13 internal standard at 25 ng/ml (B). Upper limit of quantification shows peaks corresponding to [Pyr1]apelin-13 (C) and [Pyr1]apelin-13 internal standard at 25 ng/ml (D). MRM = multiple reaction monitoring.
Plasma concentrations of [Pyr1]apelin-13 in healthy human volunteer samples
In samples obtained before infusion of [Pyr1]apelin-13, no chromatographic peak was observed for the peptide (Fig. 2A,B). Samples obtained at the end of the infusion (t = 120 minutes) showed strong peaks at 3.68 minutes corresponding to [Pyr1]apelin-13 (Fig. 2C,D). The measured concentration of [Pyr1]apelin-13 in these samples after 120 minutes was 58.3 ± 10.5 ng/ml. Additionally, data from the six donor control samples that did not receive [Pyr1]apelin infusion showed that the endogenous levels of [Pyr1]apelin in these samples were below the LLOQ (see Supplementary Fig. 1). The peak height obtained from the chromatogram of these donor samples had a maximum height that was 19.8% of that seen in the LLOQ and so was considered as blank for quantitative purposes based on the FDA method validation guidelines for demonstrating selectivity of an LC-MS methodology30.
Figure 2.

Representative chromatogram for [Pyr1]apelin-13 and its internal standard in volunteer samples. (A,B) chromatograms for samples obtained at t = 0 minutes; (C,D) chromatograms for samples obtained at t = 120 minutes. (A) no [Pyr1]apelin-13 was detected; (B) [Pyr1]apelin-13 internal standard chromatogram showing 3.67 minutes retention time; (C) [Pyr1]apelin-13 chromatogram showing 3.68 minutes retention time; (D) [Pyr1]apelin-13 internal standard. MRM = multiple reaction monitoring.
Identification of potential C-terminal metabolites of [Pyr1]apelin-13
In order to identify potential metabolites of [Pyr1]apelin-13 generated during the 120 minutes infusion period, samples were re-analysed using a high resolution mass spectrometer. Full scan LC-MS data were interrogated for potential [Pyr1]apelin-13 derived metabolites by comparing extracted ion chromatograms for each analyte in the 0 and 120 minute samples in the Qualbrowser software package (Thermofisher). Peptides that were identified in the 120 minute samples were mainly generated by the loss of C-terminal amino acids (Fig. 3A). Their relative abundance in the samples are displayed in Fig. 3B. Notably, the most abundant fragments were [Pyr1]apelin-13(1–12) (known to be biologically active24), [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6). Other metabolites identified, although at lower levels (<10% of parent [Pyr1]apelin-13) included [Pyr1]apelin-13(1–8), [Pyr1]apelin-13(1–7) and [Pyr1]apelin-13(1–5) that are likely to be biologically inactive. The chromatographic spectra corresponding to each of these metabolites are shown in Fig. 4A–G. In addition to [Pyr1]apelin-13(1–12), the most abundant metabolite identified, [Pyr1]apelin-13(1–10) (Fig. 5) and [Pyr1]apelin-13(1–6) (Fig. 6) were present at a sufficient level to generate suitable product ion spectra allowing experimentally acquired fragments to be matched against theoretical fragments from the peptide sequence. The relative mass accuracy of all potential metabolites were generated and the experimental values were all within 1 ppm of expected values, whilst the mass accuracy of the parent peptide had the highest value of 1.3 ppm.
Figure 3.
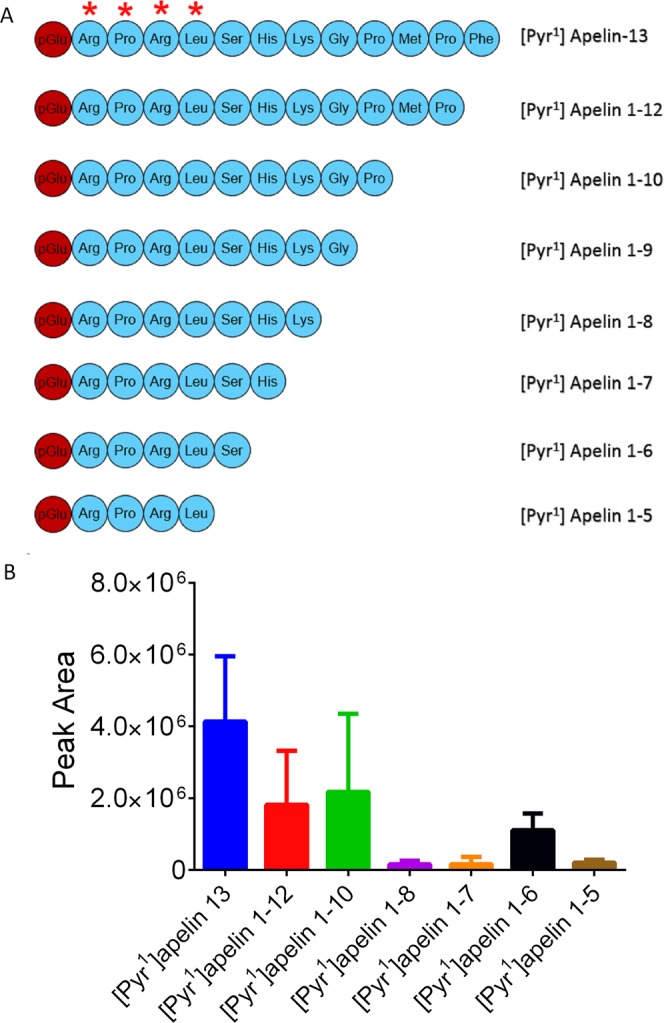
Relative abundance of [Pyr1]apelin-13 C-terminal metabolites identified from human plasma identified by LC-MS/MS. (A) peptide sequences of [Pyr1]apelin-13 metabolites identified in human plasma (the RPRL motif required for receptor binding was indicated by (*); (B) relative peak area of the metabolites (n = 6, data represent mean ± SD).
Figure 4.

Representative chromatogram of [Pyr1]apelin-13 and its C-terminal metabolites identified in human plasma. The extracted ion chromatogram for [Pyr1]apelin-13 shows the m/z for the first 13C ion, as the extracted chromatogram for the monoisotopic peak had significant background noise throughout. (A) [Pyr1]apelin-13 with 8.80 minutes retention time; (B) [Pyr1]apelin-13(1–12) with 6.26 minutes retention time; (C) [Pyr1]apelin-13(1–10) with 4.29 minutes retention time; (D) [Pyr1]apelin-13(1–8) with 3.25 minutes retention time; (E) [Pyr1]apelin-13(1–7) with 4.22 minutes retention time; (F) [Pyr1]apelin-13(1–6) with 5.09 retention time; (G) [Pyr1]apelin-13(1–5) with 5.88 minutes retention time. These data were acquired using Orbitrap Mass spectrometer used for metabolite identification. The mass accuracy of the experimentally acquired monoisotopic peak was calculated for each potential metabolite, and is included along with the 13C isotopic cluster for each peptide with their corresponding chromatogram.
Figure 5.

Product ion mass spectrum of [Pyr1]apelin-13(1–10). The major ions identified are shown in brackets.
Figure 6.

Product ion mass spectrum of [Pyr1]apelin-13(1–6). The major ions identified are shown in brackets.
Oxidation of the methionine residue in [Pyr1]apelin-13 was identified, however since this modification was also observed in the extracted standards, it could not be ascertained if they occurred in vivo or as an artefact of the extraction process.
Identification of potential N-terminal metabolites of [Pyr1]apelin-13
Using the same approach described above, several N-terminal metabolites of [Pyr1]apelin-13 were identified (Fig. 7). Of note, the peak areas of these fragments were lower compared to those observed for the C-terminal fragments. The most abundant N-terminal fragments observed were [Pyr1]apelin-13(6–13), [Pyr1]apelin-13(11–13), [Pyr1]apelin-13(7–13) and [Pyr1]apelin-13(10–13) (Fig. 7A,B). Other fragments present but low in abundance include [Pyr1]apelin-13(3–13), [Pyr1]apelin-13(4–13) and [Pyr1]apelin-13(8–13). The mass accuracy of the experimentally acquired monoisotopic m/z for these metabolites are shown in Fig. 8, and were all within 0.9 ppm of expected values.
Figure 7.

N-terminal metabolites of [Pyr1]apelin-13 identified from human plasma. (A) sequence of [Pyr1]apelin-13 N-terminal fragments identified, (B) relative abundance of N-terminal metabolites (n = 6, data represent mean ± SD). Star on the [Pyr1]apelin-13 residues indicate amino acid residues critical for receptor binding.
Figure 8.

Representative chromatogram of the N-terminal metabolites of [Pyr1]apelin-13 identified in human plasma. (A) [Pyr1]apelin-13(6–13) with retention time of 8.01 minutes, (B) [Pyr1]apelin-13(7–13) with retention time of 8.08 minutes, (C) [Pyr1]apelin-13(8–13) with retention time of 9.35 minutes, (D) [Pyr1]apelin-13(10–13) with retention time of 10.45 minutes, (E) [Pyr1]apelin-13(11–13) with retention time of 9.32 minutes. The mass accuracy of the experimentally acquired monoisotopic peak was calculated for each potential metabolite, and is included along with the 13C isotopic cluster for each peptide with their corresponding chromatogram.
Discussion
We have developed and validated a high resolution LC-MS/MS method for detection and quantification of [Pyr1]apelin-13 and relative quantification of its metabolites in vivo in human plasma. We have shown that this method was robust, reproducible and had a high sensitivity for [Pyr1]apelin-13 with an LLOQ of 1 ng/ml. Using this method, we have quantified the intact peptide after constant infusion for 120 minutes into healthy volunteers and showed for the first time that in humans in vivo, [Pyr1]apelin-13 was cleaved from both the N- and C-termini, with the C-terminus being the most susceptible to proteolytic activity. The most abundant metabolite identified by this method was [Pyr1]apelin-13(1–12) but very high levels of [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6) were also detected, the sequences of which were confirmed using tandem mass spectrometry and manual product ion matching.
The discovery that [Pyr1]apelin-13 was cleaved from both ends was unexpected since to date only cleavage from the C-terminus has been described23,24. Our findings may therefore better explain the extremely unstable nature of apelin peptides in plasma6,28,31. It is worth noting that the C-terminus was more susceptible to proteolytic activity than the N-terminus, whose metabolites were present at approximately 20-fold lower levels. This may partly be explained by the pyroglutamylation of the N-terminus which may protect this region from enzymatic activity to some degree. The N-terminus of [Pyr1]apelin-13 also contains the RPRL motif critical for binding to the apelin receptor32, hence any cleavage from this direction is likely to profoundly affect the affinity of these N-terminal fragments for the receptor. A previous study showed in vitro that neprilysin cleaves [Pyr1]apelin-13 between Arg4 and Leu5 and between Leu5 and Ser6 amino acids25, thereby making neprilysin the first enzyme identified to date that completely inactivates the peptide. Importantly, we have now shown in this study the presence of one of these proposed neprilysin cleavage products, [Pyr1]apelin-13(6–13), in humans in vivo with additional evidence for cleavage of the scissile bond between Leu5 and Ser6 given by the detection of the C-terminal fragment, [Pyr1]apelin-13(1–5). To date very few studies have investigated the metabolism of peptides in vivo in humans. Interestingly, like [Pyr1]apelin-13, arginine vasopressin was also proposed to be cleaved in vivo from both the C- and N- termini, with carboxypeptidase and post-proline enzymes cleaving the C-terminus of arginine vasopressin, while aminopeptidases cleaved the N-terminal region33–35. In contrast, other in vivo studies of this nature identified only a single terminus cleavage of Peptide YY36, growth hormone-releasing hormone37, liraglutide, a glucagon-like peptide-1 (GLP-1) analogue38 and big endothelin-139.
Previous in vitro studies in plasma, suggested that [Pyr1]apelin-13(1–12) was a metabolite of [Pyr1]apelin-13 produced by the enzymatic activity of ACE2 resulting in the removal of the C-terminal phenylalanine20,22–24,40. However, it was unclear whether this metabolite retained biological activity at the apelin receptor. One study argued that [Pyr1]apelin-13(1–12) had reduced biological activity compared to the native [Pyr1]apelin-13 as measured by its hypotensive effects in mice23. However, Yang et al.24 demonstrated that the ACE2 metabolite, [Pyr1]apelin-13(1–12) contracted human saphenous vein with sub-nanomolar potency and was a potent positive inotrope in paced mouse and human heart ex vivo. The authors demonstrated [Pyr1]apelin-13(1–12) was present endogenously in the endothelium of human heart and lungs, and went on to show that it was biologically active in vivo in humans and rodents24. Similarly, a previous study reported that [Pyr1]apelin-13 was cleaved to [Pyr1]apelin-13(1–12) in vivo in rats22. Consistent with these studies, our work now provides clear evidence that [Pyr1]apelin-13(1–12) is produced endogenously in human plasma in vivo possibly via the activity of ACE2.
[Pyr1]apelin-13 was also cleaved between Pro10 and Met11 and between Ser6 and His7 resulting in generation of [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6) but the enzyme responsible for producing these metabolites remains unknown. The corresponding N-terminal fragments of these C-terminal metabolites [Pyr1]apelin-13(11–13) and [Pyr1]apelin-13(7–13), were also identified. These data were consistent with a previous in vivo study in male rats which also identified the C-terminal metabolites22. The authors reported on the accumulation of [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6) signals with time as [Pyr1]apelin-13 and [Pyr1]apelin-13(1–12) signals decreased, suggesting that following ACE2 cleavage of [Pyr1]apelin-13 to [Pyr1]apelin-13(1–12), other unidentified enzymes subsequently cleave both [Pyr1]apelin-13 and [Pyr1]apelin-12 into [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6). These metabolites [Pyr1]apelin-13(1–10) and [Pyr1]apelin-13(1–6), retained the RPRL motif required for binding32, although it is unclear if they retain biological activity. Taken together, these findings may suggest that there is at least some common metabolic pathways for [Pyr1]apelin-13 in rats and humans in vivo. Further studies are required to identify the specific proteases involved.
Inhibition of degradative enzymes is a well-established strategy to generate therapeutic agents. ACE2 is an important member of the renin-angiotensin system that converts angiotensin-II to angiotensin 1–7, with the latter mediating vasodilatation, anti-proliferation, anti-apoptosis and anti-fibrotic effects41. In addition, ACE2 has been implicated in heart failure42,43, diabetic nephropathy44,45, acute lung failure46, lung injury induced by the lethal avian influenza A H5N1 virus47, respiratory syncytial virus48 and severe acute respiratory syndrome (SARS)46. Recently, GSK developed a recombinant human ACE2, GSK2586881 for treatment of acute respiratory distress syndrome (ARDS) and showed that this molecule was well-tolerated in clinical trials49. Corroborating on this, apelin signalling induces ACE2 expression in failing hearts12, and protects against lung injury in experimental models of acute respiratory distress syndrome50, possibly by inhibiting the NF-κB pathway and components of the inflammasome51. Furthermore, apelin counteracts the effects of angiotensin-II signalling, which is negatively regulated by ACE2, suggesting that targeting ACE2 and apelin could be a potentially novel therapeutic strategy for treatment of lung injury related pathologies and heart failure.
The beneficial effects of apelin in heart failure are well characterised. Plasma apelin levels have been suggested to increase in early stages5 of heart failure but decrease in late stages of the disease52–54. In support of this, heart failure therapies such has cardiac resynchronisation therapy used to treat refractory chronic heart failure were shown to increase plasma apelin suggesting that increased apelin levels are associated with improved therapeutic benefit54. Apelin administration increased stroke volume and contractility in failing hearts11, thereby improving the performance of the failing heart. Similarly, neprilysin inhibitors have emerged as a pivotal therapeutic strategy for clinical management of heart failure due to the role of neprilysin in the degradation of vasoactive peptides including natriuretic peptides and bradykinin55. Indeed, neprilysin inhibitors such as sacubitril are used for clinical management of heart failure56. Our data may therefore suggest that an additional benefit of neprilysin inhibitors in heart failure is to reduce apelin inactivation resulting in beneficial vasodilation, increased contractility and cardiac output. Building on these findings, further studies could substitute the amino acids at the neprilysin cleavage sites in [Pyr1]apelin-13 with unnatural amino acids to improve its resistance to degradation. Indeed, it was recently shown that infusion of neprilysin resistant apelin-17 in an established mice model of abdominal aortic aneurysm ameliorated the adverse aortic remodelling and aneurysm formation27. Such a strategy was also demonstrated to significantly increase the resistance of [Pyr1]apelin-13 and apelin-17 to ACE2 activity22,23, suggesting that this could potentially be a mechanism to improve plasma stability of apelin-based therapeutics for clinical indications. We have recently published on an another approach to stabilise apelin peptides in human blood using albumin domain (AlbudAb) antibody conjugated to an apelin analogue, MM202 and showed that this peptide was resistant to degradation yet retained biological activity at the human apelin receptor in vitro and in vivo9. Therefore, these strategies could in the near future result in the development of the first apelin-based therapeutics for treatment of human diseases.
In conclusion, apelin peptides have protective roles in cardiovascular diseases, however, any potential therapeutic use is impaired by the poor plasma stability of the peptide. In this study, we have developed a highly sensitive method for detection and quantification of [Pyr1]apelin-13 in human plasma. For the first time in humans in vivo we have identified as the most abundant metabolite of [Pyr1]apelin-13, the ACE2 cleavage product, [Pyr1]apelin-13(1–12) that we have previously demonstrated retains significant biological activity in addition to the putative neprilysin metabolites [Pyr1]apelin-13(4–13) and [Pyr1]apelin-13(6–13). Combined inhibition of ACE2 and neprilysin may be a novel strategy to enhance endogenous apelin levels in conditions in which the peptide is downregulated. Additionally, these data will inform the design of more stable apelin peptides for therapeutic use.
Material and Method
Materials
[Pyr1]apelin-13 was custom synthesised by Severn Biotech (Kidderminster, England), and analysed by mass spectrometry and purity by HPLC analysis (99.2%) dispensed under sterile conditions. Pharmacological activity of [Pyr1]apelin-13 was confirmed using in vitro and in vivo assays (Supplementary Figs. 2 and 3). Peptides were stored below −40 °C in a monitored freezer until use2. Stable isotope labelled [Pyr1]apelin-13 (pGlu-R-[U-13C5, 15N-Pro]-R-[U-13C6, 15N-Leu]-SHKGPMPF-acid) was custom synthesised by Cambridge Research Biochemicals (Billingham, England). Protein LoBind Eppendorf tubes (Cat No.: 0030108094) and 1 ml protein LoBind 96-well plate (Cat No.: 0030504216) were purchased from Eppendorf (Stevenage, UK), and Oasis HLB Prime µ-Elution 96-well plates were purchased from Waters (Waters, Wilmslow, UK; Cat No.: 186008052). Acetonitrile (ACN) (Cat. No.: 270717) and glacial acetic acid (Cat. No.: 33209-1L) were purchased from Sigma Aldrich (Saint Louis, USA), methanol (Cat. No.: 10675112) and 0.1% formic acid (FA) in water (%v/v) (Cat. No.: LS118-212) were purchased from Fisher Scientific (New Hampshire, USA). ACQUITY UPLC HSS T3 1.8 μm 2.1 × 50 mm column (Cat No.: 186003538) used for the LC-MS/MS analysis was obtained from Waters (Wilmslow, UK).
Study protocol
This study was registered on Clinicaltrials.gov (NCT03449251) and carried out with ethical approval from the Yorkshire & The Humber – Sheffield Research Ethics Committee (REC reference 18/YH/0010). All participants gave written informed consent and studies adhered to the Declaration of Helsinki. Six healthy volunteers (3 male and 3 female, mean age 43.8 ± 6.9, with body mass index within the normal range of 23.0 ± 1.0) were recruited for infusion. Volunteers were fasted and were lying supine with their heads supported in a quiet, temperature controlled (23–25 °C) room for the duration of the study. Following a period of acclimatisation, the first sample of venous blood was obtained from the arm contralateral to the arm used for infusion of apelin. Vials containing [Pyr1]apelin-13 were allowed to warm to room temperature and diluted with physiological saline to produce stock solutions, that were then filtered using a 0.2 µm Portex flat filter (Portex, UK) before undergoing serial dilutions with 0.9% sterile saline. There was no loss of apelin following this filtration procedure. Volunteers were infused with a concentration of 135 nmol/min of [Pyr1]apelin-13, at a rate of 1 ml/min for 120 minutes, using a syringe pump, equipped with a 50 ml syringe and 16 gauge catheter. The second venous sample was obtained immediately after the end of the infusion. Blood samples were collected into 2.6 ml EDTA tubes, immediately put on wet ice and centrifuged for 7 minutes at ~4 °C, 4000 rpm and stored a −70 °C, prior to analysis. A previous study had used a concentration of up to 100 nmol/min for systemic infusion, where they obtained a therapeutic response in patients with pulmonary arterial hypertension and the highest dose was well tolerated15. The dose chosen of 135 nmol/min of [Pyr1]apelin-13, was slightly higher in order to identify possible metabolites. Additional control samples were obtained from 6 donors (3 male and 3 female) within a similar age group who did not receive the apelin infusion for comparison.
[Pyr1]apelin-13 LC-MS/MS and SRM based detection method development
An LC-MS/MS method was developed for [Pyr1]apelin-13 and its stable isotope labelled [Pyr1]apelin-13 analogue. LC-MS/MS instrumentation used for the quantitation of [Pyr1]apelin-13 included a H-Class Acquity (Waters) attached to a TQ-XS triple quadrupole mass spectrometer (Waters). Peptides were separated using a 2.1 × 50 mm 1.8 mm particle HSS T3 Acquity column held at 60 °C and flowing at 350 µl/minute. Gradient starting conditions were 95% A (0.1% FA in water v/v) and 5% B (0.1% FA in ACN). Starting conditions were held for 0.2 minutes before raising to 25% B over 4 minutes. The column was flushed with 90% B for 0.8 minutes before returning to starting conditions. The total time of each analysis was 7 minutes, with the first 1.2 minutes and last 2.8 minutes diverted to waste. The source parameters used included a positive electrospray voltage of 3.0 kV, gas flow of 1000 L/hour, desolvation temperature of 600 °C and a cone voltage of 40 V.
A full scan analysis of the peptide showed that the [M + 4 H]4+ charge state was the predominant ion in the spectrum as previously described by Mesmin et al.31 in their LC-MS/MS analysis of [Pyr1]apelin-13. Therefore this was selected for fragmentation. A product ion spectrum was collected over a range of 100 to 1600 m/z and two ions were selected for SRM optimisation (m/z 424.6 and 408.55). The 424.6 ion corresponded to the b11 fragment and the 408.55 ion was derived from the loss of a methyl-sulphide group from the methionine on the b11 ion, as previously described by Mesmin et al.31. Optimal conditions for the two SRM transitions for [Pyr1]apelin-13 were 384.2/408.55, 384.2/424.6 with collision energy values of 14 and 12 eV respectively. The internal standard used the same collision energy but targeted transitions of 387.45/412.88 and 387.45/428.26. Peptide peak areas were integrated using the TargetLynx program associated with Masslynx V 4.2 (Waters), and peak area ratios were generated against the corresponding stable isotope-labelled internal standard peptide peak.
Extraction of [Pyr1]apelin-13 from human plasma
Plasma samples were thawed on ice and 50 µl transferred into protein LoBind Eppendorf tubes containing 25 µl GuHCl (6 M). A 300 µl aliquot of 80% ACN in water (containing 25 ng/ml internal standard) was added to all plasma samples and vortexed before centrifuging at 12000 x g for 5 minutes to precipitate plasma proteins. The supernatant was transferred to a 1 ml protein LoBind 96-well plate and evaporated. Samples were reconstituted in 500 μl 0.1% FA (v/v) and loaded onto an Oasis HLB Prime µ-elution 96-well plate (Waters, Wilmslow, UK) and slowly extracted on a positive pressure manifold (Waters). The columns were washed with 200 µl of 5% methanol in water with 1% acetic acid (v/v) and eluted from the cartridge using 2 × 50 µl of 60% methanol in water with 10% acetic acid (v/v). The eluate was evaporated to dryness and reconstituted in 150 µl 0.1% FA (v/v) in water and 10 µl was injected onto the LC-MS/MS system.
Precision and accuracy of the extraction method
Blank plasma was pre-incubated at 37 °C for at least 2 hours, to degrade any endogenous [Pyr1]apelin-13 and used to generate an eight point calibration line of custom synthesised [Pyr1]apelin-13 over a range of 1–1000 ng/ml. A 50 µl aliquot of each calibration standard (1, 2, 5, 10, 50, 100, 900, and 1000 ng/ml) was extracted using the SPE method described above. Four levels of QC were also generated (1, 3, 100 and 800 ng/ml) and extracted six times in order to assess the precision and accuracy of the method. Calibration line followed a linear fit, and 1/x2 weighting was applied. Recovery of the [Pyr1]apelin-13 from plasma was assessed by analysing spiked solution before and after extraction at a concentration of 100 ng/ml. Plasma samples from six individuals were also extracted to assess the selectivity of the LC-MS/MS method.
Peptide Identification using high-resolution mass spectrometry
Samples were reanalysed on a high resolution mass spectrometer to identify potential metabolites from the administered [Pyr1]apelin-13 peptide. A full scan analysis was performed using a ThermoScientific Ultimate 3000 LC system connected to a ThermoScientific Orbitrap Q-Exactive Plus mass spectrometer. Solvents used for the separation were A: 0.1% FA in water (v/v) and B: 0.1% FA in ACN (v/v). A volume of 30 μl of extract was injected onto a HSS T3 UPLC™column (2.1 × 50 mm; Waters, Elstree, UK) held at 60 °C and with a flow rate of 300 µL/min. A starting condition of 1% B was used to capture the more hydrophilic peptide metabolites, and these were eluted using a linear gradient up to 30% B over 16 minutes. The column was washed for 2 minutes at 90% B and returned to starting conditions for 2 minutes, totalling a run time of 20 minutes. Mass spectrometry was performed using positive electrospray mode with a needle voltage of 3 kV, gas settings of 55 and 10 for sheath gas and aux gas flow rates. The temperature of the gas was set at 350 °C and the transfer capillary at 350 °C and a s‐lens value of 70 V. Full scan data were acquired over an m/z range of 250–1000, using a resolution of 70,000 and a maximum fill time of 100 ms. Acquired LC-MS data were interrogated for potential [Pyr1]apelin-13 metabolites by searching for all potential cleavage products from the parent peptide in the RAW data files using the Qualbrowser software package (Thermofisher). The m/z values for these peptides at multiple charge states are displayed in Supplementary Table 1. The potential [Pyr1]apelin-13 metabolites that were manually identified were subsequently characterised, where 30 μl of sample was reinjected using a targeted MS/MS analysis. The potential [Pyr1]apelin-13(1–6) and [Pyr1]apelin-13(1–10) peptides were targeted using precursor ion m/z values of 370.214 (collision energy of 30) and 290.417 (collision energy of 22) respectively. The MS/MS analysis involved the same LC separation, but MS/MS data were acquired at 17,500 resolution with an AGC of 1e6 ions, lowest m/z value of 100 and a max fill time of 200 ms.
Supplementary information
Supplementary information for Development and validation of an LC-MS/MS method for detection and quantification of in vivo derived metabolites of [Pyr1]apelin-13 in humans
Acknowledgements
We thank the following for support: Wellcome Trust [WT107715/Z/15/Z to APD, JJM] and Programme in Metabolic and Cardiovascular Disease, Astra-Zeneca, Cambridge Biomedical Research Centre Biomedical Resources Grant, University of Cambridge, Cardiovascular Theme, RG64226. Research in the laboratory of FG and FR was supported by the MRC [MRC_MC_UU_12012/3], Wellcome Trust [106262/Z/14/Z, 106263/Z/14/Z]. The LC-MS/MS analysis was supported by the MRC “Enhancing UK clinical research” grant (MR/M009041/1).
Author contributions
D.N., R.G.K. designed and performed laboratory experiments, analysed data, wrote manuscript, R.E.K. performed laboratory experiments, P.S. and J.C. performed the clinical study, P.A., L.J., F.R., provided resources and funding, F.M.G. provided resources, funding and comments on manuscript, J.J.M., A.P.D. provided experimental design, data analysis, wrote manuscript, supervision of project and funding.
Data availability
All data generated or analysed during this study are included in this published article.
Competing interests
P.A. and L.S. are employees of AstraZeneca (UK) Plc. APD holds a research grant from Astra Zeneca.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-56157-9.
References
- 1.Tatemoto K, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 2.De Mota N, Lenkei Z, Llorens-Cortès C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology. 2000;72:400–407. doi: 10.1159/000054609. [DOI] [PubMed] [Google Scholar]
- 3.Habata Y, et al. Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim. Biophys. Acta - Mol. Cell Res. 1999;1452:25–35. doi: 10.1016/S0167-4889(99)00114-7. [DOI] [PubMed] [Google Scholar]
- 4.Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: Vasoactive mechanisms and inotropic action in disease. Hypertension. 2009;54:598–604. doi: 10.1161/HYPERTENSIONAHA.109.134619. [DOI] [PubMed] [Google Scholar]
- 5.Chen MM, et al. Novel Role for the Potent Endogenous Inotrope Apelin in Human Cardiac Dysfunction. Circulation. 2003;108:1432–1439. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- 6.Zhen EY, Higgs RE, Gutierrez JA. Pyroglutamyl apelin-13 identified as the major apelin isoform in human plasma. Anal. Biochem. 2013;442:1–9. doi: 10.1016/j.ab.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 7.El Messari S, et al. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J. Neurochem. 2004;90:1290–1301. doi: 10.1111/j.1471-4159.2004.02591.x. [DOI] [PubMed] [Google Scholar]
- 8.Japp AG, et al. Vascular Effects of Apelin In Vivo in Man. J. Am. Coll. Cardiol. 2008;52:908–913. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Read, C. et al. Apelin peptides linked to anti-serum albumin domain antibodies retain affinity in vitro and are efficacious receptor agonists in vivo. Basic Clin. Pharmacol. Toxicol., 10.1111/bcpt.13227 (2019). [DOI] [PubMed]
- 10.Salcedo A, et al. Apelin effects in human splanchnic arteries. Role of nitric oxide and prostanoids. Regul. Pept. 2007;144:50–55. doi: 10.1016/j.regpep.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Pitkin SL, Maguire JJ, Bonner TI, Davenport AP. International Union of Basic and Clinical Pharmacology. LXXIV. Apelin receptor nomenclature, distribution, pharmacology, and function. Pharmacol. Rev. 2010;62:331–342. doi: 10.1124/pr.110.002949. [DOI] [PubMed] [Google Scholar]
- 12.Sato T, et al. Apelin is a positive regulator of ACE2 in failing hearts. J. Clin. Invest. 2013;123:5203–5211. doi: 10.1172/JCI69608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang P, et al. A novel cyclic biased agonist of the apelin receptor, MM07, is disease modifying in the rat monocrotaline model of pulmonary arterial hypertension. Br. J. Pharmacol. 2019;176:1206–1221. doi: 10.1111/bph.14603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Read C, et al. International Union of Basic and Clinical Pharmacology. CVII. Structure and Pharmacology of the Apelin Receptor with a Recommendation that Elabela/Toddler Is a Second Endogenous Peptide Ligand. Pharmacol. Rev. 2019;71:467–502. doi: 10.1124/pr.119.017533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brash L, et al. Short-Term Hemodynamic Effects of Apelin in Patients With Pulmonary Arterial Hypertension. JACC Basic to Transl. Sci. 2018;3:176–186. doi: 10.1016/j.jacbts.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nyimanu D, et al. Apelin-36-[L28A] and Apelin-36-[L28C(30kDa-PEG)] peptides that improve diet induced obesity are G protein biased ligands at the apelin receptor. Peptides. 2019;121:170139. doi: 10.1016/j.peptides.2019.170139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galon-Tilleman H, et al. Apelin-36 Modulates Blood Glucose and Body Weight Independently of Canonical APJ Receptor Signaling. J. Biol. Chem. 2017;292:1925–1933. doi: 10.1074/jbc.M116.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsault Eric, Llorens‐Cortes Catherine, Iturrioz Xavier, Chun Hyung J., Lesur Olivier, Oudit Gavin Y., Auger‐Messier Mannix. The apelinergic system: a perspective on challenges and opportunities in cardiovascular and metabolic disorders. Annals of the New York Academy of Sciences. 2019;1455(1):12–33. doi: 10.1111/nyas.14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harford-Wright E, et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain. 2017;140:2939–2954. doi: 10.1093/brain/awx253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vickers C, et al. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 21.Murza Alexandre, Sainsily Xavier, Coquerel David, Côté Jérôme, Marx Patricia, Besserer-Offroy Élie, Longpré Jean-Michel, Lainé Jean, Reversade Bruno, Salvail Dany, Leduc Richard, Dumaine Robert, Lesur Olivier, Auger-Messier Mannix, Sarret Philippe, Marsault Éric. Discovery and Structure–Activity Relationship of a Bioactive Fragment of ELABELA that Modulates Vascular and Cardiac Functions. Journal of Medicinal Chemistry. 2016;59(7):2962–2972. doi: 10.1021/acs.jmedchem.5b01549. [DOI] [PubMed] [Google Scholar]
- 22.Murza A, Belleville K, Longpré J-M, Sarret P, Marsault É. Stability and degradation patterns of chemically modified analogs of apelin-13 in plasma and cerebrospinal fluid. Biopolymers. 2014;102:297–303. doi: 10.1002/bip.22498. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, et al. Angiotensin-Converting Enzyme 2 Metabolizes and Partially Inactivates Pyr-Apelin-13 and Apelin-17: Physiological Effects in the Cardiovascular System. Hypertension. 2016;68:365–377. doi: 10.1161/HYPERTENSIONAHA.115.06892. [DOI] [PubMed] [Google Scholar]
- 24.Yang P, et al. [Pyr1]Apelin-13(1–12) Is a Biologically Active ACE2 Metabolite of the Endogenous Cardiovascular Peptide [Pyr1]Apelin-13. Front. Neurosci. 2017;11:92. doi: 10.3389/fnins.2017.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKinnie SMK, et al. The Metalloprotease Neprilysin Degrades and Inactivates Apelin Peptides. ChemBioChem. 2016;17:1495–1498. doi: 10.1002/cbic.201600244. [DOI] [PubMed] [Google Scholar]
- 26.Fischer C, et al. Plasma kallikrein cleaves and inactivates apelin-17: Palmitoyl- and PEG-extended apelin-17 analogs as metabolically stable blood pressure-lowering agents. Eur. J. Med. Chem. 2019;166:119–124. doi: 10.1016/j.ejmech.2019.01.040. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, et al. Apelin protects against abdominal aortic aneurysm and the therapeutic role of neutral endopeptidase resistant apelin analogs. Proc. Natl. Acad. Sci. 2019;116:13006–13015. doi: 10.1073/pnas.1900152116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onorato JM, et al. Linking (Pyr)1apelin-13 pharmacokinetics to efficacy: Stabilization and measurement of a high clearance peptide in rodents. Anal. Biochem. 2019;568:41–50. doi: 10.1016/j.ab.2018.12.022. [DOI] [PubMed] [Google Scholar]
- 29.DeSilva B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm. Res. 2003;20:1885–900. doi: 10.1023/B:PHAM.0000003390.51761.3d. [DOI] [PubMed] [Google Scholar]
- 30.Center for Drug Evaluation and Research. Bioanalytical Method Validation Guidance for Industry Bioanalytical Method Validation. FDA Guid. Ind. 1–22 (2013).
- 31.Mesmin C, Dubois M, Becher F, Fenaille F, Ezan E. Liquid chromatography/tandem mass spectrometry assay for the absolute quantification of the expected circulating apelin peptides in human plasma. Rapid Commun. Mass Spectrom. 2010;24:2875–2884. doi: 10.1002/rcm.4718. [DOI] [PubMed] [Google Scholar]
- 32.Langelaan DN, Bebbington EM, Reddy T, Rainey JK. Structural Insight into G-Protein Coupled Receptor Binding by Apelin †. Biochemistry. 2009;48:537–548. doi: 10.1021/bi801864b. [DOI] [PubMed] [Google Scholar]
- 33.Carone FA, Christensen EI, Flouret G. Degradation and transport of AVP by proximal tubule. Am. J. Physiol. 1987;253:F1120–1128. doi: 10.1152/ajprenal.1987.253.6.F1120. [DOI] [PubMed] [Google Scholar]
- 34.Argent NB, Burrell LM, Goodship TH, Wilkinson R, Baylis PH. Osmoregulation of thirst and vasopressin release in severe chronic renal failure. Kidney Int. 1991;39:295–300. doi: 10.1038/ki.1991.36. [DOI] [PubMed] [Google Scholar]
- 35.Miao DC, Velaphi SC, Roy T, Despain K, Rosenfeld CR. Metabolism and synthesis of arginine vasopressin in conscious newborn sheep. Am. J. Physiol. Endocrinol. Metab. 2008;295:E672–677. doi: 10.1152/ajpendo.90441.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toräng S, et al. In vivo and in vitro degradation of peptide YY3–36 to inactive peptide YY3–34 in humans. Am. J. Physiol. Integr. Comp. Physiol. 2016;310:R866–R874. doi: 10.1152/ajpregu.00394.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frohman L A, Downs T R, Williams T C, Heimer E P, Pan Y C, Felix A M. Rapid enzymatic degradation of growth hormone-releasing hormone by plasma in vitro and in vivo to a biologically inactive product cleaved at the NH2 terminus. Journal of Clinical Investigation. 1986;78(4):906–913. doi: 10.1172/JCI112679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malm-Erjefalt M, et al. Metabolism and Excretion of the Once-Daily Human Glucagon-Like Peptide-1 Analog Liraglutide in Healthy Male Subjects and Its In Vitro Degradation by Dipeptidyl Peptidase IV and Neutral Endopeptidase. Drug Metab. Dispos. 2010;38:1944–1953. doi: 10.1124/dmd.110.034066. [DOI] [PubMed] [Google Scholar]
- 39.Hemsén A, Ahlborg G, Ottosson-Seeberger A, Lundberg JM. Metabolism of Big endothelin-1 (1–38) and (22–38) in the human circulation in relation to production of endothelin-1 (1–21) Regul. Pept. 1995;55:287–297. doi: 10.1016/0167-0115(94)00119-I. [DOI] [PubMed] [Google Scholar]
- 40.Liu P, et al. A Fluorometric Method of Measuring Carboxypeptidase Activities for Angiotensin II and Apelin-13. Sci. Rep. 2017;7:45473. doi: 10.1038/srep45473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai H, Jiang L, Xiao Z, Guang X. ACE2–angiotensin-(1–7)–Mas axis might be a promising therapeutic target for pulmonary arterial hypertension. Nat. Rev. Cardiol. 2015;12:374–374. doi: 10.1038/nrcardio.2015.6-c1. [DOI] [PubMed] [Google Scholar]
- 42.Crackower MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto K, et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertens. (Dallas, Tex. 1979) 2006;47:718–26. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 44.Oudit GY, et al. Loss of Angiotensin-Converting Enzyme-2 Leads to the Late Development of Angiotensin II-Dependent Glomerulosclerosis. Am. J. Pathol. 2006;168:1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tikellis C, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 46.Imai Y, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou Z, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014;5:3594. doi: 10.1038/ncomms4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu H, et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016;6:19840. doi: 10.1038/srep19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan A, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care. 2017;21:234. doi: 10.1186/s13054-017-1823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X-F, et al. The Apelin-APJ Axis Is an Endogenous Counterinjury Mechanism in Experimental Acute Lung Injury. Chest. 2015;147:969–978. doi: 10.1378/chest.14-1426. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, et al. Apelin-13 Administration Protects Against LPS-Induced Acute Lung Injury by Inhibiting NF-κB Pathway and NLRP3 Inflammasome Activation. Cell. Physiol. Biochem. 2018;49:1918–1932. doi: 10.1159/000493653. [DOI] [PubMed] [Google Scholar]
- 52.Földes G, et al. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Biochem. Biophys. Res. Commun. 2003;308:480–485. doi: 10.1016/S0006-291X(03)01424-4. [DOI] [PubMed] [Google Scholar]
- 53.Chong KS, Gardner RS, Morton JJ, Ashley EA, McDonagh TA. Plasma concentrations of the novel peptide apelin are decreased in patients with chronic heart failure. Eur. J. Heart Fail. 2006;8:355–60. doi: 10.1016/j.ejheart.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 54.Francia P, et al. Cardiac resynchronization therapy increases plasma levels of the endogenous inotrope apelin. Eur. J. Heart Fail. 2007;9:306–309. doi: 10.1016/j.ejheart.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Bavishi C, Messerli FH, Kadosh B, Ruilope LM, Kario K. Role of neprilysin inhibitor combinations in hypertension: insights from hypertension and heart failure trials. Eur. Heart J. 2015;36:1967–1973. doi: 10.1093/eurheartj/ehv142. [DOI] [PubMed] [Google Scholar]
- 56.Velazquez EJ, et al. Angiotensin–Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019;380:539–548. doi: 10.1056/NEJMoa1812851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information for Development and validation of an LC-MS/MS method for detection and quantification of in vivo derived metabolites of [Pyr1]apelin-13 in humans
Data Availability Statement
All data generated or analysed during this study are included in this published article.