

Abstract
Glycolytic enzyme fructose-bisphosphate aldolase A is an emerging therapeutic target in cancer. Recently we have solved the crystal structure of murine aldolase in complex with naphthalene-2,6-diyl bisphosphate (ND1) that served as a template of the design of bisphosphate-based inhibitors. In this work, a series of ND1 analogs containing difluoromethylene (−CF2), methylene (−CH2) or aldehyde substitutions were designed. All designed compounds were studied using MD simulations with the AMOEBA force field. Both energetics and structural analyses have been done to understand the calculated binding free energies. The average distances between ligand and protein atoms for ND1 were very similar to the ND1 crystal structure, which indicates our MD simulation is sampling the correct conformation well. CF2 insertion lowers the binding free energy by 10~15 kcal/mol while CF2 substitution slightly increases the binding free energy, which matches the experimental measurement. In addition, we found that NDB with two CF2 insertions, the strongest binder, is entropically driven, while others including NDA with one CF2 insertion are all enthalpically driven. This work provides insights into the mechanisms underlying protein-phosphate binding and enhances the capability of applying computational and theoretical frameworks to model, predict and design diagnostic strategies targeting cancer.
Graphical Abstract
Introduction
For most cells, glycolysis is critical for generating energy and supplying metabolic intermediates for cellular biomass. One of the hallmarks of cancer is the altered metabolism preferential dependence on glycolysis in an oxygen-independent manner instead of oxidative phosphorylation, known as the “Warburg effect”.1 Recently, a novel feed-forward mechanism for hypoxic cancer has been identified. While HIF-1 upregulates transcription of glycolytic enzymes, the glycolysis under inadequate oxygen supply, in turn, increases HIF1a transcriptional activity and stimulates tumor growth.2 (Figure 1). Tumor glycolysis has been actively studied and serves as a potential target for cancer therapy.3–4
Figure 1.

Glycolysis acts as a feed-forward mechanism for HIF-1 action.
A leading candidate for this target is the fructose-bisphosphate aldolase A (ALDOA), a central enzyme in glycolysis.5 ALDOA is responsible for converting fructose-1,6-biphosphate (FDP) into glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP). The generally accepted catalytic mechanism for ALDOA is shown in Figure S1. The reaction proceeds with the formation of a Schiff base intermediate between LYS229 of the active site and the carbonyl group of the substrate FDP.6–7 The inhibition of ALDOA has been shown to block the glycolysis, decrease HIF-1 activity and break the feed-forward loop mechanism in cells. Thus aldolase A has prospects for controlling cancer proliferation.2, 8–10
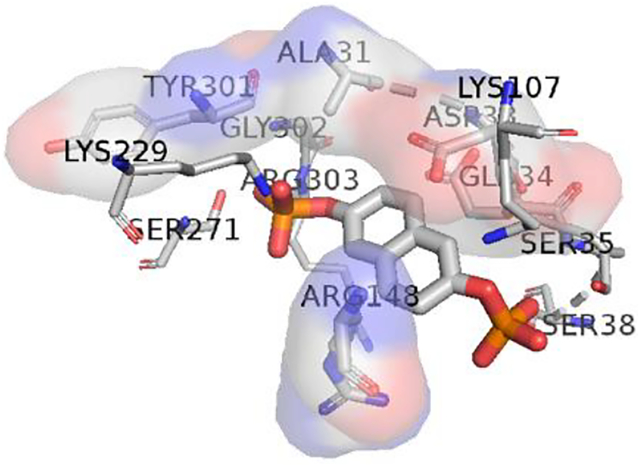
Aldolase inhibitors have been designed to mimic the substrate of FDP by probing the nature of the active site.6, 11 General principles of drug design involve keeping the strong electrostatic interactions with residues in the active site while maintaining hydrophobic interactions in the linkage. Aldolase A has been co-crystallized with naphthalene-2,6-diyl bisphosphate (ND1), an active site substrate-mimetic. Figure 2 shows the 2D structure of the ND1 and highlights the key residues in the binding pocket. Figure S2 shows the same graphic enlarged. H-bonds have been found between the two negatively charged phosphate groups and the polar and positive charged residues including SER35, SER38, SER271, LYS229 and LYS107 as well as the neutral GLY272 and GLY302. These interactions include binding to both residues’ backbone O and N, and sidechain −OH and −NH2. Besides, hydrophobic interactions are marked in red involving LEU270, ALA31, ASP33, and TRY301. Note that negatively charged Asp33 interacts with the naphthalene ring, not the phosphate groups. Although ND1 is a potent inhibitor, with two polar phosphate groups, it is easy to be hydrolyzed and hard to deliver in vivo.
Figure 2.

2D plot of the binding pocket of ND1 in crystal structure2 generated using LigPlot+. Left: Key residues include LYS107, SER35, SER38, SER271, GLY272, GLY302, LEU270, ALA31, ASP33, and Try301. Right: Intermolecular interactions around negatively charged phosphate groups are marked in green with distances while those hydrophobic ones involved aromatic systems are marked in red. These interactions include binding to both residues’ backbone O and N, and sidechains’ −OH and −NH2.
Molecular dynamics (MD) simulations are a powerful tool for understanding the driving forces underlying molecular recognition, accelerating drug discovery, and guiding molecular design.12–18 Classical force fields such as AMBER19, CHARMM20, OPLS-AA21, or GROMOS22 are computationally efficient and sufficiently accurate for many applications.14, 23–25 However, for highly charged species like phosphates-containing ligands, the actual charge distributions of atoms and their changes in response to the environment’s electric field is complicated and challenging to model and simulate.26–32 Recently, polarizable force fields have shown encouraging results for depicting these complicated interactions.33–34 As a physics-grounded force field, AMOEBA depicts molecular polarizability and electrostatic potential terms by using mutual atomic dipole-dipole induction along with permanent atomic point multipoles up to quadrupole.35–36
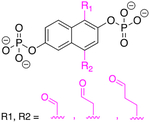
In this work, we explored a series of ND1 analogs for both covalent and non-covalent inhibitors using MD simulations with AMOEBA. For the non-covalent inhibitors, the difluoromethylene (−CF2) and methylene (−CH2) groups have been inserted or substituted targeting the phosphate bridging oxygen. For the covalent ones, aldehyde substitutions have been done on the naphthalene rings. (Figure 3) For the non-covalent inhibitors, we dissected the roles of entropy and enthalpy in binding for each system based on the calculated binding free energy. We also conducted a structural analysis of the distances changing in the key interactions to further compare the ligand binding modes. For the covalent inhibitors, we conducted non-covalent binding simulations to investigate the potential sites for bond formation. This work provides insights into the mechanisms underlying protein-phosphate binding and enhance the capability of applying computational and theoretical frameworks to model, predict and design diagnostic strategies targeting caner.
Figure 3.

Structures of non-covalent and covalent inhibitors. Experimentally measured Ki values for ND1 and ND5 are 0.89 μM and 11.22 μM, respectively. Only ND1 has crystal structure2 co-crystalized with ALDOLASE A protein. The third row contains three covalent inhibitors ND-C, ND-D, and ND-E, while all the others are noncovalent inhibitors. The bridging Os are marked in red. Substitutions and insertions of −CF2 and −CH2 are marked in purple and the aldehyde groups are marked in magenta.
Methods
Parameterization:
Ab initio quantum mechanics calculations (QM) were performed using Gaussian 0937 and PSI4 program.38–40 All molecular mechanics (MM) force field-based calculations were performed using TINKER 8 Software.36, 41 The parameters for ND1 and its derivatives were derived by using POLTYPE program.42 The structures were optimized at MP2/cc-pVTZ level with Polarizable Continuum Model (PCM)43 and the single point energy were calculated at the MP2/6–311++G(2d,2p) level. Atomic multipole moments were initially assigned from QM electron density calculated at the MP2/6–311G** level via Stone’s distributed multipole analysis44 and the optimizations were done with Tinker’s POTENTIAL program to fit electrostatic potentials around molecules. The Van der Waals parameters were optimized to capture the ligand-water interaction energy at different orientations calculated at the MP2/Aug-cc-pVTZ/QZ, extrapolated to CBS level. The torsional parameters were derived to reproduce the QM conformational energy profile at MP2/6–311++G** level. For interaction energy calculations, the dimer structures were optimized using MP2/cc-pVTZ with PCM in Gaussian4 and the interaction energies were calculated using MP2/aug-cc-pVT/QZ in Psi45 and extrapolated to complete basis set. All parameters for water, protein and ions were adopted from the current AMOEBA force field.15, 45–46
| (1) |
| (2) |
| (3) |
Molecular dynamics simulations:
The structure of the ND1-ALDOLASE complex was taken from chain A in the crystal structure.2 All other derivatives were aligned to ND1 by the naphthalene ring in the same protein pocket. The complexes were then solvated in periodic boxes of 82.48×82.48×82.48 Å3 with NaCl added to yield 0.15 M salt concentration. All molecular dynamics simulations were run using the Tinker-OpenMM program on GPU47 with a RESPA integrator48, Bussi thermostat49, Berendsen barostat,50 and 3.0 fs time step with hydrogen-mass repartition (heavy-hydrogen keyword). The van der Waals (vdW) iterations used a 12.0 Å cutoff, while the electrostatic interactions used a 7.0 Å cutoff. The systems were gradually heated up to 298 K and sequentially relaxed from water and ions first, and then protein-ligand complex before free energy production simulations. The non-covalent binding free energies were calculated for all the ligands by the double-decoupling method (Figure S3).15, 47 A mixed potential was defined to calculate the free energy difference between the end states that were connected analytically (Eq. 1). The free energy changes from one state to the other is thus given by Eq. 2. The energy difference between adjacent states was estimated by the Bennett acceptance ratio (BAR) method.51 For the entropy and enthalpy decomposition, the enthalpy of binding was calculated first by averaging the potential energy from each dynamic step. The entropy contribution will be obtained from the difference between free energy and enthalpy (Eq. 3). Detailed methodology and perturbation schedules can be found in previous papers.52–53 Energies reported in this work are relative to ND1 in kcal/mol. It is noted that all of these systems do have a net charge of −4, which can cause some error in the absolute binding free energies. However, here we are only concerned with relative binding free energy changes to ND1. Error cancelation is expected to compensate for the charged system. Charge corrections schemes using a Poisson calculation with periodic boundary conditions could be used to correct this if absolute binding free energy was important54–55.
Structure analysis:
Interaction distances between protein and ligands were calculated over all frames of molecular dynamics trajectories. Then a heatmap were generated to visualize the interaction distances for all derivatives. Amino acids were chosen from being within a 4-angstrom distance of any ligand atom from the first MD trajectory frame. The heatmap has been sorted to show the interactions that are most conserved among the derivatives. For atoms that share the same force field atom type, such as O−s in [PO3], Fs in [CF2], and Hs in [CH2], the heatmap only shows interactions of the atom that has the closest distance with a given protein residue. The standard deviation of distances is typically 0.1–0.4 angstroms, with few exceptions of 0.4–1 angstroms. The detailed statistics are listed in Table S3 and S4.
Experimental details:
For the ligand ND5, the ALDOA activity was measured using standard NADH coupled enzymatic assay by measuring the absorbance at 340nm. The assay uses glycerol-3-phosphate dehydrogenase to catalyze the conversion of dihydroxyacetone phosphate (product of ALDOA substrate cleavage) to glyceraldehyde-3 phosphate in an NADH oxidation-dependent manner. The reaction was carried out in 50 mM TEA-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 0.01% Triton-X 100, 40 u M NADH, excess of 1.7 U of glycerol-3-phosphate dehydrogenase/triosephosphate isomerase (-GDH and TPI) (Sigma-Aldrich, St. Louis, MO) and varying concentrations of fructose 1,6-bisphosphate, at 30 °C. For ND1, the chemical assay has been published previously.9 All the detailed measurements of the Ki value can be found in FigureS4.
Results and Discussions
From chemical assays, we determined that ND1 is a strong inhibitor of ALDOA with the Ki value of 0.89 μM while ND5 is ~1.49 kcal/mol weak relative to the ND1 with the Ki value of 11.22 μM. From MD simulations using AMOEBA, the ligands ranked by calculated binding free energies from tightest to weakest are NDB, NDA1, ND7 and ND1, NDA2, ND5 for the non-covalent competitive inhibitors. All the aldehyde substituted derivatives designed to mimic the covalent inhibitors are weaker binder compared to ND1. ND5 has more positive binding free energy compare to ND1 and the relative binding free energies of ND5 to ND1 is ~2.98 kcal/mol, which matches the experimental measurement. By comparing NDA_2 and NDB, we observed that the free energy decreases when inserting another CF2 group. Neither inserting CH2 groups in ND7 nor substituting the bridge O with CF2 group in ND5 improve the binding. All the experimental details are described in the Methods section.
Most of the ligands have symmetric structures with one functional group on each side. NDA was designed to study the interaction of single functional group with different side of the binding pocket. Two simulations were set up: NDA_1 with the CF2 site facing the protein pocket (close to LYS229 and SER271) and NDA_2 with the CF2 toward the water (close to SER38 and SER35). Similarly, the interactions of ND5 at two different sides were studied by mutating only the functional group at one side. The binding free energies of the single functionalization with two different orientations were compared with those of double functionalization in Figure 4 and Table 1. ΔΔG are positive for ND5 with both orientations, while it is smaller when the CF2 group faces the water side. The sum of ΔΔG for two CF2 groups of ND5 is slightly higher than the total ΔΔG of ND5. For NDA, ΔΔG is close to zero when the CF2 group faces the water side (NDA_2) and much lower when CF2 faces the protein side (NDA_1). The sum of ΔΔG for NDA is significantly higher than ΔΔG of NDB, which indicates a cooperative effect of the two CF2 groups. The results of ND5 and NDA agree with each other in that modifications on ligand interacting with the protein side have a large effect.
Figure 4.

Relative binding free energy ΔΔG of all ligands to ND1 in kcal/mol. Blue ones represent that ligands that bind weaker than ND while the oranges ones represent that ligand bind tighter than ND1. For all ligands, the non-covalent binding free energies were computed. All energies are in kcal/mol and the values are listed in Table S1.
Table 1.
Relative binding free energies ΔΔG (kcal/mol) of mutating the functional group on each side of ND5/NDB separately compared to ND1. The functional groups are on the 2, 7 positions on the naphthalene ring. In ALDOA, the protein side represents the side facing the protein binding pocket (close to LYS229 and SER271) while the water side represents the side that is close to SER38 and SER35, more exposed to water. The uncertainties are in parenthesis.
| Water side | Protein side | Both sides | ||
|---|---|---|---|---|
| ND5 | 0.9 (0.3) | 2.9 (0.3) | ND5 | 3.0 (1.1) |
| NDA | 1.2 (1.1) | −10.1 (1.1) | NDB | −16.1 (1.1) |
To explore the driving force underlying ND series inhibitions, we examined the enthalpy and entropy contributions of the binding free energy. Figure 5 lists the calculated binding enthalpy and entropy for non-covalent ligand. The binding enthalpies are vastly different. There appears to be no simple relationship between the binding thermodynamics and the size or the length of the ligands. Comparing ND5 with NDB and NDAs with NDB, we find that the binding enthalpy does not correlate with the inserting 1 or 2 atoms in the phosphate. For the two orientations of NDA, NDA_1 with the CF2 pointing to the protein side has a much lower binding enthalpy. Interestingly, the strongest binder NDB is entropically driven, while NDA_1, which also has the CF2 insertion and a negative ΔΔG, is enthapically driven. The weak binders including ND7, NDA_2 and ND5 have favorable binding enthalpies which are compensated by entropy contributions. These results suggest the importance of enthalpy-entropy compensation: either enthalpy or entropy itself cannot guide the design of potent inhibitors.
Figure 5.

Relative energy decomposition results of selected non-covalent inhibitors to ND1 including the relative enthalpy (ΔΔH) in blue, relative entropy (TΔΔS) in green, and the relative binding free energy in yellow. All energies are in kcal/mol and the uncertainties are in Table S2.
In an attempt to understand what interactions make each derivative binding specific, we computed the average distance for interactions between protein and ND1 derivatives (Figure 6). With the exception of a few interactions such as ARG303-N-O_P1, ALA31-CB-O_P1, SER38-OG-G2, the crystal structure of ND1 is very close to the averaged MD trajectory of ND1. NDA_1, while keeping most of the interactions found in ND1, has an additional salt bridge between Lys107 and the PO3 group. Some of the conserved interactions across most derivatives, i.e. the interactions of the PO3 head inside the pocket with Ser271 and Arg303 are not observed in NDB. Instead, the CF2 group interact with these residues. The missing of the salt bridge in NDB binding may explain its unfavorable binding enthalpy.
Figure 6.

Heatmap of distances for conserved interactions between protein and ND1 derivatives. Interactions that are conserved on protein pocket side are defined with a 1 in their label, while atoms on the water exposed side are defined with 2 in their label. Any oxygen not on a phosphate group is defined by OB, where B refers to bridging oxygen. CF refers to the carbon attached to a Fluorine. CH refers to the carbon attached to a hydrogen. G represents the naphthalene ring. The bar on the right presents distances ranging from 0.2 nm in black to 0.4 nm in white. Blue indicates that an interaction does not exist for a given derivative. ND1CRY, refers to the crystal PDB of ND1.
The design of a covalent inhibitor targets the hydrophobic naphthalene on ND1 by adding functional groups to enhance the inhibitive properties of ND1. As designed, those substitution groups can form covalent bonds with polar residues in the binding pocket including Lys107, Lys229, and Ser38. The inhibition mechanisms can be categorized as covalent reversible or covalent irreversible. 12 designed covalent inhibitors in 4 categories are listed in Table 2. Since ND1 has a symmetric structure, two positions on the naphthalene ring (shown as R1, R2) have been selected. The proposed reactions are listed in the second column with residues in the binding pocket, modifications, original ND1 colored in blue, pink, and black respectively. Only one mechanism for each category is listing as an illustration while the rest of the reactions in the same row should follow the same mechanism. The second and third columns show the designed covalent reversible inhibitors (design #1.1~1.3 and design #2.1~2.3). By adding and aldehydes and difluoroketone, the new inhibitors could react with the −OH in Ser38 and form covalent bond with the binding pocket. The last two rows show the covalent irreversible designs (design #3.1~3.3 and design #4.1~4.3). Two phosphates serve as selectivity group while the aldehydes and methyl acetimidate cations are the affinity groups/warheads. The most interesting design is the last one that was inspired by the chemical reagent inhibition. Because of the intrinsic reactivity, lysine forms covalent bonds with methyl acetimidate cations.
Table 2.
Covalent inhibitors design. 4 types of inhibitors have been designed, where two of them are categorized into covalent reversible while the other two are covalent irreversible inhibitors. Two hydrogens on the naphthalene ring were designed to be substituted by R1 or R2 highlighted in pink. The reaction site in the binding pocket has been simplified as the key residues highlighted in blue. The proposed inhibition mechanisms are illustrated by using one of the possible derivatives listed in column #2.
| Designed inhibitors | Proposed Inhibition mechanism | Category |
|---|---|---|
 (design #1.1~#1.3) |
covalent reversible, aldehydes | |
 (design #2.1~#2.3) |
covalent reversible, difluoroketone | |
 (design #3.1~#3.3) |
covalent irreversible, “classic” affinity label | |
 (design #4.1~#4.3) |
covalent irreversible, |
From the design of covalent inhibitors, three aldehyde derivatives, i.e. NDC, NDD and NDE were chosen for MD simulations to investigate the potential reaction site. Figure 7 shows the distribution of distances between all covalent inhibitors with all three Lysine in the binding pocket. The close interactions with LYS107 in the binding pocket were found among all designs of the covalent inhibitors. This indicates a potential covalent bond formation while LYS146 and LYS229 are unlikely to react with the aldehyde groups. Refering back to Figure 4, the calculated binding free energies for design NDC, NDD and NDE are 0.3, 1.1 and 8.3 kcal/mol compared to ND1, respectively. The three positive values represent weaker binding after the modification. This might be due to the limitations of MD simulations, which can mimic the intermolecular interactions accurately without new bond formation in one continued simulation. Thus, without reactions, the addition of aldehyde makes the hydrophobic ring bulky and pushes some of the residues away from the inhibitor, and results in weak inhibitors.
Figure 7.

Distribution of distances between all covalent inhibitors with all three Lysine in the binding pocket. Close distances were found between aldehyde O and LYS N, which indicates a potential formation of the covalent bonds.
Conclusion
In this work, a series of inhibitors for ALDOA have been designed in both non-covalent and covalent categories to improve the stability of ND1 and investigate the effects of CF2 group. MD simulations with the AMOEBA force field were used to study the structures and free energies of the inhibitor binding. We observed that the average distances between ligand and protein atoms for ND1 were very similar to the ND1 crystal structure, which indicates our MD simulation is sampling the correct conformation. This gives credence to the derivatives sampling the correct conformation distribution. We found the most potent non-covalent inhibitor to be NDB with two CF2 insertions, followed by NDA with one CF2 insertion, while CF2 substitutions and CH2 insertion do not improve the binding affinity. Biochemical assays also showed that CH2 insertion weakens the inhibition effect.
Besides, it was found that CF2 substitution or insertion interacting with the protein side of the pocket has a larger contribution to free energy change than that interacting with the other side of the pocket. Notably, NDA with CF2 pointing to the protein side lowers the free energy by ~10 kcal/mol while it has little effect when CF2 points to the water side.
Thermodynamic analyses revealed nontrivial enthalpy-entropy compensation. NDB binding is entropy-driven while NDA binding is enthalpy-driven. This indicates the importance of using free energy calculations instead of energy calculation on minimized structures for lead optimization.
For the covalent inhibitors, we observed that LYS107 is a potential site for forming covalent bonds with ND1-aldehyde derivatives. This work suggests that the inhibitors containing insertions of CF2 have potential as chemotherapeutic drug leads, pending synthesis.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for support by the National Institutes of Health (Nos. R01GM106137 and R01GM114237), the Robert A. Welch Foundation (No. F-1691) and CPRIT(RP160657). We thank Dr. C. Liu for careful reading of the manuscript.
Funding Sources
NIH R01GM106137 and R01GM114237; CPRIT RP160657; Welch Foundation F-1691
Footnotes
The authors declare no competing financial interest.
Supporting Information
The SI document contains figures and tables that elucidate more clearly, the reaction pathway for Aldolase, the most important interactions in the binding pocket, the thermodynamic cycle used to obtain binding free energy as well as tabulated values of the binding free energies relative to ND1. Additional informatin in SI includes, experimental Ki values as a function of subtrate concentration for ND1 and ND5. A table of the matrix used to generate the heatmap (Figure 6) is also included.
REFERENCES
- 1.Vander Heiden MG; Cantley LC; Thompson CB, Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324 (5930), 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grandjean G; de Jong PR; James B; Koh MY; Lemos R; Kingston J; Aleshin A; Bankston LA; Miller CP; Cho EJ, et al. , Definition of a novel feed-forward mechanism for glycolysis-hif1alpha signaling in hypoxic tumors highlights aldolase a as a therapeutic target. Cancer Res 2016, 76 (14), 4259–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang SH; Cho A; Yun M; Choi YD; Rha SY; Kang WJ, Prognostic value of pretreatment metabolic tumor volume and total lesion glycolysis using 18f-fdg pet/ct in patients with metastatic renal cell carcinoma treated with anti-vascular endothelial growth factor-targeted agents. Clin Nucl Med 2017, 42 (5), e235–e241. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y; Cai WS; Chen L; Wang G, Molecular dynamics simulation reveals how phosphorylation of tyrosine 26 of phosphoglycerate mutase 1 upregulates glycolysis and promotes tumor growth. Oncotarget 2017, 8 (7), 12093–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang YC; Yang YC; Tien CP; Yang CJ; Hsiao M, Roles of aldolase family genes in human cancers and diseases. Trends Endocrinol Metab 2018, 29 (8), 549–559. [DOI] [PubMed] [Google Scholar]
- 6.Gefflaut T; Blonski C; Perie J; Willson M, Class i aldolases: Substrate specificity, mechanism, inhibitors and structural aspects. Prog Biophys Mol Biol 1995, 63 (3), 301–40. [DOI] [PubMed] [Google Scholar]
- 7.Garrabou X; Beck T; Hilvert D, A promiscuous de novo retro-aldolase catalyzes asymmetric michael additions via schiff base intermediates. Angew Chem Int Ed Engl 2015, 54 (19), 5609–12. [DOI] [PubMed] [Google Scholar]
- 8.Chang YC; Chan YC; Chang WM; Lin YF; Yang CJ; Su CY; Huang MS; Wu ATH; Hsiao M, Feedback regulation of aldoa activates the hif-1alpha/mmp9 axis to promote lung cancer progression. Cancer Lett 2017, 403, 28–36. [DOI] [PubMed] [Google Scholar]
- 9.Cho EJ; Devkota AK; Stancu G; Edupunganti R; Powis G; Dalby KN, A fluorescence-based high-throughput assay for the identification of anticancer reagents targeting fructose-1,6-bisphosphate aldolase. SLAS Discov 2018, 23 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- 10.Han X; Zhu X; Hong Z; Wei L; Ren Y; Wan F; Zhu S; Peng H; Guo L; Rao L, et al. , Structure-based rational design of novel inhibitors against fructose-1,6-bisphosphate aldolase from candida albicans. J Chem Inf Model 2017, 57 (6), 1426–1438. [DOI] [PubMed] [Google Scholar]
- 11.Heron PW; Abellan-Flos M; Salmon L; Sygusch J, Bisphosphonate inhibitors of mammalian glycolytic aldolase. J Med Chem 2018, 61 (23), 10558–10572. [DOI] [PubMed] [Google Scholar]
- 12.Gohlke H; Klebe G, Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed Engl 2002, 41 (15), 2644–76. [DOI] [PubMed] [Google Scholar]
- 13.Houk KN; Leach AG; Kim SP; Zhang X, Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes. Angew Chem Int Ed Engl 2003, 42 (40), 4872–97. [DOI] [PubMed] [Google Scholar]
- 14.Buch I; Giorgino T; De Fabritiis G, Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci U S A 2011, 108 (25), 10184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao D; Golubkov PA; Darden TA; Ren P, Calculation of protein-ligand binding free energy by using a polarizable potential. Proc Natl Acad Sci U S A 2008, 105 (17), 6290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dror RO; Dirks RM; Grossman JP; Xu H; Shaw DE, Biomolecular simulation: A computational microscope for molecular biology. Annu Rev Biophys 2012, 41 (1), 429–52. [DOI] [PubMed] [Google Scholar]
- 17.Yang W; Gao YQ; Cui Q; Ma J; Karplus M, The missing link between thermodynamics and structure in f1-atpase. Proc Natl Acad Sci U S A 2003, 100 (3), 874–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Efficient drug lead discovery and optimization. Acc Chem Res 2009, 42 (6), 724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Case DA; Berryman JT; Betz RM; Cerutti DS; T.E. Cheatham I; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW, et al. Amber 2015; University of California, San Francisco: 2015. [Google Scholar]
- 20.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I, et al. , Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J Comput Chem 2010, 31 (4), 671–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson MJ; Tirado-Rives J; Jorgensen WL, Improved peptide and protein torsional energetics with the oplsaa force field. J Chem Theory Comput 2015, 11 (7), 3499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reif MM; Hunenberger PH; Oostenbrink C, New interaction parameters for charged amino acid side chains in the gromos force field. J Chem Theory Comput 2012, 8 (10), 3705–23. [DOI] [PubMed] [Google Scholar]
- 23.Modi N; Benz R; Hancock RE; Kleinekathofer U, Modeling the ion selectivity of the phosphate specific channel oprp. J Phys Chem Lett 2012, 3 (23), 3639–45. [DOI] [PubMed] [Google Scholar]
- 24.Noskov SY; Berneche S; Roux B, Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 2004, 431 (7010), 830–4. [DOI] [PubMed] [Google Scholar]
- 25.Modi N; Barcena-Uribarri I; Bains M; Benz R; Hancock RE; Kleinekathofer U, Tuning the affinity of anion binding sites in porin channels with negatively charged residues: Molecular details for oprp. ACS Chem Biol 2015, 10 (2), 441–51. [DOI] [PubMed] [Google Scholar]
- 26.Gruber MF; Wood E; Truelsen S; Ostergaard T; Helix-Nielsen C, Computational design of biomimetic phosphate scavengers. Environ Sci Technol 2015, 49 (16), 9469–78. [DOI] [PubMed] [Google Scholar]
- 27.Rao L; Cui Q; Xu X, Electronic properties and desolvation penalties of metal ions plus protein electrostatics dictate the metal binding affinity and selectivity in the copper efflux regulator. J Am Chem Soc 2010, 132 (51), 18092–102. [DOI] [PubMed] [Google Scholar]
- 28.Dudev T; Lim C, Competition among metal ions for protein binding sites: Determinants of metal ion selectivity in proteins. Chem Rev 2014, 114 (1), 538–56. [DOI] [PubMed] [Google Scholar]
- 29.Mackerell AD Jr., Empirical force fields for biological macromolecules: Overview and issues. J Comput Chem 2004, 25 (13), 1584–604. [DOI] [PubMed] [Google Scholar]
- 30.Ponder JW; Case DA, Force fields for protein simulations. Adv Protein Chem 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- 31.Cieplak P; Dupradeau FY; Duan Y; Wang J, Polarization effects in molecular mechanical force fields. J Phys Condens Matter 2009, 21 (33), 333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopes PE; Roux B; Mackerell AD Jr., Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability. Theory and applications. Theor Chem Acc 2009, 124 (1–2), 11–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jing Z; Liu C; Cheng SY; Qi R; Walker BD; Piquemal JP; Ren P, Polarizable force fields for biomolecular simulations: Recent advances and applications. Annu Rev Biophys 2019, 48, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemkul JA; Huang J; Roux B; MacKerell AD Jr., An empirical polarizable force field based on the classical drude oscillator model: Development history and recent applications. Chem Rev 2016, 116 (9), 4983–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y; Xia Z; Zhang J; Best R; Wu C; Ponder JW; Ren P, The polarizable atomic multipole-based amoeba force field for proteins. J Chem Theory Comput 2013, 9 (9), 4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA Jr., et al. , Current status of the amoeba polarizable force field. J Phys Chem B 2010, 114 (8), 2549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomperts R; Frisch M; Scalmani G; Leback B, Current status of the project to enable gaussian 09 on gpgpus. Abstr Pap Am Chem S 2014, 247. [Google Scholar]
- 38.Hohenstein EG; Sherrill CD, Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J Chem Phys 2010, 133 (1), 014101. [DOI] [PubMed] [Google Scholar]
- 39.Parker TM; Burns LA; Parrish RM; Ryno AG; Sherrill CD, Levels of symmetry adapted perturbation theory (sapt). I. Efficiency and performance for interaction energies. J Chem Phys 2014, 140 (9), 094106. [DOI] [PubMed] [Google Scholar]
- 40.Turney JM; Simmonett AC; Parrish RM; Hohenstein EG; Evangelista FA; Fermann JT; Mintz BJ; Burns LA; Wilke JJ; Abrams ML, et al. , Psi4: An open-source ab initio electronic structure program. Wires Comput Mol Sci 2012, 2 (4), 556–565. [Google Scholar]
- 41.Rackers JA; Wang Z; Lu C; Laury ML; Lagardere L; Schnieders MJ; Piquemal JP; Ren P; Ponder JW, Tinker 8: Software tools for molecular design. J Chem Theory Comput 2018, 14 (10), 5273–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu JC; Chattree G; Ren P, Automation of amoeba polarizable force field parameterization for small molecules. Theor Chem Acc 2012, 131 (3), 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mennucci B; Tomasi J; Cammi R; Cheeseman JR; Frisch MJ; Devlin FJ; Gabriel S; Stephens PJ, Polarizable continuum model (pcm) calculations of solvent effects on optical rotations of chiral molecules. J Phys Chem A 2002, 106 (25), 6102–6113. [Google Scholar]
- 44.Stone AJ, Distributed multipole analysis: Stability for large basis sets. J Chem Theory Comput 2005, 1 (6), 1128–32. [DOI] [PubMed] [Google Scholar]
- 45.Ren P; Ponder JW, Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B 2003, 107 (24), 5933–5947. [Google Scholar]
- 46.Jing Z; Liu C; Qi R; Ren P, Many-body effect determines the selectivity for ca(2+) and mg(2+) in proteins. Proc Natl Acad Sci U S A 2018, 115 (32), E7495–E7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harger M; Li D; Wang Z; Dalby K; Lagardere L; Piquemal JP; Ponder J; Ren P, Tinker-openmm: Absolute and relative alchemical free energies using amoeba on gpus. J Comput Chem 2017, 38 (23), 2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuckerman M; Berne BJ; Martyna GJ, Reversible multiple time scale molecular-dynamics. J Chem Phys 1992, 97 (3), 1990–2001. [Google Scholar]
- 49.Bussi G; Donadio D; Parrinello M, Canonical sampling through velocity rescaling. J Chem Phys 2007, 126 (1), 014101. [DOI] [PubMed] [Google Scholar]
- 50.Berendsen HJC; Postma JPM; Vangunsteren WF; Dinola A; Haak JR, Molecular-dynamics with coupling to an external bath. J Chem Phys 1984, 81 (8), 3684–3690. [Google Scholar]
- 51.Bennett CH, Efficient estimation of free-energy differences from monte-carlo data. J Comput Phys 1976, 22 (2), 245–268. [Google Scholar]
- 52.Bell DR; Qi R; Jing Z; Xiang JY; Mejias C; Schnieders MJ; Ponder JW; Ren P, Calculating binding free energies of host-guest systems using the amoeba polarizable force field. Phys Chem Chem Phys 2016, 18 (44), 30261–30269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qi R; Jing Z; Liu C; Piquemal JP; Dalby KN; Ren P, Elucidating the phosphate binding mode of phosphate-binding protein: The critical effect of buffer solution. J Phys Chem B 2018, 122 (24), 6371–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rocklin GJ; Mobley DL; Dill KA; Hunenberger PH, Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: An accurate correction scheme for electrostatic finite-size effects. J Chem Phys 2013, 139 (18), 184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simonson T; Roux B, Concepts and protocols for electrostatic free energies. Mol Simul 2016, 42 (13), 1090–1101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.