

Abstract
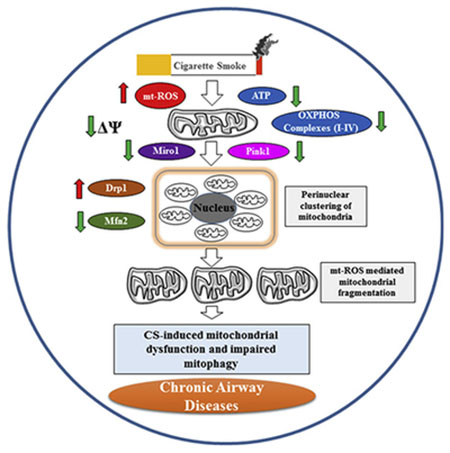
Cigarette smoke (CS) is known to cause mitochondrial dysfunction leading to cellular senescence in lung cells. We determined the mechanism of mitochondrial dysfunction by CS in lung epithelial cells. CSE treatment differentially affected mitochondrial function, such as membrane potential, mitochondrial reactive oxygen species (mtROS) and mitochrondrial mass as analyzed by FACS, and were associated with altered oxidative phosphorylation (OXPHOS) proteins levels (Complexes I-IV) in primary lung epithelial cells (SAEC and NHBE), and (complexes I and II) in BEAS2B cells. There were dose- and time-dependent changes in mitochondrial respiration (oxygen consumption rate parameters i.e. maximal respiration, ATP production and spare capacity, measured by the Seahorse analyzer) in control vs. CSE treated BEAS2B and NHBE/DHBE cells. Electron microscopy (EM) analysis revealed perinuclear clustering by localization and increased mitochondrial fragmentation by fragement length analysis. Immunoblot analysis revealed CS-mediated increase in Drp1 and decrease in Mfn2 levels that are involved in mitochondrial fission/fusion process. CS treatment reduced Miro1 and Pink1 abundance that play a crucial role in the intercellular transfer mechanism and mitophagy process. Overall, these findings highlight the role of Miro1 in context of CS-induced mitochondrial dysfunction in lung epithelial cells that may contribute to the pathogenesis of chronic inflammatory lung diseases.
Keywords: Miro1, Cigarette smoke, Epithelial cells, Mitochondrial dysfunction, Oxidative stress
Graphical Abstract
1. Introduction
Cigarette smoke (CS), the most important etiological risk factor for the development of Chronic Obstructive Pulmonary Disease (COPD)/emphysema, causes lung injuries and damage responses. These effects include mitochondrial dysfunction (i.e., reduced mitochondrial membrane potential and increased mitochondrial reactive oxygen species [mtROS] generation), and defective mitophagy (removal of damaged mitochondria from a cell prior to cell death) (Lerner et al., 2016). We have previously shown that defective mitophagy is associated with CS-induced perinuclear localization of dysfunctional mitochondria and disruption of telomere-shelterin complex (a complex which protects telomeres from DNA damage) in lung cells (Ahmad et al., 2015; Ahmad et al., 2017). However, the cellular and molecular mechanisms for CS-induced mitophagy impairment and mitochondrial translocation, as well as their roles in mitochondrial dyfunction by CS are not known.
Damaged or dysfunctional mitochondria are normally cleared from the cell by mitophagy, whereby defective mitochondria are loaded into the autophagosomes followed by lysosomal degradation (Ashrafi and Schwarz, 2013; Cloonan and Choi, 2016; Lerner et al., 2016; Scarffe et al., 2014; Zhu et al., 2013). Mitophagy process is initiated by mitochondrial membrane depolarization followed by stabilization of Pink1 [PTEN-induced putative kinase 1 (Pink1), which is involved in mitochondrial quality control through fission] on mitochondrial outer membranes. Pink1 then recruits an E3 ubiquitin ligase called Parkin (a protein involved in mitochondrial quality control), from the cytosol (Ashrafi and Schwarz, 2013; Chen and Dorn, 2013; Scarffe et al., 2014; Zhu et al., 2013). Once recruited to mitochondria, Parkin leads to degradation of mitofusin2 (Mfn2, a core protein involved in mitochondrial fusion), which initiates localization of damaged mitochondria with the isolation membranes containing protein microtubule-associated protein light chain 3 (LC3) and formation of autophagosome, where damaged mitochondria are cleared from the cell.
In general, mitochondria elongation is induced by either Drp1 (dynamin-1-related protein that regulates mitochondrial fission) inhibition and/or Mfn2 overexpression. This in turn is regulated by coordinated action of Pink1 and Parkin during mitophagy. Mitochondria elongation has been shown to prevent mitochondria from degradation (Gomes et al., 2011). On the other hand GTPase Miro1 and 2 are crucial in maintainance of mitochondrial shape and trafficking (Nemani et al., 2018).
We have shown that CS extract (CSE) increases mitochondria elongation/fragmentation and mtROS, and reduces ATP levels in lung epithelial cells and fibroblasts (Ahmad et al., 2015). Furthermore, we observed increased mitochondria mass, reduced mitochondria membrane potential, and decreased Parkin mitochondrial translocation following CS exposure. Accumulation of damaged or dysfunctional mitochondria, due to impaired mitophagy, is associated with many chronic lung diseases including COPD (Chen and Chan, 2009; Cloonan and Choi, 2016; Ito et al., 2015; Mizumura et al., 2014; Wiegman et al., 2015). CSE induces mitochondrial fission in human airway smooth muscle cells and bronchial epithelial cells, which may contribute to cell death during COPD/emphysema (Aravamudan et al., 2014; Ballweg et al., 2014; Hara et al., 2013; Hoffmann et al., 2013). Similarly, long-term CSE treatment causes elongated mitochondria with increased mitochondrial fusion activity in lung epithelial cell lines and fibroblasts (Ahmad et al., 2015; Hoffmann et al., 2013). This suggests that mitochondrial structural changes and/or its dysfunction occur due to CS exposure leading to impaired mitophagy. However, the molecular mechanism of these phenomena of mitochondrial dysfunction by CS in lung epithelial cell is not known.
In light of above, we hypothesize that CS-induced mitochondrial dysfunction leads to defective mitophagy by reducing Miro1 levels in lung epithelial cells. We, therefore, studied the effect of CS exposure in primary lung epithelial cells leading to mitochondrial dysfunction (i.e., reduced mitochondrial membrane potential and increased mitochondrial reactive oxygen species [mtROS] generation) as a result of altered mitochondrial dynamics, oxidative phosphorylation (OXPHOS) protein complexes, and defective mitophagy via Miro1 reduction.
2. Materials and Methods
2.1. Cell culture and treatments
Human small airway epithelial cells (SAEC), normal human bronchial epithelial cells (NHBE) from healthy non-smoking subjects and diseased human bronchial epithelial cells (DHBE) from patients with COPD were obtained from Lonza (Walkersville, MD), and grown as per the recommendations of the supplier in appropriate primary cell culture growth media. Human bronchial epithelial cells (BEAS2B; ATCC, Manassas, VA), grown in the DMEM-Ham’s F12 50:50 mixture (DMEM-F12; Mediatech, Manassas, VA) supplemented with 5% FBS, 15 mM HEPES, penicillin (100 U/mL), and streptomycin (100 μg/mL). Before treatment with CSE, cells were cultured to 85%–90% confluence under 5.0% CO2 in six-well plates.
CSE preparation was performed as described earlier (Ahmad et al., 2015). In brief, one cigarette (3R4F, University of Kentucky, KY) was bubbled into 10 ml of culture media without FBS which is represented as 10% CSE stock solution. CSE is widely used to model the effects of cigarette smoke exposure in an in vitro model system using human lung epithelial cells in culture. Different concentrations of CSE (0.25–1.0%) were used in SAEC, NHBE, DHBE and BEAS2B cells. SAEC, NHBE and BEAS2B cells were cultured at 37°C with 5% CO2 (Tri-Gas HERA150I, Thermo Scientific) and treated with diluted CSE (0.25%) for 15 days every alternate day to induce cellular senescence as reported previously from our laboratory (Ahmad et al., 2015).
2.2. Mitochondrial content, membrane potential and matrix oxidant burden by fluorescence-activated cell sorting analysis (FACS)
We utilized FACS analysis of SAEC and NHBE cells from control and CSE treated groups for relative quantification of mitochondrial content, membrane potential and matrix oxidant burden using mitochondrial-localized fluorescent dye intensity (Dingley et al., 2012). Mitochondrial content was measured by MitoTracker green staining (cat #M-7514, Life Technologies). Cells were treated with mitogreen (25 nM) for 10 min before the FACS measurement. Mitochondrial membrane potential was measured using tetramethylrhodamine ethyl ester (TMRM, cat #T-668, Life Technologies). Cells were incubated in TMRM (1 μM) for 15 min before performing the FACS analysis. Similarly, mitochondrial reactive oxygen species/matrix oxidant burden was measured by MitoSOX Red (5 μM, cat #M36008, Life Technologies). Cells were stained with MitoSOX Red for 15 min before performing the FACS measurments as described previously (Ahmad et al., 2015; Dingley et al., 2012).
2.3. Mitochrondrial Bioenergetics using the Seahorse XFp analyzer
Prior to CSE treatments, cell density and dose optimizations for various mitochondrial complex inhibitors were conducted as per the XFp user’s manual (Agilent Technologies, Santa Clara, CA). BEAS2B cells and primary lung epithelial cells (NHBE/DHBE) were plated at a density of 20,000 cells/well and 25,000 cell/well respectively in a XFp 8 well plate using appropriate growth medium. Out of 8 wells, 2 were used as the blank background wells. CSE treatments were performed in DMEM + F12 media without any growth factors and supplements as described earlier. At the end of the treatment period (2 hr or 24 hrs), the wells were replaced with prewarmed XF basal medium containing glutamine (2 mM), pyruvate (1 mM) and glucose (10 mM) and placed for 1 hr in a non–CO2 incubator at 37 °C. All medium and solutions were made freshly and adjusted to pH 7.4 on the day of performing the assay. XFp sensor cartridges were hydrated overnight with XF calibrant at 37°C. Three of the four injection ports were loaded with 10x concentration of mitochondrial inhibitors as per the instruction from Seahorse XFp Cell Mito Stress Test kit (cat-103010-100; Agilent Technologies). After establishing the baseline, sequential injection of oligomycin (2μM), FCCP (1μM), rotenone and antimycin A (0.5μM) were done. All the data were analyzed using WAVE software version 2.6.0.
2.4. Electron microscopy
SAEC and NHBE cells cultured in chamber slides or cover glass post CSE treatment were fixed with a combination of paraformaldehyde and glutaraldehyde (2%) fixative. Cells were then processed and sectioned with a diamond knife on copper grids. Grids were examined with a Hitachi (Tokyo, Japan) 7100 electron microscope and images were captured using a Megaview III digital camera (Soft Imaging System, Lakewood, CO) (Ahmad et al., 2015). Representative images were provided for mitochondrial localization and structural changes. Mitochondrial fragment length was analyzed manually from several images (1 μm magnification) that were captured from control and CSE treated SAEC and NHBE cells using ImageJ software.
2.5. Immunoblot analyses of electron transport chain (ETC) protein complexes and mitochrondial proteins
Whole cell lysates (WCL) from control and CSE treated SAEC, NHBE and BEAS2B cells were measured using a BCA Protein Assay kit (Thermo Fisher). Equal amounts of protein from each sample were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. WCL for OXPHOS immunoblot analysis were denatured and reduced using SDS (1%) and 2- mercaptoethanol (200 mM) respectively (no heat-denaturation).The membrane was incubated with anti-OXPHOS antibody cocktail (Abcam, 1:2000 dilution), anti-Rhot1 antibody (Novus, 1:1000), anti-Drp1 antibody (Abcam, 1:1000), anti-Pink1 antibody (Cell Signaling Technology, 1:1000) and anti-β-actin antibody (Santa-Cruz, 1:1000 dilution) overnight at 4 °C. This primary antibody incubation was followed by incubation with HRP-conjugated anti-mouse or anti-rabbit or anti-goat antibody (1:10,000 dilution) as the secondary antibody for 1 hr at room temperature. The chemiluminescence was detected using the Bio-Rad ChemiDoc MP imaging system (Bio-Rad, Hercules, CA). Densitometric analyses of the band intensities were performed using ImageJ software.
2.6. Statistical analysis
Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA) The results were shown as the mean ± SEM. Comparisons were made using the 2-tailed Student’s t-test for two different treatment groups. P < 0.05 was considered as statistically significant.
3. Results
3.1. CSE treatment alters mitochondrial function in primary human lung epithelial cells
We have previously shown that CSE treatment led to an increase in mitochondrial ROS and mitochondrial mass but decreased membrane potential in human lung fibroblasts (HFL1 cells) in vitro cellular senescence model (Ahmad et al., 2015). Here, we comprehensively investigated mitochondrial function in control and CSE-treated SAEC and NHBE cells by analyzing mitochondrial content, membrane potential and mt-ROS/matrix oxidant burden by fluorescence-activated cell sorting analysis (FACS). Primary lung epithelial cells were treated with CSE (0.25%) for 24 hrs or every alternate day for 15 days. SAEC cells treated with CSE for 24 hrs shows non-significant reduction in TMRE-membrane potential, but significant increase mitochondrial ROS (mt-ROS) as measured by MitoSOX red (Fig. 1A). Similarly, when NHBE cells were treated with CSE for 24 hrs showed significant decrease in TMRE-membrane potential but not significant increase in mt-ROS (Fig. 1B). Using an in vitro lung epithelial cellular senescence model, CSE treatment every alternate day for 15 days in SAEC cells showed significantly reduced mitochondrial membrane potential (TMRE), and increased Mito green (mitochondrial mass), and MitoSOX red staining (mtROS) confirmed by FACS-enabled flow cytometry (Fig. 1C). These findings summarize that short-term (24 hrs) and long-term (15 days) CSE treatment causes mitochondrial dysfunction in primary lung epithelial cells in vitro.
Fig. 1.

CSE treatment causes mitochondrial dysfunction as well as mtROS generation in primary human lung epithelial cells. SAEC and NHBE were treated with CSE (0.25%) for 24 h or every alternate day for 15 days. (A) Fluorescence-activated cell sorting (FACS) analysis of SAEC and NHBE cells stained with tetramethylrhodamine ethyl ester (TMRE; Δψm) for quantitative assessment of mitochondrial membrane potential, MitoTracker green (mitochondrial mass), and MitoSOX red for detection of mtROS. Data are shown as mean ± SEM with n=4–6/group. *P < 0.05, **P < 0.01, ***P < 0.001, vs control.
3.2. CSE treatment alters mitochondrial OXPHOS protein complexes in primary human lung epithelial cells
Regulation of oxidative phosphorylation is a key mechanism by which mitochondria mediates protection against oxidative stress. Hence, we analyzed the protein abundance of five different OXPHOS systems i.e., Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), Complex IV (COX II) and Complex V (ATP5A) in control and CSE treated SAEC, NHBE and BEAS2B cells in 15 day CSE exposure model by immunoblotting. Interestingly, both SAEC and NHBE cells treated with CSE for 15 days showed significant reduction in the levels of Complexes I, II, III and IV (Fig. 2A–B). Surprisingly, only the protein abundance of complex V remained unaffected in both SAEC and NHBE cells (Fig. 2A–B). However, BEAS2B cells treated with CSE for 15 days showed significant reduction in complexes I and II, but all the other complexes remain unaltered (Fig. 3). These results support that CS-induced mitochondrial dysfuntion in primary human lung epithelial cells affects individual OXPHOS complexes that are part of the mitochondrial electron transport chain.
Fig. 2.

CSE altered mitochondrial OXPHOS capacity in primary human lung epithelial cells. SAEC and NHBE cells were treated with CSE (0.25%) every alternate day for 15 days. (A) Representative Western blot images for Total OXPHOS showing altered levels of mitochondrial Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), Complex IV (COX II) with no change in Complex V (ATP5A) relative to β-actin loading control. (B) Densitometry of the corresponding mitochondrial complexes (Complex I–V) was normalized with β-actin as loading control. Data are shown as mean ± SEM with n = 4–6 per group. *P < 0.05, **P < 0.01, ***P < 0.001, vs control.
Fig. 3.

CSE altered mitochondrial OXPHOS proteins in human lung epithelial cells. BEAS2B cells were treated with CSE (0.25%) every alternate day for 15 days. Representative Western blot images for Total OXPHOS showing altered levels of mitochondrial Complex I (NDUFB8) and Complex II (SDHB), with no change in Complex III (UQCRC2), Complex IV (COX II) or Complex V (ATP5A) relative to β-actin loading control. Densitometry of the corresponding mitochondrial complexes (Complexes I–V) was normalized with β-actin as loading control. Data are shown as mean ± SEM with n= 3 per group. **P < 0.01 vs control.
3.3. CSE treatment alters mitochondrial respiration in BEAS2B and primary human lung epithelial cells
Previously, we have shown that CSE treatment can cause signficiant increase in mt-ROS and decreased ATP production in human lung fibroblasts (HFL1) (Ahmad et al., 2015). To determine whether CSE dose- and time-dependently affects ATP production and bioenergetic functions in bronchial epithelial cells we used a Seahorse XFp analyzer. BEAS2B, NHBE and DHBE cells were treated with CSE (0.5–1.0%) for 1–2 hrs or 24 h. Then, the mitochondrial oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were evaluated. BEAS2B cells treated with CSE showed a significant reduction in mitochrondrial OCR, but not ECAR (Fig. 4A–B). The changes in maximal respiration, ATP production and spare capacity were significant at 24 hrs post treatment compared to what was observed in 1 h post CSE treatment (Fig. 4A–B). NHBE cells treated with CSE for 2 hrs (0.5% CSE) showed changes in OCR and ECAR associated with a significant reduction in ATP production (Fig. 4C). Similarly, when we compared NHBE and DHBE cells treated with CSE (0.5%) for 2 hrs, DHBE cells showed significant changes in OCR parameters such as reduced maximal respiration and ATP production (Fig. 4D). Additionally, DHBE cells showed very low basal repiration compared to NHBE cells without CSE treatment at baseline. Overall, these data suggest that CSE treatment as early as 2 hrs–24 hrs affects OCR, but not ECAR demonstrating altered mitochondrial respiration that may contribute to mitochondrial dysfunction observed in lung epithelial cells.
Fig. 4.

CSE reduces mitochondrial function as measured by oxygen consumption rate (OCR) an indicator of oxidative phosphorylation (OXPHOS) in human lung epithelial cells. BEAS2B, NHBE and DHBE cells were treated with CSE (0.5–1.0%) for 1–2 hrs or 24 h. Mitochondrial oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) was evaluated by using a Seahorse XFp analyzer in (A–B) BEAS2B, (C–D) NHBE and DHBE cells. CS-induced suppression of basal OCR, ECAR, maximal respiration, proton leak, ATP production and spare capacity. Data are mean ± SEM, n = 2–3 per group. *P < 0.05, **P < 0.01, vs respective controls.
3.4. CSE treatment alters mitochondrial structure and localization in primary human lung epithelial cells
CS exposure has direct effect on the morphology of mitochondria as shown by the loss of the normal, connected and networked structures in human bronchial/alveolar epithelial cells, airway smooth muscle cells and fibroblasts (Ahmad et al., 2015; Aravamudan et al., 2014; Ballweg et al., 2014; Hoffmann et al., 2013). SAEC cells treated with CSE (0.25%) for 15 days showed an increase in perinuclear accumulation/clustering of mitochondria (Fig. 5A). Semi-quantitative analysis of mitochondrial fragment length using electron microscopy images revealed CSE-induced significant reduction in mitochondrial fragment length in both SAEC and NHBE cells compared to controls (Fig. 5B–C). These findings suggest that mitochondrial fragmentation by CSE treatment in primary lung epitheial cells could be due to increased mt-ROS generation and alteration in mitochondrial functional markers that play an important role in mitochondrial fission/fusion process.
Fig. 5.

CSE causes mitochondrial dysfunction, perinuclear mitochondria clustering and fragmentation in primary human lung epithelial cells. SAEC and NHBE cells were treated with CSE (0.25%) for 15 days. (A) Representative electron microscopy (EM) images from control and CSE treated SAEC showing perinuclear mitochondria clustering around the nucleus. Red arrows indicates the nuclear membrane and white circle with dotted lines indicates perinuclear mitochondria. (B-C) Representative EM images of SAEC and NHBE cells treated with CSE showing fragmented mitochondria. Mitochondrial fragment length were determined from same magnification images obtained in multiple cells from both control and CSE treated group using Image J. Scale bars; 1 μm. Data are shown as mean ± SEM with n = 3-4. *P < 0.05, **P < 0.01, vs control.
3.5. CSE treatment alters mitochondrial markers/proteins involved in transport and mitophagy process in primary human lung epithelial cells
Miro1 is an important integral mitochondrial motor protein that is part of the microtubular network responsible for mitochondrial transport within the cells (Chang et al., 2011). To determine the mechanisms by which CSE alters mitochondrial function in primary human lung epithelial cells, we examined the expression of proteins involved in mitochondrial transport and mitophagy. SAEC and NHBE cells treated with CSE (0.25%) for 15 days showed significant reduction in Miro1 protein abundance (Fig. 6A). CSE treatment to SAEC cells altered mitochondrial morphology (perinuclear accumulation and fragmentation) in primary lung epithelial cells as shown by EM was further supported by increased expression of Drp1 protein that is known to regulate mitochondrial fission (Fig. 6B). Similarly, CSE treatment in NHBE cells showed non-significant reduction in the expression of Mfn2 protein that is involved in mitochondrial fusion (Fig. 6C). Finally, CSE treatment in NHBE cells also show reduced protein abundance of Pink1 (PTEN induced putative kinase 1: a mitochondrial serine/threonine-protein kinase) which plays a crucial role in the removal of damaged or dysfunction mitochondrial from the cells (Fig. 6D). These results revealed that CSE treated primary lung epithelial cells showed defective protein turnover in mitochondrial intercellular transport machinery and fission/fusion process including mitophagy that may directly contribute to CS-induced mitochondrial dysfuction.
Fig. 6.

CSE reduces Miro1 and Pink1 abundance and augments Drp1 levels in primary lung epithelial cells. SAEC and NHBE cells were treated with CSE (0.25%) every alternate day for 15 days. (A) Representative Western blot images for SAEC and NHBE cells showing reduced Miro1 levels and (B) SAEC cells showing increased Drp1 and (C–D) NHBE cells showing reduced Mfn2 and Pink1 levels. Densitometry of the corresponding protein band was normalized with β-actin as loading control. Data are mean ± SEM, n = 3–5 per group. **P < 0.01, ***P < 0.001, vs control.
4. Discussion
We and others have shown that human lung cells (e.g., airway and alveolar epithelial cells, fibroblasts, airway smooth muscle cells) exposed to CSE causes mitochondrial injury, dysfunction, and morphological alterations (Ahmad et al., 2015; Aravamudan et al., 2014; Ballweg et al., 2014; Hara et al., 2013; Hoffmann et al., 2013). However, the role of Miro1 in CSE-induced mitochondrial dysfunction in primary lung epithelial cells is not known. In this study, we treated primary human lung epithelial cells (SAEC and NHBE) and bronchial epithelial cells (BEAS2B) with CSE to determine the levels of Miro1 during CSE-induced mitochondrial dysfunction/cellular senescene.
It has been shown that mitochondrial dysregulation plays an important role in the pathogenesis of age-related lung diseases including COPD and IPF (Ahmad et al., 2015; Bueno et al., 2015; Lerner et al., 2016; Mizumura et al., 2014; Wiegman et al., 2015). The term mitochondrial dysfunction is refered to as a complex display of cellular events. Mitochondrial injury causes change in mitochondrial morphology, impaired oxidative phosphorylation, reduced membrane potential, altered metabolic activity (e.g. glycolysis vs. fatty acid β-oxidation), increased mitochondrial superoxide levels including alterations in intracellular Ca2+ flux (Cloonan and Choi, 2016; Lerner et al., 2016). Mitochondria is a dynamic organelle that changes shape rapidly with the help of proteins that regulate the fission (fragmentation) and fusion (elongation) process. The major proteins MFNs (membrane-anchored proteins: Mfn1 and 2) and OPA-1 (optic atrophy 1) promote mitochondrial fusion by interacting with the mitochondrial outer and inner membranes. Reduction in fusion proteins can result in mitochondrial fragmentation (Chen and Dorn, 2013). Further, we found time- and dose-dependent changes in the protein abundance of OPA1 four hrs (2% CSE) and 15 days (0.25% CSE) post-CSE treatment in BEAS2B cells (unpublished observations). Mitochondrial fission is mediated by cytosolic dynamin-related protein 1 (Drp1), Fission 1 protein (Fis-1), Mitochondrial Fission Factor (MFF) and other proteins (Gomes et al., 2011; Kim et al., 2011). Our data show that CSE treatment affects the balance of protein involved in mitochondrial fission (increase Drp1) and fusion (reduced Mfn2) in primary human lung epithelial cells.
During prolonged oxidant stress, mitochondrial morphology does not actually correlate with the mitochondrial function. Several factors, such as the type of stress, duration of stress and the ability of the mitochondrial quality control mechanism to modulate the stress response (mitophagy) may play an important role in the measured outcomes. Recent reports from our laboratory and others have shown that mitochondrial morphology varies (elongated vs. fragmented) as a determinant of cell fate following CSE treatment associated with reduced mitochondrial function, mitophagy and increase ROS production (Ahmad et al., 2015; Hara et al., 2013). Prior reports have shown that mitophagy is the quality control mechanism in mitochondria associated with several cellular processes, such as cellular senescence, apoptosis and necroptosis in a stress- and cell-type dependent manner (Ahmad et al., 2015; Hara et al., 2013; Ito et al., 2015;Mizumura et al., 2014). During oxidative stress-induced mitochondrial dysfunction, mitochondrial membrane depolarization stabilizes PINK1, which then recruits PARK2 (Parkin: an E3-ubiquitin ligase) to the mitochondria (Ashrafi and Schwarz, 2013; Vincow et al., 2013). PARK2 ubiquitinates mitochondrial proteins such as MFNs, thereby prevent mitochondrial fusion. Thus, it activates the localization of damaged mitochondria to microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), resulting in the formation of autophagosomes through the adaptor protein SQSTM1/p62 (Ni et al., 2015). Hence, autophagosomes containing dysfunctional mitochondria is fused with the lysosomes and removed from the cells (Ashrafi and Schwarz, 2013; Chen and Dorn, 2013; Ni et al., 2015).
Our data corroborate with the previous report that show increase in CS-induced ROS from damaged mitochondria leading to cellular senescence in primary lung epithelial cells (Hara et al., 2013; Ito et al., 2015). Prior evidence from in vitro studies on mitochondrial morphology demonstrates varied outcomes. For example, low dose of CSE induced mitochondrial elongation that was accompanied by heightened metabolic activity without any change in mitochondrial function and ROS production in mouse alveolar epithelial cells (Ballweg et al., 2014). Earlier reports from BEAS2B (exposed to CS for 1 week) and airway smooth muscle cells (exposed to CSE for 24 h) showed an increase in mitochondrial ROS (Aravamudan et al., 2014; Malinska et al., 2018). Mitochondrial elongation was observed in other cell types such as human fibroblast and lung alveolar epithelial cells treated with low dose CSE (Ahmad et al., 2015; Ballweg et al., 2014). In contrast, prior reports support that increasing dose of CSE or long-term CSE treatment induced mitochondrial fragmentation in primary lung epithelial cells as shown in this study (Hara et al., 2013; Ito et al., 2015). In another study, human bronchial epithelial cells exposed to CSE for 6 months demonstrated mitochondrial elongation (Hoffmann et al., 2013). Studies using human primary lung epithelial cells and tissues from patients with COPD showed both elongated and fragmented mitochondria (Hara et al., 2013; Hoffmann et al., 2013). ASM cells from patients with COPD show reduced membrane potential, ATP content, complex expression levels associated with changes in basal and maximal respiration, and respiratory spare capacity including increased mt-ROS compared to healthy controls (Wiegman et al., 2015). Thus, our data on CSE-induced mitochondrial dysfunction in primary lung epithelial cells corroborate with findings reported in ASM cells from patients with COPD.
We observed a dose- and time-dependent effect in the mitochondrial dysfunction parameters measured in SAEC vs. NHBE cells (24 hrs vs. 15 days post CSE treatment). In a recent study, similar to what we found in primary lung epithelial cells, BEAS2B cells when exposed to 1 week with the reference cigarettes (3R4F) show decreased mitochondrial respiration (basal and maximal respiration) accompanied with a decrease in the content of selected mitochondrial OXPHOS protein complexes I and II (Malinska et al., 2018). Our findings on CSE treatment induced changes in abundance of respiratory chain complexes I-IV partly correlate with the above mentioned study (Malinska et al., 2018). Hoffmann et al., found increased levels of specific repiratory chain subunits in long-term CSE-treated BEAS2B cells which was in contrast to what we observed in this study (Hoffmann et al., 2013). In a recent study, 1-week exposure outcomes were conflicting to 12-weeks exposure data from BEAS2B cells to 3R4F in terms of mitochondrial respiration (increased basal, maximal respiration and proton leak) (Malinska et al., 2018). This could be explained as possible adaptive mechanisms, that allow survival of cells during conditions of chronic stress. These discrepancies in the literature following CSE treatment could be due to different protocols of exposures followed, treatment regeims, dose and time duration used, and cell-types tested by different investigators.
Both Pink1 and Parkin target Miro1 for phosphorylation, ubiquitination, and degradation, which quarantine damaged mitochondria for mitophagy (Birsa et al., 2014; Kane and Youle, 2011; Kazlauskaite et al., 2014; Liu et al., 2012; Wang et al., 2011). Reduction in Pink1/Parkin may be associated with impaired mitophagy via Miro1 in response to CS. Our result showed that Miro1 abundance was decreased in primary human lung cells (SAEC and NHBE) treated with CSE. Immunoblot analysis showed the involvement of important mitochondrial proteins, such as Rho GTPase1 (Miro1), dynamin-related protein 1 (Drp1), mitofusin2 (Mfn2), and Pink1 in mitochondrial dysfunction caused by CSE. We speculate Miro1 reduction observed in our model of mitochondrial dysfunction may be caused as a result of Parkin activation-mediated ubiquitination and degradation of Miro1 as reported recently (Safiulina et al., 2019a), or due to oxidative postransational modifications of Miro1 (unpublished data). Current studies have provided new insights into the role of Miro1 (Rhot1) and Miro2 (Rhot2) in PINK1/Parkin-dependent mitophagy (López-Doménech et al., 2018; Safiulina et al., 2019a, b), which requires further investigation in the context of CS/CSE-induced mitochondrial dysfunction models (in vitro and in vivo). Mitochondrial transfer is shown to occur in co-culture cellular system (Spees et al., 2006). Manipulation of this process may hold promise as a novel therapeutic tool for diseases wherein mitochondrial dysfunction is a critical aspect of progression and etiology (Ahmad et al., 2014; Islam et al., 2012;Li et al., 2014; Otsu et al., 2009; Plotnikov et al., 2008). Miro1 plays an impotant role in regulating mitochondrial morphogenesis and tracking along microtubules (Desai et al., 2013; Morlino et al., 2014). However, it remains unknown whether Miro1 enhances mitochondria transfer into lung epithelial (airway/alveolar) cells and increased protection against chronic CS stress-induced injurious responses.
5. Conclusion
In summary, we show that CSE treatment in primary lung epithelial cells causes mitochondrial dysfunction associated with increased mitochondrial ROS and mitochondrial mass and reduced membrane potential. CSE treatment affects abundance of OXPHOS protein complexes which correlated with alterations in mitochondrial respiratory chain as evidenced by reduced basal and maximal respiration, ATP production and spare capacity ananlyzed by Seahorse as early as 2 hrs–24 hrs in BEAS2B and primary human lung epithelial cells (SAEC and NHBE). CSE treatment for 15 days in primary lung epithelial cells showed increase in mitochondrial fragmentation and perinuclear mitochondrial clustering around the nucleus. These changes in mitochondrial structure were linked to increased Drp1 and reduced Mfn2 protein abundance which play crucial role in mitochondrial fission/fusion. Additionally, we found CSE treatment significantly reduced the protein levels of GTPase Miro1 that is involved in mitochondrial trafficking and mitophagy marker Pink1 in primary lung epithelial cells. Currently, we are examining the role of Miro1 in mitochondrial transfer mechanisms that can be used to restore mitochondrial function in damaged lung epithelial cells (airway and alveolar type II) following CSE treatment in the context of mitophagy and cellular senescence. Hence, understanding the mechanism of Miro1-mediated mitochondrial transfer will provide a rationale for developing novel autologous MSC/MSC-derived extracellular vesicles-based translational strategies for treatment of a variety of conditions including chronic lung diseases associated with dysfunctional mitochondria.
Acknowledgements
This study was supported by the NIHR01 HL135613, R21 ES028006, R01 HL133404, and R01 HL137738.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A, 2014. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 33, 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, Rahman I, 2015. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 29, 2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Sundar IK, Tormos AM, Lerner CA, Gerloff J, Yao H, Rahman I, 2017. Shelterin telomere protection protein 1 reduction causes telomere attrition and cellular senescence via sirtuin 1 deacetylase in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol 56, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YS, 2014. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol 306, L840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL, 2013. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballweg K, Mutze K, Konigshoff M, Eickelberg O, Meiners S, 2014. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol 307, L895–907. [DOI] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Wauer T, Mevissen TE, Wu HC, Foltynie T, Bhatia K, Hirst WD, Komander D, Plun-Favreau H, Kittler JT, 2014. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem 289, 14569–14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL, 2015. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest 125, 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Niescier RF, Min KT, 2011. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. U. S. A 108, 15456–15461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2009. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet 18, R169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW 2nd, 2013. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan SM, Choi AM, 2016. Mitochondria in lung disease. J. Clin. Invest 126, 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SP, Bhatia SN, Toner M, Irimia D, 2013. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J 104, 2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley S, Chapman KA, Falk MJ, 2012. Fluorescence-activated cell sorting analysis of mitochondrial content, membrane potential, and matrix oxidant burden in human lymphoblastoid cell lines. Methods Mol. Biol 837, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L, 2011. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kamiya N, Odaka M, Morikawa T, Kaneko Y, Nakayama K, Kuwano K, 2013. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell Mol. Physiol 305, L737–746. [DOI] [PubMed] [Google Scholar]
- Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, Ten Hacken NH, van der Want JJ, van Oosterhout AJ, Heijink IH, 2013. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res 14, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J, 2012. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med 18, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, Kojima J, Shimizu K, Numata T, Kawaishi M, Odaka M, Morikawa T, Harada T, Nishimura SL, Kaneko Y, Nakayama K, Kuwano K, 2015. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Youle RJ, 2011. PINK1 and Parkin flag Miro to direct mitochondrial traffic. Cell 147, 721–723. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, Muqit MM, 2014. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 4, 130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA, 2011. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner CA, Sundar IK, Rahman I, 2016. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol 81, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, Ip MS, Tse HF, Mak JC, Lian Q, 2014. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol 51, 455–465. [DOI] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, Takahashi R, Hattori N, Imai Y, Lu B, 2012. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8, e1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Doménech G, Covill-Cooke C, Howden JH, Birsa N, Morfill C, Brandon NJ, Kittler JT, 2018. Miro ubiquitination is critical for efficient damage-induced PINK1/Parkin-mediated mitophagy. bioRxiv, 414664. [Google Scholar]
- Malinska D, Szymanski J, Patalas-Krawczyk P, Michalska B, Wojtala A, Prill M, Partyka M, Drabik K, Walczak J, Sewer A, Johne S, Luettich K, Peitsch MC, Hoeng J, Duszynski J, Szczepanowska J, van der Toorn M, Wieckowski MR, 2018. Assessment of mitochondrial function following short- and long-term exposure of human bronchial epithelial cells to total particulate matter from a candidate modified-risk tobacco product and reference cigarettes. Food Chem. Toxicol 115, 1–12. [DOI] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM, 2014. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest 124, 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlino G, Barreiro O, Baixauli F, Robles-Valero J, Gonzalez-Granado JM, Villa-Bellosta R, Cuenca J, Sanchez-Sorzano CO, Veiga E, Martin-Cofreces NB, Sanchez-Madrid F, 2014. Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Mol. Cell. Biol 34, 1412–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani N, Carvalho E, Tomar D, Dong Z, Ketschek A, Breves SL, Jana F, Worth AM, Heffler J, Palaniappan P, Tripathi A, Subbiah R, Riitano MF, Seelam A, Manfred T, Itoh K, Meng S, Sesaki H, Craigen WJ, Rajan S, Shanmughapriya S, Caplan J, Prosser BL, Gill DL, Stathopulos PB, Gallo G, Chan DC, Mishra P, Madesh M, 2018. MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca(2+) stress. Cell Rep. 23, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Ding WX, 2015. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J, 2009. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 113, 4197–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov EY, Khryapenkova TG, Vasileva AK, Marey MV, Galkina SI, Isaev NK, Sheval EV, Polyakov VY, Sukhikh GT, Zorov DB, 2008. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J. Cell. Mol. Med 12, 1622–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina D, Kuum M, Choubey V, Gogichaishvili N, Liiv J, Hickey MA, Cagalinec M, Mandel M, Zeb A, Liiv M, Kaasik A, 2019a. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina D, Kuum M, Choubey V, Hickey MA, Kaasik A, 2019b. Mitochondrial transport proteins RHOT1 and RHOT2 serve as docking sites for PRKN-mediated mitophagy. Autophagy 15, 930–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM, 2014. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ, 2006. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U. S. A 103, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ, 2013. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U. S. A 110, 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL, 2011. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM, Copdmap, 2015. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol 136, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wang KZ, Chu CT, 2013. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9, 1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]