

Abstract
Spinal muscular atrophy (SMA) is a devastating infantile genetic disorder caused by the loss of survival motor neuron (SMN) protein that leads to premature death due to loss of motor neurons and muscle atrophy. The approval of an antisense oligonucleotide therapy for SMA was an important milestone in SMA research; however, effective next-generation therapeutics will likely require combinatorial SMN-dependent therapeutics and SMN-independent disease modifiers. A recent cross-disease transcriptomic analysis identified Stathmin-1 (STMN1), a tubulin-depolymerizing protein, as a potential disease modifier across different motor neuron diseases, including SMA. Here, we investigated whether viral-based delivery of STMN1 decreased disease severity in a well-characterized SMA mouse model. Intracerebroventricular delivery of scAAV9-STMN1 in SMA mice at P2 significantly increased survival and weight gain compared to untreated SMA mice without elevating Smn levels. scAAV9-STMN1 improved important hallmarks of disease, including motor function, NMJ pathology and motor neuron cell preservation. Furthermore, scAAV9-STMN1 treatment restored microtubule networks and tubulin expression without affecting tubulin stability. Our results show that scAAV9-STMN1 treatment improves SMA pathology possibly by increasing microtubule turnover leading to restored levels of stable microtubules. Overall, these data demonstrate that STMN1 can significantly reduce the SMA phenotype independent of restoring SMN protein and highlight the importance of developing SMN-independent therapeutics for the treatment of SMA.
Introduction
Spinal muscular atrophy (SMA) is an infantile genetic disorder that causes the loss of spinal motor neurons and leads to progressive muscle atrophy, paralysis and eventually premature death. SMA is caused by reduced levels of survival motor neuron (SMN) protein due to homozygous deletion of the SMN1 gene (1). While SMN protein is encoded by both SMN1 and SMN2, only SMN1 produces 100% SMN protein (2–4). In contrast, SMN2 carries a C to T transition in exon 7 that disrupts an exonic splicing enhancer, causing the alternative splicing of the SMN2 exon 7 transcript leading to ~10% full-length and ~90% of a truncated and dysfunctional SMNΔ7 protein (4,5). Due to the low levels of SMN protein produced by SMN2, and the correlation between disease severity and SMN levels (6,7), SMN2 is considered a fundamental disease modifier. Thus, an attractive therapeutic approach for SMA is to increase SMN levels by targeting SMN2 exon 7 inclusion. The first SMA approved drug, SpinrazaTM, employs this mechanistic approach to increase production of the full-length SMN transcript from SMN (8). The most recent FDA approved therapeutic, Zolgensma, in contrast, employs a gene replacement approach in order to restore SMN expression (9). However, continuing efforts should be made to identify alternative therapeutics that could potentially be combined to target a wider range of SMA patients and disease manifestations.
Current therapeutics in clinical trials for SMA include SMN viral gene replacement (10), modulation of SMN2 splicing (11–14), increased SMN protein stability (15–19), SMN-independent neuroprotectants (20) and muscle activators (21). However, current data from clinical trials and animal model studies indicate that SMN-dependent or SMN-independent strategies alone might not be sufficient to fully prevent or reverse SMA pathogenesis (22–24); therefore it is important to explore alternative approaches that could extend the therapeutic window or increase the patient population that responds to SMN-targeting therapeutics. Combinatorial approaches are currently being examined in SMA mouse models. Plastin-3 (PLS3), a potent disease modifier identified in discordant SMA families (25), significantly improved disease phenotype when combined with a sub-optimal antisense oligonucleotide (ASO) treatment over treatment of PLS3 or ASO alone (26–28) in animal models of SMA. Similarly, treatment of a severe SMA mouse model with Azithromycin, an FDA-approved read-through drug, in combination with a sub-optimal ASO dose significantly improved disease phenotype over either treatment alone (29). An important pipeline for complementary therapeutic pathways revolves around the identification of potent disease modifiers.
Although SMN is ubiquitously expressed, a key feature of SMA that is not fully understood is the selective pathology within motor neurons (30). A major step forward in the understanding of selective vulnerability to pathology was the identification of specific motor units that are selectively resistant or vulnerable to pathology, as defined by denervation of the neuromuscular junction (NMJ) (31–33). Differentially vulnerable motor neuron populations have also been observed in other genetically distinct, but phenotypically similar, motor neuron diseases (MNDs) including amyotrophic lateral sclerosis (ALS) (34,35), and spinal and bulbar muscular atrophy (SBMA) (36). This suggests that there may be specific genetic components that are intrinsic to particular motor neuron pools that confer resistance to pathology.
Independent transcriptional profiling analyses of neurologically normal motor neurons, which are differentially vulnerable in each disease, revealed a large number of differentially regulated transcripts (34–40). Recently, comparisons of some of the independent transcriptional profilings across all three diseases generated a discrete list of uniformly dysregulated genes, a population that could be important genetic contributors to the pathology-resistant phenotype (41). Alpha synuclein (SNCA) was one of these genes that were downregulated in each disease context. When SNCA was delivered to the central nervous system of SMA mouse models via viral vector, SNCA expression significantly reduced disease severity, prolonged lifespan and ameliorated NMJ pathology (41). Importantly, the initial basis for this screen was motor neuron pathology, and the demonstration that AAV9-SNCA significantly decreased motor neuron pathology validated the screen as a means to identify functionally relevant disease modifiers. Another important candidate in the list of top targets commonly downregulated across MNDs is Stathmin-1 (STMN1) (41). STMN1 is a ubiquitously expressed phosphoprotein involved in regulating microtubule dynamics by preventing protofilament lateral association (42). Given the involvement of STMN1 in neuronal cytoskeleton dynamics, it is a target with high potential for modifying the SMA phenotype.
In this study, we investigated whether reintroduction of STMN1 could improve the phenotype and pathology in an intermediate mouse model of SMA. AAV-mediated delivery of STMN1 in SMA (Smn2B/−) mice significantly increased survival and birth-to-peak weight gain compared to untreated SMA mice. Motor function was also significantly improved indicated by improved time-to-right (TTR) performance. NMJ pathology, a hallmark of SMA disease progression, was significantly reduced and accompanied with a reduced loss of lumbar motor neuron cell bodies. Collectively, this work emphasizes the importance of SMN-independent pathways and sheds light upon alternative mechanisms, including tubulin polymerization, that can contribute to the complex SMA pathology.
Results
Strategy for identification of potential disease modifying genes
Identifying genes that have the potential to provide resistance or modify disease phenotypes can be challenging as gene expression patterns shift dramatically during the course of disease and development. To streamline the identification process, we previously utilized transcriptomic data derived from the analysis of differential gene expression between vulnerable and resistant motor neurons (41). Motor neuron cell bodies that were resistant or vulnerable to disease pathology were excised via laser microdissection, and RNA was extracted (41). RNAseq identified differentially expressed genes, and an enriched list of candidate genes was identified by cross-referencing to similar screens in related disease contexts, including ALS and SBMA. Lead candidates, including STMN1, were cloned and packaged into recombinant AAV9 virus and delivered to neonatal mouse models of SMA to test their efficacy at modifying disease phenotype (Fig. 1). Stmn1 was found to be significantly downregulated in motor neurons that were vulnerable to pathology compared to resistant motor neuron, suggesting that its expression correlates with resistance to pathology.
Figure 1.
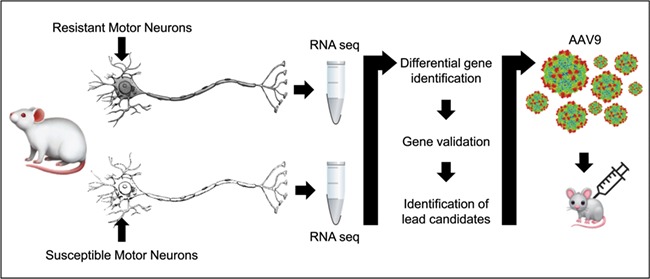
Strategy for identifying potential disease gene modifiers. The first step is to identify pools of motor neurons that are resistant and vulnerable to pathogenesis, which is determined by the vulnerability to denervation of target muscle fibers. Then by laser microdissection, motor neuron cell bodies are isolated and RNA is extracted from both pools. Transcriptomic analysis is performed on both pools, and differentially expressed genes are identified and validated to generate a list of potential genes. These genes are then packaged into AAV9 virus and used to treat neonatal mouse models of the disease via ICV or IV injection. The effects of the gene modifier are then evaluated.
STMN1 treatment significantly extended survival and decreased disease severity in SMA mice
To investigate the potential of STMN1 to decrease the SMA phenotype, 1 × 1011 viral particles of scAAV9-STMN1 were delivered via a single intracerebroventricular (ICV) injection at postnatal day 2 (P2) to the intermediate Smn2B/− SMA mouse model. Treatment resulted in a significant (28%) increase in mean survival of treated over untreated SMA mice, as well as an extension in the maximal life span from 24 days to beyond 50 days (Fig. 2A). While an overall increase in survival was observed, average weight gain did not increase in the treated cohort from birth to day 25 in treated SMA mice compared to untreated SMA mice (Fig. 2B). However, the birth-to-peak (overall percent increase) weight gain in STMN1-treated SMA mice was significantly increased compared to untreated SMA mice (Fig. 2C). Moreover, overt phenotype appearance of scAAV9-STMN1-treated SMA mice (rightmost mouse) appeared improved as compared to SMA-untreated mice (leftmost mouse) at P20 (Fig. 2D).
Figure 2.
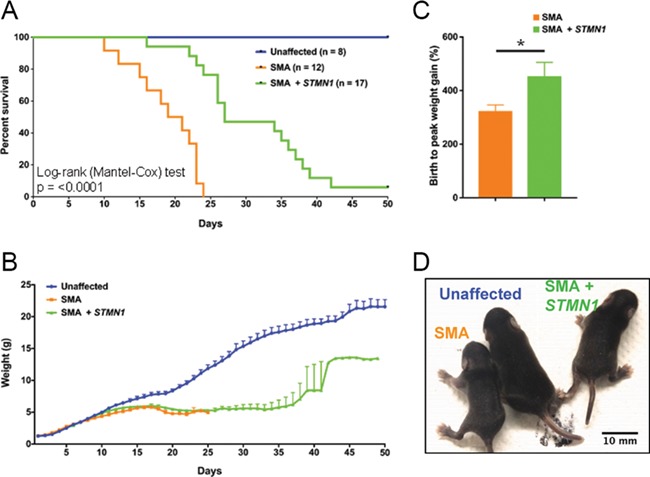
scAAV9-STMN1 treatment increases lifespan and birth-to-peak weight gain in SMA mice. Phenotypic analyses in SMA mice after ICV injection of 1.0 × 1011 scAAV9-STMN1 viral injection at P2. (A) Survival was significantly increased (P < 0.001) in treated SMA mice compared to untreated SMA, healthy controls survived past 50 days. (B) Average weight gain was not different in treated SMA mice compared to untreated SMA up to day 25. (C) Birth-to-peak weight gain was significantly increased (P = 0.0486) in treated SMA mice compared to untreated SMA. (D) Representative image showing health pup at P25 and improvement in overt appearance of treated SMN mice compared to untreated SMA mice. N = 8, healthy control; n = 13, untreated SMA; n = 17, treated SMA. Data expressed as mean ± SEM.
We next investigated if STMN1 also improved motor function in SMA mice. Analysis of the righting reflex demonstrated that the percent of scAAV9-STMN1-treated SMA mice able to right themselves from P5 to P21 was similar to that of healthy controls, while the percent of untreated SMA remained below STMN1 the treated cohort from P7 to P21 (Fig. 3A). Moreover, STMN1-treated mice showed a significant improvement in the average TTR from P6 to P16 compared to untreated SMA mice (Fig. 3B). In contrast, grip strength, motor performance and balance analyses at P18 showed no significant difference between treated and untreated SMA mice (Supplemental Fig. 1). Collectively, the significant increase in lifespan, birth-to-peak weight gain and improved motor performance indicate that scAAV9-STMN1 treatment ameliorated the SMA phenotype in SMA mice.
Figure 3.
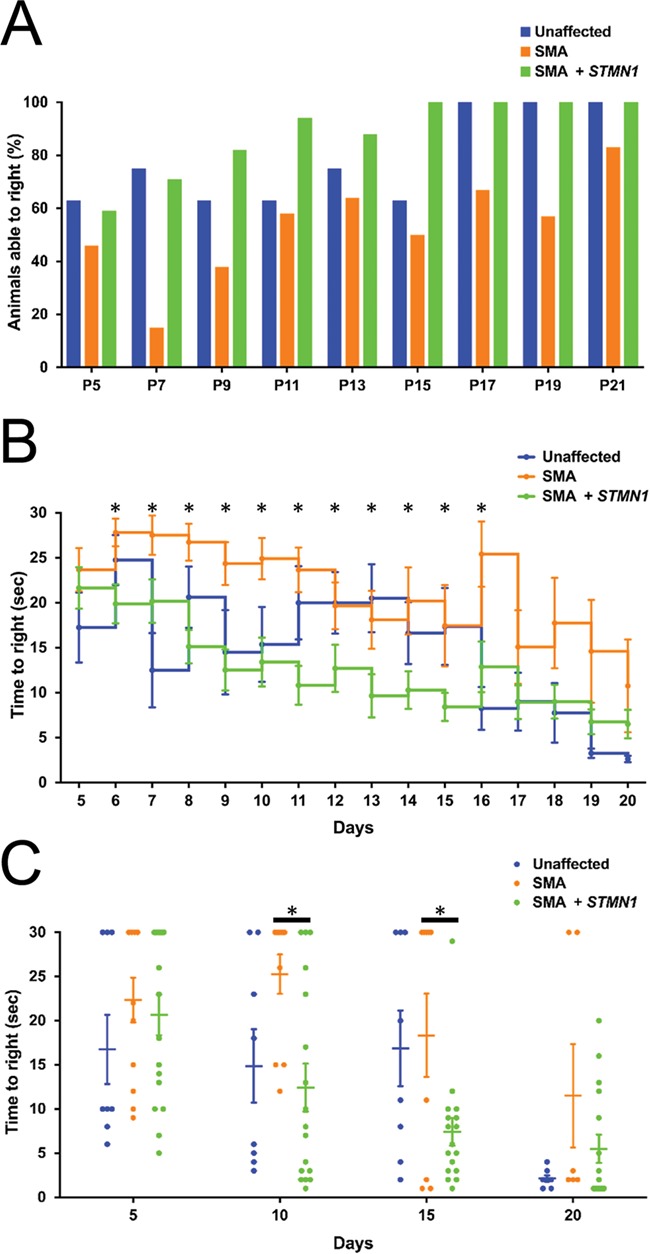
Motor performance was improved in SMA mice following STMN1 treatment. TTR analysis in unaffected, untreated SMA and scAAV9-STMN1-treated SMA mice from P5 to P25. (A) Percent of animals able to right at each specified time point shows an improvement of treated SMA mice compared to untreated SMA mice. (B) Daily average TTR (s) shows that treated SMA mice are able to right significantly faster than untreated SMA mice from Days P6 to P16 (P < 0.05). Moreover, SMA mice treated from P17 to P20 perform better than untreated SMA mice, although not statistically significantly. Moreover, no statistical significance was found between unaffected and treated SMA mice from P17 to P20. TTR comparisons were analyzed by a two-way-ANOVA followed by a Holm–Sidak post hoc test for multiple comparisons. *P < 0.05. N = 8, healthy control; n = 13, untreated SMA; n = 17, treated SMA. Data expressed as mean ± SEM.
scAAV9-STMN1 treatment increases STMN1 protein expression without altering SMN protein levels
To confirm the expression of the vector-derived transgene, STMN1 protein expression in brain tissue and spinal cord were analyzed by western blot at P18. In brain, western blot analysis demonstrated a 1.5-fold increase above untreated samples (Fig. 4A). In spinal cord, western blot revealed no increase in STMN1 expression after treatment (Fig. 4B). However, immunohistochemistry analysis of spinal cord sections demonstrated that STMN1 expression was dramatically upregulated in motor neuron cell bodies (Fig. 4C). Moreover, STMN1 expression extended to dendrites and axons of motor neurons in scAAV9-STMN1-treated animals compared to untreated SMA and unaffected controls (Fig. 4C). Since we were observing a positive impact upon the SMA phenotype, we wanted to confirm that SMN levels were not increased in scAAV9-STMN1-treated animals. Using the same tissues from the STMN1 blots, SMN levels were determined to be unchanged in both brain and spinal cord tissue (Supplementary Fig. 2). These data demonstrate that in scAAV9-STMN1-treated animals, the improvement in the SMA phenotype occurred independent of SMN modulation and that STMN1 can reduce disease severity in intermediate SMA mice.
Figure 4.
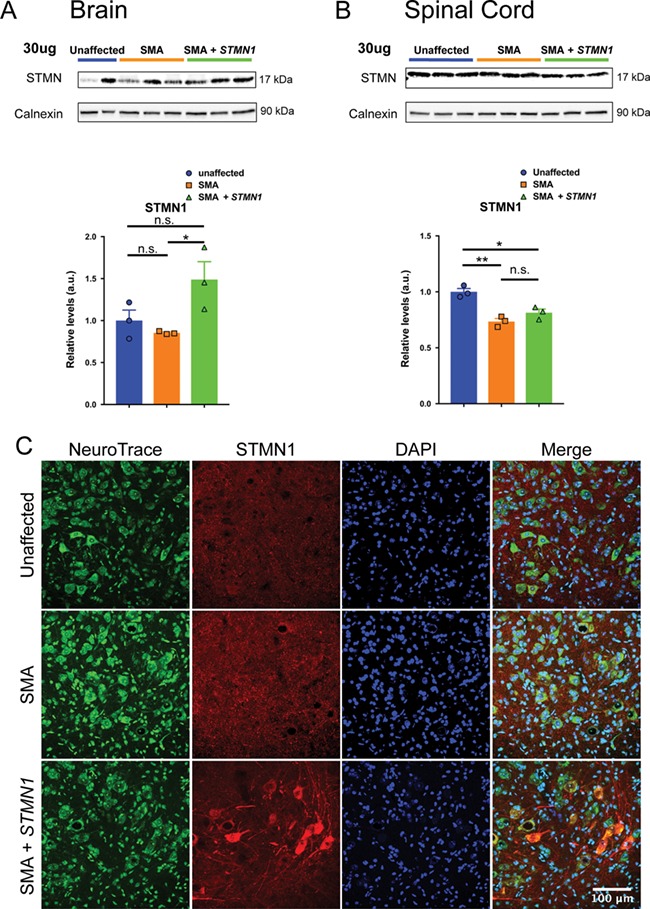
scAAV9-STMN1 treatment results in a significant increase in STMN1 protein expression in peripheral tissues of SMA mice. Western blot analysis from brain tissue show that ICV injection of scAAV9-STMN1 leads to significant upregulation of STMN1 protein expression. (A) Western blots show upregulation of STMN1 protein in treated SMA mice compared to untreated SMA mice. (B) Quantification of blots demonstrate a significant increase in STMN1 protein expression in treated SMA mice compared to untreated mice in peripheral tissues. Comparisons were analyzed by Student t-test. *P < 0.05. Data expressed as mean ± SEM. n = 3 animals per treatment.
scAAV9-STMN1 treatment improves NMJ pathology in highly vulnerable muscles
Differentially vulnerable motor neurons in SMA mouse models were defined by the level of pathology present at the NMJ (32,37). To determine if STMN1 improved NMJ pathology in treated SMA mice, P18 muscles were analyzed by immunofluorescence staining of vulnerable (transversus abdominis (TVA), rectus abdominis (RA) and external oblique (EO)) and resistant (levator auris longus (rostral), auricularis superior (AS) and abductor auris longus (AAL)) muscle groups (37,43). Antibodies against neurofilament heavy (NF-H) and synaptic vesicle protein 2 (SV2) were used to label the axon and pre-synaptic terminals, while Alexa-594-conjugated alpha-bungarotoxin was used to label the motor endplate. Representative images of each muscle were taken and analyzed for denervation. To score the degree of pathology, we assessed the state of innervation for each terminal: (1) complete overlap between axon terminal and the endplate indicated full innervation; (2) partial overlap indicated partial innervation; and (3) empty endplates indicated full denervation (Fig. 5A). As expected, control unaffected muscles showed no defects in NMJ innervation (Fig. 5A, left panel) while untreated SMA muscles displayed evidence of partially and fully denervated endplates (Fig. 5A, middle panel). In contrast, STMN1-treated SMA tissues showed a decrease in the frequency of denervated endplates compared to untreated SMA tissues (Fig. 5A, right panel). Quantification of NMJ innervation profiles showed that treated SMA mice contained significantly more fully innervated NMJs in highly vulnerable muscles, including the TVA, RA and EO muscles compared to untreated SMA mice (Fig. 5A–D). Moreover, NMJ innervation profiles of resistant muscle groups were not changed in STMN1-treated SMA mice compared to untreated SMA mice (Fig. 5E–G), demonstrating that while improvements are observed in vulnerable populations, there was no significant impact upon muscles that were already resistant to disease development.
Figure 5.
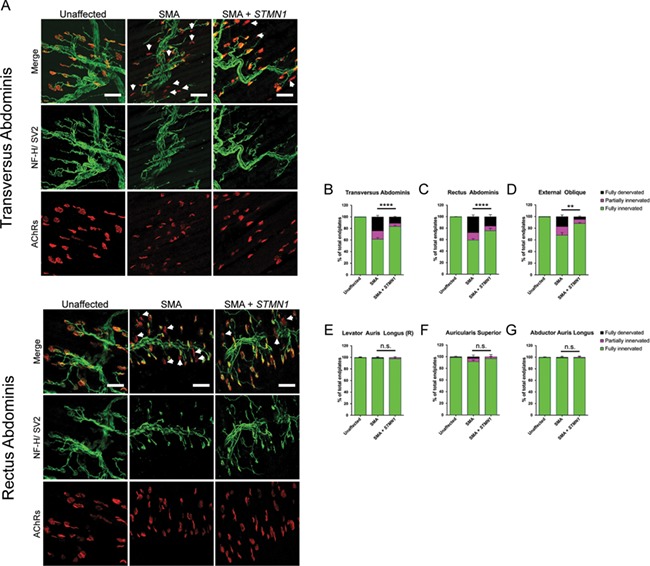
scAAV9-STMN1 treatment improves NMJ pathology in vulnerable muscles of SMA mice. Immunohistochemistry analysis of NMJ innervation in vulnerable (TVA, RA and EO) and resistant (levator auris longus (rostral), AS and AAL) muscles from unaffected, untreated SMA and STMN1-treated SMA mice. Muscles were immunostained to label the axon (NF-H), axon terminal (SV) and endplate (AChRs). (A) Representative images of PND18 TVA and RA (vulnerable) muscles showing reduced frequency of denervated endplates in scAAV9-STMN1-treated SMA mice (right panel) compared to untreated SMA mice (middle panel). Unaffected controls show no denervated endplates (left panel). Maximum projection confocal microscope images taken at ×20 magnification. White arrows point to the location of denervated endplates. (B, C and D) Quantification of NMJ denervation showing percentages of fully innervated, partially innervated and fully denervated endplates in vulnerable muscles. (E, F and G) Quantification of NMJ denervation in resistant muscles showing percent fully innervated, partially innervated and fully denervated NMJs. Denervation analysis shows that STMN1 treatment increases frequency of fully innervated endplates in SMA vulnerable muscles without affecting innervation of resistant muscles. For ease of presentation, only statistical comparisons of fully innervated NMJ percentages are presented. Data analyzed by a two-way ANOVA followed by a Tukey post hoc test for multiple comparisons. Data expressed as mean ± SEM. ****P < 0.0001, ***P < 0.001, n.s. = not significant. n = 3 animals per treatment.
Treatment with STMN1 improves ventral horn (L3–L5) motor neuron pathology
To further understand how STMN1 improved the SMA phenotype, P18 lumbar spinal cords were cross-sectioned and immunostained with Nissl stain (NeuroTrace) to label all neuronal cells and with ChAT to detect motor neuron cell bodies. As expected, a significant difference was observed in the average number of motor neuron cell bodies per 16 μm section between wild-type (Fig. 6A, left panel) and untreated SMA mice (Fig. 6A, middle panel). Consistent with the NMJ data, STMN1-treated SMA samples showed an increase in motor neuron cell body numbers per section compared to untreated SMA mice (Fig. 6A right panel, B). Similarly, morphological measurements of cell bodies within the L3–L5 region of the spinal column revealed that STMN1-treated SMA neurons had a significant increase in cell body area (Fig. 6C) and perimeter (Fig. 6D) compared to untreated SMA neurons. Overall, STMN1 treatment resulted in an increase in motor neuron cell body number as well as improved motor neuron cell morphometrics that resembled those of unaffected control neurons.
Figure 6.
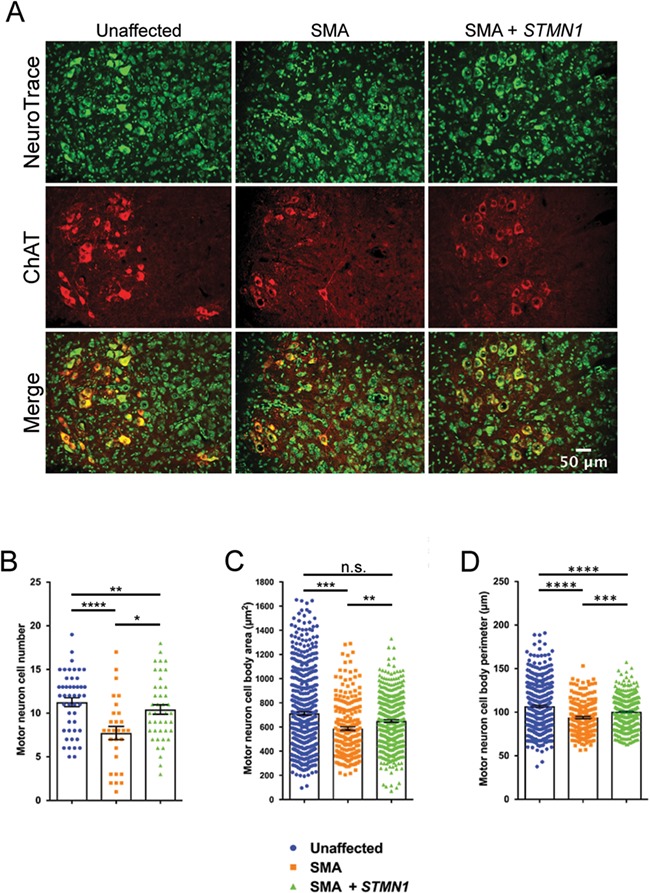
scAAV9-STMN1 treatment prevents motor neuron cell body pathology in SMA mice. Cytological analysis of motor neuron cell body in L3–L5 spinal cord from PND18 treatment groups. (A) Representative images of unaffected (left panel), untreated SMA (middle panel) and treated SMA (right panel) lumbar motor neurons immunostained with Nissl stain (neurotrace) to label neuronal cell bodies and ChAT to label specifically motor neurons. ChAT stain revealed an increased number of motor neurons in STMN1-treated SMA compared to untreated SMA mice. Fluorescent microscope images taken at ×40 magnification. (B) Quantification of motor neuron cell bodies showed a significant increase in cell numbers in STMN1-treated SMA mice compared to untreated SMA. However, STMN1-treated motor neuron cell numbers were still significantly lower than unaffected healthy controls. (C) Morphometric analysis showed a significant improvement in motor neuron cell body area (μm2) of STMN1-treated SMA mice compared to untreated SMA mice. (D) Analysis of cell body perimeter also showed significant improvement in SMTN1-treated SMA motor neurons as compared to untreated SMA. However, these improvements were still significantly lower than unaffected control. Data were analyzed by a one-way ANOVA followed by a Tukey post hoc test for multiple comparisons. Data expressed as mean ± SEM. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n.s. = not significant. n = 3 animals per treatment (n > 400, cells measured per treatment).
AAV9-STMN1 restored microtubule networks in ventral spinal cord motor neurons in SMA mice
Neuronal cell body morphology is dictated by the cells’ cytoskeleton networks, which are made up of microtubules, neurofilaments and microfilaments. As the best described function of STMN1 is to promote microtubule depolymerization (42), we investigated whether STMN1 treatment was functionally impacting microtubule networks surrounding neuronal cell bodies. Spinal cords from all treatment groups at P18 were cross-sectioned and immunostained using antibodies against βIII-tubulin to label microtubules, and Nissl stain (neurotrace) to label neuronal cell bodies. Super-resolution confocal imaging showed detection of discernible filamentous networks in unaffected spinal cords (Fig. 7A, left panel). However, filamentous networks were obviously reduced in untreated SMA spinal cords (Fig. 7A, middle panel). Interestingly, an improvement in the filamentous nature of the tubulin networks was observed in STMN1-treated SMA spinal cords. Filament microtubule density quantification revealed a significant increase in filament density in STMN1-treated spinal cords over both unaffected controls and untreated SMA spinal cords (Fig. 7B). These analyses showed that STMN1 treatment induced a robust restoration of filamentous tubulin networks and correction of phenotype at the cellular level in spinal cords of SMA mice.
Figure 7.
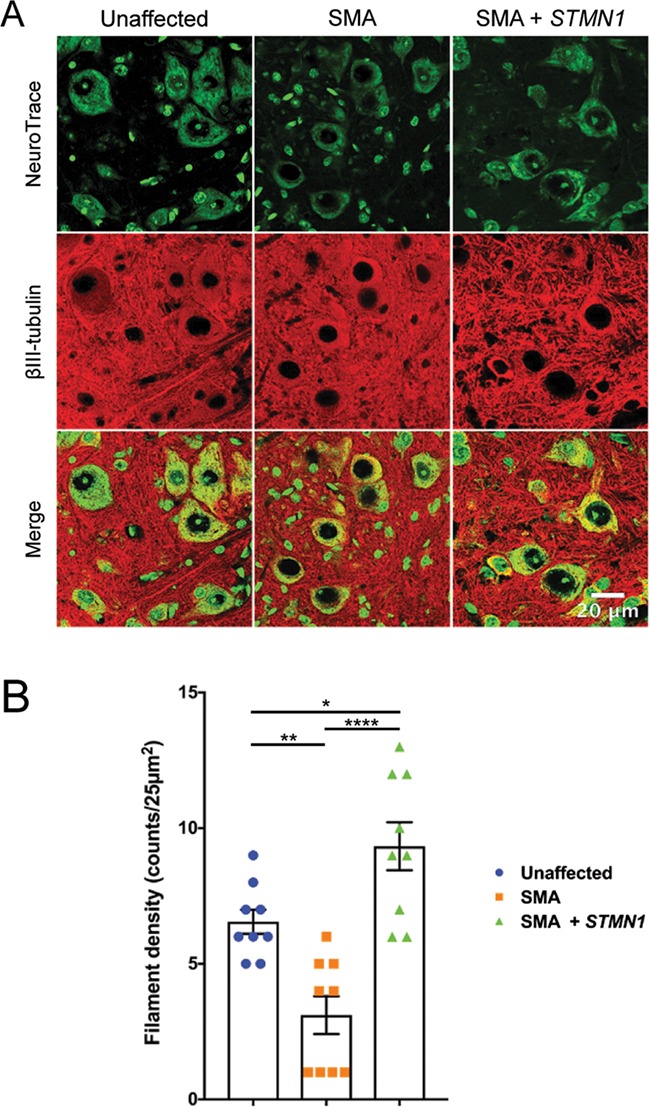
scAAV9-STMN1 treatment restores microtubule filamentous networks in SMA mice. Tubulin filament immunohistochemistry from L3–L5 spinal cords of treatment groups. (A) Representative images of lumbar spinal cord ventral horn motor neurons stained with Nissl stain (neurotrace) and anti-BIII-tubulin antibodies conjugated to Alexa Fluor 594 to label microtubule filaments. Unaffected control tissues showed distinct filamentous networks (left panel), which were absent in untreated SMA ventral horn spinal cords (middle panel). STMN1 treatment restored filamentous networks in SMA lumbar spinal cords to a greater extent compared to unaffected controls. Maximum projection high-resolution confocal microscope images taken at ×63 magnification. (B) Quantifications of filaments per 25 μm2 showed a significant increase in filament density in treated SMA spinal cord compared to untreated and unaffected controls. Data were analyzed by a one-way ANOVA followed by a Tukey post hoc test for multiple comparisons. Data expressed as mean ± SEM. ****P < 0.0001, **P < 0.01, *P < 0.05, n.s. = not significant.
STMN1 treatment does not alter microtubule stability in SMA mice
Microtubules undergo several posttranslational modifications that affect cytoskeleton dynamics (44). Microtubule acetylation is a microtubule posttranslational modification correlated with microtubule stability (45,46). To investigate whether microtubule stability was affected by STMN1 treatment, we analyzed the levels of acetyl-α-tubulin expression in nervous tissue. Immunoblots from P18 brain homogenate showed no detectable change in α-tubulin levels between STMN1-treated SMA tissue compared to untreated SMA and healthy controls (Fig. 8A). Similarly, no change in acetylated-α-tubulin was detected at P18 between any of the cohorts (Fig. 8B). Interestingly, immunoblot quantifications from the P18 spinal cord homogenate showed a slight, but statistically significant, decrease in α-tubulin levels in the untreated SMA spinal cord compared to unaffected controls. However, in the STMN1-treated SMA spinal cord the levels of α-tubulin were restored to those of unaffected controls (Fig. 8C). Similarly, immunoblots for acetylated-α-tubulin showed significantly reduced levels in untreated SMA tissue, but in STMN1-treated SMA tissues these levels were restored to unaffected levels (Fig. 8D). Given that restoration of acetylated-α-tubulin levels mirrors the restoration of α-tubulin levels in STMN1-treated SMA spinal cords, these results indicate that STMN1 treatment does not affect microtubule stability, but restores overall α-tubulin to unaffected control levels.
Figure 8.
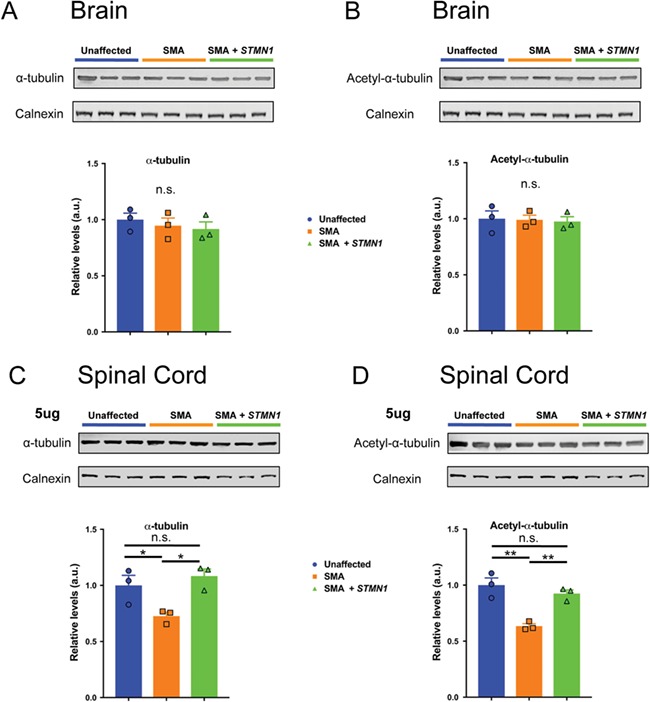
scAAV9-STMN1 treatment restores microtubule levels without affecting microtubule stability in the spinal cord of SMA mice. Western blot analyses of tubulin expression and tubulin stability in brain and spinal cord of PND8-unaffected, untreated SMA and STMN1-treated SMA mice. (A and B) Protein extracts from brain immunoblotted with antibodies specific for α-tubulin (A, top) and acetylated-α-tubulin (B, top) and blot quantifications showing no difference in α-tubulin (A, bottom) or acetylated-α-tubulin (B, bottom) expression in untreated SMA or treated SMA mice compared to unaffected controls. (C and D) Protein extracts form lumbar spinal cord immunoblotted for α-tubulin (C, bottom) and acetylated-α-tubulin (D, top) and quantifications showed significant decrease in α-tubulin (C, bottom) and acetylated-α-tubulin (D, bottom) in untreated SMA spinal cords compared to unaffected controls. Moreover, quantifications show restoration of both α-tubulin and acetylated-α-tubulin (C and D, bottom) in STMN1-treated SMA spinal cords compared to unaffected controls. Data were analyzed by a one-way ANOVA followed by a Tukey post hoc test for multiple comparisons. Data expressed as mean ± SEM. **P < 0.01, *P < 0.05, n.s. = not significant. n = 3 animals per treatment.
Discussion
The search for SMA therapeutics has resulted in the development SpinrazaTM and Zolgensma, the only approved SMA drugs, and a significant pipeline with several exciting compounds that are currently in clinical trials (47). A significant area of emphasis in the SMA field is to expand therapeutic options to enhance the efficacy of existing SMN-dependent therapeutics with the goal of increasing the number of patients who respond to drugs. The severity of SMA has so far indicated that there is a relatively short therapeutic window to achieve a maximal effect; however, adjunctive or combinatorial strategies may expand this window, thereby increasing the number and the types of patients that respond to drugs (26–29,48–54).
Transcriptomic analyses can generate vast amounts of data. However, a significant hurdle often revolves around narrowing the data set and identifying functionally relevant candidates. To identify STMN1 and other putative targets, a prior RNAseq analysis was performed between resistant and vulnerable neuronal populations in a SMA mouse model (37). These results were subsequently cross-referenced against similar data sets generated from resistant/vulnerable neuronal populations in other disease models, including ALS, SBMA and SMA (36,38,39,41). In each instance, Stmn1 was shown to be downregulated in the vulnerable neurons, suggesting that an increase in Stmn1 could provide protection from disease development. Given the roles of STMN1 in cellular functions, such as cell growth, motility and intracellular transport, it had the potential to be a significant disease modifier (55). An important proof of concept for this approach was whether or not STMN1 would improve the NMJ pathology since this was the initial basis for identification in the resistant versus susceptible neuronal populations (31,37). Consistent with this notion, STMN1 significantly reduced NMJ pathology in susceptible neurons in SMA mice. Moreover, our data indicate that vector-mediated delivery of STMN1 improved the SMA phenotype, reduced motor neuron cell body pathology and also improved microtubule filament networks. Further studies will be required to determine if improved microtubule networks directly correlate with improved motor neuron survival and thus improved SMA phenotype. Importantly, delivery of STMN1 did not alter SMN protein expression in SMA mice, demonstrating that the improvement in phenotype involves a mechanism independent of an increase in SMN function.
This work demonstrates that the strategy utilized for identifying SMA disease modifiers is possible through cross-disease transcriptomic analysis and validates its potential use in other genetic diseases, in particular those disease models that were utilized in the cross-referencing analysis: ALS and SBMA. Additionally, investigating the effectiveness of the remaining potential genetic modifiers identified in the cross-disease screen (41) would further validate the utility of this genetic analysis to identify functionally relevant disease modifying genes. To date, two of the genes identified in the cross-disease screen, STMN1 and alpha-synuclein, have been evaluated in similar experimental contexts and are capable of significantly improving the SMA phenotype in intermediate SMA mice. In particular, each of these genes improves the NMJ pathology, an important consideration, as these factors were identified in large part due to aberrant NMJ structures and maturation. Collectively, these two genetic modifiers may not be restricted to functioning solely within the SMA context, but may be useful in a broader disease context of neurodegeneration and neuropathology.
Our data are consistent with previous reports showing that deletion of Stmn1 in mice leads to reduced nerve conduction velocity and central and peripheral axon degeneration attributed to disorganized microtubule networks (56,57). Our studies demonstrate that STMN1 significantly improved microtubule filamentous networks, possibly through increased microtubule turnover, which directly influences cellular morphology growth and prevent motor neuron cell death. In contrast to the positive impact that we observed with Stmn1, another report showed that in a severe SMA mouse, Stmn1 levels were slightly elevated in spinal cord extracts (58). While homozygous deletion of Stathmin did not improve survival or the SMA phenotype (58), a hemizygous Stmn/SMA model showed improvements in NMJ pathology and a reduction in neuroinflammation (58). These differences could be attributed to the fact that the original tissues analyzed were laser-captured motor neuron cell bodies compared to the total cellular extract of the spinal cord and that each study utilized very different mouse models. Taken together, our data show that in an intermediate mouse model of SMA, overexpression of STMN1 significantly reduced the SMA pathology and extended survival without altering SMN expression. Thus, STMN1 could be a candidate for combinatorial therapeutic approaches in the future.
Materials and Methods
Animals, animal procedures and injection
The intermediate Smn2B/− mouse model of SMA and healthy littermate Smn2B/+ controls are referred to herein as SMA and unaffected, respectively. Smn2B/− mice of the C57/BL6 background were a gift from Dr Rashmi Kothary at the University of Ottawa, Canada. Smn2B/2B and Smn+/− mice were kept as separate colonies, bred and genotyped as previously described (59).
All animal procedures were carried out in accordance with procedures approved by the NIH and MU Animal Care and Use Committee. Smn2B/− animals were genotyped at P1 according to the genotyping procedure previously described (59). Mice underwent ICV (53,60,61) injection on PND 2 1.0 × 1011 scAAV9-STNM1 viral genomes. At PND18, mice were anesthetized with 2.5% isoflurane and then perfused transcardially with cold 0.1 M PBS (pH = 7.4) followed by 4% paraformaldehyde in PBS. Mice were postfixed for 48 h at 4°C in 4% paraformaldehyde (PFA). After postfixing, animals were placed in 0.1 M PBS and stored at 4°C until used for spinal cord dissection. For NMJ analyses, the mice were sacrificed with 2.5% isoflurane and muscles were dissected fresh, fixed for 20 min in 4% PFA at room temperature and stored in PBS until stained.
Generation of scAAV9-STMN1 virus
To induce overexpression of STMN1, we utilized a vector-based delivery system. Self-complimentary AAV has been used in several disease models to induce a robust and rapid trans-gene expression. We utilized the self-complimentary scAAV9 vector to induce overexpression of STMN1 in a wide variety of cells. Chicken-β-actin (CBA) was used to drive the ubiquitous expression of STMN1 along with an optimized intron within the 5′ leader sequence as well as a synthetic polyA site (Supplementary Fig. 3). Viral particles were generated, purified and delivered via ICV injection as previously described (62,63).
Motor performance tests
Motor function was assayed by time to right (TTR) assay from PND7, time at which unaffected controls begin to turn themselves over. Each pup was turned on its back, and the time it takes for them to turn over and stabilize on all four paws was recorded with a maximum attempt time of 60 s. The TTR was measured (s) every day from PND6 to PND25, and averages were calculated for each treatment per day. Motor activity and coordination were measured using a rotarod treadmill for mice (IITC Rotarod Series 8, IITC Life Science, CA) with gradual speed acceleration from 0 to 40 rpm over 5 min. At P20, pups were trained to balance and walk on a 3¾-inch-diameter drum and the time and distance it took for the mice to fall off were measured and averaged from three separated trials with at least 20 min of rest between trials. Averages for each metric were obtained for each treatment. For grip strength analysis, a grasping response test was used. At P20, each animal was placed on a wire mesh (1-cm2 grids) that is attached to a force transducer (Bioseb). The animals were allowed to grasp the mesh, then, gently, the animal was pulled horizontally from the tail until the grip was lost. The maximum force in grams (g) was recorded and averaged for each mouse from five separate trials. Averages per treatment were calculated and compared.
Western blots
For western blot analyses, brain and spinal cords were harvested at P18 and immediately flash-frozen in liquid nitrogen. Tissue was then lysed using JLB buffer [50 mm Tris pH = 8.0, 150 mm NaCl, 10% glycerol, 20 mm NaH2PO4, 50 mm NaF, 2 mm EDTA, 5 mm NaVO3] supplied with cOmplete™, Mini Protease Inhibitor Cocktail (Roche). Following SDS-PAGE, protein was transferred to Immobilon®-P transfer membrane (MilliporeSigma) and probed with the following primary antibodies: anti-SMN (1:10000; 610 647, BD Transduction Laboratories™) and anti-Calnexin (1:2000; catalog C4731, anti-tubulin (1:2000; catalog T7451, Sigma), anti-acetylated tubulin (1:2000; catalog T6793, Sigma) and anti-STMN1(Ab-38) (1:1000; catalog SAB4300477, Sigma). Horseradish peroxidase-conjugated secondary antibodies were used (Jackson ImmunoResearch Laboratories). Immunoblots were visualized using (BioSpectrum® 815 Imaging Systems, UVP, LLC). Densitometry analysis was performed using ImageJ software (NIH).
Immunohistochemistry of NMJs
P18-untreated Smn2B/+ (unaffected), untreated Smn2B/− (SMA) and scAAV9-STMN1-treated Smn2B/− (SMA + STMN1) mice were used for NMJ pathology analyses. Whole mount preparations of previously fixed vulnerable (TVA, RA and EO) and resistant (levator auris longus rostral (LALR), AS, AAL) muscle groups. Muscles were stained using specific antibodies, including anti-NF-H (1:2000; catalog AB5539, Chemicon, EMD Millipore) and anti-SV2 (1:200, catalog YE269, Life Technologies). Acetylcholine receptors were labeled with Alexa Fluor 594-conjugated α-bungarotoxin (1:200; Life Technologies). Representative muscle images were obtained using a laser scanning confocal microscope (×40 objective; Leica TCS SP8, Leica Microsystems Inc.). NMJ analysis was performed in a double-blinded manner on at least three randomly selected fields of view per muscle per mouse (×20 objective; Leica DM5500 B, Leica Microsystems Inc.). On average, each field of view contains between 30 and 50 NMJs that were used for endplate denervation. Images were analyzed using freely available Fiji Software (NIH). Endplates missing overlapping nerve terminal staining were considered fully denervated. Endplates with partial overlap were considered partially denervated, and endplates with complete overlap were considered fully innervated.
Motor neuron immunohistochemistry
For motor neuron immunohistochemistry analyses, P18-untreated Smn2B/+, untreated Smn2B/− and scAAV9-Stmn1-treated Smn2B/− mice were used. The animals were sacrificed, perfused with ice-cold 4% PFA and postfixed in 4% PFA for 24 h at 4°C. Spinal lumbar spinal cord was dissected, cryoprotected in 30% sucrose overnight. Cryoprotected spinal cords were embedded in OCT and cryosectioned at 16 μm thickness. Every 10th section from the L3–L5 spinal cord were collected and processed for immunohistochemistry. Sections were labeled with NeuroTrace Nissl and ChAT for motor neuron identification. Digital images were collected using a Leica DM5500 B fluorescent microscope (Leica Microsystems Inc.) under ×20 magnification. Motor neuron counts, cell diameter and perimeter measurements were performed in a double-blinded manner from 16 sections per mouse.
STMN1 localization
For STMN1 immunohistochemistry, P18 spinal cord cross sections were obtained similarly to sections for motor neuron immunohistochemistry. Sections were stained using anti-STMN1 (Ab-38) (1:400; catalog SAB4300477) primary and anti-rabbit-alexa-594 secondary antibodies to detect STMN1 localization. Sections were counter stained with Nissl stain (neurotrace) to identify motor neuron cell bodies. Images were obtained using a laser scanning confocal microscope (×40 objective; Leica TCS SP8, Leica Microsystems Inc.).
Tubulin immunohistochemistry
For tubulin immunohistochemistry, P18 spinal cord cross sections were obtained similarly to sections for motor neuron immunohistochemistry. Sections were stained using an antibody against β-III tubulin to detect microtubule filaments and Nissl stain (neurotrace) to identify motor neuron cell bodies. For quantifying microtubule density, three sections of 25 μm2 were selected per spinal cord image at random (three spinal cord images were used per animal), and using NeuronJ plugin (64) for ImageJ software (NIH), filament tracings were made to label all visible filaments in the selected image (Supplemental Fig. 4). Then, the tracings were counted in every 25-μm2 section and averaged for every genotype. The density of the tracings was calculated for every treatment and compared.
Statistics
All analyses were performed in a double-blinded manner and analyzed for statistical significance using GraphPad Prism version 7.0 (GraphPad Software Inc.). Survival was analyzed by a log-rank (Mantel-Cox) test. NMJ denervation, TTR and analyzed by two-way ANOVA with a Tukey post hoc test for multiple comparisons, and for ease of explanation, only comparisons between fully innervated percentages were presented. Motor neuron cytological changes, microtubule network studies, western blots, grip strength and rotarod studies were analyzed by a one-way ANOVA followed by a Tukey post hoc test for multiple comparisons.
Supplementary Material
Acknowledgements
We would like to thank Madeline Simon for technical assistance. We also appreciate the services rendered by MU Molecular Cytology Core and Electron Microscopy Core.
Conflict of Interest statement. C.L.L. is the co-founder and Chief Scientific Officer of Shift Pharmaceuticals, LLC.
Funding
National Institutes of Health (R21NS106490 to C.L.L.). C.E.S. is supported by a National Institutes of Health training grant [T32 GM008396].
Author Contributions
E.V. and R.A.K. conducted the experiments. C.C.L., E.V. and R.A.K. designed the experiments. L.M.M performed preliminary studies to identify STMN1 as a potential genetic modifier. E.V., C.E.S., E.O., S.O. and Z.C.L. analyzed the data. E.V. and C.C.L. wrote the manuscript.
References
- 1. Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 2. Kolb S.J. and Kissel J.T. (2011) Spinal muscular atrophy: a timely review. Arch. Neurol., 68, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burghes A.H. and Beattie C.E. (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci., 10, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lorson C.L., Hahnen E., Androphy E.J. and Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A., 96, 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H. and McPherson J.D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet., 8, 1177–1183. [DOI] [PubMed] [Google Scholar]
- 6. Feldkotter M., Schwarzer V., Wirth R., Wienker T.F. and Wirth B. (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet., 70, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G. and Melki J. (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet., 16, 265–269. [DOI] [PubMed] [Google Scholar]
- 8. Hua Y., Sahashi K., Hung G., Rigo F., Passini M.A., Bennett C.F. and Krainer A.R. (2010) Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev., 24, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoy S.M. (2019) Onasemnogene Abeparvovec: first global approval. Drugs, in press. [DOI] [PubMed] [Google Scholar]
- 10. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. et al. (2017) Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med., 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- 11. Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De Vivo D.C., Yamashita M., Rigo F., Hung G., Schneider E. et al. (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet, 388, 3017–3026. [DOI] [PubMed] [Google Scholar]
- 12. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E. et al. (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med., 377, 1723–1732. [DOI] [PubMed] [Google Scholar]
- 13. Chiriboga C.A., Swoboda K.J., Darras B.T., Iannaccone S.T., Montes J., De Vivo D.C., Norris D.A., Bennett C.F. and Bishop K.M. (2016) Results from a phase 1 study of nusinersen (ISIS-SMN (Rx)) in children with spinal muscular atrophy. Neurology, 86, 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palacino J., Swalley S.E., Song C., Cheung A.K., Shu L., Zhang X., Van Hoosear M., Shin Y., Chin D.N., Keller C.G. et al. (2015) SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol., 11, 511–517. [DOI] [PubMed] [Google Scholar]
- 15. Swoboda K.J., Scott C.B., Crawford T.O., Simard L.R., Reyna S.P., Krosschell K.J., Acsadi G., Elsheik B., Schroth M.K., D'Anjou G. et al. (2010) SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS ONE, 5, e12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kissel J.T., Scott C.B., Reyna S.P., Crawford T.O., Simard L.R., Krosschell K.J., Acsadi G., Elsheik B., Schroth M.K., D'Anjou G. et al. (2011) SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE, 6, e21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kissel J.T., Elsheikh B., King W.M., Freimer M., Scott C.B., Kolb S.J., Reyna S.P., Crawford T.O., Simard L.R., Krosschell K.J. et al. (2014) SMA valiant trial: a prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Mus. & Nerve, 49, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Darbar I.A., Plaggert P.G., Resende M.B., Zanoteli E. and Reed U.C. (2011) Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurol., 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krosschell K.J., Kissel J.T., Townsend E.L., Simeone S.D., Zhang R.Z., Reyna S.P., Crawford T.O., Schroth M.K., Acsadi G., Kishnani P.S. et al. (2018) Clinical trial of L-Carnitine and valproic acid in spinal muscular atrophy type I. Mus. & Nerve, 57, 193–199. [DOI] [PubMed] [Google Scholar]
- 20. Bertini E., Dessaud E., Mercuri E., Muntoni F., Kirschner J., Reid C., Lusakowska A., Comi G.P., Cuisset J.M., Abitbol J.L. et al. (2017) Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol., 16, 513–522. [DOI] [PubMed] [Google Scholar]
- 21. Hwee D.T., Kennedy A.R., Hartman J.J., Ryans J., Durham N., Malik F.I. and Jasper J.R. (2015) The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J. Pharm. Exper. Therap., 353, 159–168. [DOI] [PubMed] [Google Scholar]
- 22. Kariya S., Obis T., Garone C., Akay T., Sera F., Iwata S., Homma S. and Monani U.R. (2014) Requirement of enhanced survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Invest., 124, 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F. and Krainer A.R. (2011) Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature, 478, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou H., Meng J., Marrosu E., Janghra N., Morgan J. and Muntoni F. (2015) Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet., 24, 6265–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oprea G.E., Krober S., McWhorter M.L., Rossoll W., Muller S., Krawczak M., Bassell G.J., Beattie C.E. and Wirth B. (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science, 320, 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaifer K.A., Villalon E., Osman E.Y., Glascock J.J., Arnold L.L., Cornelison D.D. and Lorson C.L. (2017) Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight, 2, e89970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hosseinibarkooie S., Peters M., Torres-Benito L., Rastetter R.H., Hupperich K., Hoffmann A., Mendoza-Ferreira N., Kaczmarek A., Janzen E., Milbradt J. et al. (2016) The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet., 99, 647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng Z., Ling K.K., Zhao X., Zhou C., Karp G., Welch E.M., Naryshkin N., Ratni H., Chen K.S., Metzger F. et al. (2016) Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet., 25, 964–975. [DOI] [PubMed] [Google Scholar]
- 29. Osman E.Y., Washington C.W., Simon M.E., Megiddo D., Greif H. and Lorson C.L. (2017) Analysis of azithromycin monohydrate as a single or a combinatorial therapy in a mouse model of severe spinal muscular atrophy. J. Neuromuscul. Dis., 4, 237–249. [DOI] [PubMed] [Google Scholar]
- 30. Monani U.R. (2005) Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron, 48, 885–896. [DOI] [PubMed] [Google Scholar]
- 31. Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K. and Gillingwater T.H. (2008) Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 17, 949–962. [DOI] [PubMed] [Google Scholar]
- 32. Ling K.K., Gibbs R.M., Feng Z. and Ko C.P. (2012) Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet., 21, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thomson S.R., Nahon J.E., Mutsaers C.A., Thomson D., Hamilton G., Parson S.H. and Gillingwater T.H. (2012) Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLoS ONE, 7, e52605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Comley L.H., Nijssen J., Frost-Nylen J. and Hedlund E. (2016) Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol., 524, 1424–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nijssen J., Comley L.H. and Hedlund E. (2017) Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta. Neuropathol., 133, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hedlund E., Karlsson M., Osborn T., Ludwig W. and Isacson O. (2010) Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain, 133, 2313–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murray L.M., Beauvais A., Gibeault S., Courtney N.L. and Kothary R. (2015) Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/−) mouse model of spinal muscular atrophy. Acta. Neuropathol. Commun., 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brockington A., Ning K., Heath P.R., Wood E., Kirby J., Fusi N., Lawrence N., Wharton S.B., Ince P.G. and Shaw P.J. (2013) Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta. Neuropathol., 125, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaplan A., Spiller K.J., Towne C., Kanning K.C., Choe G.T., Geber A., Akay T., Aebischer P. and Henderson C.E. (2014) Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron, 81, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boyd P.J., Tu W.Y., Shorrock H.K., Groen E.J.N., Carter R.N., Powis R.A., Thomson S.R., Thomson D., Graham L.C., Motyl A.A.L. et al. (2017) Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet., 13, e1006744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kline R.A., Kaifer K.A., Osman E.Y., Carella F., Tiberi A., Ross J., Pennetta G., Lorson C.L. and Murray L.M. (2017) Comparison of independent screens on differentially vulnerable motor neurons reveals alpha-synuclein as a common modifier in motor neuron diseases. PLoS Genet, 13, e1006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta K.K., Li C., Duan A., Alberico E.O., Kim O.V., Alber M.S. and Goodson H.V. (2013) Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc. Natl. Acad. Sci. U S A., 110, 20449–20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murray L.M., Beauvais A., Bhanot K. and Kothary R. (2013) Defects in neuromuscular junction remodelling in the Smn(2B/−) mouse model of spinal muscular atrophy. Neurobiol. Dis., 49, 57–67. [DOI] [PubMed] [Google Scholar]
- 44. Janke C. and Bulinski J.C. (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol., 12, 773–786. [DOI] [PubMed] [Google Scholar]
- 45. Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S. et al. (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J., 21, 6820–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tran A.D., Marmo T.P., Salam A.A., Che S., Finkelstein E., Kabarriti R., Xenias H.S., Mazitschek R., Hubbert C., Kawaguchi Y. et al. (2007) HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J. Cell Sci., 120, 1469–1479. [DOI] [PubMed] [Google Scholar]
- 47. Parente V. and Corti S. (2018) Advances in spinal muscular atrophy therapeutics. Ther. Adv. Neurol. Disord., 11, 1756285618754501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Farrelly-Rosch A., Lau C.L., Patil N., Turner B.J. and Shabanpoor F. (2017) Combination of valproic acid and morpholino splice-switching oligonucleotide produces improved outcomes in spinal muscular atrophy patient-derived fibroblasts. Neurochem. Int., 108, 213–221. [DOI] [PubMed] [Google Scholar]
- 49. Riessland M., Kaczmarek A., Schneider S., Swoboda K.J., Lohr H., Bradler C., Grysko V., Dimitriadi M., Hosseinibarkooie S., Torres-Benito L. et al. (2017) Neurocalcin Delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am. J. Hum. Genet., 100, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. d'Ydewalle C., Ramos D.M., Pyles N.J., Ng S.Y., Gorz M., Pilato C.M., Ling K., Kong L., Ward A.J., Rubin L.L. et al. (2017) The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron, 93, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harris A.W. and Butchbach M.E. (2015) The effect of the DcpS inhibitor D156844 on the protective action of follistatin in mice with spinal muscular atrophy. Neuromuscul. Disord., 25, 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nizzardo M., Simone C., Salani S., Ruepp M.D., Rizzo F., Ruggieri M., Zanetta C., Brajkovic S., Moulton H.M., Muehlemann O. et al. (2014) Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Delta7 mouse model phenotype. Clin. Ther., 36, 340–356 e345. [DOI] [PubMed] [Google Scholar]
- 53. Shababi M., Glascock J. and Lorson C.L. (2011) Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther., 22, 135–144. [DOI] [PubMed] [Google Scholar]
- 54. Kwon D.Y., Motley W.W., Fischbeck K.H. and Burnett B.G. (2011) Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum. Mol. Genet., 20, 3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rubin C.I. and Atweh G.F. (2004) The role of stathmin in the regulation of the cell cycle. J. Cell. Biochem., 93, 242–250. [DOI] [PubMed] [Google Scholar]
- 56. Liedtke W., Leman E.E., Fyffe R.E., Raine C.S. and Schubart U.K. (2002) Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am. J. Pathol., 160, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strey C.W., Spellman D., Stieber A., Gonatas J.O., Wang X., Lambris J.D. and Gonatas N.K. (2004) Dysregulation of stathmin, a microtubule-destabilizing protein, and up-regulation of Hsp25, Hsp27, and the antioxidant peroxiredoxin 6 in a mouse model of familial amyotrophic lateral sclerosis. Am. J. Pathol., 165, 1701–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wen H.L., Ting C.H., Liu H.C., Li H. and Lin-Chao S. (2013) Decreased stathmin expression ameliorates neuromuscular defects but fails to prolong survival in a mouse model of spinal muscular atrophy. Neurobiol. Dis., 52, 94–103. [DOI] [PubMed] [Google Scholar]
- 59. Bowerman M., Murray L.M., Beauvais A., Pinheiro B. and Kothary R. (2012) A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord., 22, 263–276. [DOI] [PubMed] [Google Scholar]
- 60. Glascock J.J., Shababi M., Wetz M.J., Krogman M.M. and Lorson C.L. (2012) Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem. Biophys. Res. Commun., 417, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shababi M., Feng Z., Villalon E., Sibigtroth C.M., Osman E.Y., Miller M.R., Williams-Simon P.A., Lombardi A., Sass T.H., Atkinson A.K. et al. (2016) Rescue of a Mouse Model of spinal muscular atrophy with respiratory distress type 1 by AAV9-IGHMBP2 is dose dependent. Mol. Ther., 24, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shababi M., Osman E.Y. and Lorson C.L.. (2015) Bo, X. and Verhaagen, J. (eds.) In Gene Del. and Ther. Neurol. Dis. Springer New York, New York, NY, pp. 297–320. [Google Scholar]
- 63. Glascock J.J., Osman E.Y., Coady T.H., Rose F.F., Shababi M. and Lorson C.L. (2011) Delivery of therapeutic agents through intracerebroventricular (ICV) and intravenous (IV) injection in mice. J. Vis. Exp., (56), 2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meijering E., Jacob M., Sarria J.C., Steiner P., Hirling H. and Unser M. (2004) Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A, 58, 167–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.