

Abstract
Single germline or somatic activating mutations of mammalian target of rapamycin (mTOR) pathway genes are emerging as a major cause of type II focal cortical dysplasia (FCD), hemimegalencephaly (HME) and tuberous sclerosis complex (TSC). A double-hit mechanism, based on a primary germline mutation in one allele and a secondary somatic hit affecting the other allele of the same gene in a small number of cells, has been documented in some patients with TSC or FCD. In a patient with HME, severe intellectual disability, intractable seizures and hypochromic skin patches, we identified the ribosomal protein S6 (RPS6) p.R232H variant, present as somatic mosaicism at ~15.1% in dysplastic brain tissue and ~11% in blood, and the MTOR p.S2215F variant, detected as ~8.8% mosaicism in brain tissue, but not in blood. Overexpressing the two variants independently in animal models, we demonstrated that MTOR p.S2215F caused neuronal migration delay and cytomegaly, while RPS6 p.R232H prompted increased cell proliferation. Double mutants exhibited a more severe phenotype, with increased proliferation and migration defects at embryonic stage and, at postnatal stage, cytomegalic cells exhibiting eccentric nuclei and binucleation, which are typical features of balloon cells. These findings suggest a synergistic effect of the two variants. This study indicates that, in addition to single activating mutations and double-hit inactivating mutations in mTOR pathway genes, severe forms of cortical dysplasia can also result from activating mutations affecting different genes in this pathway. RPS6 is a potential novel disease-related gene.
Introduction
Single germline or somatic, often brain confined, activating mutations of mammalian target of rapamycin (mTOR) pathway genes are the main known cause of type II focal cortical dysplasia (FCD), hemimegalencephaly (HME), and tuberous sclerosis complex (TSC). These neurodevelopmental disorders are all characterized by areas of abnormal lamination of the cerebral cortex, with ectopic subcortical and giant, dysmorphic neurons, and, inconstantly, by balloon cells. The main associated clinical features include intractable epilepsy and intellectual disability (reviewed by D’Gama and Walsh (1)). In a limited number of patients with TSC or FCD, it has been documented that, in addition to the autosomal dominant mechanism, a cumulative effect of a primary germline mutation in one allele and a secondary somatic hit affecting the other allele of the same negative regulator of the mTOR pathway (either TSC1/TSC2 or DEPDC5) is also possible (2–5).
Using different next-generation sequencing (NGS) approaches in paired DNA samples extracted both from blood and dysplastic hemimegalencephalic brain tissue surgically removed to treat drug-resistant epilepsy, we identified two somatic variants affecting two different genes of the mTOR pathway (ribosomal protein S6 [RPS6] p.R232H and MTOR p.S2215F). RPS6 encodes the ribosomal protein S6, a component of the 40S ribosomal subunit, which acts as a downstream effector of PI3K/Akt/TSC/Rheb/mTORC1/S6K and Ras/Raf/MEK/ERK/RSK pathways (6). A number of studies have demonstrated that this protein is involved in 5' terminal oligopyrimidine tract (TOP) messenger RNAs (mRNAs) translation as well as in the regulation of cell size and proliferation (6). Variants in RPS6 have not been associated with human disease yet. MTOR encodes the mammalian (or mechanistic) target of rapamycin, a highly conserved atypical member of the phosphoinositide 3-kinase-related kinase family which is ubiquitously expressed in eukaryotic cell types, including neural cells (7). Acting through two large biochemical complexes (mTORC1 and mTORC2), MTOR regulates several processes including cell growth, proliferation, motility and survival; protein synthesis; autophagy and transcription (7, 8). The MTOR variant we identified had previously been related to type II FCD and hemimegalencephhaly (4, 9–12).
Independently expressing either variant in animal models, we demonstrated that MTOR p.S2215F caused neuronal migration delay and cytomegaly, while RPS6 p.R232H prompted increased cell proliferation. Double mutants exhibited a more severe phenotype, pointing to a synergistic effect of the variants.
This study indicates that, in addition to single activating mutations and double-hit inactivating mutations in mTOR pathway genes, HME can result from activating mutations affecting different genes of this pathway. Our data also indicate RPS6 as a potential novel disease-related gene.
Results
Clinical findings
The patient we studied is a 9-year-old girl, the second child of consanguineous healthy parents. At 6 months of gestation, her mother referred abnormal fetal movements that were subsequently interpreted as in utero seizures. Level 2 fetal ultrasound revealed left HME that was confirmed by fetal magnetic resonance imaging (MRI). Delivery was at 40 gestational weeks by cesarean section. Birth weight was 3.2 kg (47th centile), length was 50 cm (67th centile) and cccipitofrontal circumference (OFC) was 37 cm (>99th centile).
Seizures started in the first day of life and were promptly documented by neonatal video electroencephalography (video-EEG) recordings showing sub-continuous discharges on the left hemisphere, or at times bilateral, accompanied by right-sided or bilateral jerking and asymmetric repetitive spasms. Brain MRI scan at day 3 confirmed left HME (Fig. 1A and B).
Figure 1.

Patient’s neuroimaging, cutaneous and anatomopathological findings. (A) and (B) Axial T2 and proton density (PD)-weighted MRI sections showing left HME, with enlarged ventricles, rightward deviation of the brain midline, coarse cortical gyri, with cortical thickening, abnormal with matter signal (decreased in T2 and increased in PD) blurring of the gray-white matter junction. (C) Hypochromic patches on the patient’s face (white arrows) and neck (white circle). (D) H&E and immunohistochemical staining of the dysplastic brain tissue. Dysmorphic neurons (white arrows) and balloon cells (black arrows) exhibit a marked hyperexpression of anti-PS6. Anti-NFs staining demonstrates neurofilament protein accumulation in dysmorphic neurons. Anti-GS and anti-GFAP immunoreactivity is present in balloon cells and reactive astrocytes but not in dysmorphic neurons. Anti-NEU-N staining shows cortical dyslamination. Scale bar for H&E, PS6, NFs, GS and GFAP = 50 μm. Scale bar for NEU-N = 500 μm.
In view of the electroclinical and imaging findings, accompanied by right hemiparesis, functional hemispherectomy was performed at 2 months of age. The procedure had limited benefits on seizure severity, prompting a second operation at 12 months of age, which included left temporal lobectomy, severing of the left hippocampal commissure and complete parietal and occipital lobectomy. Following transient benefit lasting several weeks, seizures resumed pre-operation frequency, reaching up to 20 episodes per day.
At 4 years of age, a third surgical procedure was performed, with completion of the left hemispherectomy. Postoperartively seizures manifested in clusters and became polymorphic, with asymmetric tonic posturing, oroalimentary automatisms and either right- or left-sided eyelid fluttering and eye deviation. These latter episodes were recorded and were correlated with ictal discharges arising on either side.
The patient is currently under treatment with phenytoin, lacosamide, and clonazepam and has not presented seizure clusters in the last year.
Clinical examination shows severe intellectual disability, absent speech, axial hypotonia, right spastic hemiplegia, bilateral ankle clonus, erratic eye movements, OFC at the 75th percentile and hypochromic patches on face and neck (Fig. 1C).
Neuropathological findings
Neuropathological evaluation of surgically removed brain specimens revealed cortical dyslamination with dysmorphic neurons exhibiting neurofilament protein accumulation, and balloon cells exhibiting anti-glutamine synthetase (GS) and anti-glial fibrillary acidic protein (GFAP) reactivity (Fig. 1D). Cytoarchitectural abnormalities were overall consistent with HME.
Immunohistochemistry against phosphorylated RPS6Ser240–244 (PS6) revealed an increased immunosignal in dysmorphic neurons and balloon cells (Fig. 1D), a finding which is in line with the known hyperexpression of phospho-RPS6 in these cells subtypes (13).
Identification of MTOR and RPS6 somatic variants
Massive parallel sequencing of a panel of 54 mTOR pathway genes revealed a missense variant of unknown significance (c.695G > A; p.R232H) in the RPS6 gene (RefSeq accession number NM_001010.2), affecting a highly conserved residue (Supplementary Material, Fig. S1) and present as somatic mosaicism at ~15.1% (GS Junior sequencing validation: 14.2–16%) in dysplastic brain tissue and ~11% in blood. RPS6 p.R232H was classified as damaging by in silico predictors Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) Mutation Taster (http://www.mutationtaster.org/), SIFT (https://sift.bii.a-star.edu.sg/) and M-CAP (http://bejerano.stanford.edu/mcap/); it was not present in the gnomAD database (http://gnomad.broadinstitute.org/, accessed on November 25, 2018), while it has been identified as somatic mosaicism in one endometrioid carcinoma sample in the Catalogue of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic, accessed on November 25, 2018, variant ID: COSM1107652).
According to Gene Constraint prediction reported in the gnomAD database (http://gnomad.broadinstitute.org/gene/ENSG00000137154, accessed on November 25, 2018), the RPS6 gene is mildly intolerant to missense variants (number of expected missense variants = 153.7, number of observed missense variants = 106, Z score = 1.39). To statistically evaluate the pathogenicity of p.R232H, we performed a variant-specific chi-squared test with Yates correction for a 2 × 2 contingency table. p.R232H reached a significant value (P < 0.0001) in the chi-squared test performed using the 123 136 gnomAD exomes as controls, as well as using 3582 ethnicity matched controls (Supplementary Material, Table S3). These findings suggest that the association between p.R232H and the patient’s phenotype was not related to chance occurrence.
The panel analysis revealed, in this same patient, a second missense, pathogenic variant in MTOR (RefSeq accession number NM_004958.3, c.6644C > T:p.S2215F), detected as ~8.8% mosaicism (single-molecule molecular inversion probes [smMIPs] sequencing validation: 8.06–13.44%; droplet digital polymerase chain reaction [ddPCR]: 4.85–8.87%) in the dysplastic brain tissue, but not in blood. Somatic mosaic occurrence of MTOR p.S2215F has already been reported in several patients with type II FCD (9–12) and in a single patient with HME (4). Whole exome sequencing (WES) performed in paired DNA samples extracted from dysplastic brain tissue and blood confirmed the findings identified through panel analysis.
RPS6 p.R232H and MTOR p.S2215 variants affect different specific steps of cortical development
To investigate the impact of each variant on cortical development, we electroporated rat embryos at E15 with pCAG-RPS6-wild-type (WT) or p.R232H-IRES-green fluorescent protein (GFP) plasmids (RPS6 WT or RPS6 R232H), pCAG-MTOR-WT or p.S2215F-IRES-GFP plasmids (MTOR WT or MTOR S2215F) or pCAG-IRES-GFP (IRES-GFP; control) plasmids and performed analyses at E16.5 and E20.
For neuroprogenitors proliferation analyses, we performed immunostaining on E16.5 brains with an anti-phospho-histone H3 (PH3) antibody and determined the mitotic index calculating the number of electroporated PH3 positive cells (PH3+; GFP+) divided by the number of electroporated cells (GFP+) in the ventricular/subventricular zone (VZ/SVZ) (Fig. 2A and B). Neuroprogenitors electroporated with RPS6 R232H plasmid exhibited an increased mitotic index, whereas MTOR S2215F electroporated neuroprogenitors showed a mitotic index similar to that observed in neurons electroporated with IRES-GFP, RPS6 WT or MTOR WT plasmids (Fig. 2B).
Figure 2.

Neuroprogenitors proliferation analysis on embryonic rat brains. (A) Confocal microphotographs of E16.5 brain coronal sections from rat embryos electroporated at E15 with IRES-GFP, RPS6 WT, RPS6 R232H, MTOR WT or MTOR S2215F plasmids. Immunostaining was performed with anti-PH3 and anti-GFP antibodies. Sections were counterstained with Hoechst. Arrows indicate PH3+ and GFP+ neuroprogenitors. Scale bar = 30 μm. (B) Quantification of the percentage of mitotic neuroprogenitors (PH3+, GFP+) among transfected cells (GFP+) in the VZ/SVZ of electroporated brains. Values are given as mean ± SEM. One-way ANOVA and Tukey’s multiple comparison tests, ****P < 0.0001; IRES-GFP, RPS6 WT, RPS6 R232H, MTOR WT, MTOR S2215F, n = 10; IRES-GFP/IRES-TOMATO, RPS6 WT/MTOR WT, RPS6R232H/MTOR S2215F, n = 8. Statistical elements are provided in Supplementary Material, Table S4. (C) Confocal microphotographs of E16.5 brain coronal sections from rat embryos electroporated at E15 with IRES-GFP, RPS6 WT, RPS6 R232H, MTOR WT or MTOR S2215F plasmids. Immunostaining was performed with anti-Ki67 and anti-GFP antibodies. Sections were counterstained with Hoechst. Arrows indicate Ki67+ and GFP+ neuroprogenitors. Scale bar: 30 μm. (D) Quantification of the percentage of cycling neuroprogenitors (Ki67+, GFP+) among transfected cells (GFP+) in the VZ/SVZ of electroporated brains. Values are given as mean ± SEM. One-way ANOVA and Tukey’s multiple comparison tests, ****P < 0.0001, ***P = 0.003; IRES-GFP, n = 7; RPS6 WT, MTOR WT, IRES-GFP/IRES-TOMATO, RPS6 WT/MTOR WT, RPS6 R232H/MTOR S2215F, n = 8; RPS6 R232H, MTOR S2215F, n = 9. Statistical elements are provided in Supplementary Material, Table S5.
We then performed immunostaining with an anti-Ki67 antibody to quantify the percentage of cycling electroporated cells (Ki67+; GFP+) among electroporated cells (GFP+) in the VZ/SVZ (Fig. 2C and D). Ki67 is expressed during all phases of the cell cycle, and the percentage of Ki67+ cells reflects the proportion of neuroprogenitors reentering or exiting the cell cycle. As for the mitotic index, neuroprogenitors electroporated with the RPS6 R232H plasmid exhibited an increased percentage of cycling cells, whereas for those electroporated with MTOR S2215F plasmid the percentage of cycling cells matched that observed in neurons electroporated with IRES-GFP, RPS6 WT or MTOR WT plasmids (Fig. 2D).
Increase in mitotic index and percentage of cycling cells observed in RPS6 R232H electroporated brains were not paralleled by an increased phosphorylation of RPS6 Ser240–244 residues (Supplementary Material, Fig. S2). Supernumerary neuroprogenitors did not die due to apoptosis, as demonstrated by anti-cleaved caspase 3 staining (Supplementary Material, Fig. S3).
For neuronal migration analyses, we determined the relative position of electroporated neurons in the cortical thickness on E20 brains (Fig. 3A and B). Most IRES-GFP and RPS6 or MTOR WT electroporated neurons reached the cortical plate (CP), indicating that overexpression of RPS6 WT or MTOR WT did not affect neuronal migration. Neurons electroporated with RPS6 R232H also reached the CP and were normally distributed, whereas only two thirds of those electroporated with MTOR S2215F reached the CP, mostly remaining distributed in the lower region of the CP and at the border with the intermediate zone (IZ). The remaining MTOR S2215F electroporated neurons stayed in the IZ.
Figure 3.

Neuronal migration analysis on embryonic rat brains. (A) Upper panels: Confocal microphotographs of E20 brain coronal sections immunostained for GFP from rat embryos electroporated at E15 with IRES-GFP, RPS6 WT, RPS6 R232H, MTOR WT or MTOR S2215F plasmids. Lower panels: Confocal microphotographs of E20 brain coronal sections immunostained for GFP and TdTomato from rat embryos electroporated at E15 with IRES-GFP/IRES-TOMATO, RPS6 WT/MTOR WT or RPS6 R232H/MTOR S2215F plasmids. Sections were counterstained with Hoechst. White dotted lines delimitate CP, IZ and VZ/SVZ. Scale bars: 350 μm. (B) Quantification of the percentage of GFP+ neurons in VZ/SVZ (left), IZ (middle) and CP (right) of electroporated brains for each condition. Values are given as mean ± SEM. One-way ANOVA and Tukey’s multiple comparison tests, ****P < 0.0001; IRES-GFP, IRES-GFP/IRES-TOMATO, n = 9; RPS6 WT, RPS6 R232H, MTOR WT, n = 10; MTOR S2215F, RPS6 WT/MTOR WT, RPS6 R232H/MTOR S2215F, n = 11. Statistical elements are provided in Supplementary Material, Table S6.
These in vivo studies suggest that, when overexpressed alone, RPS6 p.R232H increases neuroprogenitors proliferation as evidenced by the increased mitotic index and percentage of Ki67+ cycling neuroprogenitors, whereas MTOR p.S2215F delays neuronal migration.
RPS6 p.R232H and MTOR p.S2215 variants coexpression worsens the phenotype observed at embryonic stages
Since both variants affect genes involved in the same signaling pathway and could in theory coexist in the same neurons, we investigated whether their coexpression would worsen the defects induced by each of them. We coelectroporated E15 rat embryos with pCAG-RPS6 WT-IRES-tdTomato and pCAG-MTOR WT-IRES-GFP (RPS6 WT/MTOR WT), pCAG-RPS6 p.R232H-IRES-tdTomato and pCAG-MTOR p.S2215F-IRES-GFP (RPS6 R232H/MTOR S2215F) or pCAG-IRES-tdTomato (IRES-TOMATO) and IRES-GFP controls plasmids and performed neuroprogenitors proliferation and neuronal migration analyses as described above. Both the mitotic index and the percentage of cycling RPS6 R232H/MTOR S2215F coelectroporated neuroprogenitors were slightly increased compared to those of RPS6 R232H electroporated neuroprogenitors (Fig. 2B and D). At E20, IRES-GFP/IRES-TOMATO or RPS6 WT/MTOR WT coelectroporated neuroprogenitors exhibited a similarly normal neuronal migration profile, with most neurons reaching the CP, whereas RPS6 R232H/MTOR p.S2215F coelectroporated neurons manifested a migration delay which was more severe than that elicited by the MTOR p.S2215F variant alone (Fig. 3A and B).
Altogether, these experiments show that RPS6 p.R232H and MTOR p.S2215 variants can act synergistically and worsen the proliferation and the migration phenotypes induced independently by either of them.
RPS6 p.R232H and MTOR p.S2215F coexpression partially phenocopies cytological anomalies observed in the hemimegalencephalic brain tissue
Neuropathological evaluations in the hemimegalencephalic brain tissue revealed dysmorphic giant neurons and balloon cells hyperexpressing phospho-RPS6 protein. To investigate the role of RPS6 p.R232H and MTOR p.S2215 variants in the development of these cells in vivo, we electroporated rat embryos at E15 with IRES-GFP, RPS6 R232H, MTOR S2215F or RPS6 R232H/MTOR S2215F plasmids and performed hematoxylin–eosin (H&E) and immunohistochemical staining in P28 brains.
In brains electroporated with RPS6 R232H, neuronal size was comparable with that observed in brains electroporated with IRES-GFP, whereas in MTOR S2215F electroporated brains, neurons exhibited a statistically significant increase of soma size (Fig. 4A and B). This increase in cell size was alike that observed after brain electroporation with RPS6 R232H/MTOR S2215F plasmids (Fig. 4A and B). In both MTOR S2215F and RPS6 R232H/MTOR S2215F conditions, cytomegalic neurons showed phospho-RPS6 protein hyperexpression (Fig. 4C).
Figure 4.
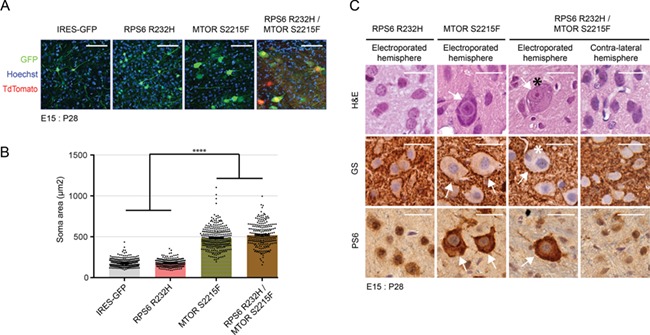
Cell size quantification and morphological characterization of electroporated neurons at postnatal stages. (A) Confocal microphotographs of P28 brain coronal sections from rat embryos electroporated at E15 with IRES-GFP, RPS6 R232H, MTOR S2215F or RPS6 R232H/MTOR S2215F plasmids. All sections were immunostained for GFP and counterstained with Hoechst. RPS6 R232H/MTOR S2215F electroporated sections were additionally stained for tdTomato. Scale bar = 100 μm. (B) Quantification of the soma area (μm2) of electroporated neurons for each condition. Values are given as mean ± SEM. Kruskal Wallis and Dunn’s multiple comparisons tests, ****P < 0.0001; IRES-GFP, n = 356; RPS6 R232H, n = 256; MTOR S2215F, n = 294; RPS6 R232H/MTOR S2215F, n = 222. Statistical elements are provided in Supplementary Material, Table S7. (C) H&E and immunohistochemical staining of P28 brain coronal sections from rat embryos electroporated at E15 with RPS6 R232H, MTOR S2215F or RPS6 R232H/MTOR S2215F plasmids. Immunohistochemistry was performed with anti-PS6 and anti-GS antibodies. White arrows indicate cytomegalic neurons. Black and white asterisks indicate cytomegalic neurons with small cytoplasmic vacuoles/eccentric nuclei and binucleated cells, respectively. Control neurons from the contralateral hemisphere of a RPS6 R232H/MTOR S2215F electroporated brain are shown for comparison. Scale bars: 30 μm.
Cytomegalic neurons showed accumulation of Nissl aggregates displaced to the membrane (Fig. 4C). In addition, a subset of double RPS6 R232H/MTOR S2215F cytomegalic cells showed a more severe morphological phenotype with eccentric nuclei and binucleation, which are typical features of balloon cells (14). However, these cells did not stain with anti-GS, a marker usually expressed by balloon cells.
Altogether, these experiments show that, at postnatal stages, MTOR p.S2215F expression is sufficient for generating cytomegalic neurons that hyperexpress the phospho-RPS6 protein. Although RPS6 p.R232H alone does not seem to influence cell morphology, its coexpression with MTOR p.S2215F is necessary for the development of cytomegalic cells morphologically similar to balloon cells.
Discussion
Using NGS, we identified two somatic variants affecting two different genes within the mTOR signaling pathway in a patient with HME, severe intellectual disability, intractable seizures and hypochromic skin patches. Using in vivo experimental models, we demonstrated that RPS6 p.R232H and MTOR p.S2215F act synergistically, worsening the defects induced by each single variant.
The RPS6 p.R232H variant was present with a similar percentage of mosaicism in dysplastic brain tissue and blood cells (~15.1% in brain and 11% in blood), indicating its origin in early embryonic stages. Variants in this gene had not been associated with human disease before, thus making additional studies necessary to confirm its causative role in HME.
The MTOR p.S2215F variant was present in dysplastic brain tissue only, with a percentage of mosaicism around 8.8%, which indicates that it arose in advanced embryonic stages. This substitution had already been identified, always with brain confined somatic distribution, in type II FCD, with mosaicism rates ranging from 1% to 8% (9–12), and in one patient with HME (4), exhibiting 18–20% mosaicism.
RPS6 is mainly known for its role as a downstream effector of MTOR in the regulation of cell growth and proliferation and its level of phosphorylation reflects the mTORC1 complex activation (6). Neuropathological demonstration of phospho-RPS6Ser240–244 hyperexpression in cytomegalic cells of the hemimegalencephalic tissue is in line with previous immunocytochemical and immunohistochemical assays performed to characterize the MTOR p.S2215F variant (11).
Our in utero electroporation (IUE) experiments indicate that RPS6 p.R232H overexpression selectively increases neuroprogenitors proliferation (Fig. 2), without apparently affecting other developmental steps, such as migration (Fig. 3), maturation (Fig. 4) or apoptosis (Fig. S3). Previous studies on animal and cellular models have demonstrated that conditional deletion of RPS6 in adult mice liver blocks hepatocyte proliferation without affecting cell growth (15) and that downregulation of RPS6 levels in HeLa cells decreases cell proliferation (16). Overall, these evidences indicate that RPS6 is a positive regulator of cell proliferation and suggest that RPS6 p.R232H acts as an activating mutation.
How p.R232H impacts on cell cycle regulation remains to be determined. RPS6 is a key ribosomal protein that undergoes phosphorylation in response to a wide variety of stimuli. This modification plays a critical regulatory role in multiple cellular and organismal processes (6). RPS6 phosphorylation occurs at five highly conserved and clustered serine residues (Ser 235, 236, 240, 244 and 247) (17). All these serines can be phosphorylated by the ribosomal S6 kinase (S6K) through an mTOR pathway dependent mechanism. Ser 235 and 236 can also be phosphorylated through the Ras/Raf/MEK/ERK/RSK pathway, which is mTOR independent (18). In RPS6 R232H electroporated brains, we could not observe increased phospho-RPS6Ser240–244 expression at E16.5 (Fig. S2) or cytomegalic neurons hyperexpressing phospho-RPS6Ser240–244 at P28 (Fig. 4C). These findings suggest that the p.R232H variant might exert its pathogenic effect independently of its mTOR pathway mediated phosphorylation. As RPS6 p.R232H maps three amino acids upstream the first phosphorylatable serine residue of the cluster, it is conceivable that it might interfere with Ser 235 and 236 phosphorylation/dephosphorylation, leading to increased cell proliferation mediated by the RAS/ERK/RSK cascade.
Studying embryonic fibroblasts of a knock-in mouse model in which the five phosphorylatable serines were replaced by alanines, Ruvinsky and collaborators (19) demonstrated that RPS6P−/− cells exhibited increased rate of protein synthesis, accelerated cell division, and efficient 5' TOP mRNAs translation. 5' TOP mRNAs encode various proteins associated with the function or the assembly of the translational apparatus. Their translation, which is in part regulated by their direct association with RPS6, is selectively activated when resting cells are induced to grow or proliferate (19). Therefore, p.R232H might also induce increased cell proliferation by altering the binding between RPS6 and 5' TOP mRNAs and, consequently, their translation.
In utero MTOR p.S2215F overexpression induces a neuronal migration defect with development of cytomegalic neurons but has no apparent effects on cortical neuroprogenitors proliferation. This phenotype recapitulates those observed in previous IUE studies performed to characterize other mTOR mutations (9, 20, 21) as well as the one observed in a transgenic mouse model overexpressing a hyperactive mutant mTOR (22).
Two previous studies in which combined somatic and germline mutations involving a same gene within the mTOR pathway were demonstrated (4, 5) hypothesized a double-hit pathogenic mechanism, analogous to the model proposed by Knudson for retinoblastoma (23). In particular, D’Gama and collaborators (4) demonstrated cooccurring germline and somatic biallelic mutations in TSC2 in two patients with HME, and Ribierre and collaborators (5) found one germline and one somatic mutation in DEPDC5 in a patient with FCD.
Both TSC2 and DEPDC5 are mTORC1 complex inhibitors. Deep sequencing of TSC1, an mTORC1 complex inhibitor too, has uncovered second hit events in several cortical tubers in one out of 34 patients with TSC (2). We demonstrated that RPS6 p.R232H and MTOR p.S2215F variants are pathogenic in vivo and hypothesize that the HME phenotype we observed results from their cumulative neurobiological effect.
By cooverexpressing both variants in the same cells, we observed that a) neuroprogenitors proliferation further increased compared to RPS6 p.R232H alone, b) alteration of neuronal migration was more pronounced compared to MTOR p.S2215F alone and c) a subset of cytomegalic cells exhibited morphological features (i.e. eccentric nuclei and binucleation) that are typical of balloon cells. These findings indicate that RPS6 p.R232H and MTOR p.S2215F, when cooccurring, enhance their respective functional impact through a synergistic effect, whose magnitude is of course also influenced by the proportions of alternative allele fractions. However, it cannot be ruled out that both variants arose in different cell clones, thus exerting their pathogenic effects still cumulatively but independently. In either case, the more severe phenotype observed in the double mutant could be ascribed to a global dysregulation of cell metabolism involving mTOR-related and -unrelated signaling cascades and their possible cross-talks. This hypothesis is supported by the evidence that, in cancer, RAS/ERK/RSK and mTORC1 pathways regulate each other via cross-inhibition and cross-activation mechanisms (24).
The double-hit mechanism we identified does not follow the classical Knudson model (23) as both variants are somatic and affect two different genes, although within the same signaling pathway. Our study rather uncovers a possible novel double-hit mechanism for the pathogenesis of HME, resulting from two independent activating somatic variants, each affecting a single allele of a positive regulator of the mTOR pathway. Our data also identify RPS6 as a potential novel disease-related gene whose pathogenic effect might be exerted, in part, through mTOR unrelated mechanisms.
Since RPS6 p.R232H was present at 11% mosaicism in blood and 15% in the dysplastic hemisphere, it is likely it was also present with similar proportions in the anatomically seemingly normal right hemisphere. Independent right-sided seizure onset was clearly demonstrated, although it became fully apparent only after complete removal of the redundantly epileptogenic left hemisphere. IUE experiments, which recapitulate the effects of mosaicism, demonstrated that introducing RPS6 p.R232H in a subset of developing neurons affects neuronal proliferation, with no abnormal cell types or visible migration anomalies. It is therefore conceivable that similar abnormalities in the patient’s right hemisphere be sufficient to produce epileptogenesis without resulting in an MRI visible cortical abnormality.
The possible cumulative effect of different somatic variants in different genes should be ruled out even after a known pathogenic variant has been demonstrated, either in blood or in the surgically removed brain tissue.
Materials and Methods
Patient recruitment, samples collection and DNA extraction
All procedures were conducted after written informed consent. Additional written informed consent was obtained to publish identifying information included in this article.
The patient belonged to a cohort of 55 patients with malformations of cortical development, with morphologic characteristics suggesting altered neuronal proliferation/migration, seen at the Children’s Hospital Anna Meyer from 2010 to 2016, in whom an NGS study on brain tissue removed during epilepsy surgery was performed.
We extracted DNA from dysplastic brain samples using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and from blood leukocytes using an automated DNA isolation robot (QIASymphony, Qiagen), according to manufacturer’s protocols.
This patient’s blood and brain-derived DNA was studied with both massive parallel targeted resequencing of a panel of 54 mTOR pathway genes and WES, as the study was conducted simultaneously in two different laboratories.
mTOR genes panel analysis
After reviewing the literature and performing in silico pathway predictions, using String (http://string-db.org/) and GeneMANIA (http://genemania.org), we designed a panel containing 54 genes known or predicted to belong to the mTOR pathway (Supplementary Material, Table S1).
We captured all exons and flanking intronic regions of target genes using the HaloPlex target enrichment system (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. Briefly, after enzymatic digestion of 200 ng of genomic DNA and quality check with the Agilent 2100 Bioanalyzer (Agilent Technologies), we hybridized the samples with the HaloPlex probes. Then, we captured circularized target DNA-HaloPlex probes containing biotin with the HaloPlex streptavidin-coated magnetic beads and performed a PCR amplification step of the captured target libraries. Upon purifying and quantifying enriched target DNA, we pooled samples with different indexes at an equimolar ratio and sequenced DNA libraries on a Genome Analyzer IIx (GaIIx, Illumina, San Diego, CA) in 100 base-pairs (bp) reads.
We used BWA (V0.7.7-r441, http://bio-bwa.sourceforge.net/) to map sequencing reads to the UCSC GRCh37/hg19 assembly, Picard (v1.109, http://broadinstitute.github.io/picard/) to remove duplicates, GATK (v3.1, https://www.broadinstitute.org/gatk/) to call variants and Varscan (v2.3.9, http://varscan.sourceforge.net/) to detect somatic (~5%) variants. We filtered resulting variants by removing all possible sequencing/alignment artifacts and annotated them using Annovar (v17 June 15, http://www.openbioinformatics.org/annovar/).
WES analysis
WES was performed as previously described (13) using 300× average coverage for DNA extracted from dysplastic brain tissue and 100× average coverage for DNA extracted from blood. Upon extracting DNA form dysplastic brain samples and blood using Qiagen methodology, Illumina sequencing libraries were prepared and whole-exome capture was performed with the Agilent SureSelect exome enrichment (Human All Exon 50 Mb Kit, Agilent). Libraries underwent pair-end sequencing (2 × 100 bp) on an Illumina Hiseq 2000 sequencer according to the manufacturer’s protocol.
We mapped raw data to reference genome (GRCh37d5) and processed them with GATK 3.8 best practice with indel realignment and base quality recalibration. After recalibration and realignment, we used the paired sample model of Mutect2 (https://software.broadinstitute.org/gatk/documentation/tooldocs/3.80/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2), Strelka2 (https://www.nature.com/articles/s41592-018-0051-x) and the single mode of MosaicHunter (http://mosaichunter.cbi.pku.edu.cn/) to compare data obtained in brain and blood for identifying brain confined somatic variants. Variants detected by Mutect2 and Strelka2, or by MosaicHunter were listed in the call set. Candidate variants were further filtered to reduce false positive rate: variants with gnomAD allelic frequency > 0.01, variants overlapping with UCSC SegDup or RepeatMasker regions, and variants that fell within 5 bp of a germline insertion/deletion were all excluded. Candidate variants were further annotated with the Ensembl Variant Effect Predictor (https://www.ensembl.org/info/docs/tools/vep/index.html) and the likely pathogenic variants were collected.
mTOR and RPS6 variants confirmation
To validate the somatic variant in RPS6, we used the GS Junior sequencing approach (Roche, Mannheim, Germany). We designed primers containing genome-specific sequences with Primer3 plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and added M13 sequences at 5' end of each primer to generate amplicons ranging between 290 and 320 bp in size.
Following a first PCR amplification step, we added sequencing primers with different multiplex identifier sequences (used to differentiate samples being run together on the same plate) to the mix and performed a second PCR amplification step. Then, we removed small DNA fragments using the Agencourt AMPure XP (Beckman Coulter, Beverly, MA), according to the manufacturer’s protocol, and quantified all amplicons using the Quant-iT PicoGreen dsDNA reagent (Invitrogen, Carlsbad, CA). After pooling amplicons at an equimolar ratio and an emulsion PCR amplification step using the GS Junior Titanium emPCR kit (Lib-A) (Roche), we pyrosequenced the pooled amplicons in the sense and antisense strands. We performed data analysis using the GS Amplicon Variant Analyzer version 2.7 (AVAv2.7) software (Roche).
To validate the somatic variant in MTOR, we used ddPCR and smMIPs approaches. For ddPCR, we designed primers and both probes for the WT and mutant allele. The primer and probes are provided in Supplementary Material, Table S2. The working temperature of the probe assay was assessed on a test run on a QX200 machine (Bio-Rad, Hercules, CA) with gBlocks positive control DNA. Samples were further detected with the optimized protocol. Droplets with WT and mutant signals were gated, and the 95% confidence intervals of mutant allelic fractions were estimated according to a Poisson distribution. For smMIPS analysis, MIPs were designed and experiments were carried out according to the manufacturer’s recommendations (https://www.illumina.com/science/sequencing-method-explorer/kits-and-arrays/smmip.html). Mutant allelic fractions and the 95% confidence intervals were calculated with a Bayesian model described previously (25).
Variants classification and submission to public databases
We classified variants according to the standards and guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (26).
According to the recommendations of the 2006 Human Variome Project meeting (27), we submitted the two variants we identified in our patient to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) to ensure their searchability. The uniform resource locators (URLs) to access the two variants are https://submit.ncbi.nlm.nih.gov/subs/clinvar_wizard/SUB5616740/overview for the MTOR p.S2215F variant and https://submit.ncbi.nlm.nih.gov/subs/clinvar_wizard/SUB5616389/overview for the RPS6 p.R232H variant.
Statistical validation of the RPS6 variant
To statistically evaluate variant pathogenicity, we performed a chi-squared test with Yates correction for a 2 × 2 contingency table according to an already established protocol (28). Briefly, we calculated chi-squared test, using the QuickCalcs tool (graphpad.com/quickcalcs/contingency1.cfm), comparing the frequency of RPS6 p.R232H in a cohort of 55 patients who underwent panel analysis with that reported in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/, accessed on November 25, 2018). We carried out the chi-squared test both at large and through ethnicity-matched analysis.
Neuropathology
We cut formalin-fixed, paraffin-embedded sections from brain specimens removed during epilepsy surgery at 5 μm thickness and stained them with H&E for anatomopathological evaluation.
We used additional sections to carry out immunohistochemical analysis using standard avidin–biotin complex/immunoperoxidase method and commercially available antibodies against neurofilaments (NFs) (1:20, Zymed Laboratory, San Francisco, CA; PAN clone DA2; FNP7, RMb020.11), neuronal nuclei (NEU-N) (1:100, Millipore, Billerica, MA), PS6 (1:1200, Cell Signalling Technology, Danvers, MA; #D68F8), GFAP (pre-diluted, Zymed Laboratory, San Francisco, CA; clone ZCG29) and GS (1:2000, Sigma-Aldrich St. Louis, MO).
We acquired images using an Eclipse 80i microscope (Nikon Corporation, Tokyo, Japan) with Digital Sight DS-U1 camera (Nikon Corporation).
Animals
Animals were housed in cages with ad libitum food/water, controlled temperature (21–23°C) and 12:12 h light–dark cycle (light period from 8 A.M. to 8 P.M.).
Cloning of RPS6 and mTOR WT and mutant plasmids
All constructs were generated by Genscript (Piscataway, NJ). WT DNA fragments encoding human WT RPS6 (NM_001010.2; 747 nucleotides) and MTOR (NM_004958.3; 7650 nucleotides) were synthetized and cloned into the EcoR1/Xho1 or Not1 restriction sites of the pCAGIG plasmid (pCAG-IRES-GFP (29), Addgene #11159), respectively. Mutagenesis was performed to introduce the RPS6 c.737G > A p.R232H and the MTOR c.6644C > T p.S2215F variants in the WT RPS6 and MTOR plasmids (pCAG-RPS6 WT-IRES-GFP and pCAGIG-MTOR WT-IRES-GFP) to obtain the mutated ones (pCAG-RPS6 p.R232H-IRES-GFP and pCAG-MTOR p.S2215F-IRES-GFP).
WT and mutated RPS6 DNA fragments were afterwards transferred into the pCAG-IRES-tdTomato plasmid (gift from MC Tiveron) to create the pCAG-RPS6 WT-IRES-tdTomato and pCAG-RPS6 p.R232H-IRES-tdTomato plasmids used in coelectroporation experiments with IRES-GFP plasmids. All plasmids were checked by Sanger sequencing.
In utero electroporation
Timed pregnant Wistar rats (Janvier Labs, Le Genest-Saint-Isle, France) received buprenorphine at 0.03 mg/kg and were anesthetized with sevoflurane (4%) 30 min later. Uterine horns were exposed, and plasmids were microinjected with a PV 820 Pneumatic PicoPump (World Precision Instruments, Sarasota, FL) into the right lateral ventricle of embryonic day 15 embryos (E15), with 0.2% Fast green (F7252, Sigma-Aldrich) dye. Electroporations were performed by delivering 40 V voltage pulses using a BTX ECM 830 electroporator (BTX Harvard Apparatus, Holliston, MA) across tweezer-type electrodes (Nepa Gene Co, Chiba, Japan) pinching the head of each embryo through the uterus.
Single plasmid electroporations were performed with WT or mutant RPS6 plasmids (pCAG-RPS6 WT-IRES-GFP; pCAG-RPR6 p.R232H-IRES-GFP; 1.5 μg/μl), WT or mutant MTOR plasmids (pCAG-MTOR WT-IRES-GFP; pCAG-MTOR p.S2215F; 3.0 μg/μl) or empty plasmids (pCAG-IRES-GFP; 0.5 μg/μl).
Coelectroporations were performed with WT RPS6 and MTOR plasmids (pCAG-RPS6 WT-IRES-tdTomato and pCAG-MTOR WT-IRES-GFP; 1.5 and 3.0 μg/μl respectively), mutant RPS6 and MTOR plasmids (pCAG-RPS6 p.R232H-IRES-tdTomato and pCAG-MTOR p.S2215F-IRES-GFP; 1.5 and 3.0 μg/μl, respectively) or empty plasmids (pCAG-IRES-tdTomato and pCAG-IRES-GFP; 0.5 μg/μl each).
Fluorescent immunohistochemistry and image analysis
E16.5 and E20 brains were dissected out and fixed with AntigenFix (Diapath, Martinengo, Italy) overnight at 4°C. P28 brains were dissected out after transcardial perfusion with AntigenFix (Diapath) and post-fixed overnight at 4°C. Embryonic and P28 brains were sectioned (100 μm) with a vibratome (Leica VT 1000S, Leica, Nussloch, Germany) and processed for immunohistochemistry as free-floating sections. Sections were permeabilized in phosphate-buffered saline (PBS) with 0.3% Triton X-100 for 15 min at room temperature, blocked in PBS with 0.3% Triton X-100 and 10% normal goat serum for 1 h at room temperature and incubated in PBS with 0.3% Triton X-100 and 5% normal goat serum overnight at 4°C with the following primary antibodies: chicken anti-GFP (1/1000; Thermo Fisher Scientific, Waltham, MA; #A10262), rabbit anti-DsRed (1/1000; Takara Bio Nojihigashi, Japan; #632496), rabbit anti-PH3 (1/500; Millipore, #06-570) and anti-Ki-67 (1/300; Millipore, #AB9260). Sections were washed in PBS and incubated for 2 h at room temperature with the following secondary antibodies: goat anti-chicken Alexa Fluor 488 (1/500; Invitrogen, #A11039), goat anti-rabbit Alexa Fluor 555 (1/500; Invitrogen, #A21428) and goat anti-rabbit Alexa Fluor 647 (1/500; Invitrogen, #A21244). Sections were counterstained with Hoechst 33342 (1/1000, Thermo Fisher Scientific), washed in PBS and mounted in Fluoromount (Thermo Fisher Scientific). All images were acquired on a LSM800 Zeiss confocal microscope (Zeiss, Jena, Germany) and analyzed with the Fiji software.
For proliferation analyses on E16.5 brains, images were acquired with a 40× objective, and the quantifications were made with the Image J (https://imagej.nih.gov/ij/) Cell Counter plugin, on maximum intensity projection (MIP) images. (PH3+; GFP+) or (Ki-67+; GFP+) and GFP+ cells in the VZ/SVZ were counted. For each embryo, counting was performed on three sections, and the percentage of (PH3+; GFP+)/GFP+ or (Ki-67+; GFP+)/GFP+ cells was calculated. Eight to 10 brains were analyzed for each experimental condition, from two to three different litters.
For neuronal migration analyses on E20 brains, images were acquired with a 10× objective, and the quantifications were made with the Image J Cell Counter plugin, on two different optical plans (separated by four optical plans or 24 μm) from one image. GFP+ positive cells were counted in CP, IZ and VZ/SVZ defined by the Hoechst 33342 staining and the percentage of GFP+ cells in each of these regions calculated. Nine to 11 brains were analyzed for each experimental condition, from two to three litters.
For neuronal soma area analysis on P28 brains, mosaic images covering the whole thickness of the cerebral cortex (from the white matter to the pial surface) were acquired with a 20× objective. Soma area of GFP+ neurons was measured on MIP images. Soma area from 222 to 356 neurons was determined, and 2 to 3 brains were analyzed for each experimental condition, from one to two litters.
Histology and visible light immunohistochemistry
Paraffin-embedded sections (7 μm) of antigen-fixed rat P28 electroporated brains were stained with H&E. Immunohistochemistry on paraffin sections was performed using a BenchMark XT immunostainer (Roche) with the following primary antibodies: mouse anti-GS (1:200, Millipore, #MAB302) or rabbit anti-PS6 (1:200, Cell Signalling Technology, #2215). Sections were then incubated with secondary horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies in the presence of peroxide/diaminobenzidine (Sigma-Aldrich, #D8001). Images were acquired using an Olympus BX40 microscope (Olympus, Tokyo, Japan).
Statistical analyses
All statistical analyses for in vivo studies were performed using Prism 6 (Graphpad). Normality of the data distributions was systematically tested using d'Agostino & Pearson and Shapiro–Wilk tests. Comparison of groups was subsequently tested with unpaired Student’s t-tests for normal data sets (comparison between two data sets) or with one-way analysis of variance (ANOVA) and Tukey’s multiple comparison tests for normal data sets or Kruskal–Wallis and Dunn’s multiple comparison tests for non-normal data sets (comparisons between data sets >2). All values are given as mean ± SEM. All tests were two-tailed, and the level of significance was set at P < 0.05. `n’ refers to the number of brains except for the quantification of neuronal soma area at P28 where it refers to the number of neurons.
Supplementary Material
Acknowledgements
The authors thank the INMED platforms (PPGI, PBMC and InMagic).
Conflict of Interest statement
None of the authors reports conflicts of interest in relation to the contents of this article.
Funding
European Commission, 7th Framework programme (Health-F2-602531-2013) to R.G. and A.R.; French National Center for Scientific Research to A.R.; Howard Hughes Medical Institute to J.G.G.; US National Institutes of Health (U01MH108898 and R01NS083823); Italian Ministry of Health and the Tuscany Region (RF-2013-02355240 to R.G.).
References
- 1. D'Gama A.M. and Walsh C.A. (2018) Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci., 21, 1504–1514. [DOI] [PubMed] [Google Scholar]
- 2. Qin W., Chan J.A., Vinters H.V., Mathern G.W., Franz D.N., Taillon B.E., Bouffard P. and Kwiatkowski D.J. (2010) Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol., 20, 1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baulac S., Ishida S., Marsan E., Miquel C., Biraben A., Nguyen D.K., Nordli D., Cossette P., Nguyen S., Lambrecq V. et al. (2015) Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol., 77, 675–683. [DOI] [PubMed] [Google Scholar]
- 4. D'Gama A.M., Woodworth M.B., Hossain A.A., Bizzotto S., Hatem N.E., LaCoursiere C.M., Najm I., Ying Z., Yang E., Barkovich A.J. et al. (2017) Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep., 21, 3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ribierre T., Deleuze C., Bacq A., Baldassari S., Marsan E., Chipaux M., Muraca G., Roussel D., Navarro V., Leguern E. et al. (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest., 128, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyuhas O. (2008) Physiological roles of ribosomal protein S6: one of its kind. Int. Rev. Cell Mol. Biol., 268, 1–37. [DOI] [PubMed] [Google Scholar]
- 7. Lipton J.O. and Sahin M. (2014) The neurology of mTOR. Neuron, 84, 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hay N. and Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev., 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- 9. Lim J.S., Kim W.-I., Kang H.C., Kim S.H., Park A.H., Park E.K., Cho Y.W., Kim S., Kim H.M., Kim J.A. et al. (2015) Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med., 21,395–400. [DOI] [PubMed] [Google Scholar]
- 10. Nakashima M., Saitsu H., Takei N., Tohyama J., Kato M., Kitaura H., Shiina M., Shirozu H., Masuda H., Watanabe K. et al. (2015) Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann. Neurol., 78, 375–386. [DOI] [PubMed] [Google Scholar]
- 11. Mirzaa G.M., Campbell C.D., Solovieff N., Goold C.P., Jansen L.A., Menon S., Timms A.E., Conti V., Biag J.D., Olds C. et al. (2016) Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol., 73, 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Møller R.S., Weckhuysen S., Chipaux M., Marsan E., Taly V., Martina Bebin E., Hiatt S.M., Prokop J.W., Bowling K.M., Mei D. et al. (2016) Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol. Genet., 2, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee J.H., Huynh M., Silhavy J.L., Kim S., Dixon-Salazar T., Heiberg A., Scott E., Bafna V., Hill K.J., Collazo A. et al. (2012) De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet., 44, 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luan G., Gao Q., Zhai F., Zhou J., Liu C., Chen Y. and Li T. (2015) Adenosine kinase expression in cortical dysplasia with balloon cells: analysis of developmental lineage of cell types. J. Neuropathol. Exp. Neurol., 74, 132–147. [DOI] [PubMed] [Google Scholar]
- 15. Volarevic S., Stewart M.J., Ledermann B., Zilberman F., Terracciano L., Montini E., Grompe M., Kozma S.C. and Thomas G. (2000) Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science, 288, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 16. Hagner P.R., Mazan-Mamczarz K., Dai B., Balzer E.M., Corl S., Martin S.S., Zhao X.F. and Gartenhaus R.B. (2011) Ribosomal protein S6 is highly expressed in non-Hodgkin lymphoma and associates with mRNA containing a 5′ terminal oligopyrimidine tract. Oncogene, 30, 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biever A., Valjent E. and Puighermanal E. (2015) Ribosomal protein S6 phosphorylation in the nervous system: from regulation to function. Front. Mol. Neurosci., 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., Sonenberg N. and Blenis J. (2007) RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem., 282, 14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruvinsky I., Sharon N., Lerer T., Cohen H., Stolovich-Rain M., Nir T., Dor Y., Zisman P. and Meyuhas O. (2005) Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev., 19, 2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park S.M., Lim J.S., Ramakrishina S., Kim S.H., Kim W.K., Lee J., Kang H.C., Reiter J.F., Kim D.S., Kim H. et al. (2018) Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron, 99, 83–97.e7. [DOI] [PubMed] [Google Scholar]
- 21. Tarkowski B., Kuchcinska K., Blazejczyk M. and Jaworski J. (2019) Pathological mTOR mutations impact cortical development. Hum. Mol. Genet., 28, 2107–2119. [DOI] [PubMed] [Google Scholar]
- 22. Kassai H., Sugaya Y., Noda S., Nakao K., Maeda T., Kano M. and Aiba A. (2014) Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep., 7, 1626–1639. [DOI] [PubMed] [Google Scholar]
- 23. Knudson A.G. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A., 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mendoza M.C., Er E.E. and Blenis J. (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci., 36, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang X., Liu A., Xu X., Yang X., Zeng Q., Ye A.Y., Yu Z., Wang S., Huang A.Y., Wu X. et al. (2017) Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci. Rep., 7, 15677–15689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cotton R.G.H., Appelbe W., Auerbach A.D., Becker K., Bodmer W., Boone D.J., Boulyjenkov V., Brahmachari S., Brody L., Brookes A. et al. (2007) Recommendations of the 2006 Human Variome Project meeting. Nat. Genet., 39, 433–436. [DOI] [PubMed] [Google Scholar]
- 28. Fassio A., Esposito A., Kato M., Saitsu H., Mei D., Marini C., Conti V., Nakashima M., Okamoto N., Olmez Turker A. et al. (2018) De novo mutations of the ATP6V1A gene cause developmental encephalopathy with epilepsy. Brain, 141, 1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuda T. and Cepko C.L. (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A., 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.