

SUMMARY
Combined immunotherapy targeting the immune checkpoint receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1), or CTLA-4 and the PD-1 ligand (PD-L1) exhibits superior anti-tumor responses compared with single-agent therapy. Here, we examined the molecular basis for this synergy. Using reconstitution assays with fluorescence readouts, we found that PD-L1 and the CTLA-4 ligand CD80 heterodimerize in cis but not trans. Quantitative biochemistry and cell biology assays revealed that PD-L1:CD80 cis-heterodimerization inhibited both PD-L1:PD-1 and CD80:CTLA-4 interactions through distinct mechanisms but preserved the ability of CD80 to activate the T cell co-stimulatory receptor CD28. Furthermore, PD-L1 expression on antigen-presenting cells (APCs) prevented CTLA-4-mediated trans-endocytosis of CD80. Atezolizumab (anti-PD-L1), but not anti-PD-1, reduced cell surface expression of CD80 on APCs, and this effect was negated by co-blockade of CTLA-4 with ipilimumab (anti-CTLA-4). Thus, PD-L1 exerts an immunostimulatory effect by repressing the CTLA-4 axis; this has implications to the synergy of anti-PD-L1 and anti-CTLA-4 combination therapy.
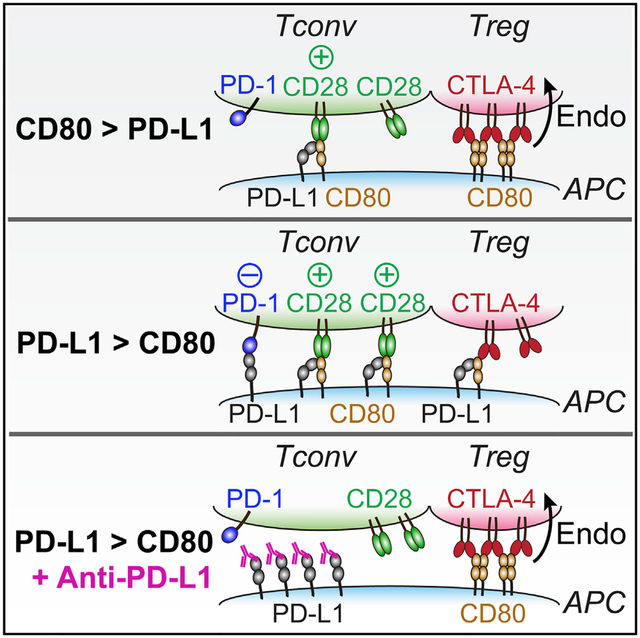
Graphical Abstract
In Brief
Combined immunotherapy targeting the checkpoint receptors CTLA-4 and PD-1, or CTLA-4 and the PD-1 ligand (PD-L1) results in superior anti-tumor responses. Zhao et al. show that PD-L1 heterodimerizes with CD80, a shared ligand for CTLA-4 and CD28, to selectively weaken CD80:CTLA-4 interaction but not CD80:CD28 interaction. Thus, PD-L1 can repress the CTLA-4 axis; this has implications to the synergy observed in combination immunotherapies.
INTRODUCTION
Programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) are co-inhibitory receptors that restrict T cell activity (Nishimura et al., 1999; Tivol et al., 1995; Waterhouse et al., 1995) and as such are referred to as immune checkpoints. Antibodies that block CTLA-4, PD-1, or the PD-1 ligand PD-L1 have impressive clinical activities against an array of human cancers (Hodi et al., 2010; Powles et al., 2014; Ribas and Wolchok, 2018; Rizvi et al., 2015; Topalian et al., 2012). The combination of anti-CTLA-4 and anti-PD-1, or anti-CTLA-4 and anti-PD-L1, is more effective than either agent alone in clinical trials (Callahan et al., 2015; Larkin et al., 2015) or pre-clinical models (Duraiswamy et al., 2013; Lim et al., 2016), and the former combo is approved by the US Food and Drug Administration for treatment of human melanoma and renal cancer patients (Callahan et al., 2015; Larkin et al., 2015). Nevertheless, durable response to immune checkpoint blockade therapy is restricted to a minority of patients and cancer indications (Wei et al., 2018). Therefore, a better mechanistic understanding of anti-PD-1, anti-PD-L1, anti-CTLA-4, and their crosstalk, is needed for rational design of mono- and combination therapies.
Binding of T cell PD-1 with its ligand PD-L1 (Dong et al., 1999; Freeman et al., 2000) on antigen-presenting cells (APCs) triggers tyrosine phosphorylation of PD-1 and recruitment of SHP2, a phosphatase that dephosphorylates the T cell receptor (TCR) and CD28 co-stimulatory signaling components (Hui et al., 2017; Yokosuka et al., 2012). CTLA-4 outcompetes CD28 for their shared ligands, CD80 and CD86, in part because of its higher binding affinity to these ligands (Krummel and Allison, 1995; Linsley et al., 1991; van der Merwe et al., 1997). Additionally, CTLA-4 depletes CD80 and CD86 from APCs via trans-endocytosis to indirectly inhibit CD28 signaling (Qureshi et al., 2011; Wing et al., 2008). PD-L1 and CD80 bind each other (Butte et al., 2007; Butte et al., 2008), suggesting another layer of crosstalk among PD-1, CTLA-4, and CD28 pathways. Mechanistic understanding of PD-L1:CD80 interaction is challenging because of a complex network consisting of two ligands (PD-L1 and CD80) and three receptors (PD-1, CD28, and CTLA-4). Indeed, the biochemical nature and cell biological consequence of PD-L1:CD80 interaction remain debated. The original reports suggested that PD-L1 and CD80 on different cells bind in trans, but more recent evidence indicated that they interact in cis on the same cells (Chaudhri et al., 2018) and that the PD-L1:CD80 cis-interaction inhibits PD-L1:PD-1 axis both in vitro and in vivo (Haile et al., 2011; Sugiura et al., 2019). However, it is less clear whether PD-L1:CD80 cis-interaction affects the abilities of CD80 to interact with CD28 and CTLA-4.
Here, we examined the PD-L1:CD80:PD-1:CD28:CTLA-4 signaling network by using in vitro reconstitution, engineered cell lines, and immune checkpoint inhibitors. We found that PD-L1:CD80 cis-interaction inhibits both PD-1 and CTLA-4 axes while fully preserving the co-stimulatory CD80:CD28 interaction. Thus, PD-L1 restricts CTLA-4 activity; this has mechanistic implications as to the effects of anti-PD-L1 treatment and combination therapies that block CTLA-4 function.
RESULTS
PD-L1 and CD80 Interact in Cis but Not in Trans
We first asked whether PD-L1 binds to CD80 in trans. To this end, we utilized a membrane adhesion assay (Zhao et al., 2018), in which trans-interaction between two proteins is measured as the association of two model membranes. Specifically, we reconstituted (1) His-tagged ectodomain of PD-L1 (PD-L1-His) to BODIPY-PE containing large unilamellar vesicles (LUVs) and (2) His-tagged ectodomain of its binding partner, CD80 or PD-1, to a supported lipid bilayer (SLB) through the use of a His-tag chelating lipid DGS-NTA-Ni. Incubation of PD-L1 LUVs with the PD-1 SLB resulted in a number of SLB-associated LUVs, as visualized by total internal reflection fluorescence (TIRF) microscopy. In contrast, CD80 SLB captured 99% fewer PD-L1 LUVs than PD-1 SLB, similar to SLB containing CD86 (Figure 1A), a negative control with no reported PD-L1 binding activity. Thus, PD-L1 does not bind CD80 in trans, consistent with a recent study (Chaudhri et al., 2018).
Figure 1. PD-L1 Binds CD80 in Cis, and Atezolizumab Disrupts this Interaction.

(A) Representative TIRF images of PD-L1 LUVs captured by PD-1 SLB, CD80 SLB, or CD86 SLB; each LUV is registered as a green spot. Bar graph summarizes the fluorescence intensity (FI) of the LUV channel under indicated conditions, normalized to the intensity of the condition with PD-1 SLBs. Data are means ± SEM, n = 3. Scale bars, 5 μm.
(B) A FRET assay showing PD-L1:CD80 cis-interaction on cell membranes. Cartoons on the left depict a HEK293T cell co-expressing PD-L1 (labeled with CS547, donor) and either CD80 or CD86 (labeled with SSAF647, acceptor). On the immediate right are pre- and post-bleaching confocal images of a representative cell at the indicated channels. Further right are calculated FRET efficiency images (pseudo-color; the yellow to purple spectrum denotes strong to weak FRET) and the differential interference contrast (DIC) images. Rightmost are bar graphs summarizing the FRET efficiencies as mean ± SEM, n > 25 cells from 3 independent experiments. Scale bars, 10 μm.
(C) Same as (B) except replacing PD-L1 with PD-L2.
(D) On the left is a cartoon depicting an LUV FRET assay for probing PD-L1:CD80 cis-interaction and atezolizumab (Atezo) effects. SC505 (donor) labeled SNAP-PD-L1-His was pre-bound to LUVs via DGS-NTA-Ni. Subsequently added TMR (acceptor) labeled SNAP-CD80-His bound to the LUVs and interacted with PD-L1 in cis, causing FRET and SC505 quenching (black trace). On the right are time courses of normalized SC505 fluorescence under the indicated conditions. Color coding is as follows: blue, same as black except plus atezolizumab; magenta, same as black except using TMR*CD80 lacking a His tag; orange, same as black except replacing PD-L1 with PD-L2; gray, same as black except presenting TMR*CD80 in trans. Data are representative of 3 independent replicates. Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001.
We next determined whether PD-L1 and CD80 bind in cis by using Förster resonance energy transfer (FRET) (Zhao et al., 2018). To this end, we co-transfected HEK293T cells with CLIP-tagged PD-L1 and SNAP-tagged CD80 and labeled them with CLIP-Surface 547 (CS547) (energy donor) and SNAP-Surface Alexa Fluor 647 (SSAF647) (energy acceptor), respectively. Photobleaching of SSAF647*CD80 increased the fluorescence of CS547*PD-L1 (Figure 1B, top), indicative of FRET. Replacement of CD80 with CD86 (Figure 1B, bottom) or of PD-L1 with PD-L2 decreased the FRET signal (Figure 1C). These data suggest that PD-L1 associates with CD80 in cis on cell membranes. We next examined this cis-interaction quantitatively in an LUVs reconstitution assay. Specifically, we attached DGS-NTA-Ni containing LUVs with PD-L1-His labeled with energy donor (SNAP-Cell-505 [SC505]). Subsequent addition of energy acceptor (tetramethyl rhodamine [TMR])-labeled CD80–His (25 nM), but not CD80 lacking the membrane-targeting His-tag or CD80-His presented on trans-LUVs, quenched PD-L1 fluorescence (Figure 1D; black, magenta, and gray). Thus, despite the weak interactions in solution (Kd: 17.8 μM) (Cheng et al., 2013), PD-L1 and CD80 can strongly interact in cis on membranes. CD80-His also induced a reproducible, but much weaker quenching of LUV-bound PD-L2 (Figure 1D; orange), because of a molecular crowding effect. These results demonstrate that PD-L1 and CD80 bind directly in cis. Furthermore, atezolizumab, a therapeutic PD-L1 antibody reported to block both PD-L1:PD-1 interaction and PD-L1:CD80 interaction in solution (Herbst et al., 2014), decreased the PD-L1:CD80 FRET to a level similar to the negative control PD-L2:CD80 pair (Figure 1D; blue). Thus, atezolizumab blocks PD-L1:CD80 cis-interaction, presumably underpinning an additional activity of anti-PD-L1 not engaged by anti-PD-1.
PD-L1:CD80 Cis-Interaction Blocks PD-L1:PD-1 Signaling
Having established that CD80 binds PD-L1 in cis, we next determined whether CD80 inhibits PD-L1:PD-1 interaction. To examine PD-L1:PD-1 interaction in the absence of cis-CD80, we generated CD80 deleted, PD-L1-mCherry transduced (CD80−PD-L1-mCherry+) Raji B cells. Soluble PD-1-human-Fc fusion protein (PD-1-huFc) stained this type of Raji cell in a dose-dependent manner (Figure 2A; black). Co-expression of CD80 (3.5-fold excess to PD-L1) substantially decreased PD-1-huFc staining (Figure 2A; blue) (Raji [CD80hiPD-L1-mCherry+]), see Figures S1A–S1E for quantitation of PD-L1 and CD80 levels), indicating that PD-L1:CD80 cis-interaction inhibits PD-L1:PD-1 interaction. Notably, antibody staining of PD-L1 was much weaker for Raji (CD80hiPD-L1-mCherry+) cells than for Raji (CD80−PD-L1-mCherry+) cells (Figure S1A, left), despite the nearly identical expressions of PD-L1-mCherry in these two cell lines (Figure S1A, right). This discrepancy indicates that cis-CD80 can block at least some clones of PD-L1 antibodies. Thus, immunohistochemistry staining might underestimate PD-L1 amounts on CD80+ cells.
Figure 2. CD80 Inhibits PD-1 Signaling through Neutralizing PD-L1 in Cis.

(A) Representative flow-cytometry histograms of PD-1-huFc staining of the indicated types of Raji cells. Bound PD-1-huFc was labeled by AF647 anti-human IgG Fc, the mean fluorescence intensity (MFI) of which was plotted against PD-1-huFc concentration (means ± SEM, n = 3). In gray is the staining of Raji (CD80−PD-L1-mCherry+) by isolated huFc domain.
(B) A T-cell-SLB assay showing cis-CD80 effects on PD-L1-induced PD-1 microclusters. On the left is a cartoon for a PD-1-mCherry transduced OT-1 cell interacting with an SLB containing pMHC, intercellular adhesion molecule(ICAM) (not shown), and PD-L1. On the immediate right are representative TIRF images of PD-1-mCherry (rendered in green) and TCR stained by an AF647 anti-TCR-β antibody (rendered in magenta) 30 s after cells contacted the pMHC- and ICAM-containing SLB supplemented with the indicated ligands. The bar graph shows the clustering indices of PD-1 and TCR under each condition (means ± SEM, n ≥18 cells from 3 independent replicates). Scale bars, 5 μm.
(C)AT-cell-APC conjugate assay showing Raji CD80 inhibits the synaptic enrichment of Jurkat PD-1. The leftmost cartoons depict Jurkat (PD-1-mGFP+) forming conjugates with 3 types of Raji with similar PD-L1 amounts but increasing CD80 amounts: CD80−, CD80lo, and CD80hi. On the immediate right are confocal images of the cell conjugate acquired 2 min after cell-cell contact. The bar graph summarizes the synaptic enrichment indices of the 3 conditions(means ± SEM, n = 38 conjugates from 3 independent experiments). Scale bars, 10 μm.
(D) On the left are representative immunoblots (IB) showing the phosphorylation and the bound SHP2 of PD-1-mGFP, immunoprecipitated (IP) from the lysates of the indicated Jurkat-Raji co-cultures, with the time points of lysis denoted. On the right are quantification bar graphs of blots (means ± SEM, n = 3). See also Figure S1.
Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001. See Table S3 for genotypes of cells related to this figure.
To study the cis-CD80 effects on PD-L1:PD-1 trans-interaction, we then employed a T-cell-SLB system in which a cytotoxic T cell interacts with an SLB containing peptide-bound major-histocompatibility-complex (pMHC) and PD-L1. As reported (Hui et al., 2017; Yokosuka et al., 2012; Zhao et al., 2018), interaction of PD-1 on T cells with SLB-attached PD-L1 in trans led to the formation of PD-1 microclusters at the cell-bilayer interface. Notably, addition of CD80-His (3.0-fold excess to PD-L1) to the SLB abolished PD-1 microclusters but with no effect on TCR microclusters (Figure 2B). By contrast, equal amounts of CD86-His did not affect PD-1 clustering (Figure 2B). These data suggest that cis-CD80 inhibits PD-L1:PD-1 trans-interaction.
We next extended our investigation to a T-cell-APC co-culture system consisting of PD-1-mGFP transduced Jurkat T cells and PD-LI-mCherry transduced Raji B cells. We created three Raji lines expressing similar numbers of PD-L1-mCherry (~1,700 molecules per μm2) but increasing amounts of CD80: (1) Raji (CD80−PD-L1-mCherry+), (2) Raji (CD80loPD-L1-mCherry+) (~600 CD80 molecules per μm2), and (3) Raji (CD80hiPD-L1-mCherry+) (~6,000 CD80 molecules per μm2) (Figures 2C, 2D, and S1A–S1E). These PD-L1 and CD80 amounts are comparable to those on human monocyte-derived dendritic cells (DCs) (Figure S1F). Using confocal microscopy, we found that conjugation of superantigen SEE-loaded Raji (CD80−PD-L1-mCherry+) cells with Jurkat (PD-1-mGFP+) cells enriched both PD-L1 and PD-1 to the Raji-Jurkat interface. Raji (CD80loPD-L1-mCherry+) cells, which express 66% lower CD80 than PD-L1 (Figures S1A–S1E), induced a similar degree of PD-1 enrichment. Raji (CD80hiPD-L1-mCherry+) cells, which express ~3.5-fold higher CD80 than PD-L1, decreased PD-1 enrichment (Figure 2C), phosphorylation, and SHP2 recruitment (Figure 2D). Collectively, these results indicate that besides its well-established function in triggering CD28, CD80 stimulates T cell activity by neutralizing an inhibitory ligand, consistent with prior reports (Haile et al., 2011; Sugiura et al., 2019). In the case of (CD80loPD-L1-mCherry+) cells, the inability of cis-CD80 to reduce PD-L1:PD-1 signaling was likely due to the much higher amount of Raji PD-L1 than that of Jurkat PD-1 (~700 molecules per μm2).
PD-L1:CD80 Cis-Heterodimer Preserves the Ability of CD80 to Bind CD28
In the reciprocal experiment, we asked how PD-L1:CD80 cis-interaction might affect CD80:CD28 interaction. First, CD28-human-Fc fusion protein (CD28-huFc) stained Raji (CD80+CD86−) cells in a dose-dependent manner (Figure 3A; black), because of CD80:CD28 interaction. A similar degree of CD28-huFc staining was observed for Raji (CD80+CD86−PD-L1-mCherry+) cells (Figure 3A; blue), which express 5.9-fold excess PD-L1 to CD80 (Figures S1G and S1H). These results indicate that cis-PD-L1 does not affect CD80:CD28 interaction. Consistent with this result, disruption of PD-L1:CD80 cis-heterodimerization on Raji (CD80+CD86−PD-L1-mCherry+) cells using atezolizumab (Figure 1D) did not affect the staining of CD28-mouse-Fc fusion protein (CD28-moFc) (Figure 3B; black versus blue). The use of a moFc tag allowed us to stain this protein with fluorescently labeled anti-moFc antibody, with minimal crosstalk with atezolizumab, a humanized antibody. Isolated huFc and moFc domains also weakly stained the Raji cells at high doses, perhaps due to non-specific binding (Figure 3A and 3B; gray).
Figure 3. Cis-PD-L1 Does Not Affect CD80:CD28 Interaction.

(A) Representative flow-cytometry histograms of CD28-huFc staining of the indicated types of Raji cells. Bound CD28-huFc was labeled by AF647 anti-human IgG Fc, the MFI of which was plotted against (CD28-huFc. Shown in gray are Raji (CD80+CD86−) cells stained by isolated huFc domain. Means ± SEM, n = 3.
(B) Representative flow-cytometry histograms of CD28-moFc staining of Raji (CD80+CD86−PD-L1-mCherry+) cells with or without atezolizumab (Atezo) (20 μg/mL). Bound moFc was labeled by AF647 anti-mouse IgG Fc, the MFI of which was plotted against (CD28-moFc). Shown in gray are atezolizumab-treated Raji (CD80+CD86−PD-L1-mCherry+) cells stained by isolated moFc domain. Means ± SEM, n = 3.
(C) A T-cell-SLB assay showing cis-PD-L1 effects on CD80-induced CD28 microclusters. On the left is a cartoon for a CD28-mGFP transduced OT-1 cell interacting with an SLB containing pMHC, ICAM (not shown), and CD80. On the immediate right are TIRF images of CD28-mGFP (rendered in green) and TCR stained by AF647-labeled H57–597 TCR-β antibody(rendered in magenta) 30 s after cells contacted the pMHC- and ICAM-containing SLB supplemented with the indicated ligands. The bar graph shows the clustering indices of CD28 and TCR under each condition (means ± SEM of ≥ 18 cells from 3 independent experiments). Scale bars, 5 μm.
(D) A T-cell-APC conjugate assay probing how cis-PD-L1 affects CD80:CD28 interaction. The leftmost cartoons depict a Jurkat (CD28-mCherry+) cell forming a conjugate with a Raji (CD80-mGFP+CD86+) cell, a Raji (CD80-mGFP+CD86+CLIP-PD-L1+) cell, or a Raji (CD86+CLIP-PD-L1+) cell. On the immediate right are confocal images of a cell conjugate acquired 2 min after Jurkat-Raji contact. The bar graph summarizes the synaptic enrichment indices of the 3 conditions with CD28 rendered in magenta, CD80 in green, and PD-L1 in blue (means ± SEM, n = 30 conjugates from 3 independent experiments). Scale bars, 10 μm.
(E) At the top is a representative IB showing CD28:p85 co-IP from the lysates of the indicated co-cultures, with the times of lysis denoted. On the bottom is a quantification bar graph (means ± SEM, n = 3).
(F) Same as (E) except CD86 was deleted from all 3 types of Raji cells. See also Figures S1 and S2.
Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001. See Table S3 for genotypes of cells related to this figure.
We further confirmed the lack of effect of cis-PD-L1 on CD80:CD28 interaction in two independent assays. First, in a T-cell-SLB system, addition of cis-PD-L1 (3.0-fold excess to CD80) did not inhibit the formation of CD28 microclusters or TCR microclusters (Figure 3C). Moreover, in the Raji-Jurkat conjugate assay, PD-L1− and PD-L1+ Raji cells led to indistinguishable degrees of CD28 and CD80 interface enrichment and CD28 phosphorylation (measured by co-immunoprecipitated p85) (Figures 3D–3F). We also noted that PD-L1 was enriched to the Raji-Jurkat interface in a CD80-dependent manner, suggesting that PD-L1, CD80, and CD28 form a tripartite complex that triggers CD28 signaling. Finally, deletion of CD80 from Raji significantly decreased the interface enrichment of both PD-L1 and CD28 (Figure 3D, bottom), as well as CD28-mediated p85 recruitment (Figure 3E). The residual p85 recruitment appeared to be due to CD86:CD28 interaction, because deletion of both CD80 and CD86 abolished this signal (Figure 3F). Collectively, data presented in this section demonstrate that cis-PD-L1 does not affect CD80:CD28 trans-interaction and PD-L1:CD80 heterodimer is fully capable of activating CD28.
PD-L1:CD80 Cis-Interaction Inhibits CD80:CTLA-4 Interaction
We next asked whether the PD-L1:CD80 cis-interaction affects CD80:CTLA-4 interaction. CTLA-4-human-Fc fusion protein (CTLA-4-huFc) stained Raji (CD80+CD86−) cells in a dose-dependent fashion (Figure 4A; black), despite at a much lower concentration range than CD28-huFc, consistent with its stronger interaction with CD80 (Collins et al., 2002; van der Merwe et al., 1997). Contrasting to the lack of effect on CD28 binding, co-expression of PD-L1 (5.9-fold excess to CD80) (Figures S1G and S1H) decreased CTLA-4-huFc staining at a wide range of concentrations (Figure 4A; blue) (Raji [CD80+CD86−PD-L1-mCherry+] cells). Consistent with this result, atezolizumab, which disrupts PD-L1:CD80 cis-interaction (Figure 1D), significantly increased CTLA-4-moFc staining of this type of Raji cells (Figure 4B; black versus blue). These data suggest that cis-PD-L1 inhibits CD80:CTLA-4 interaction.
Figure 4. Cis-PD-L1 Inhibits CD80:CTLA-4 Interaction through Disrupting CD80 Homodimers.

(A) Representative flow-cytometry histograms of CTLA-4-huFc staining of the indicated types of Raji cells. Bound CTLA-4-huFc was labeled by AF647 anti-human IgG Fc, the MFI of which was plotted against (CTLA-4-huFc). Shown in gray are Raji (CD80+CD86−) cells stained by isolated huFc domain. Means ± SEM, n ≥ 3.
(B) Representative flow-cytometry histograms of CTLA-4-moFc staining of Raji (CD80+CD86−PD-L1-mCherry+) cells with or without atezolizumab (Atezo) (20 μg/mL). Bound moFc was labeled by AF647 anti-mouse IgG Fc, the MFI of which was plotted against (CTLA-4-moFc). Shown in gray are atezolizumab-treated Raji (CD80+CD86−PD-L1-mCherry+) cells stained by isolated moFc domain. Means ± SEM, n ≥ 3.
(C) Representative flow-cytometry histograms of CTLA-4-GCN4*SC647 staining of Raji (CD80+CD86−PD-L1-mCherry+) cells with or without atezolizumab and of Raji (CD80−CD86−) cells with atezolizumab. MFI of SC647 was plotted against the input concentration (means ± SEM, n ≥ 3).
(D) At the top are flow-cytometry histograms showing both PD-L1 and CD80 amounts on a population of Raji (CD80wdCD86−PD-L1-mCherry+) cells with tight PD-L1 expression and a wide range of CD80 expression. The cells were stained with either phycoerythrin (PE) anti-CD80, PE anti-PD-L1, or PE isotype, and the 3 histograms overlaid. On the bottom is a flow-cytometry dot plot showing CTLA-4-GCN4*SC647 staining of Raji (CD80wdCD86−PD-L1-mCherry+) cells with or without atezolizumab. Gray dots correspond to control signals of unstained cells. CD80+ cells were gated by the vertical dash line, determined by the mGFP signal of parental Raji (CD80−CD86−) cells.
(E) A FRET assay probing CD80:CD80 homodimerization on cell membranes. In the first row, the leftmost cartoon depicts a HEK293T cell expressing SNAP-CD80, with a subpopulation labeled with SS549 (donor) and the rest labeled with SSAF647 (acceptor). On the immediate right are pre- and post-bleaching confocal images of a representative cell. Further on the right is the calculated pseudo-color FRET efficiency image (yellow to purple spectrum denotes strong to weak FRET) and the DIC image. The second and third rows are the same as the first row except replacing SNAP-CD80 with SNAP-CD80 (I92R) or with SNAP-CD86. The fourth row is the same as the first row except with co-expressed unlabeled PD-L1. The fifth row is the same as fourth row except in the presence of atezolizumab. The bar graph summarizes the FRET efficiencies as mean ± SEM, n > 22 cells from 3 independent experiments. Scale bars, 10 μm.
(F) An LUV FRET assay for probing CD80:CD80 homodimerization and PD-L1 effects. Shown is a representative time course of normalized FI of LUV-bound SC505*CD80-His, challenged by TMR*CD80-His and then by indicated concentrations of unlabeled PD-L1-His, with or without atezolizumab (Atezo) (20 μg/mL).
(G) An LUV FRET assay showing that a single point mutation in CD80 disrupts both CD80:CD80 homodimerization and PD-L1:CD80 heterodimerization. Each indicated SC505 (energy donor)-labeled protein was pre-coupled to DGS-NTA-Ni containing LUVs through its His-tag, and challenged with TMR (energy acceptor)-labeled proteins as indicated. Shown are representative time courses of 3 independent replicates.
(H) Representative TIRF images of Raji (CD80-mGFP+CD86−PD-L1-SNAP+) cells stained with CTLA-4-GCN4*SC647, at the indicated channels, under the depicted conditions. Rightmost are FI profiles along the dashed line at the overlaid images. Scale bars, 5 μm. See also Figures S1–S4.
Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001. See Table S3 for genotypes of cells related to this figure.
Both CTLA-4 and CD28 are homodimers on cell membranes because of a disulfide bond at the extracellular stalk region (Linsley et al., 1995). Soluble CTLA-4-Fc and CD28-Fc proteins used in the foregoing staining assays were also dimeric (Figure S2) due to the disulfide-linked Fc domain. However, a fluorescently labeled anti-Fc antibody was needed to detect the bound Fc-fusion protein on Raji cells. This step might introduce artifacts because of antibody-mediated crosslinking. To directly assess the cis-PD-L1 effect on CD80:CTLA-4 binding without the use of anti-Fc, we produced a homemade fluorescently labeled CTLA-4 dimer, in which the ectodomain of CTLA-4 was fused with an N-terminal SNAP tag and a C-terminal GCN4 leucine zipper motif (SNAP-CTLA-4-GCN4), and labeled with SNAP-Cell-647-SiR (SC647). This home-made CTLA-4 dimer, as confirmed by gel filtration chromatography (Figure S2), bound to Raji cells in a CD80-dependent manner (Figure 4C; gray versus blue) and this binding was strongly enhanced by atezolizumab (Figure 4C; black versus blue). The atezolizumab-mediated increase in CTLA-4 binding was observed at a wide range of CD80 amounts (Figure 4D). In contrast, atezolizumab had no effect on the binding of the analogous dimeric CD28 protein SC647*SNAP-CD28-GCN4 (Figures S2 and S3), consistent with data obtained with CD28-Fc proteins (Figure 3). Collectively, data presented in this section demonstrate that cis-PD-L1 restricts CD80:CTLA-4 interaction.
Cis-PD-L1 Inhibits CD80:CD80 Dimerization and CD80:CTLA-4 Co-clustering
We next examined why cis-PD-L1 inhibits CD80:CTLA-4 but not CD80:CD28 interaction even though CTLA-4 and CD28 bind to CD80 via the same conserved motif (Evans et al., 2005; Peach et al., 1994; Stamper et al., 2001). We noted that CTLA-4 binds to CD80 more potently than CD28 mainly due to an avidity effect. CD80 dimerizes non-covalently through its N-terminal immunoglobulin (Ig)-like domain (Ikemizu et al., 2000) and exists in a dynamic equilibrium between monomers and dimers on cell membranes (Bhatia et al., 2005; Girard et al., 2014). CTLA-4 dimers interact with CD80 dimers, in a zipper-like fashion, to form a lattice array that reduces the off-rate of CTLA-4 by two orders of magnitude (Collins et al., 2002; Evans et al., 2005; Ostrov et al., 2000; Stamper et al., 2001). In contrast, CD28, despite being a covalent dimer, interacts with CD80 in a monovalent fashion (Collins et al., 2002; Evans et al., 2005). In light of these findings, we asked how cis-PD-L1 might affect CD80 dimerization and therefore the avidity of its interaction with CTLA-4.
We first established a FRET assay to probe CD80 cis-homodimerization on cell membranes. We transfected SNAP-CD80 to HEK293T cells and labeled a subpopulation of this protein with SNAP-Surface-549 (SS549) (energy donor), and the rest with SNAP-Surface-Alexa Fluor-647 (SSAF647) (energy acceptor). Photobleaching of SSAF647 significantly restored the SS549 fluorescence, indicative of CD80:CD80 FRET (Figure 4E, first row). A point mutation (I92R) that disrupts the CD80 dimerization interface (Bhatia et al., 2005; Ikemizu et al., 2000) decreased the CD80:CD80 FRET signal (Figure 4E, second row) to a similar level as the FRET between CD86 (Figure 4E, third row), a monomeric membrane protein. These data demonstrate that at least a subpopulation of CD80 molecules existed as homodimers. Furthermore, we found that co-expression of unlabeled PD-L1 decreased the CD80:CD80 FRET signal (Figure 4E, fourth row), and this effect was reversed by atezolizumab (Figure 4E, fifth row), which disrupts PD-L1:CD80 cis-interaction. These results indicate that PD-L1:CD80 cis-heterodimerization inhibits CD80:CD80 cis-homodimerization.
Analogously, cis-PD-L1 inhibited CD80:CD80 homodimerization in an LUV reconstitution system. We conducted this experiment as in Figure 1D except replacing SC505*PD-L1-His with SC505*CD80-His to report CD80:CD80 homodimerization. Consistent with the cellular FRET assay, LUV-attached SC505*CD80-His was partially quenched by TMR*CD80-His, suggesting that TMR*CD80 bound to SC505*CD80 on LUVs, evidence of CD80 homodimerization (Figure 4F). Consistent with the model that cis-PD-L1 inhibits CD80 homodimerization, the fluorescence of SC505*CD80-His was restored by unlabeled PD-L1-His (Figure 4F, left), but not by atezolizumab-treated PD-L1-His (Figure 4F, right), in a dose-dependent manner. Additionally, the I92R mutation of CD80 abrogated both CD80:CD80 FRET and CD80:PD-L1 FRET (Figure 4G), indicating that CD80:PD-L1 heterodimerization interface at least partially overlaps with CD80:CD80 homodimerization interface.
On the basis of the previous model that high avidity CD80:CTLA-4 interaction occurs via a lattice array of bridged homodimers (Collins et al., 2002; Stamper et al., 2001) and our result that cis-PD-L1 inhibits CD80:CD80 homodimerization, we predicted that cis-PD-L1 would inhibit CD80:CTLA-4 lattice formation. To test this, we visualized the spatial patterns of CD80 and CTLA-4 on CTLA-4-GCN4-stained Raji cells. Using TIRF imaging, we found that soluble, dimeric CTLA-4-GCN4 weakly stained the Raji (CD80-mGFP+CD86−PD-L1-SNAP+) cells; both CD80 and CTLA-4 were largely diffusive on the cell membrane (Figure 4H, first row). Moreover, disruption of PD-L1:CD80 cis-interaction by atezolizumab enhanced CTLA-4 binding, consistent with the flow-cytometry results (Figures 4A–4C), and caused co-clustering of CD80 and CTLA-4 (Figure 4H, second row), evidence of multivalent interactions. Atezolizumab treatment alone did not cluster CD80 (Figure 4H, third and fourth rows), thus CD80 clustering was a consequence of CTLA-4 crosslinking. These results demonstrate that cis-PD-L1 dissolves CD80 homodimers to prevent high-avidity CD80:CTLA-4 interactions.
We further tested whether PD-L1 and CTLA-4 directly compete for CD80 at the level of monovalent interactions. To exclude avidity effects, we prepared a CTLA-4 monomer protein (Figure S4A) and asked whether this protein inhibits CD80:PD-L1 interaction, at CD80 concentrations well below its dimerization Kd (20–50 μM) (Ikemizu et al., 2000) in order to keep CD80 monomeric (Figure S4A). Using surface plasmon resonance (SPR), we found that PD-L1-coated chip captured CD80 from solution in a dose-dependent fashion (Figure S4B). Moreover, inclusion of CTLA-4 monomer did not inhibit, but rather enhanced the SPR signal (Figure S4C). This was not due to direct binding of CTLA-4 to PD-L1, given that no CTLA-4 binding was detected in the absence of CD80 (Figure S4D). Conceivably, the PD-L1-bound CD80 recruited CTLA-4 from solution, further increasing the SPR signal, which depends on the mass but not the identities of the bound species. These data suggest that PD-L1 and CTLA-4 can simultaneously bind to a CD80 monomer. Collectively, data presented in this section suggest that PD-L1 inhibits CD80:CTLA-4 interaction largely through an avidity effect rather than directly blocking the CTLA-4 binding site on CD80.
Cis-PD-L1 Protects CD80 from CTLA-4-Mediated Trans-Endocytosis
We had attempted to further examine the PD-L1 effect on CD80:CTLA-4 interaction in the T-cell-SLB assay by probing CTLA-4 plasma membrane microclusters, but this effort was precluded by the predominantly intracellular localization of CTLA-4, consistent with previous reports (Alegre et al., 1996; Linsley et al., 1996; Qureshi et al., 2012; Valk et al., 2008). Indeed, the endocytic property of CTLA-4 allows it to trans-endocytose CD80 and CD86 (Hou et al., 2015; Qureshi et al., 2011), resulting in their depletion from APCs and therefore weaker CD28 activation. On the basis of our finding that cis-PD-L1 inhibits CD80:CTLA-4 interaction, we predicted that cis-PD-L1 would protect CD80 from CTLA-4-mediated depletion. To test this, we established a CTLA-4-mediated trans-endocytosis assay: CTLA-4-mGFP transduced Jurkat (CTLA-4-mGFP+) cells, but not wild-type (WT) Jurkat cells lacking CTLA-4, decreased CD80 amounts on Raji (CD80+) cells upon 0.5 h of Jurkat-Raji contact (Figure 5A), indicating that CTLA-4 trans-endocytosed CD80 from Raji APCs. In addition, co-expression of PD-L1-mCherry on Raji cells significantly inhibited the depletion of CD80 (Figure 5A) (Raji [CD80+PD-L1-mCherry+]) and sustained CD80 amounts through 24 h of Jurkat-Raji co-culture (Figure S5). In contrast, WT Raji (CD80+CD86+) cells lost ~94% of CD80 after 12 h co-culture with Jurkat (CTLA-4-mGFP+) cells (Figure S5). These data uncovered a role of cis-PD-L1 in protecting CD80 from CTLA-4-mediated depletion.
Figure 5. Cis-PD-L1 Protects CD80 from CTLA-4 Mediated Trans-Endocytosis.

(A) A Jurkat-Raji co-culture assay analyzing how PD-L1 interferes with CTLA-4-mediated CD80 depletion. Cartoons on the left depict the co-cultured cells. On the immediate right are representative flow-cytometry histograms of CD80 expression (anti-CD80 allophycocyanin) on Raji cells before (0 h) and after co-culture (0.5 h). Further on the right are representative confocal images for the Jurkat-Raji conjugate (scale bars, 10 μm). Rightmost is a bar graph showing CD80 MFI of Raji at 0.5 h, normalized to CD80 MFI at 0 h (mean ± SEM, n = 4).
(B) An independent Jurkat-Raji conjugation assay examining how anti-PD-L1 and anti-CTLA-4 affect CD80 amounts. Experiments were conducted as in (A) except pretreating the indicated cell type with atezolizumab (Atezo) or ipilimumab (Ipi), as depicted in the cartoons. On the immediate right are representative flow-cytometry histograms of CD80 expression and confocal images for the Jurkat-Raji conjugate (scale bars, 10 μm). Rightmost is a bar graph showing CD80 MFI of Raji at 0.5 h, normalized to CD80 MFI at 0 h (mean ± SEM, n = 5).
(C) Representative flow-cytometry histograms of CD80 and CD86 surface expressions on mouse splenic DCs co-cultured with either Tconv or Treg cells with or without the indicated checkpoint inhibitors for 16 h. Bar graphs summarize the CD80 and CD86 MFI (mean ± SEM, n = 3).
(D) Bar graph summarizing the IL-2 production from the indicated Jurkat-Raji cocultures in the presence of indicated checkpoint inhibitors (mean ± SEM, n = 3). See also Figures S5 and S6.
Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001. See Table S3 for genotypes of cells related to this figure.
Anti-PD-L1 Depletes CD80 on APCs in a CTLA-4-Dependent Manner
A key prediction from our finding is that atezolizumab, which blocks PD-L1:CD80 cis-interaction (Figure 1D), would deplete CD80 from APCs in a CTLA-4-dependent manner. Consistent with this notion, we found that atezolizumab treatment decreased CD80 amounts on Raji (CD80+CD86+PD-L1-mCherry+) cells in the presence of Jurkat (CTLA-4-mGFP+) cells, and this effect was substantially reversed by the CTLA-4 blockade antibody ipilimumab (Figure 5B). Longer co-culturing potentiated the effects of these checkpoint inhibitors (Figure S5). Acquisition of CD80 by Jurkat (CTLA-4-mGFP+) cells was also evident: after the incubation with Raji (CD80-mApple+) cells, Jurkat (CTLA-4-mGFP+) cells became fluorescent in the mApple channel (Figure S6A). Moreover, co-expression of PD-L1 with CD80-mApple on Raji cells decreased the mApple signal in Jurkat (CTLA-4-mGFP+) cells. As expected, blocking PD-L1 by atezolizumab increased the mApple signal in Jurkat (CTLA-4-mGFP+) cells, and co-blockade of CTLA-4 by ipilimumab over-rode the atezolizumab effect, inhibiting mApple transferto Jurkat cells (Figure S6A). We also noted that Jurkat (CTLA-4-mGFP+) cells express low amounts of PD-L1 (Figure S6B), raising the question of whether the atezolizumab effect on CD80 amounts was because of the blockade of Jurkat PD-L1. We ruled out this possibility by using WT Raji (CD80+) cells, which completely abolished the atezolizumab effect (Figure S6C).
We further tested the PD-L1 effect on trans-endocytosis in a co-culture system containing primary mouse DCs and regulatory T (Treg) cells, a type of suppressive T cell that depletes CD80 and CD86 in vivo through CTLA-4-mediated trans-endocytosis (Hou et al., 2015; Schmidt et al., 2009). Indeed, Treg cells, but not conventional T (Tconv) cells, decreased CD80 amounts on DCs. The presence of anti-PD-L1 further decreased the CD80 amounts, and co-treatment of anti-CTLA-4 restored the CD80 amounts. By contrast, CD86 amounts were unaffected by anti-PD-L1 treatment (Figure 5C).
Next, we examined the potential functional consequence of anti-PD-L1 induced depletion of CD80 from APCs. For this purpose, we co-cultured Jurkat (CTLA-4-mGFP+) cells with Raji (CD80-mApple+PD-L1-SNAP+) cells and determined how atezolizumab affects interleukin-2 (IL-2) secretion; pembrolizumab (anti-PD-1) was used as a control. Although pembrolizumab had little effect on IL-2 production, as expected due to the marginal expression of PD-1 on Jurkat (CTLA-4-mGFP+) cells (data not shown), atezolizumab significantly decreased IL-2 amounts (Figure 5D, gray bars). This is likely because atezolizumab-induced, CTLA-4-mediated CD80 depletion from Raji cells compromised CD28 co-stimulation in Jurkat (CTLA-4-mGFP+) cells. Consistent with this model, co-blockade of CTLA-4 with ipilimumab restored IL-2 amounts (Figure 5D, gray bars). Moreover, replacement of Jurkat (CTLA-4-mGFP+) cells with Jurkat (WT) cells abrogated the IL-2 suppression effect of atezolizumab (Figure 5D, black bars), providing additional evidence that atezolizumab can inhibit T cell function by promoting CTLA-4-mediated CD80 trans-endocytosis.
Anti-PD-L1, but not Anti-PD-1, Depletes CD80 on Tumor-Infiltrating APCs
Finally, we asked how in vivo administration of anti-PD-L1 affects the CD80 expression amounts on APCs. Treatment of 4T1 murine breast cancer model implanted BALB/c mice with anti-PD-L1 led to a significant decrease of CD80, but not CD86 amounts on tumor-infiltrating DCs and macrophages (Figure 6A). By contrast, administration of anti-PD-1 did not affect CD80 or CD86 amounts on these cells (Figure 6A). We also detected a similar anti-PD-L1-mediated CD80 depletion on tumor-infiltrating APCs in CT26 murine colon carcinoma (Figure 6B). Furthermore, co-administration of anti-CTLA-4 was able to recover the CD80 amounts on multiple types of CT26-infiltrating DCs in anti-PD-L1-treated animals (Figure 6C). In conjunction with cell culture assays, these in vivo experiments demonstrate a distinction between anti-PD-L1 and anti-PD-1 antibodies and a rationale to combine anti-PD-L1 and anti-CTLA-4 in cancer immunotherapy.
Figure 6. Anti-PD-L1, but Not Anti-PD-1 Reduces CD80 Amounts on Tumor Infiltrating APCs.

(A) On the left are flow-cytometry histograms of CD80 and CD86 surface levels on DCs and macrophages (Macs) isolated from tumor tissues of 4T1 implanted BALB/C female mice treated with anti-PD-L1 (magenta traces), anti-PD-1 (cyan traces), or control IgG (black traces). On the right are bar graphs summarizing the MFI of CD80 and CD86 staining under the indicated conditions. Data are shown as mean ± SEM, n ≥ 3 mice.
(B) Same as (A), except using CT26-implanted mice. Data are shown as mean ± SEM, n ≥ 3 mice.
(C) At the top are flow-cytometry histograms of CD80 expression amounts on the indicated types of DCs isolated from CT26 tumor tissues, 24 h after subcutaneous injection of anti-PD-L1 plus either anti-CTLA-4 (green traces) or IgG control (black traces). On the bottom is a bar graph summarizing the MFI of CD80 staining histogram under the indicated conditions. Shown is the mean ± SEM of n ≥ 3 mice. Unpaired two-tailed Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Combining the use of cell-free reconstitution, cell cultures, and checkpoint inhibitors, we showed that PD-L1:CD80 bind exclusively in cis but not trans and that this cis-interaction on APCs simultaneously restricts both PD-L1:PD-1 and CD80:CTLA-4 co-inhibitory axes while leaving the co-stimulatory CD80:CD28 interaction unaffected.
The original report that CD80-expressing cells bind a PD-L1-Ig-coated surface (Butte et al., 2007) led the field to assume a trans-interaction model. However, Ostrand-Rosenberg and colleagues reported that the overexpression of CD80 on PD-L1+ tumor cell lines blunts the tumor protection role of PD-L1 (Haile et al., 2011), indicating that CD80 can interact with PD-L1 in cis to block PD-L1:PD-1 trans-interaction. Moreover, Freeman and colleagues provided more concrete evidence that PD-L1 and CD80 bind only in cis but not trans (Chaudhri et al., 2018). Here, we confirmed this exclusive cis-nature of PD-L1:CD80 interaction by using orthogonal assays and further examined its effects on PD-1, CD28, and CTLA-4 axes.
Cis-interactions involving PD-L1 have emerged as a mechanism to restrict PD-L1:PD-1 signaling. We recently showed that PD-L1:PD-1 cis-interaction can block PD-L1:PD-1 trans-interaction and PD-1 signaling in cell culture systems (Zhao et al., 2018). Moreover, Sugiura et al., (2019) reported that point mutations that disrupt CD80:PD-L1 cis-interaction on the APCs inhibits T cell activity in both autoimmunity and tumor models, demonstrating the biological significance of PD-L1:CD80 cis-interaction. Here, using quantitative biochemistry and cell biology, we showed that 3.5-fold excess of cis-CD80 can block PD-L1:PD-1 interaction by 80%. Thus, by sequestering PD-L1 in cis, APC-intrinsic CD80 and PD-1 might function as rheostats for PD-L1:PD-1 signaling. We propose CD80 as a biomarker for PD-1-targeted immunotherapy. PD-L1 expressions on tumor cells and tumor-infiltrating APCs have been used to predict patient response for PD-L1 and PD-1 inhibitors (Herbst et al., 2014; Kluger et al., 2017; Lin et al., 2018; Tang et al., 2018). However, many PD-L1+patients fail to respond to PD-1 or PD-L1 inhibitors. Although tumor mutational burden and immune infiltrates are key factors to consider (Zappasodi et al., 2018; Zou et al., 2016), additional mechanisms likely contribute. The ability of cis-CD80 to neutralize PD-L1 indicates that patients with high CD80 expression on APCs would have less ongoing PD-1 signaling. We also note that cis-CD80 can block at least some clones of PD-L1 antibodies, consistent with a recent report (Chaudhri et al., 2018). Thus, PD-L1 immunohistochemistry might report the levels of CD80-free, PD-1 binding competent PD-L1 rather than total PD-L1. A reliable measurement of PD-L1 amounts on CD80-expressing cells would require a careful selection of PD-L1-staining antibodies.
In addition to CD80-mediated blockade of PD-L1:PD-1 signaling, we also described a role of PD-L1 in attenuating CD80:CTLA-4 interaction, independent from its well-established role in triggering PD-1 in trans. We note that a recent study found no effect of cis-PD-L1 on CD80:CTLA-4 interaction (Sugiura et al., 2019), but this study used a CTLA-4 pentamer, the artificially high avidity of which might have masked the cis-PD-L1 effects. Here, using CTLA-4 dimers that resemble its physiological state, we found that cis-PD-L1 inhibits CD80:CTLA-4 interaction. We further showed that PD-L1:CD80 cis-heterodimerization disrupts CD80 homodimerization, thereby preventing the formation of high-avidity CD80:CTLA-4 lattice. Thus, PD-L1:CD80 cis-interaction inhibits PD-1 and CTLA-4 pathways through distinct mechanisms. For the former, cis-CD80 directly competes with PD-1 for an overlapping interface on PD-L1 (Chaudhri et al., 2018; Sugiura et al., 2019). For the latter, cis-PD-L1 disrupts CD80 homodimerization to decrease the avidity of CD80:CTLA-4 interactions. Unlike CTLA-4, CD28 binds monovalently to CD80 (Collins et al., 2002; Evans et al., 2005), rendering CD80:CD28 interaction resistant to cis-PD-L1 inhibition. Therefore, by dissolving CD80 homodimers to selectively weaken CTLA-4 binding, cis-PD-L1 might “steer” CD80 from CTLA-4 to the much more abundantly expressed CD28. We speculate that compounds or point mutations that abrogate PD-L1:CD80 cis-interaction (Sugiura et al., 2019) might promote both PD-1 and CTLA-4 activities.
Conceivably, a substantial population of PD-L1 molecules on APCs exist as cis-heterodimers with CD80 and vice versa. PD-L1:CD80 heterodimers are defective in either PD-1 or CTLA-4 interactions but bind and activate CD28 equally well as free CD80. When CD80 is in large excess of PD-L1, CD80 would block the PD-L1:PD-1 pathway, and the excess free CD80 would allow CTLA-4 to function as a dominant immune checkpoint, either through trans-endocytosis or a cell-intrinsic mechanism (Kong et al., 2014; Lee et al., 1998). Conversely, when PD-L1 is in excess of CD80, those in cis-complex with CD80 would mitigate CTLA-4 and Treg functions by protecting CD80 from trans-endocytosis, and those CD80-free PD-L1 would trigger PD-1 in trans. Therefore, the relative abundance of PD-L1 and CD80 might help predict the functions of APCs and the relative strength of PD-1 and CTLA-4 signaling in the associated T cells, thereby serving as a potential biomarker for choosing anti-CTLA-4 versus anti-PD-1 versus anti-PD-L1 therapies.
PD-L1 blockade antibodies have achieved some clinical success in part attributed to their known ability to block the inhibitory PD-L1:PD-1 interaction. Here, we identified an opposing “side effect” of anti-PD-L1: by disrupting PD-L1:CD80 heterodimers, anti-PD-L1 licenses high-avidity CD80:CTLA-4 interactions to unleash Treg-mediated depletion of CD80 from APCs, thereby inhibiting CD28 co-stimulation. We speculate that the net activities of PD-L1 and anti-PD-L1 depend on the relative amounts of PD-1 and CTLA-4. Our model predicts that as PD-1 amounts increase on T cells, PD-L1 would switch from a positive regulator (CD80 protecting) to a negative regulator (PD-1 triggering).
Because CD80 depletion by anti-PD-L1 depends on CTLA-4 activity, this “side effect” can be prevented by CTLA-4 blockade. This finding, in conjunction with other established crosstalk between the PD-1 and CTLA-4 axes (Wei et al., 2018), provide a rationale for co-blockade of PD-L1 and CTLA-4 in cancer immunotherapies. In addition, despite the general assumption that anti-PD-1 and anti-PD-L1 have similar therapeutic mechanisms, we show that anti-PD-L1 but not anti-PD-1 depletes CD80 on APCs. Even though clinical trials to directly compare PD-1 inhibitors and PD-L1 inhibitors are unavailable, meta-analysis of clinical trials suggest that anti-PD-1 has a significantly higher overall response rate than anti-PD-L1 (Brahmer et al., 2012; Passiglia et al., 2018; Zhang et al., 2018). Altogether, the present study suggests that compounds that selectively block PD-L1:PD-1 trans-interactions, but not PD-L1:CD80 cis-interactions, might prove more effective in stimulating the immune system. As these compounds emerge for research and clinical use, future studies will test the efficacy of selective blockade of PD-L1 trans or cis interaction versus blocking both interactions in immunotherapies.
Finally, whereas the present study focused on cis-interaction on APCs, PD-L1 and CD80 are also expressed on antigen-experienced T cells (Keir et al., 2008). Although we and Sugiura et al., (2019) both report immunostimulatory functions of PD-L1:CD80 cis-interaction, earlier studies indicate inhibitory roles of this interaction (Butte et al., 2007; Latchman et al., 2004; Park et al., 2010). This discrepancy might be attributed to PD-L1, CD80, and their cis-interactions on the T cell side. The overall function of PD-L1:CD80 interaction likely depends on the spatio-temporal dynamics of surface expressions and cell types that mediate the immune response, a topic that warrants further investigation.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Enfu Hui (enfuhui@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Cultures
HEK293T cells and Raji B cells were obtained from R. Vale (University of California San Francisco), Jurkat T cells from A. Weiss (University of California San Francisco), HEK293F cells from Dr. Andrew Ward (Scripps Research). HEK293T cells were maintained in DMEM medium (Genesee Scientific, 25–501) supplemented with 10% fetal bovine serum (Omega Scientific, FB-02), 100 U/mL of Penicillin (GE Healthcare, SV30010), and 100 μg/mL of Streptomycin (GE Healthcare, SV30010)) at 37°C, 5% CO2. Jurkat T cells and Raji B cells were maintained in RPMI medium (corning, 10–041-CM) supplemented with 10% fetal bovine serum, 100 U/mL of Penicillin, and 100 μg/mL of Streptomycin) at 37°C, 5% CO2. HEK293F cells were maintained in FreeStyle 293 Expression Medium (Thermo Fisher Scientific, 12338018) at 37°C, 8% CO2. OT-1 splenocytes were harvested from C57BL/6-Tg (TcraTcrb) 1100Mjb/J (OT-1) mice (Jackson Laboratory) and maintained in OT-1 culture medium (RPM11640 supplemented with 10% fetal bovine serum, 1 mM Sodium Pyruvate (Corning, 25–000-CI), 50 μM β-mecaptoethanol (Fisher Scientific, ICN19024283), 100 U/mL of Penicillin, and 100 μg/mL of Streptomycin) at 37°C, 5% CO2. Human peripheral blood CD14+ monocytes (iXCells, 10HU-008) were cultured in RPMI medium supplemented with 50 ng/mL of granulocyte-macrophage colony-stimulating factor (GM-CSF) and 50 ng/mL of interleukin-4 (IL-4) in a 37°C, 5% CO2 incubator.
4T1 tumor cells, a metastasizing mammary adenocarcinoma triple-negative mouse cell line, were maintained as monolayer cultures in DMEM supplemented with 10% FBS, sodium pyruvate, nonessential amino acids, L-glutamine and vitamins (Life Technologies, GIBCO BRL, Gaithersburg, MD). CT26 was obtained from the ATCC and cultured in RPM11640 supplemented with 10% FBS, sodium pyruvate, nonessential amino acids, L-glutamine, β-mercaptoethanol, penicillin, streptomycin, and sodium bicarbonate (all from GIBCO). Cultures were rendered mycoplasma free upon initiation from outside source using ciprofloxacin. Mycoplasma free cell cultures were incubated at 37°C, 5% CO2. Tumor cells were harvested with 0.25% trypsin-0.02% EDTA. Trypsin was neutralized with medium containing 10% FBS, washed and suspended in PBS for injection. Cells were used for injections only if the viability was greater than 90% as determined by Trypan Blue (Sigma).
METHOD DETAILS
Recombinant Proteins
Recombinant His-tagged mouse and human PD-L1, PD-L2, CD80, CD86, and ICAM used in OT-1–SLB assays and LUVs-SLB adhesion assays were purchased from Sino Biological. For Raji cells staining, recombinant PD-1–huFc, CD28–moFc, CTLA-4–moFc, and isolated moFc domain were purchased from ACROBiosystems. Recombinant CD28–huFc, CTLA-4–huFc, and isolated huFc domain were purchased from Sino Biological and the aggregate fractions were removed by gel filtration. For FRET assay using LUVs, the extracellular portion (ectodomain) of human PD-L1 (aa 19–239), PD-L2 (aa 20–220), CD80 (aa 35–242), or CD80 mutant (aa 35–242, I92R) was expressed in HEK293F cells, as described previously (Murin et al., 2014). The N terminus of each ectodomain was fused with the signal peptide of HIV envelope glycoprotein gp120 followed by a twinstrep tag (amino acids sequence: WSHPQFEKGGGSGGGSGGSAWSHPQFEK) and a SNAP-tag, and the C terminus of each fused to a decahistidine tag. GCN4-tagged huCTLA-4 (aa 36–161) and huCD28 (aa19–152) were constructed in a similar manner except replacing the decahistidine tag with a GCN4 homodimerization motif fused with a hexahistidine tag. Monomeric human CTLA-4 (aa 36–161) and CD28 (aa19–152) were produced using the same expression plasmid except removing the GCN4 sequence. For the SPR experiments, monomeric huCTLA-4 was constructed and produced in a similar manner except lacking the twinstrep tag. All His-tagged proteins were purified from the cell culture medium using HisTrap Excel column (GE Healthcare, 17371206) and eluted using buffer containing 50 mM HEPES-NaOH, pH 8.0, 150 mM NaCl, 0.5 M imidazole. For His-tag free huCD80 used in Figure 1D, the His-tag coding sequence was removed from the expression construct, and the expressed protein purified with a StrepTrap HP column (GE Healthcare, 28907547) in 100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 8.0 and eluted with the same buffer containing 2.5 mM desthiobiotin (Sigma-Aldrich, D1411). The ectodomain of mouse MHC-I molecule H2-Kb was produced as a disulfide-stabilized single chain trimer with a covalently linked ovalbumin (OVA) peptide SIINFEKL (Mitaksov et al., 2007), and a C-terminal His-tag, using the Bac-to-Bac baculovirus expression system, as previously described (Hui et al., 2017). All affinity-purified proteins were gel filtered using a Super-dex 200 Increase 10/300 GL column (GE Healthcare, 28990944) in HEPES buffered saline (50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 10% glycerol). Gel filtered proteins were labeled with either SNAP-Cell 505 (NEB, S9103S), SNAP-Cell TMR (NEB, S9105S) or SNAP-Cell 647-SiR (NEB, S9102S) following manufacturer’s instructions. Free dyes were then removed using Zeba Spin Desalting Columns (Thermo Fisher Scientific, P187769). All proteins were quantified by SDS-PAGE and Coomassie blue staining, using bovine serum albumin (BSA, Thermo Scientific, 23209) as a standard.
Cell Lines
Genotypes of Raji and Jurkat cells used in this study are summarized in Table S3. To generate CD80-deleted Raji (CD80−CD86+) cells, two PX330-GFP vectors coding different CD80 sgRNAs were electroporated into WT Raji (CD80+CD86+) cells using Cell Line Nucle-ofector Kit V (LONZA, VACA-1003). Electroporated cells were recovered for 2 days at 37°C, 5% CO2. GFP-positive cells were then sorted by flow cytometry and maintained in culture medium for 1 week, after which the cells were stained with allophycocyanin anti-CD80 (Biolegend, 305220) and CD80-negative cells were sorted by flow cytometry. Raji (CD80+CD86−) cells were generated in the same manner as CD80-deleted Raji cells except using CD86 sgRNAs, and sorted for CD86-negative cells after BV421 anti-CD86 (Biolegend, 305425) staining. Raji (CD80−CD86−) cells were generated by deleting CD80 from Raji (CD80+CD86−) cells. CD80 and CD86 double negative cells were sorted after co-staining with allophycocyanin anti-CD80 and BV421 anti-CD86.
Each gene of interest was introduced into Jurkat and Raji cells via lentiviral transduction, as described previously (Zhao et al., 2018). All transduced genes were driven by SFFV promoter except specified otherwise. For Figures 2 and S1A–S1E, Raji (CD80−PD-L1–mCherry+) and Raji (CD80loPD-L1–mCherry+) cells were generated by transducing PD-L7–mCherry to Raji (CD80−CD86+) cells and WT Raji (CD80+CD86+) cells, respectively. Raji (CD80hiPD-L1–mCherry+) cells were generated by further transducing Raji (CD80loPD-L1–mCherry+) cells with CD80–CLIP. Jurkat (PD-1-mGFP+) cells were generated previously by transducing PD-7–mGFP into WT Jurkat (CD28+/+) cells (Hui et al., 2017). For Figures 3A, 3B, 3F, 4A–4C, S1G, S1H, and S3, Raji (CD80+ CD86−PD-L1–mCherry+) cells were generated by transducing PD-L7–mCherry into Raji (CD80+CD86−) cells. Jurkat (CD28–mCherry+) cells used in Figure 3D were generated by transducing WT Jurkat (CD28+/+) cells with CD28–mCherry. Raji (CD80–mGFP+ CD86+), Raji (CD80–mGFP+CD86+CLIP–PD-L1+), and Raji (CD80−CD86+CLIP–PD-L1+) cells were generated by transducing dSV40-promoter-driven CD80–mGFP and/or SFFV-promoter-driven CLIP–PD-L7 into CD80−Raji (CD80−CD86+) cells. For Figure 3E, Raji (CD80+CD86+PD-L1–mCherry+) cells were the same as Raji (CD80loPD-L1–mCherry+) used in Figure 2 that express low, endogenous levels of both CD80 and CD86. For Figure 4D, to achieve a wide range of CD80 expression, Raji (CD80−CD86−) cells were transduced with both PD-L7–mCherry and CD80–mGFP but only sorted for high PD-L1 expression. For Figure 4H, Raji (CD80-mGFP+CD86−PD-L1–SNAP+) was made by sequentially transducing Raji (CD80−CD86−) with dSV40-promoter-driven CD80–mGFP and SFFV-promoter-driven PD-L1–SNAP. Jurkat (CTLA-4–mGFP+) cells used in trans-endocytosis assays and IL-2 assays in Figures 5, S5, and S6 were generated by transducing dSV40-promoter-driven CTLA-4–mGFP to WT Jurkat (CD28+/+) cells. Raji (CD80+PD-L1–mCherry+) cells used in Figures 5A, 5B, S5, and S6 were generated by transducing PD-L1–mCherry to WT Raji (CD80+) cells. Raji (CD80–mApple+) cells used in Figure S6A were generated by transducing dSV40-promoter-driven CD80–mApple into Raji (CD80−CD86−) cells. Raji (CD80–mApple+PD-L1–SNAP+) cells used in Figures 5D and S6 were generated by transducing PD-L1–SNAP to Raji (CD80–mApple+) cells.
Confocal Microscopy Based FRET Assay with HEK293T Cells
For Figure 1B and 1C, pHR plasmid encoding CLIP-tagged full length human PD-L1 (CLIP–PD-L1) or PD-L2 (CLIP–PD-L2) was co-transfected with pHR encoding either SNAP-tagged full length human CD80 (SNAP–CD80) or CD86 (SNAP–CD86) into HEK293T cells using polyethylenimine (Fisher Scientific, NC1014320). 72 h post transfection, cells were trypsinized and seeded on poly-D-lysine (Sigma-Aldrich, P6407) treated 96 wells plate with a glass bottom (Dot Scientific, MGB096-1-2-LG-L). 24 h later, cells were labeled with CS547 (NEB, S9233S) and SSAF647 (NEB, S9136S) at 37°C, 5% CO2 for 30 min, and washed 3 times with 1x PBS (pH 7.4). Labeled cells were then fixed with 4% paraformaldehyde (PFA, Fisher Scientific, 50980494) and used for the FRET assay. Images were acquired with an Olympus FV1000 confocal microscope by exciting CS547 (energy donor) at 543 nm and SSAF647 (energy acceptor) at 635 nm. For Figure 4E, SNAP-tagged full length CD80 (SNAP–CD80), CD80 mutant (SNAP–CD80 I92R) or CD86 (SNAP–CD86) was transfected to either HEK293T cells or CLIP–PD-L1 transduced HEK293T cells using polyethylenimine. 24 h post transfection, cell was stained with a 1:1 mixture of SS549 (energy donor, NEB S9112S) and SSAF647 (energy acceptor) at 37°C, 5% CO2 for 30 min, and washed 3 times with 1x PBS (pH 7.4). For the atezolizumab treated condition, 20 μg/mL atezolizumab was included in the staining solution. After staining, cells were washed 3 times, fixed by 4% PFA and used for the FRET assay. Images were acquired with an LEICA SP8 confocal microscope by exciting SS549 at 561 nm and SSAF647 at 633 nm. Donor images before and after acceptor photobleaching were acquired for FRET analysis using ImageJ (Fiji) with the AccPbFRET plugin, as described (Roszik et al., 2008).
FRET Assays with Protein-Reconstituted LUVs
Synthetic 1,2-dioleyl-sn-glycero-3-phosphocholine (POPC, 850457C) and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt, DGS-NTA-Ni, 790404C) were purchased from Avanti Polar Lipids. LUVs consisting of 80% POPC and 20% DGS-NTA-Ni were generated by extrusion, as described (Hui and Vale, 2014). Briefly, desired lipids were mixed in chloroform, dried under a nitrogen stream and desiccated in a vacuum container for 1 h. The desiccated lipid film was resuspended in 1x PBS and extruded for 20 times through a pair of polycarbonate filters containing pores of 200 nm diameter. For Figure 1D, 0.23 nM LUVs in PBS containing 1.5 mg/mL BSA and 1 mM TCEP were incubated with 25 nM SC505-labeled PD-L1–His alone, with 25 nM SC505*PD-L1–His and 75 nM atezolizumab combined, or with 25 nM SC505*PD-L2–His alone, in a 96-well solid white microplate (Greiner Bio-One, 655075), during which the SC505 fluorescence was monitored in real time using a plate reader (Tecan Spark 20) with 504-nm excitation and 540-nm emission. Following 1 h incubation, TMR-labeled CD80–His (TMR*CD80–His) or TMR*CD80 lacking a His-tag was injected and SC505 fluorescence monitored for an additional 1 h. Forthe trans-interaction control, TMR*CD80–His was pre-attached to a different set of LUVs prior to injection to the SC505*PD-L1 coupled LUVs. For Figure 4F, 0.23 nM LUVs in PBS containing 1.5 mg/mL BSA and 1 mM TCEP were incubated with 50 nM SC505*CD80–His for 1 h, and challenged sequentially with 50 nM TMR*CD80–His and indicated concentrations of unlabeled PD-L1–His, with the SC505 fluorescence monitored over the entire time course. For atzeolizumab treatment, PD-L1–His was incubated with atezolizumab (three-fold molar excess) for 30 min before added to the reaction. For Figure 4G, 0.23 nM LUVs were incubated with 50 nM of either SC505-labeled CD80–His, CD80 (I92R)–His, or PD-L1–His for 1 h, followed by injection of 50 nM TMR-labeled CD80–His or CD80 (I92R)–His. Data were normalized by the mean fluorescence intensity of the last 10 data points before the addition of TMR*CD80–His or TMR*CD80 (no His) and plotted with GraphPad Prism 5.0. All experiments described in this section were conducted at room temperature.
LUVs–SLB Adhesion Assay
To form SLB, a glass bottom 96-well plate was incubated with 5% Hellmanex III (Helma Analytics, Z805939) overnight on a 50°C heat pad, thoroughly rinsed with ddH2O and sealed with a Nunc sealing tape (Thermo Fisher Scientific, 232698). The desired wells were washed twice with 5 M NaOH (30 min each), and three times with 500 μL ddH2O followed by equilibration with PBS. Small unilamellar vesicles (SUVs, consisting of 97.5% POPC, 2% DGS-NTA-Ni and 0.5% PEG5000 PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-5000], ammonium salt) (Avanti Polar Lipids, 880230C) were prepared as described previously (Hui et al., 2017), added to the cleaned wells containing 200 μL 1x PBS, and incubated for 90 min at 50°C, followed by 30 min at 37°C to induce SLB formation. The SLBs were then rinsed thoroughly with PBS to remove excess SUVs, and blocked with 1 mg/mL BSA in 1x PBS for 30 min at 37°C. The SLBs were then overlaid with 200 μL of either 3 nM human PD-1-His (Sino Biological, 10377-H08H), 3 nM human CD80–His (Sino Biological, 10698-H08H), or 3 nM human CD86–His (Sino Biological, 10699-H08H). After 1 h incubation at 37°C, the unbound proteins were washed away using excess 1x PBS containing 1 mg/mL BSA. The plate was incubated at 37°C for another 30 min and washed again with PBS containing 1 mg/mL BSA to remove dissociated proteins, leaving the SLB with stably bound proteins (Nye and Groves, 2008). Bodipy-PE LUVs (lipids composition: 89.7% POPC + 10% DGS-NTA-Ni + 0.3% Bodipy-PE (N-(4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Propionyl)-1,2-Di-hexadecanoyl-sn-Glycero-3-Phosphoethanolamine, Triethylammonium Salt (Thermo Fisher Scientific, D3800)) were prepared by the aforementioned extrusion method. 0.23 nM Bodipy-PE LUVs were then incubated with 8.3 nM human PD-L1–His (Sino Biological, 10084-H08H) for 90 min in 1x PBS with 1 mg/mL BSA at room temperature, to ensure stable, 100% protein binding. The lack of free proteins in solution was confirmed by SDS-PAGE. The protein-bound LUVs were then added onto either PD-1, CD80, or CD86 functionalized SLBs. After 10-min incubation, unbound LUVs were washed away with excess 1x PBS and the SLB-captured LUVs visualized and recorded by a Nikon Eclipse Ti TIRF microscope equipped with a 100x Apo TIRF 1.49 NA objective, controlled by the Micro-Manager software (Edelstein et al., 2014). The fluorescence intensity of LUVs from the Bodipy (488 nm) channel in the TIRF field was calculated using the ImageJ software.
OT-1–SLB TIRF Microscopy Assay
OT-1 primary T cells were retrovirally transduced with either mouse PD-1–mCherry or mouse CD28–mGFP. Retroviruses were produced as described previously (Hui et al., 2017). Freshly harvested OT-1 splenocytes were stimulated with 10 nM SIINFEKL peptide (Anaspec, AS-60193–1) in OT-1 culture medium supplemented with 100 U/mL mouse recombinant IL-2 (Thermo Fisher Scientific, 14802164) at 37°C, 5% CO2. 36 h later, cells were resuspended in retrovirus supernatants containing 8 μg/mL Lipofectamine and 100 U/mL mouse recombinant IL-2, spin-infected at 35°C, 1000x g for 120 min, and incubated at 37°C, 5% CO2 overnight. The virus supernatant was replaced with fresh OT-1 culture medium supplemented with 10 nM SIINFEKL peptide and 100 U/mL mouse recombinant IL-2 the second day and cells incubated for another 48–96 h before microscopy. For TIRF microscopy, the 96-well plate was treated with SUVs to form SLB as described above. For Figure 2B, SLB was functionalized by a mixture of 5 nM pMHC-I-His, 2 nM mouse ICAM–His (Sino Biological, 50440-M08H) and either 3 nM mouse PD-L1–His (Sino Biological, 50010-M08H), 3 nM mouse PD-L1–His plus 9 nM mouse CD80–His (Sino Biological, 50446-M08H), or 3 nM mouse PD-L1–His plus 9 nM mouse CD86–His (Sino Biological, 50068-M08H). For Figure 3C, SLB was functionalized by a mixture of 5 nM pMHC-I–His, 2 nM mouse ICAM–His and either 3 nM mouse CD80–His, 3 nM mouse CD80–His plus 9 nM mouse PD-L1–His, or 3 nM mouse CD80–His plus 9 nM mouse PD-L2–His (Sino Biological, 50804-M08H). Transduced OT-1 cells were harvested via centrifugation at 200x g for 4 min, incubated with 10 μg/mL AF647-labeled mouse TCRβ antibody (Biolegend, H57–597) for 30 min on ice, washed 3 times with imaging buffer, and then plated onto functionalized SLBs. TIRF images were acquired at 37°C on a Nikon Eclipse Ti microscope equipped with a 100x Apo TIRF 1.49 NA objective, controlled by the Micro-Manager software and analyzed with ImageJ. Clustering indices were calculated by dividing the fluorescence intensity of PD-1 or CD28 microclusters, identified using the “threshold” plug in in ImageJ, by the total fluorescence intensity of the respective channel of the entire cell.
Jurkat–Raji Conjugation Assay
For cell conjugation assay, Raji B cells were pre-pulsed with 30 ng/mL SEE superantigen alone (Figure 2C, Toxin Technology, ET404) or SEE together with 1 μM CLIP-Surface 647 (Figure 3D, NEB, S9234S) in RPMI medium for 30 min at 37°C. Cells were then washed twice to remove free SEE and dye. Following antigen loading, 4 × 105 antigen-loaded Raji B cells and 4 × 105 Jurkat T cells were precooled on ice and mixed in a 96-well plate. The plate was then centrifuged at 290× g for 1 min at 4°C to initiate cell–cell contact, and immediately transferred toa37°C water bath. Two min later, cells were resuspended and fixed with 1% PFAand loaded into a 96-well glass-bottom plate for confocal microscopy assays. Images were acquired with an Olympus FV1000 confocal microscope and processed, and quantified using ImageJ. Interface enrichment indices of PD-1 and CD28 on Jurkat cells and PD-L1 and CD80 on Raji cells were computed by dividing the fluorescence density at the interface by the fluorescence density of the cell membrane excluding the interface. Fluorescence density was calculated as fluorescence intensity divided by area. The interface was defined as the conjugated area between Jurkat and Raji cells based on the DIC images.
For examining receptor phosphorylation in Figures 2D, 3E, and 3F. Serum-starved Jurkat cells and SEE-loaded Raji cells (2 × 106 each) were co-pelleted as described above. The cell pellets were lysed with ice cold NP40 buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP40,1 mM EDTA, 5% glycerol, 1 mM PMSF, 10 mM Na3VO4,10 mM NaF) at indicated time points. For 0 min samples, Raji-Jurkat mixtures were lysed prior to centrifugation. Lysates were centrifuged at 15,000× g for 10 min. PD-1-mGFP or CD28 were immunoprecipitated by using GFP-Trap (Chromotek, gta-20) or anti-CD28 antibody (Bio X Cell, BE0291) coated Protein G Dynabeads (Thermo Fisher Scientific, 10004D), respectively. Equal fractions of the immunoprecipitates were subjected to SDS-PAGE and blotted with anti-p85 antibody (Cell Signaling Technology, 4292), anti-phosphotyrosine antibody (Sigma-Aldrich, P4110), anti-SHP2 antibody (gift from A. Veillette, Montreal Clinical Research Institute), or GFP antibody (Thermo Fisher Scientific, A6455). Whole cell lysates were blotted with anti-GAPDH antibody (Proteintech, 10494–1-AP).
For IL-2 assay in Figure 5D, Raji B cells were pre-loaded with 30 ng/mL SEE for 30 min at 37°C. 2 × 105 serum-starved Jurkat T cells were co-pelleted with 1 × 105 SEE-loaded Raji B cells in a 96-well plate in triplicate wells and the supernatants were collected after 6 h. For antibody treatment conditions, 20 μg/mL pembrolizumab or ipilimumab were preincubated with Jurkat cells, and 20 μg/mL atezolizumab was preincubated with Raji cells for 30 min before mixing the 2 types cells together. All antibodies were kept in the medium during the co-culture. IL-2 concentrations were quantified by ELISA using Human IL-2 ELISA MAX Deluxe kit (BioLegend, 431804).
Raji B Cell Staining Assay
For cell staining in Figures 2, 3, and 4, Raji B cells were incubated with huFc- or moFc-fusion proteins, or the corresponding isolated Fc domain, at indicated concentrations for 35 min on ice, washed twice with PBS containing 2% FBS, and stained with AF647-labeled anti-human IgG Fc antibody (Biolegend, 409320) or AF647-labeled anti-mouse IgG Fc antibody (Biolegend, 405322). For cell staining with home-made CTLA-4-GCN4 and CD28-GCN4 in Figures 4 and S3, cells were incubated with SNAP-Cell-647-labeled CTLA-4–GCN4 and CD28–GCN4 on ice for 35 min and washed twice with PBS containing 2% FBS. For atezolizumab treated conditions, cells were pre-incubated with 20 μg/mL atezolizumab on ice for 30 min before the addition of Fc-fusion proteins and CTLA-4–GCN4. Cells were then analyzed by flow cytometry on LSRFortessa analyzer (BD Biosciences). Data were analyzed by FlowJo and plotted by GraphPad Prism 5. In Figures 3B and 4B, nonspecific binding between atezolizumab and the anti-mouse IgG Fc antibody was calculated based on the zero CD28–moFc and the zero CTLA-4–moFc conditions, and used to correct the staining signals of all atezolizumab-treated samples.
SPR Assay for PD-L1:CD80 Interaction
SPR experiments was conducted in a SensiQ Pioneer instrument. SNAP–CTLA-4–His6 (human CTLA-4 ectodomain fused with an N-terminal SNAP tag and a C-terminal hexahistidine tag) were expressed in the HEK293F cells, and purified using a HisTrap column. This home-made SNAP–CTLA-4–His6 and human CD80–His, purchased from Sino Biological, were subjected to gel filtration chromatography, and monomeric fractions of each collected and used in the SPR assays. PD-L1 (R&D Systems, 9049-B7) was biotinylated with EZ-Link NHS-LC-Biotin (Thermo Fisher, 21335) for 1 h at room temperature, and excess NHS-LC-Biotin removed by column centrifugation. A COOH5 sensor chip was functionalized with 7,500 response units (RU) neutravidin on each channel, and then captured 200 RU of biotinylated PD-L1. CD80 alone, CTLA-4 alone, or CD80 and CTLA-4 mixtures prepared in sample buffer (50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 10% glycerol), was injected at a rate of 5 μL/min for 3 min at 25°C, followed by 2 to 4 min of dissociation phase. The chip was regenerated using 50 mM sodium glycine, pH 9.5 for 30 s. Data was analyzed by using a double reference method with the Qdat software. Reference channel contained the identical level of neutravidin.
CD80:CTLA-4 Co-clustering Assay
For detecting CD80:CTLA-4 clusters on cell membrane in Figure 4H, Raji (CD80–mGFP+CD86−PD-L1–SNAP+) cells were incubated with 1 μg/mL SC647-labeled CTLA-4-GCN4 on ice for 35 min, with or without the presence of 20 μg/mL atezolizumab, followed by 2 washes with 1x PBS plus 2% FBS. Cells were then dropped on a poly-D-lysine treated 96-well plate for TIRF microscopy. Images were acquired at 37°C on a Nikon Eclipse Ti microscope equipped with a 100x ApoTIRF 1.49 NA objective, controlled by the Micro-Manager software. Microscopy images were then analyzed by ImageJ.
Quantification of PD-L1 and CD80 Expression
For flow cytometry based quantification in Figure S1, PD-L1 and CD80 were stained by PE anti-PD-L1 (eBioscience, 14-5983-82) and PE anti-CD80 (Biolegend, 305208) and their expression levels were quantified using the QUANTUM R-PE MESF kit (Bangs Laboratories Inc, 827), following manufacturer’s instructions. For immunoblot-based quantifications, total cell lysates were subjected to SDS-PAGE, transfected to a nitrocellulose membrane. Afterward, PD-L1 was probed by PE anti-PD-L1 (eBioscience, 14–598382) and detected by a Typhoon 5 Biomolecular Imager; CD80 was sequentially probed by anti-CD80 (Novus Biologicals, NBP2–25255) and DyLight488 anti-mouse IgG (Biolegend, 405310), then detected by Typhoon 5 Biomolecular Imager. Molecular densities were calculated assuming the following diameters:13 μm for Raji B cells (Hui et al., 2017) and 12.5 μm for DCs (Dumortier et al., 2005).
Flow Cytometry Analysis of Human Monocyte-Derived DCs
Human peripheral blood CD14+ monocytes were isolated from normal human peripheral blood (iXCells, 10HU-008), and cultured in RPMI medium supplemented with 50 ng/mL of GM-CSF (PeproTech, 300–03) and 50 ng/mL of IL-4 (PeproTech, 200–04) in 37°C, 5% CO2. After 5-day incubation, when the majority of monocytes differentiated to immature DCs, cells were re-cultured in RPMI medium supplemented with 50 ng/mL tumor necrosis factor alpha (TNF-a) (PeproTech, 300–01A) for an additional two days to generate mature DCs. PD-L1 and CD80 expression levels on both immature and mature DCs were measured by flow cytometry. Briefly, cells were pre-incubated with Human TruStain FcX (BioLegend, 422301) to block Fc receptors, and then incubated with the viability dye Ghost Dye Violet 450 (Tonbo Biosciences, 10140–978), followed by an antibody mixture containing PerCP/Cy5.5 anti-CD1a (Biolegend, 300129), FITC anti-CD14 (Biolegend, 301804), and PE anti-PD-L1 (eBioscience, 14-5983-82) or PE anti-CD80 (Biolegend, 305208). Stained cells were processed on a BD LSRFortessa cell analyzer and flow cytometry data were analyzed by FlowJo software.
Trans-endocytosis Assay
For analyzing CD80 trans-endocytosis in Figure 5, CTLA-4-mGFP expressing Jurkat or WT Jurkat cells were co-cultured with either PD-L1-mCherry expressing Raji or WT Raji cells. Briefly, 4 × 105 SEE-loaded Raji B cells and 4 × 105 Jurkat T cells were mixed and co-pelleted by centrifugation at 290 × g for 1 min, and incubated at 37°C, 5% CO2 for 30 min. After incubation, cells were resuspended and stained with allophycocyanin anti-CD80, PE anti-CD3 (Biolegend, 317308), and PE/Cy7 anti-CD20 (Biolegend, 302311) for flow cytometry with a BD LSRFortessa cell analyzer. CD3 and CD20 were used to gate Raji cells out from cell mixture. CD80 expression levels on Raji cells were then analyzed. For Figures S5 and S6C, experiments were done as in Figure 5 except pre-labeling Jurkat with 405-SE. ViaFluor SE (Biotium, 30068-T) before the co-culture to allow easier discrimination of Raji and Jurkat cells. For Figure S6A, Jurkat (CTLA-4–mGFP+) cells were co-cultured with either SEE-loaded Raji (CD80–mApple+) cells or SEE-loaded Raji (CD80–mApple+PD-L1–SNAP+) cells. For gating purpose, Raji cells were also pre-labeled with 405-SE. ViaFluor SE prior to the co-culture. mApple fluorescence on Jurkat cells was analyzed with a BD FACSAria flow cytometer. For blockade treatment in Figures 5B, S5, and S6A, Jurkat (CTLA-4–mGFP+) cells and SEE-loaded Raji (CD80+PD-L1–mCherry+) cells and Raji (CD80–mApple+PD-L1–SNAP+) cells were treated with 20 μg/mL of either atezolizumab or ipilimumab for 15 min at room temperature prior to mixing, and the blockade antibodies were kept in the co-culture until the staining step. For Figure S6C, Jurkat (CTLA-4–mGFP+) cells were pre-treated with 20 μg/mL of atezolizumab for 15 min at room temperature, washed twice to remove unbound antibodies, and then mixed with WT Raji (CD80+) cells. CD80 levels on Raji cells at indicated time points were normalized to that of time zero. For confocal microscopy, stained cells were plated on a poly-D-lysine treated 96-well plate and images acquired with an FV1000 confocal microscope in GFP and allophycocyanin channels.
DCs–Treg Cells Co-Culture Assay
Tconv cells and Treg cells were isolated using magnetic bead isolation system. In brief, total CD4+ T cells were first isolated from Foxp3Thy1.1 reporter mice (Liston et al., 2008) by using mouse CD4 T cell isolation kit (Biolegend, 48006), and stained with PE anti-Thy1.1 antibody (eBioscience, 12-0900-83). Next, Treg cells were further separated with Tconv cells by using anti-PE beads (Miltenyi Biotec, 130-048-801). DCs were isolated from Ly5.1+ mice by using CD11c isolation kit (Miltenyi Biotec, 130-108-338). 5 × 103 DCs were cultured with either 5 × 104 Tconv cells or 5 × 104 Treg cells in the presence or absence of 50 μg/mL anti-PD-L1 (Bio X Cell, BE0101) and 50 μg/mL anti-CTLA-4 (Bio X Cell, BE0164) in RPMI 1640 supplemented with 10% fetal bovine serum, 50 μM β-mecaptoethanol, 100 U/mL Penicillin, and 100 μg/mL Streptomycin, 0.5 μg/mL Anti-CD3 (Bio X Cell, BE0001–1) and 1 μg/mL lipopolysaccharide (LPS) (Enzo Life Sciences, ALX-581–008-L002). After 16 h, cells were first stained with Ghost Dye Red 780 (Tonbo Biosciences, 13–0865-T100), followed by surface antibody staining for Ly5.1 (eBioscience, 11-0453-82); Thy1.1 (eBioscience, 120900–83); CD11c (eBioscience, 45-0114-82); CD80 (Biolegend, 104733); CD86 (eBioscience, 17-0862-82); and PD-L1 (Biolegend, 124315). An LSRFortessa analyzer (BD Biosciences) was used for data collection, and FlowJo software was used for data analysis.
Animals
Female BALB/c mice at 8–10 weeks were purchased from Centro de Modelos Biológicos Experimentais (CeMBE) – PUCRS or Charles Rivers. Mice were bred and housed under pathogen-free conditions at CeMBE animal facility (PUCRS, Brazil) or at the University of California San Diego (UCSD) with ad libitum access to food and water. In all procedures, mice were anesthetized intraperitoneally with 100 μl 1x PBS containing 16.7 mg/mL ketamine (Cristália) and 3.3 mg/mL Xilazine (Syntec). All procedures were previously reviewed and approved by the Ethics Committee for the Use of Animals of Pontifícia Universidade Católica do Rio Grande do Sul (CEUA-PUCRS), under protocol CEUA 13/00379 or the UCSD IACUC under protocol S06201.
Treatment of CT26 and 4T-1 Tumor Bearing Mice with Immune Checkpoint Inhibitors
For CT26 experiments, 8–12 weeks old female BALB/C mice were transplanted with 5 × 106 CT26 cells in the subcutaneous space. Five days later, when the tumor was about 5 × 5 mm in diameter, 400 μg of anti-PD-1 (Bio X Cell, BE0273), anti-PD-L1 (Bio X Cell, BE0101), anti-CTLA-4 (Bio X Cell, BE0164), or control IgG (Bio X Cell, BE0090) were intraperitoneally injected. 24 h after antibody injection, the tumors were harvested from all animals, homogenized, and stained for surface expression of CD80 or CD86 on live (7AAD) cells obtained from the tumor. Antibodies used for flow cytometry included anti-CD11b (Biolegend, 101225), anti-CD11c (Biolegend, 117317), anti-F4/80, (Biolegend, 123113), anti-CD301b (Biolegend, 146803), anti-MHC class II (Thermo Fisher Scientific, 115321–82), anti-CD80 (Biolegend, 104713), anti-CD86 (Biolegend, 105005), and anti-PD-L1 (Biolegend, 124307). An FACSCantoII analyzer (BD Biosciences) was used for data collection. FlowJo software was used for data analysis.
For 4T-1 experiments, 6–8 weeks old BALB/C female mice received 1 × 106 4T-1 cells via subcutaneous injection adjacent to the mammary gland. Four days later, mice received an intraperitoneal injection of 10 μg lipopolysaccharide (Sigma). 200 μg anti-PD-L1 (Bio X Cell, BE0101), anti-PD-1 (Bio X cell, BE0146) or IgG control (Bio X cell, BE0089) was intraperitoneally injected about 10 days after the injection of tumor cells, when the mean tumor volume (V) reached about 100 mm3, estimated using: V = ab2/2, in which a is the longer diameter and b is the shorter diameter. 24 h after the antibody injection, tumors were harvested from all animals, homogenized. Fc receptors of single cell suspensions were blocked using 50 μl of 24G2 hybridoma cell supernatant supplemented with 10% mouse serum and 10% rat serum on ice for 20 min. Cells were subsequently stained for viability using a fixable viability dye (eBioscience, 650865) and with antibodies against CD45 (Biolegend, 103130), CD11c (Biolegend, 117317), CD11b (BD Biosciences, 550993), F4/80 (Biolegend, 123113), PD-L1 (BD Biosciences, 558091), CD80 (Biolegend, 104713) and CD86 (BD Biosciences, 553691), each conjugated to a different fluorochrome. Cells were acquired on FACSCanto II (BD Biosciences) by BD FACSDiva software (BD Biosciences). Gating was performed excluding doublets, followed by gating on live, CD45+ cells, from which DCs were identified as CD11c+ and Macs as CD11b+F4/80+. Data obtained were analyzed using FlowJo software (version 10, TreeStar).
QUANTIFICATIONS AND STATISTICAL ANALYSES
Unless otherwise indicated, data were reported as mean ± SEM, and number of replicates were indicated in figure legends. Curve fitting and normalization were performed in GraphPad Prism 5. Statistical significance was evaluated by unpaired two-tailed Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001) in GraphPad Prism 5. Data with p > 0.05 are considered statistically non-significant (ns).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human CD28 antibody | Bio X Cell | Cat # BE0291; RRID: AB_2687814 |
| GFP-Trap | Chromotek | Cat # gta-20 RRID: AB_2631357 |
| PI3 Kinase p85 antibody | Cell Signaling Technology | Cat # 4292; RRID: AB_329869 |
| Phosphotyrosine antibody | Sigma-Aldrich | Cat# P4110; RRID: AB_477342 |
| Human SHP2 antibody | A. Veillette, Montreal Clinical Research Institute | N.A. |
| GFP antibody | Thermo Fisher Scientific | Cat # A6455; RRID: AB_221570 |
| GAPDH antibody | Proteintech | Cat# 10494–1-AP; RRID: AB_2263076 |
| Pembrolizumab | Selleckchem | Cat # A2005; RRID: N/A |
| Atezolizumab | Selleckchem | Cat # A2004; RRID: N/A |
| Ipilimumab | Selleckchem | Cat # A2001; RRID: N/A |
| Mouse PD-1 antibody | Bio X Cell | Cat # BE0273; RRID: AB_2687796 |
| Mouse PD-1 antibody | Bio X Cell | Cat# BE0146; RRID: AB_10949053 |
| Mouse PD-L1 antibody | Bio X Cell | Cat# BE0101; RRID: AB_10949073 |
| Mouse CTLA-4 antibody | Bio X Cell | Cat# BE0164; RRID: AB_10949609 |
| Mouse CD3 antibody | Bio X Cell | Cat # BE0001–1; RRID: AB_1107634 |
| IgG isotype antibody | Bio X Cell | Cat # BE0090; RRID: AB_1107780 |
| Alexa Fluor 647 mouse TCRβ antibody | BioLegend | Cat# 109217; RRID: AB_493347 |
| APC human CD80 antibody | BioLegend | Cat # 305220; RRID: AB_2076147 |
| BV421 human CD86 antibody | BioLegend | Cat # 305425; RRID: AB_10899582 |
| Alexa Fluor 647 human IgG Fc antibody | BioLegend | Cat # 409320; RRID: AB_2563330 |
| Alexa Fluor 647 mouse IgG Fc antibody | BioLegend | Cat # 405322; RRID: AB_2563045 |
| PE human CD80 antibody | BioLegend | Cat # 305208; RRID: AB_314504 |
| PE human PD-L1 antibody | eBioscience | Cat# 14-5983-82; RRID: AB_467784 |
| Human CD80 antibody | Novus Biologicals | Cat # NBP2–25255; RRID: N/A |
| DyLight488 mouse IgG | Biolegend | Cat #405310; RRID:AB_1575124 |
| PE isotype antibody | Biolegend | Cat #400112; RRID: N/A |
| Human TruStain FcX™ | Biolegend | Cat # 422301; RRID: N/A |
| PerCP/Cy5.5 human CD1a | Biolegend | Cat #300129; RRID:AB_2561931 |
| FITC human CD14 | Biolegend | Cat #301804; RRID:AB_314186 |
| PE human CD3 antibody | BioLegend | Cat #317308; RRID: AB_571913 |
| PE/Cy7 human CD20 antibody | BioLegend | Cat # 302311; RRID: AB_314259 |
| PE mouse Thy1.1 antibody | eBioscience | Cat # 12-0900-83; RRID: AB_465774 |
| Anti-PE Microbeads | Miltenyi Biotec | Cat # 130-048-801; RRID: AB_244373 |
| FITC mouse Ly5.1 antibody | eBioscience | Cat # 11-0453-82; RRID: AB_465058 |
| PerCP/Cy5.5 mouse CD11c antibody | eBioscience | Cat #45-0114-82; RRID: AB_925727 |
| PE/Cy7 mouse CD80 antibody | BioLegend | Cat # 305208; RRID: AB_2563112 |
| APC mouse CD86 antibody | eBioscience | Cat # 17-0862-82; RRID: AB_469419 |
| BV421 mouse PD-L1 antibody | BioLegend | Cat# 124315; RRID: AB_10897097 |
| APC/Cy7 mouse CD11b antibody | BioLegend | Cat# 101225; RRID: AB_830641 |
| PE/Cy7 mouse CD11c antibody | BioLegend | Cat # 117317; RRID: AB_493569 |
| PE/Cy7 mouse F4/80 antibody | BioLegend | Cat# 123113; RRID: AB_893490 |
| PE mouse CD301b | BioLegend | Cat # 146803; RRID: AB_2562943 |
| FITC mouse MHC class II | Thermo Fisher Scientific | Cat# 11-5321-82; RRID: AB_465232 |
| APC mouse CD80 antibody | BioLegend | Cat# 104713; RRID: AB_313134 |
| FITC mouse CD86 antibody | BioLegend | Cat # 105005; RRID: AB_313148 |
| PE mouse PD-L1 antibody | BioLegend | Cat # 124307; RRID: AB_2073557 |
| PerCP mouse CD45 antibody | BioLegend | Cat# 103130; RRID: AB_893339 |
| PerCP/Cy5.5 mouse CD11b antibody | BD Biosciences | Cat # 550993; RRID: AB_394002 |
| PE mouse PD-L1 antibody | BD Biosciences | Cat # 558091; RRID: AB_397018 |
| FITC mouse CD86 antibody | BD Biosciences | Cat # 553691; RRID: AB_394993 |
| Biological samples | ||
| Human peripheral blood CD14+ monocytes | iXCells | Cat # 10HU-008 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | Cat # 850457C |
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt, DGS-NTA-Ni) | Avanti Polar Lipids | Cat # 790404C |
| N-(4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Propionyl)-1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (Triethylammonium Salt, BODIPY-PE) | Thermo Fisher Scientific | Cat # D3800 |
| Poly-D-Lysine | Sigma-Aldrich | Cat # P6407 |
| CLIP-Surface 547 | New England Biolabs | Cat # S9233S |
| CLIP-Surface 647 | New England Biolabs | Cat # S9234S |
| SNAP-Surface Alexa Fluor 647 | New England Biolabs | Cat # S9136S |
| SNAP-Surface 549 | New England Biolabs | Cat # S9112S |
| SNAP-Cell 505-Star | New England Biolabs | Cat # S9103S |
| SNAP-Cell TMR-Star | New England Biolabs | Cat # S9105S |
| SNAP-Cell 647-SiR | New England Biolabs | Cat # S9102S |
| SEE | Toxin Technology | Cat # ET404 |
| Strep-SNAP-PD-L1-His10 | This study | N/A |
| Strep-SNAP-PD-L2-His10 | This study | N/A |
| Strep-SNAP-CD80-His10 | This study | N/A |
| Strep-SNAP-CD80 (I92R)-His10 | This study | N/A |
| Strep-SNAP-CD28-His6 | This study | N/A |
| Strep-SNAP-CD28-GCN4-His6 | This study | N/A |
| Strep-SNAP-CTLA-4-His6 | This study | N/A |
| Strep-SNAP-CTLA-4-GCN4-His6 | This study | N/A |
| SNAP-CTLA-4-His6 | This study | N/A |
| Human PD-1-His | Sino Biological | Cat# 10377-H08H |
| Human PD-L1-His | Sino Biological | Cat# 10084-H08H |
| Human PD-L1 | R&D systems | Cat # 9049-B7 |
| EZ-Link NHS-LC-Biotin | Thermo Fisher Scientific | Cat #21335 |
| Human CD80-His | Sino Biological | Cat# 10698-H08H |
| Human CD86-His | Sino Biological | Cat# 10699-H08H |
| Human PD-1-huFc | ACROBiosystems | Cat # PD1-H5257 |
| Human CD28-huFc | Sino Biological | Cat # 11524-H02H |
| Human CD28-muFc | ACROBiosystems | Cat # CD8-H52A5 |
| Human CTLA-4-huFc | Sino Biological | Cat# 11159-H02H6 |
| Human CTLA-4-muFc | ACROBiosystems | Cat # CT4-H52A4 |
| Isolated huFc | Sino Biological | Cat# 10702-HNAH |
| Isolated muFc | Sino Biological | Cat # 51094-MNAH |
| Mouse MHC-I H2Kb | Enfu Hui | N/A |
| Mouse PD-L1-His | Sino Biological | Cat # 50010-M08H |
| Mouse PD-L2-His | Sino Biological | Cat # 50804-M08H |
| Mouse CD80-His | Sino Biological | Cat # 50446-M08H |
| Mouse CD86-His | Sino Biological | Cat # 50068-M08H |
| Mouse ICAM-His | Sino Biological | Cat # 50440-M08H |
| SIINFEKL peptide | Anaspec | Cat# AS-60193–1 |
| Mouse IL-2 | Thermo Fisher Scientific | Cat# 14802164 |
| Protein G Dynabeads | Thermo Fisher Scientific | Cat# 10004D |
| Human GM-CSF | PeproTech | Cat # 300–03 |
| Human IL-4 | PeproTech | Cat # 200–04 |
| Human TNF-α | PeproTech | Cat # 300–01A |
| LPS | Enzo Life Sciences | Cat # ALX-581–008-L002 |
| Critical Commercial Assays | ||
| Human IL-2 ELISA MAX™ Deluxe | BioLegend | Cat #431804 |
| Cell Line Nucleofector® Kit V | LONZA | Cat# VACA-1003 |
| Quantum™ R-PE MESF | Bangs Laboratories Inc | Cat # 827 |
| Mouse CD4 T cell isolation kit | BioLegend | Cat # 48006 |
| CD11c isolation kit | Miltenyi Biotec | Cat# 130-108-338 |
| Experimental Models: Cell Lines | ||
| HEK 293T | Ronald Vale | N/A |
| Jurkat E6.1 T cells | Arthur Weiss | N/A |
| Raji B cells | Ronald Vale | N/A |
| HEK 293F | Andrew Ward | N/A |
| Jurkat T cells with PD-1-mGFP | Enfu Hui | N/A |
| Raji B cells with PD-L1-mCherry | Enfu Hui | N/A |
| OT-1 | Ananda Goldrath | N/A |
| 4T1 | Cristina Bonorino | N/A |
| CT26 | Jack Bui | N/A |
| Oligonucleotides | ||
| See Table S1 for the list of oligos | ||
| Recombinant DNA | ||
| See Table S2 for the list of recombinant DNA | ||
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Micro-Manager | Open Imaging, Inc. | https://micro-manager.org/ |
| AccPbFRET | (Roszik et al., 2008) | http://biophys.med.unideb.hu/accpbfret/ |
| GraphPad Prism 5 | GraphPad Software Inc. | http://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
Highlights.
Cis-CD80 blocks PD-L1:PD-1 interaction and PD-1 signaling
Cis-PD-L1 inhibits CD80:CTLA-4 interaction by disrupting CD80 homodimers
Cis-PD-L1 protects CD80 from CTLA-4-mediated trans-endocytosis
Anti-PD-L1, but not anti-PD-1, depletes CD80 on APCs in a CTLA-4-dependent manner
ACKNOWLEDGMENTS
We thank E. Bennett (UCSD) and M. Niwa (UCSD) for critically reading the manuscript; J. Wilhelm (UCSD) for allowing the use of the TIRF microscope; N. Stuurman (UCSF) for technical support with TIRF microscopy; J. Sabatini and M. Bohm for help with confocal microscopy at the UCSD School of Medicine Microscopy Core, supported by NINDS P30 Grant NS047101; and Affina Biotechnologies for conducting the SPR experiments. Y.Z. is a Cancer Research Institute Irvington Postdoctoral Fellow. C.-H.L. is supported by a UCSD Cancer Biology Postdoctoral Fellowship, R.B.G. by a CAPES fellowship, and C.B. by a CNPq Productivity Fellowship. This work was supported by a Searle Scholar Award from the Kinship Foundation to E.H., a Pew Biomedical Scholar Award from the Pew Charitable Trusts to E.H, NIH grants R37 CA239072 to E.H. and AI108651 and AI140095to L.-F.L., and a grant from the Hartwell Foundation to J.D.B.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2019.11.003.
DECLARATION OF INTERESTS
The authors declare no competing financial interest.
REFERENCES
- Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, and Thompson CB (1996). Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol 157, 4762–4770. [PubMed] [Google Scholar]
- Bhatia S, Edidin M, Almo SC, and Nathenson SG (2005). Different cell surface oligomeric states of B7–1 and B7–2: implications for signaling. Proc. Natl. Acad. Sci. USA 102, 15569–15574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366,2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, and Freeman GJ (2007). Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte MJ, Peña-Cruz V, Kim MJ, Freeman GJ, and Sharpe AH (2008). Interaction of human PD-L1 and B7–1. Mol. Immunol 45, 3567–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, and Wolchok JD (2015). CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front. Oncol 4, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, and Freeman GJ (2018). PD-L1 binds to B7–1 only in cis on the same cell surface. Cancer Immunol. Res 6, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, Huo J, Yu C, Evans EJ, Leslie AJ, et al. (2013). Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem 288, 11771–11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AV, Brodie DW, Gilbert RJC, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, and Davis SJ (2002). The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, and Chen L (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Dumortier H, van Mierlo GJD, Egan D, van Ewijk W, Toes REM, Offringa R, and Melief CJM (2005). Antigen presentation by an immature myeloid dendritic cell line does not cause CTL deletion in vivo, but generates CD8+ central memory-like T cells that can be rescued for full effector function. J. Immunol 175, 855–863. [DOI] [PubMed] [Google Scholar]
- Duraiswamy J, Kaluza KM, Freeman GJ, and Coukos G (2013). Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EJ,Esnouf RM,Manso-Sancho R,Gilbert RJC,James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, et al. (2005). Crystal structure of a soluble CD28-Fab complex. Nat. Immunol 6, 271–279. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard T, Gaucher D, El-Far M, Breton G, and Sékaly R-P (2014). CD80 and CD86 IgC domains are important for quaternary structure, receptor binding and co-signaling function. Immunol. Lett 161, 65–75. [DOI] [PubMed] [Google Scholar]
- Haile ST, Bosch JJ, Agu NI, Zeender AM, Somasundaram P, Srivastava MK, Britting S, Wolf JB, Ksander BR, and Ostrand-Rosenberg S (2011). Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J. Immunol 186, 6822–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou TZ, Qureshi OS, Wang CJ, Baker J, Young SP, Walker LS, and Sansom DM (2015). A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J. Immunol 194, 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, and Vale RD (2014). In vitro membrane reconstitution of theT-cell receptor proximal signaling network. Nat. Struct. Mol. Biol 21, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemizu S, Gilbert RJC, Fennelly JA, Collins AV, Harlos K, Jones EY, Stuart DI, and Davis SJ (2000). Structure and dimerization of a soluble form of B7–1. Immunity 12, 51–60. [DOI] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, and Sharpe AH (2008). PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger HM, Zito CR,Turcu G, Baine MK, Zhang H, Adeniran A, Sznol M, Rimm DL, Kluger Y, Chen L, et al. (2017). PD-L1 studies across tumor types, its differential expression and predictive value in patients treated with immune checkpoint inhibitors. Clin. Cancer Res. 23, 4270–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K-F, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, Becart S, Kim G, Yates JR 3rd, Kronenberg M, et al. (2014). Protein kinase C-η controls CTLA-4-mediated regulatory T cell function. Nat. Immunol 15, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, and Allison JP (1995). CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med 182, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, and Sharpe AH (2004). PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 101, 10691–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-M, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, and Bluestone JA (1998). Molecular basis of T cell inactivation by CTLA-4. Science 282, 2263–2266. [DOI] [PubMed] [Google Scholar]
- Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al. (2016). Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest 128, 1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, and Ledbetter JA (1991). CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med 174, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Nadler SG, Bajorath J, Peach R, Leung HT, Rogers J, Bradshaw J, Stebbins M, Leytze G, Brady W, et al. (1995). Binding stoichiometry of the cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). A disulfide-linked homodimer binds two CD86 molecules. J. Biol. Chem 270, 15417–15424. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, and Mittler RS (1996). Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4, 535–543. [DOI] [PubMed] [Google Scholar]
- Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, and Rudensky AY (2008). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. USA 105, 11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitaksov V, Truscott SM, Lybarger L, Connolly JM, Hansen TH, and Fremont DH (2007). Structural engineering of pMHC reagents for T cell vaccines and diagnostics. Chem. Biol 14, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murin CD, Julien JP, Sok D, Stanfield RL, Khayat R, Cupo A, Moore JP, Burton DR, Wilson IA, and Ward AB (2014). Structure of 2G12 Fab2 in complex with soluble and fully glycosylated HIV-1 Env by negative-stain single-particle electron microscopy. J. Virol 88, 10177–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Nose M, Hiai H, Minato N, and Honjo T (1999). Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151. [DOI] [PubMed] [Google Scholar]
- Nye JA, and Groves JT (2008). Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir 24, 4145–4149. [DOI] [PubMed] [Google Scholar]
- Ostrov DA, Shi W, Schwartz J-CD, Almo SC, and Nathenson SG (2000). Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science 290, 816–819. [DOI] [PubMed] [Google Scholar]
- Park J-J, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, et al. (2010). B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 116, 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passiglia F, Galvano A, Rizzo S, Incorvaia L, Listì A, Bazan V, and Russo A (2018). Looking for the best immune-checkpoint inhibitor in pre-treated NSCLC patients: An indirect comparison between nivolumab, pembrolizumab and atezolizumab. Int. J. Cancer 142, 1277–1284. [DOI] [PubMed] [Google Scholar]
- Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, and Linsley PS (1994). Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7–1.J. Exp. Med 180, 2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. (2014). MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562. [DOI] [PubMed] [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, Kenefeck R, Willox AK, Royle SJ, Rappoport JZ, and Sansom DM (2012). Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J. Biol. Chem 287, 9429–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, et al. (2015). Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 16, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszik J, Szöllosi J, and Vereb G (2008). AccPbFRET: an ImageJ plugin for semi-automatic, fully corrected analysis of acceptor photobleaching FRET images. BMC Bioinformatics 9, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EM, Wang CJ, Ryan GA, Clough LE, Qureshi OS, Goodall M, Abbas AK, Sharpe AH, Sansom DM, and Walker LS (2009). Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J. Immunol 182, 274–282. [DOI] [PubMed] [Google Scholar]
- Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS, and Mosyak L (2001). Crystal structure of the B7–1/CTLA-4 complex that inhibits human immune responses. Nature 410, 608–611. [DOI] [PubMed] [Google Scholar]
- Sugiura D, Maruhashi T, Okazaki I. m., Shimizu K, Maeda TK, Takemoto T, and Okazaki T (2019). Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science, eaav7062. [DOI] [PubMed] [Google Scholar]
- Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, et al. (2018). PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest 128, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, and Sharpe AH (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk E, Rudd CE, and Schneider H (2008). CTLA-4 trafficking and surface expression. Trends Immunol. 29, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, Bodian DL, Daenke S, Linsley P, and Davis SJ (1997). CD80 (B7–1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med 185, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, and Mak TW (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988. [DOI] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, and Saito T (2012). Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med 209, 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappasodi R, Merghoub T, and Wolchok JD (2018). Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhou H, and Zhang L (2018). Which is the optimal immunotherapy for advanced squamous non-small-cell lung cancer in combination with chemotherapy: anti-PD-1 or anti-PD-L1? J. Immunother. Cancer 6, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Harrison DL, Song Y, Ji J, Huang J, and Hui E (2018). Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep 24, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med 8, 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.