

Abstract
Objective.
Peripheral blood mononuclear cells (PBMC) from lupus patients exhibit a gene expression program (interferon signature) attributed to over-production of type I IFNs (IFN-I) by plasmacytoid dendritic cells. IFN-I were thought to play a role in the pathogenesis of lupus. This study examined an unexpected influence of macrophages/monocytes on the interferon signature.
Methods.
Proinflammatory (classical) and anti-inflammatory (non-classical) macrophages were sorted from mice and analyzed by RNA-sequencing and quantitative PCR (qPCR). Type I interferon receptor (IFNAR) expression was determined by qPCR and flow cytometry. Macrophages were stimulated in vitro with IFNα and p-Stat-1was measured.
Results.
Transcriptional profiling of peritoneal macrophages from mice with pristane-induced lupus unexpectedly indicated a strong interferon signature in classical, but not non-classical, macrophages exposed to the same IFN-I concentrations. Ifnar1 mRNA and IFNAR surface staining were higher in classical vs. non-classical macrophages (P < 0.0001 and P < 0.05 by student t test). Non-classical macrophages also were relatively insensitive to IFNα-driven Stat1 phosphorylation. Humans exhibited a similar pattern: higher IFNAR expression (P < 0.0001 by student t test) and IFNα-stimulated gene expression (P < 0.01 by paired Wilcoxon rank sum test) in classical monocytes and lower levels in non-classical monocytes.
Conclusion.
These studies reveal that the relative abundance of different monocyte/macrophage subsets helps determine the magnitude of the interferon signature. Responsiveness to IFNα signaling reflects differences in IFNAR expression in classical (high IFNAR) vs. non-classical (low IFNAR) monocytes/macrophages. Thus, the interferon signature depends on both IFN-I production and the responsiveness of macrophages/monocytes to IFNAR signaling.
Introduction
Peripheral blood mononuclear cells (PBMCs) from SLE patients exhibit high levels of gene transcripts regulated by type I interferons (IFN-I), such as IFNα and IFNβ (1, 2). In aggregate, this transcriptional program is termed the “interferon signature”. Many of the IFN-stimulated genes (ISGs) over-expressed in lupus are involved in antiviral responses. A high interferon signature correlates with anti-Sm, RNP, and dsDNA autoantibody production and lupus nephritis (3, 4). Mice with pristane-induced lupus also develop the interferon signature (5, 6).
Several lines of evidence suggest that interferon responses play a role in disease pathogenesis. Patients treated with IFNα may develop antinuclear antibodies or clinical lupus (7, 8) and anti-Sm, RNP, and dsDNA autoantibody production and lupus nephritis are greatly attenuated in lupus mice lacking the type I interferon receptor (IFNAR) (9, 10). This has prompted the search for drugs for lupus that block the IFNAR, such as the anti-IFNAR monoclonal antibody anifrolumab (11).
High interferon signatures are associated with active lupus and constitute a risk factor for SLE (4, 12). Measuring IFN-I in biological samples is technically challenging (13). Consequently IFN-I regulated gene expression in PBMCs is a widely used surrogate marker for IFN-I (1, 2, 14). Although the interferon signature has been assumed to reflect IFN-I production, its magnitude also could reflect differences in signaling through the IFNAR, which activates the kinases Jak1 and Tyk2, resulting in the phosphorylation of Stat1 and Stat2, or activation of Irf9, other Stat proteins, PI3 kinase, and/or ERK½ (15). Thus, the interferon signature could be influenced by expression of the IFNAR (Ifnar1 and Ifnar2 chains) or the expression/activity of key intermediates downstream of the receptor (16, 17).
Our laboratory studies the role of monocytes and macrophages (Mϕ) in lupus and recently found that CD138+ “non-classical” Mϕ (NCM) promoting the resolution of inflammation are deficient in pristane-induced lupus (18). In the present study, transcriptional profiling of this novel Mϕ subset revealed a much lower interferon signature than seen in “classical” Ly6Chi Mϕ (CM) (19) exposed to the same concentration of IFN-I. We show that this is because NCM are relatively insensitive to signaling through the IFNAR. Similarly, circulating human monocyte subsets exhibit differential responsiveness to IFN-I. Moreover, we show that the magnitude of the interferon signature in human PBMCs depends on the relative numbers of classical monocytes (equivalent to murine CM). These data alter how we view the interferon signature, potentially with implications for understanding the pathogenesis of lupus.
Patients and Methods
Mice.
Female C57BL/6 (B6) mice (5-10 per group, Jackson Laboratory, Bar Harbor, ME) maintained under specific pathogen free conditions were injected i.p. with 0.5 ml of either pristane (Sigma-Aldrich, St. Louis, MO) or mineral oil (MO, C.B. Fleet Co., Lynchburg, VA). Peritoneal exudate cells (PEC) were collected by lavage 3-14 days later (20). This study followed the recommendations of the Animal Welfare Act and US Government Principles for the Utilization and Care of Vertebrate Animals and was approved by the UF IACUC.
Flow cytometry, cell sorting, and RNA isolation.
Flow cytometry was performed using anti-mouse CD16/32 (Fc Block; BD Biosciences, Woburn, MA) before staining with primary antibody or isotype controls. Cells were surface-stained, fixed/permeabilized (Fix-Perm buffer, eBioscience, San Diego, CA), and then stained intracellularly. Monoclonal antibodies used for flow cytometry are listed in Table S1. For cell purification, PEC were incubated with anti-CD11b-BV421, CD138-APC, Ly6C-Alexa Fluor 488, and Ly6G-APC-Cy7 antibodies (20). Peritoneal NCM (CD11b+CD138+Ly6C−Ly6G−, which we previously have termed “CD138+ macrophages”) and CM (CD11b+CD138−Ly6ChiLy6G−, which we previously have termed Ly6Chi monocyte/macrophages) (18) were flow-sorted using a FACSAria cell sorter (3 mice/group, 30,000 cells/mouse). The gating strategy is shown in Figure 1A. Cells were lysed immediately and RNA was isolated using the RNeasy Microkit (Qiagen, Gaithersburg, MD).
Figure 1. Surface staining phenotypes of Mϕ subsets.
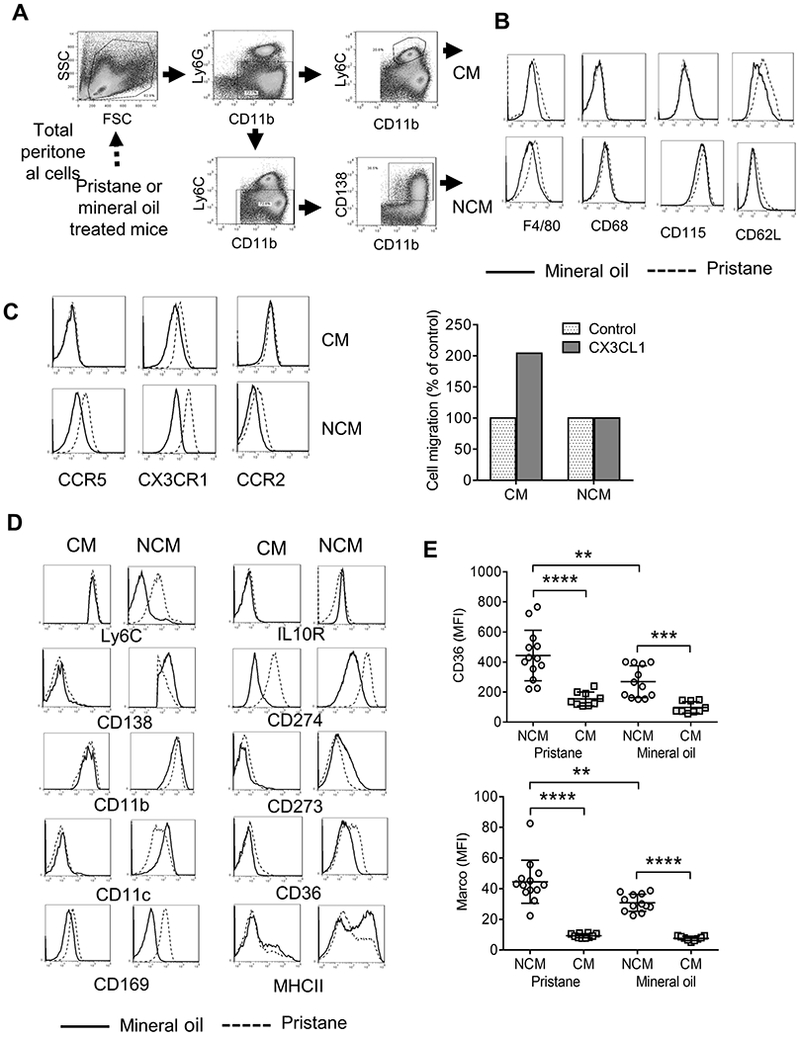
A, gating strategy for flow-sorting CM and NCM from PEC from pristane or MO-treated mice. Cells were purified based on forward scatter (FSC), side scatter (SSC), and surface staining with monoclonal antibodies specific for CD11b, Ly6G, Ly6C, and CD138. B, Mϕ marker expression on flow-sorted CM and NCM from MO (solid line) vs. pristane (dotted line) treated mice. Cells were stained with monoclonal antibodies specific for F4/80 (Emr1), CD68, CD115 (Csf1r), and CD62L (Sell). C, Left, chemokine receptor expression on CM and NCM from MO vs. pristane treated mice. Cells were stained with monoclonal antibodies specific for CCR5, CX3CR1, and CCR2. Right, transwell assay showing the migration of PEC from pristane-treated mice toward CX3CL1 (200 ng/ml, representative of 3 separate experiments). D, Cells were surface stained with monoclonal antibodies specific for Ly6C, IL-10 receptor (IL10R), CD138, CD274, CD11b, CD273, CD11c, CD36, CD169, and MHC class II (MHCII). E, Flow cytometry analysis of CD36 (up) and Marco (bottom) on the surface of NCM and CM from 14-day pristane and mineral oil treated mice. **** P < 0.0001, *** P < 0.001, ** P < 0.01 (Student t-test).
Gene expression profiling.
RNA sequencing (RNA-seq) was performed at Broad Institute using the Smart-Seq2 platform (21–23). Smart-Seq2 libraries were prepared by the Broad Institute Technology Labs and sequenced using the Broad Genomics Platform (Cambridge, MA). Transcripts were quantified using the BTL computational pipeline with Cuffquant version 2.2.1 (24). Preliminary data analysis confirmed that purity of the sorted cells was high: Ly6c2 was expressed much more highly in CM and Sdc1 (encoding syndecan-1/CD138) in NCM. Reproducibility between mice also was high (not shown). Gene set enrichment analysis (GSEA) was performed using the Broad Institute GSEA Desktop v2.2.4 software and hallmark gene sets from the Molecular Signature Database (MSigDB v.6.2) (25, 26). A false discovery rate (FDR) of q < 0.25 was considered significant (25). Heat maps were constructed using Java Treeview (27). Data and materials availability: The RNA-seq data discussed in this publication are available from the corresponding author upon request.
Quantitative PCR (qPCR).
qPCR was performed using RNA from 106 mouse PEC (TRIzol, Invitrogen, Waltham, MA). cDNA was synthesized using the Superscript II First-Strand Synthesis kit (Invitrogen). SYBR Green qPCR analysis was performed using the CFX Connect Real-Time system (Bio-Rad, Hercules, CA). Gene expression was normalized to 18S RNA and the expression level was calculated using the 2-ΔΔCt method. Primer sequences were as follows: mouse Mx1: forward 5′-GATCCGACTTCACTTCCAGATGG-3′, reverse 5′-CATCTCAGTGGTAGTCCAACCC-3′; mouse 18S: forward 5′-CGGCTACCACATCCAAGGAA-3′, reverse 5′-GCTGGAATTACCGCGGCT-3′; human LY6E: forward 5′-CAGCTCGCTGATGTG CTTCT-3′, reverse 5′-CAGACACAGTCACGCAGTAGT-3′; human CXCL10: forward 5′-GTGGCATTCAAGGAGTACCTC-3′, reverse 5′-TGATGGCCTTCGATTCTGGATT-3; human ISG15: forward 5′-CGCAGATCACCCAGAAGATCG-3′, reverse 5′-TTCGTCGCATTTGTCCACCA-3; human MX1: forward 5′-GGTGGTCCCCAGTAATGTGG-3′, reverse 5′-CGTCAAGATTCCGATGGTCCT-3 .
In vitro IFNα-stimulated Stat1 phosphorylation.
At day-15 after pristane treatment, PEC were cultured for 0-15-min in AIM-V medium in the presence or absence of IFNα4 (100 U/ml, R&D Systems, Minneapolis, MN). Cells were fixed/permeabilized for flow cytometry and stained with anti-CD11b, Ly6G, Ly6C, CD138, and phospho-Stat1 antibodies (Table S1). The mean fluorescence intensity (MFI) of phospho-Stat1 staining in the CD11b+Ly6G−Ly6ChiCD138− and CD11b+Ly6G−Ly6CloCD138+ cells was determined.
Transwell assays.
Total PEC were isolated 14-d after pristane treatment, resuspended in RPMI culture medium containing 10% fetal bovine serum, and placed in the top well of an 8 μm Falcon Fluoroblock multiwall insert (Becton-Dickinson). The lower chamber was filled with culture medium containing CX3CL1 (200 ng/ml, R&D Systems) or with culture medium alone. After 4-h, cells in the upper and lower chambers were analyzed by flow cytometry (CD11b, Ly6C, CD138, Ly6G staining). Cell migration in response to CX3CL1 (CM and NCM subsets) was calculated as a % of migration with medium alone.
Flow cytometry of human blood cells.
Heparinized whole blood was obtained from SLE patients (n=10) meeting ACR criteria (28) and healthy donors with no autoimmune disease (n=11). Monoclonal antibodies were added to 100 μl of blood as follows: anti-CD14-PerCP, anti-CD16-FITC, anti-CD64-PE, anti-CD45-BV421 and anti-IFNΑR1-APC (Table S1) and incubated for 20-min in the dark. Lysis/Fix buffer was added for 5-min, and the cells were washed and re-suspended in PBS for flow cytometry. Neutrophils, monocytes and lymphocytes were identified as CD45+ cells with high (neutrophils), low (lymphocytes), or intermediate (monocytes) side scatter (SSC). The monocyte population was gated for further analyzing the CD14++CD16− (CM), CD14++CD16+ (intermediate monocytes, IM) and CD14+CD16++ (NCM). CD14++CD16− monocytes are the human equivalent of murine CM, whereas CD14+CD16++ monocytes are the equivalent of murine NCM (29). In some experiments, CM and NCM were stained with CD169 and CD64. This study followed recommendations of the International Committee of Medical Journal Editors and was approved by the UF Institutional Review Board. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Expression of ISGs in human PBMC subsets.
PBMCs were isolated from 8 healthy controls (30-ml heparinized peripheral blood) by Ficoll-Hypaque density gradient centrifugation and cultured for 11-h in the presence of either IFNα2b (1000 U/ml, R&D Systems) or vehicle (PBS). Cells were stained with anti-CD3, CD19, CD14, and CD16 antibodies and flow-sorted using a FACSAria cell sorter. T cells (CD3+CD19−CD14−CD16−), B cells (CD3−CD19+CD14−CD16−), CM (CD3−CD19−CD14++CD16−), and NCM (CD3−CD19−CD14+CD16++) were collected and lysed immediately. RNA was isolated using the RNeasy Microkit and the expression of ISGs (LY6E, CXCL10, ISG15, and MX1) was determined by qPCR as above.
Responsiveness of human monocytes to IFN-I in vitro.
PBMCs from SLE patients (n=9) and healthy controls (n=8) were isolated by Ficoll-Hypaque density gradient centrifugation as above and cultured for 24-h with IFNα2b (1000 U/ml) or PBS followed by incubation with fluorescently-labeled monoclonal antibodies and flow cytometry. After gating on CM (CD14++CD16−), IFNAR1 and CD64 staining intensity (MFI) was determined.
Statistical analysis.
Statistical analyses were performed using Prism 6.0 (GraphPad Software). Differences between groups were analyzed by two-sided unpaired Student t test unless otherwise indicated in the figure legend. Data were expressed as mean ± SD. p < 0.05 was considered significant. Experiments were repeated at least twice.
Results
PEC from mice treated with pristane or MO contain several myeloid populations including CD11b+Ly6G−Ly6Chi CM, CD11b+Ly6CloLy6G-CD138+ NCM, and CD11b+Ly6G+ neutrophils (Fig. 1A). CM and NCM both expressed surface markers characteristic of Mϕ (F4/80, CD68, and CD115). F4/80 staining was higher in Mϕ from pristane- vs. MO-treated mice. As expected (30), CM expressed higher levels of CD62L than NCM (Fig. 1B). Both subsets expressed chemokine receptors involved in Mϕ migration (Fig. 1C). As expected (18), CM expressed more CCR2 than NCM. Pristane treatment increased the expression of CCR5, CX3CR1, and CCR2 on NCM and increased CX3CR1 expression on CM, suggesting that it may promote Mϕ recruitment to the inflamed peritoneum more potently than MO. We previously showed that the recruitment of CM to the peritoneum is dependent on CCR2/CCL2 in pristane-treated mice (31). CM from pristane-treated mice also migrated toward CX3CL1 (the ligand for CX3CR1) more efficiently than NCM (Fig. 1C, right).
CM from pristane- and MO-treated mice expressed similar levels of most surface markers, although the M1 Mϕ marker CD274 was higher in CM from pristane-treated mice (Fig. 1D). In contrast, surface staining of NCM from pristane-treated and MO-treated mice differed somewhat, with higher Ly6C and CD274 and lower CD11c, CD273, and CD36 surface staining on NCM from pristane-treated mice (Fig. 1D). However, in both pristane- and MO-treated mice, surface staining for the scavenger receptor CD36, which promotes the phagocytosis of apoptotic cells (32), was considerably higher in NCM vs. CM (Fig. 1E). The staining pattern for Marco, another scavenger receptor involved in the phagocytosis of apoptotic cells (18, 33), was similar: increased in NCM vs. CM, with higher expression on NCM from pristane- vs. MO-treated mice (Fig. 1E).
We previously showed that NCM from pristane-treated mice make more TNFα and that TNFα production and the surface phenotype become more “MO-like” after LXR agonist treatment (20). Thus, there may be more than one subset of NCM or these cells may exhibit a greater degree of phenotypic/functional plasticity than CM, potentially influencing whether peritoneal inflammation resolves or becomes chronic, resulting in autoimmune disease.
High expression of ISGs in NCM from pristane-treated mice.
To explore peritoneal Mϕ heterogeneity further, we performed transcriptional profiling of flow-sorted peritoneal Mϕ from pristane- and MO-treated mice (Fig. 2). Examination of the top 50 features differentially expressed by peritoneal NCM from pristane- vs. MO-treated mice revealed that eight of the top ten genes over-expressed in pristane-treated mice were ISGs associated with antiviral responses (Fig. 2A, red dots). Although expression of these 8 transcripts was substantially higher in NCM from pristane- vs. MO-treated mice, they also were differentially expressed in CM from pristane- vs. MO-treated mice (Fig. 2B). The highest expression levels were in CM from pristane-treated mice. Transcripts over-represented in NCM from MO-treated mice included genes reported to have an inhibitory/immunomodulatory function, including Il1r2, Lcn2, Mmp9, and NfkbIl1 (Fig. 2A, blue squares).
Figure 2. Transcriptional profiling of NCM from pristane vs. MO-treated mice.
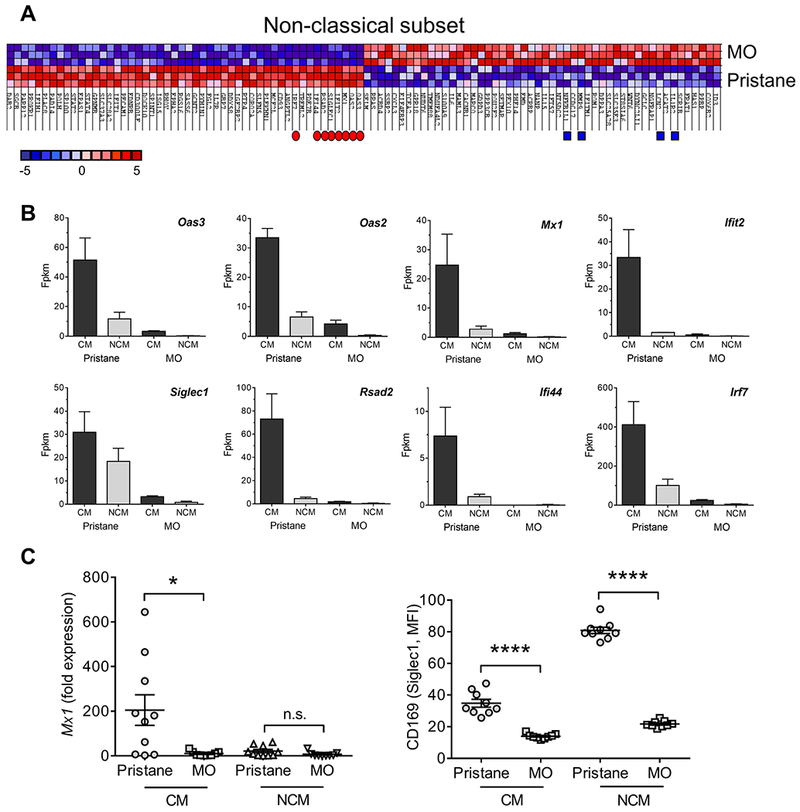
A, Heat map of the top 50 features for NCM from mineral oil (MO) vs. pristane treated mice. Of the top 10 genes over-expressed in pristane-treated mice, 8 were ISGs associated with antiviral responses (red dots). Genes reported to have an inhibitory or immunomodulatory function over-represented in NCM from MO-treated mice are indicated by blue squares. B, Expression of the 8 ISGs (red dots, Fig. 2A) in the CM and NCM from pristane vs. MO-treated mice. Fpkm, fragment per kilobase million. C, Left, Mx1 expression levels relative to 18S rRNA (qPCR) in CM vs. NCM from pristane- and MO-treated mice. Right, flow cytometry of CD169 (Siglec1) surface staining on CM and NCM from pristane vs. MO treated mice. * P=0.016, vs. CM, **** P < 0.0001 (Student t-test). n.s., not significant.
Weak expression of the ISG Mx1 in peritoneal NCM relative to CM from pristane-treated mice (Fig. 2B) was confirmed by qPCR (Fig. 2C, left). Similarly, surface staining for the IFN-I regulated protein CD169 (Siglec1) was higher on both CM and NCM from pristane- vs. MO-treated mice (Fig. 2C, right). Interestingly, Siglec1 mRNA appeared to be expressed at relatively higher levels in NCM from pristane-treated mice than other ISGs (Fig. 2B). This was reflected in high levels of Siglec1 (CD169) surface staining on NCM from pristane-treated mice (Fig. 2C).
Increased IFN-I sensitivity of CM vs. NCM.
Peritoneal injection of pristane, but not MO, induces a strong interferon signature (31). Transcriptional profiling of CM vs. NCM from pristane-treated mice revealed that many of the top 50 genes over-expressed by CM from pristane-treated mice were ISGs (Fig. 3A, red dots). GSEA confirmed that CM over-expressed members of the Hallmark IFNα Response Gene Set (25, 26) (Fig. 3B–C, Table S2). CM also up-regulated transcripts in the “IFNγ-response” and “inflammatory response” gene sets (Table S2). Expression of the top 10 ISGs (bracket, Fig. 3B) was markedly lower in CM from MO-treated mice vs. those from pristane-treated mice (Fig. 2B and 3D). The only exception was Ifitm1, which was expressed at high levels in CM from both pristane- and MO-treated mice (Fig. 3D). However, like most other ISGs, Ifitm1 was expressed only at low levels in NCM. Surprisingly, the peritoneal NCM and CM were flow-sorted from the same mice and therefore were exposed to the same concentration of IFN-I prior to isolation, suggesting that NCM from pristane-treated mice might be less sensitive to IFN-I than CM.
Figure 3. Gene expression in Mϕ subsets from pristane or MO treated mice.
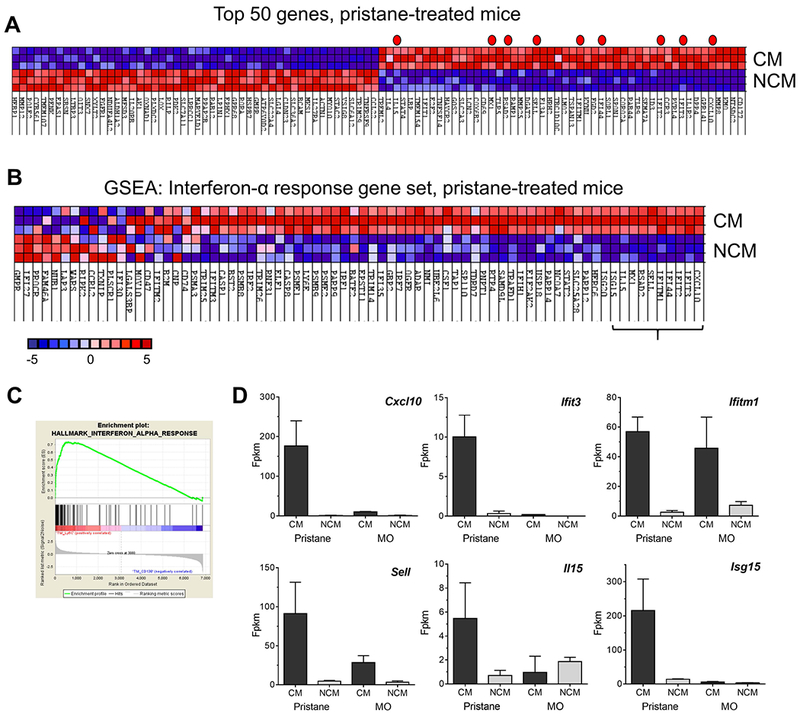
CM and NCM from pristane-treated mice were flow-sorted as in Fig. 1 and analyzed by RNA-Seq. A, heat map of the top 50 features for CM vs. NCM from pristane treated mice. Genes represented in the Hallmark IFNα Gene Set are indicated by a red dot. B, GSEA, hallmark interferon-α response gene pathway, CM vs. NCM from pristane treated mice. Expression levels of the top 10 genes (indicated by bracket) in CM and NCM are shown in Figs. 2B and 3D. C, GSEA enrichment plot, hallmark interferon-α response gene pathway. D, Expression of Cxcl10, Ifit3, Ifitm1, Sell, Il15, and Isg15 in CM vs. NCM from pristane- and MO-treated mice. Fpkm, fragment per kilobase million.
ISG expression is related to Ifnar1 levels.
RNA-sequencing suggested that Ifnar1 transcripts were less abundant in NCM vs. CM from both pristane- and MO-treated mice (Fig. 4A). The expression of Ifnar2 and other intermediates in the IFNAR signaling pathway (Tyk2 and Jak1) also appeared lower in NCM. In pristane-treated mice, expression of downstream genes encoding subunits of the transcription factor ISGF3 (Stat1, Stat2, and Irf9), was lower in NCM than in CM, but these genes are IFN-I regulated (34). We confirmed by flow cytometry that NCM from pristane-treated mice had substantially lower Ifnar1 surface staining than CM (Fig. 4B). CD11b+Ly6G+ neutrophils and “other cells” (mainly lymphocytes) from the peritoneal exudates exhibited little or no Ifnar1 staining. Thus, CM may be poised to express an interferon-regulated transcriptional program when stimulated by IFN-I. To test that possibility, PECs were isolated 15-d after pristane treatment and incubated with IFNα4 (100 U/ml) for 0-15 min, followed by measurement of phospho-Stat1 (p-Stat1) in Mϕ subsets by flow cytometry (Fig. 4C). At baseline (before IFNα treatment), p-Stat1 levels in CM were significantly higher than in NCM from the same mice (P < 0.01, Student t-test) and this pattern persisted after incubation with IFNα4. In CM, IFNα4 treatment enhanced the phosphorylation of Stat1 (P = 0.01, Student t-test), although the effect was not dramatic. This may suggest that Stat1 phosphorylation already was near-maximal in freshly isolated peritoneal CM from pristane-treated mice. Although signaling through the IFNγ receptor (IFNGR) also results in Stat1 phosphorylation, NCM and CM expressed similar levels of Ifngr1; expression of Ifngr2 mRNA may have been slightly lower in NCM (Fig. 4D). Thus, low Stat1 phosphorylation in NCM correlated with the low level of Ifnar1 mRNA and protein expressed by these cells.
Figure 4. NCM express lower levels of IFNAR than CM.
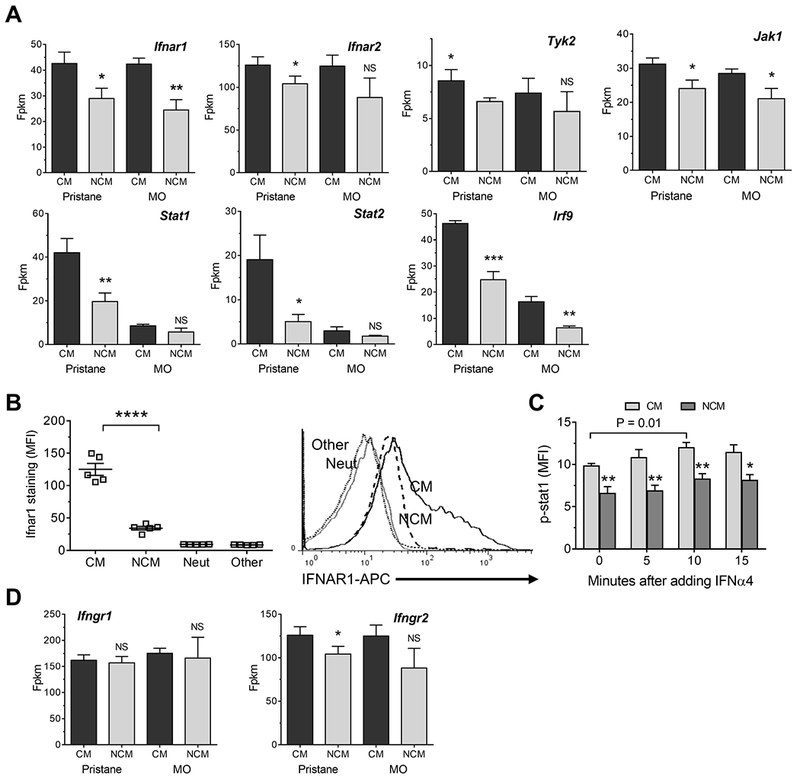
A, Expression of genes involved in IFNAR signaling (Ifnar1, Ifnar2, Tyk2, Jak1, Stat1, Stat2, and Irf9 in CM vs. NCM from pristane- and MO-treated mice. B, Ifnar surface staining of PEC subsets (CM, NCM, Ly6G+ neutrophils (Neut), and CD11b−Ly6C−CD138−Ly6G− cells (Other), primarily lymphocytes. MFI, mean fluorescence intensity. Right, representative flow cytometry plot showing IFNAR staining of CM, NCM, Neut, and Other cells. C, Intracellular phospho-Stat1 (p-stat) staining in peritoneal CM vs. NCM from pristane-treated mice 0-15 minutes after adding IFNα4. D, Expression of mRNA encoding interferon-γ receptor chains Ifngr1 and Ifngr2 in CM vs. NCM from pristane- and MO-treated mice. * P < 0.05, ** P < 0.01, **** P < 0.0001, NCM vs. CM (Student t-test). Fpkm, fragment per kilobase million.
IFNAR expression and IFN-I responsiveness of human monocyte subsets.
We examined PBMCs from SLE patients and healthy controls to see if the variable IFN-I responsiveness is also seen in human monocytes. Three human monocyte subsets have been described, CD14++CD16− (CM), CD14++CD16+ (intermediate monocytes, IM) and CD14+CD16++ (NCM) (35, 36) (Fig. 5A, R4, R5, and R6, respectively). CM and IM from peripheral blood of both healthy controls and SLE patients exhibited IFNAR1 surface staining, whereas NCM had minimal staining (Fig. 5B). CM express higher levels of the IFN-I regulated gene CD64 than NCM (37) and CD64 staining (flow cytometry) on total CD14+ cells correlates strongly with the interferon signature (38). Consistent with the IFNAR1 staining pattern, CM and IM, but not NCM, from healthy controls and SLE patients stained strongly for CD64 (Fig. 5C). Neutrophils (Neut) and lymphocytes (Lymph) (Fig. 5A, R1 and R3, respectively) had only weak IFNAR1 expression (Fig. 5B). Thus, as in mouse Mϕ, the IFNAR was expressed differentially on human CM/IM vs. NCM.
Figure 5. Flow cytometry of IFNAR and CD64 in human PBMCs.
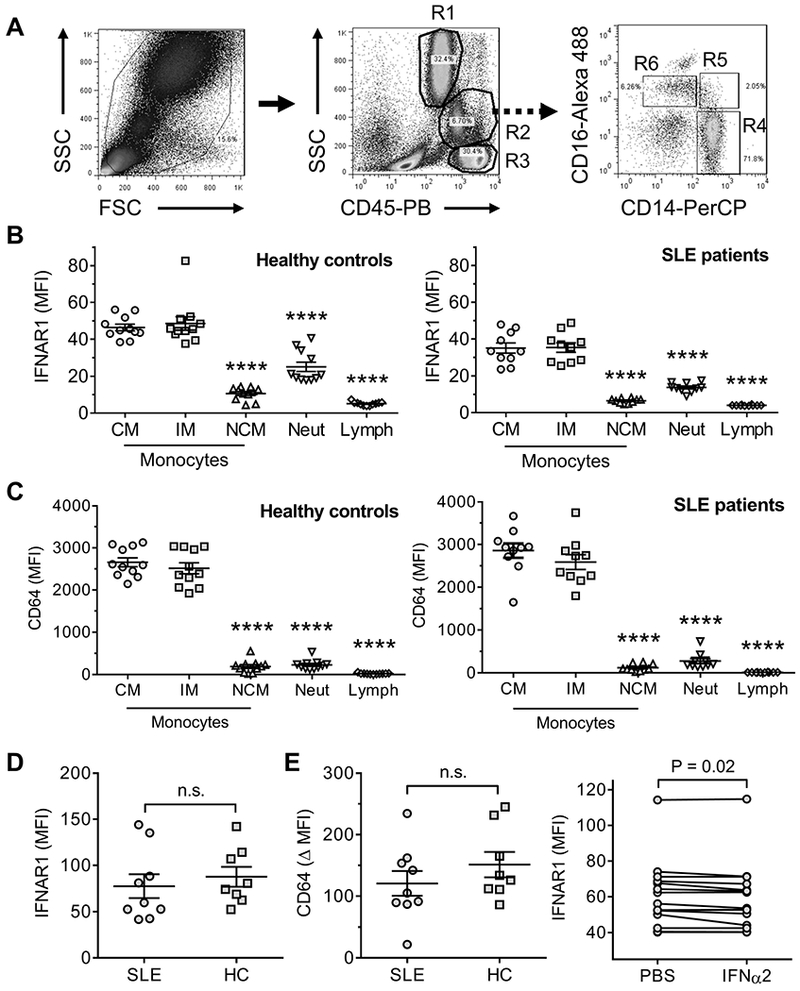
PBMCs were isolated from SLE patients (n=10), and healthy controls (n=11) and analyzed by forward scatter (FSC), side scatter (SSC), and staining with anti-CD14, CD16, CD45, CD64, and IFNAR antibodies, showing neutrophils (R1), monocytes (R2), and lymphocytes (R3). Surface staining of IFNAR and CD64 (mean fluorescence intensity, MFI) on CD14++CD16− (CM, R4), CD14++CD16+ (IM, R5), and CD14+CD16++ (NCM, R6) monocytes were quantified by flow cytometry. A, Gating strategy. B, IFNAR1 staining on monocyte subsets, neutrophils (Neut), and lymphocytes (Lymph) from peripheral blood of healthy controls (left) and SLE patients (right). C, CD64 staining on monocyte subsets, neutrophils, and lymphocytes from peripheral blood of healthy controls (left) and SLE patients (right). **** P < 0.0001 vs. CM (Student t-test). D, IFNAR staining on CM from peripheral blood of healthy controls (HC) and SLE patients. E, Left, change in CD64 surface staining (Δ MFI) on CM from SLE patients, and healthy controls (HC) after for 24-h culture with IFNα2 (1000 U/ml). n.s., not significant (Student t-test). Right, IFNAR1 expression (MFI) on CM after culture in vitro with PBS or IFNα2 (1000U/ml) for 24h (P < 0.02, paired Student t-test).
Interestingly, IFNAR1 protein expression was lower on peripheral blood CM/IM, as well as NCM and neutrophils, from SLE patients vs. healthy controls (Fig. 5B). This difference also was seen in a second cohort of SLE patients and healthy controls, although it was not statistically significant (Fig. 5D). To see if CM from SLE patients were more responsive to IFN-I, we cultured PBMC from SLE patients and healthy controls for 24-h with IFNα2b and then measured surface staining of the IFN-I regulated protein CD64. The increase of CD64 staining (ΔMFI) was similar in CD14++CD16− CM from SLE patients and healthy controls, suggesting that monocytes from SLE patients were not hypersensitive to signaling through the IFNAR (Fig. 5E, left). However, IFNAR1 surface staining was lower in CM cultured 24-h with IFNα2b than in controls cultured with PBS (Fig. 5E, right). In contrast, in CM cultured for 1.5-h with IFNα2b exhibited similar IFNAR1 staining to PBS controls (not shown).
Based on their differential IFNAR1 expression, we hypothesized that ISG expression would be higher in human CM vs. NCM, as seen in mice. CD64 and CD169 are surface markers used to assess the interferon signature in human cells by flow cytometry (38, 39). In murine Mϕ, levels of Fcgr1 (CD64) and Siglec1 (CD169) transcripts correlated and were higher in CM than NCM (Fig. 6A), consistent with other ISGs (Figs. 2 and 3). By flow cytometry, a similar pattern was seen in human CM, although the two markers were not significantly correlated in NCM (Fig. 6B). Analysis of mRNA levels in flow-sorted CM, NCM, CD3+ T cells, and CD19+ B cells for LY6E, CXCL10 (IP-10), ISG15, and MX1 by qPCR confirmed that ISG expression was higher in CM than in NCM after IFNα2b stimulation. (Fig. 6C).
Figure 6. Expression of ISGs in human PBMC subsets.
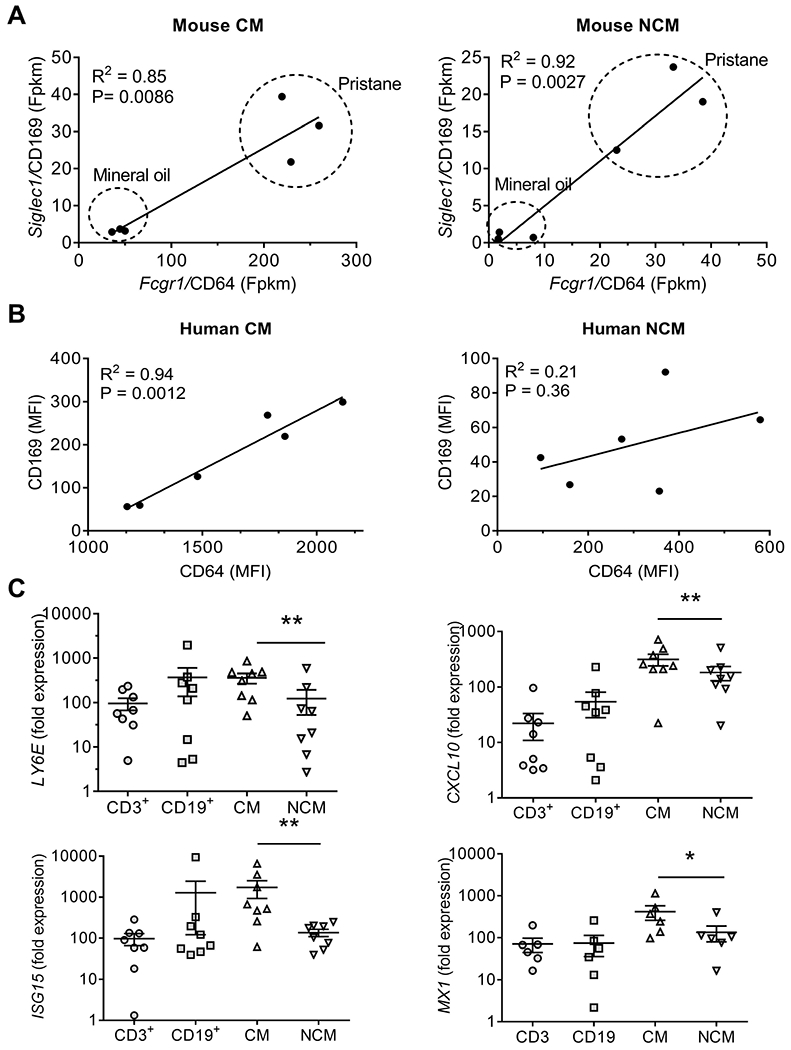
A, Correlation of Siglec1 (CD169) and Fcgr1 (CD64) mRNA levels in murine CM vs. NCM from pristane- and MO-treated mice (Fpkm, fragment per kilobase million) (Spearman rank correlation). B, Correlation of CD169 and CD64) surface staining levels on human CM and NCM. (MFI, mean fluorescence intensity, flow cytometry) (Spearman rank correlation). C, expression of ISGs (LY6E, CXCL10, ISG15) by qPCR in flow-sorted human PBMC subsets 11-h after treatment with IFNα2b (1000 U/ml). Cell subsets: T cells (CD3+), B cells (CD19+), CM, and NCM. **, P < 0.01, CM vs. NCM, paired Wilcoxon rank sum test.
Discussion
The hydrocarbon oil pristane causes non-remitting peritoneal inflammation, a strong interferon signature, and clinical features of lupus in non-autoimmune prone mice (6). Other hydrocarbon oils, such as MO, cause acute inflammation without inducing over-expression of ISGs or lupus. Resolution of peritoneal inflammation in MO-treated mice is associated with the disappearance of CM and with increasing numbers of NCM (18). Diverse mechanisms regulate the resolution of inflammation (40, 41). We show here that differential expression of the IFNAR on monocytes and Mϕ may influence whether inflammation resolves or becomes chronic. We also show that CM are the primary IFN-I responsive cell type in mouse PECs and in human PBMCs. The balance between CM and NCM plays a previously unrecognized role determining the magnitude of the interferon signature.
Differential interferon responsiveness in monocyte/Mϕ subsets.
Unexpectedly, we found that in mouse PEC, ISG expression was lower in NCM than in CM exposed to the same concentration of IFN-I (Fig. 3). The data suggest that the explanation lies in the level of IFNAR expression. Ifnar1 transcripts and IFNAR surface staining both were lower in NCM vs. CM (Fig. 4). Peritoneal lymphocytes and neutrophils also had little or no Ifnar1 surface staining. The low level of Ifnar1 may be functionally significant, as it correlated with lower Stat1 activation in NCM (Fig. 4C).
Similarly, IFNAR expression was lower on human NCM vs. CM and IM (Fig. 5). Consistent with the mouse data, there was relatively little IFNAR expression on human neutrophils and lymphocytes, suggesting that the CM are a major determinant of the interferon signature in human PBMCs. The relative expression of ISGs (LY6E, CXCL10, ISG15, and MX1) in flow-sorted PBMC subsets supports that conclusion (Fig. 6C). Thus, signaling through the IFNAR of CM may be a significant determinant of the interferon signature in human PBMCs isolated by density gradient centrifugation (containing monocytes and lymphocytes, but few neutrophils). A predominance of CM/IM over NCM may favor a strong interferon signature. Although IFNAR expression on neutrophils was lower than on CM, neutrophils are more numerous than monocytes and could influence the interferon signature in blood samples collected in PAXgene tubes.
Although the overall pattern of IFNAR1 expression was similar on murine peritoneal Mϕ and human circulating monocyte subsets (i.e. higher on CM than NCM), there was a trend toward lower IFNAR1 expression on CM from SLE patients vs. controls (Fig. 5B). Several factors may influence IFNAR expression on human monocytes. As in mice with pristane-induced lupus (31), CCR2-CCL2 mediated recruitment of CM to inflamed tissues may alter the proportions of CM and NCM in the blood of SLE patients. In addition, circulating CM in most SLE patients are exposed to higher concentrations of IFN-I than those from healthy controls. Although the lower IFNAR staining on SLE vs. control CM was not statistically significant, it was reproducible in two independent cohorts (Fig. 5B and 5D). CM from SLE patients and healthy controls displayed similar induction of ISG expression in vitro (Fig. 5E, left), suggesting that SLE monocytes were not hyper-responsive to IFNα. However, when CM from both SLE patients and healthy controls were cultured with IFNα2b, IFNAR1 surface staining was reduced (Fig. 5E, right). There are two potential explanations. The anti-IFNAR monoclonal antibody might compete with IFNα2 for binding to IFNAR1, as is true of other monoclonal anti-IFNAR1 antibodies (42, 43). Alternatively, since IFN-I binding induces IFNAR1 internalization (44), receptor expression could be modulated by endogenous IFN-I (SLE patients) or exogenous IFNα2b (in vitro). The latter explanation may be more likely, since a 1-h preincubation with IFNα2b prior to staining with anti-IFNAR antibodies did not cause a dose-dependent reduction in IFNAR staining (not shown).
The differential expression of Ifnar1 mRNA in murine peritoneal CM is under investigation. Although enhanced degradation of the IFNAR1 chain is reported in colorectal cancer and influenza infection (45, 46), the low level of Ifnar1 transcripts in NCM in murine lupus is more consistent with transcriptional regulation. However, further studies are necessary to determine the mechanisms that regulate IFNAR1 expression in lupus monocytes/Mϕ.
Clinical significance.
ISGs are expressed at high levels by PBMCs from patients with lupus (1, 2) or genetic interferonopathies (13). Although it is assumed that the interferon signature results from over-production of IFN-I, our data suggest the situation is more complex than that. First, the interferon signature critically depends on the responsiveness of monocytes/Mϕ to signaling through the IFNAR. Second, CM express high levels of the IFNAR1 and exhibit a strong interferon signature, whereas NCM and other blood cells have little IFNAR1 expression and only a weak interferon signature. Finally, even in CM, there would be no interferon signature without IFN-I production. This probably explains the low IFN-I regulated gene expression in CM from MO- vs. pristane-treated mice (Figs. 2–3) despite their comparable levels of Ifnar1, Ifnar2, Tyk2, and Jak1 mRNAs (Fig. 4). Thus, the interferon signature, which is used increasingly to monitor disease activity and/or response to therapy, is a composite measure of both IFN-I production and IFNAR expression on monocytes/Mϕ. Consequently, therapeutic intervention could abolish the interferon signature by driving monocyte/Mϕ differentiation toward an anti-inflammatory phenotype without substantially affecting the production of interferon. Recent studies suggest it may be feasible in principle to accomplish this using LXR agonists (20).
Relationship of IFNAR expression to disease severity.
The balance between CM and NCM also could influence disease severity. Monocyte/Mϕ infiltration of target organs is seen in glomerulonephritis and atherosclerosis (47, 48), both of which are associated with IFN-I production in lupus (4, 49). Lupus monocytes/Mϕ secrete higher than normal levels of proinflammatory cytokines and have an impaired capacity to take up apoptotic cells (18, 50, 51), possibly reflecting an imbalance between CM and NCM (18). In comparison with CM, NCM exhibited higher surface expression of the scavenger receptors CD36 and Marco (Fig. 1E). We showed previously that treatment with anti-Marco neutralizing antibodies or the class A scavenger receptor antagonist Poly(I) reduces in vivo phagocytosis of apoptotic cells by ~30% (18). Like Marco, the class B scavenger receptor CD36 promotes clearance of apoptotic cells (52). Increased NCM expression of CD36 and Marco may help prevent the interferon signature by reducing the generation and release of TLR7/8/9 ligands from uncleared apoptotic cells undergoing secondary necrosis (53). Thus, NCM may help limit the interferon signature in two ways: 1) by their insensitivity to signaling through the IFNAR and 2) by removing dead cells and preventing TLR-mediated IFN-I production. It will be of interest in the future to examine the relationship of IFNAR expression on tissue infiltrating monocyte/Mϕ to disease severity (lupus nephritis, accelerated atherosclerosis).
Supplementary Material
Acknowledgments
We are grateful for the assistance provided by the Broad Institute Technology Labs and to Annie Chan, R.N. for assisting with the human studies.
Supported by research grants R01-AR44731 (WR), P30-AR070253 (Joint Biology Consortium, Brigham and Women’s Hospital, PAN), R01 AR065538 (PAN), and K08-AR074562 (PYL) from NIH/NIAMS and a Lupus Research Alliance Target Identification in Lupus Grant (PAN). PYL was the recipient of a Rheumatology Research Foundation Investigator Award from the American College of Rheumatology.
Footnotes
Conflict of Interest Disclosures: The authors declare no competing financial interests
References
- 1.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100(5):2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuang H, Narain S, Sobel E, Lee PY, Nacionales DC, Kelly KM, et al. Association of anti-nucleoprotein autoantibodies with upregulation of Type I interferon-inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin Immunol. 2005;117(3):238–50. [DOI] [PubMed] [Google Scholar]
- 4.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–503. [DOI] [PubMed] [Google Scholar]
- 5.Nacionales DC, Kelly KM, Lee PY, Zhuang H, Weinstein JS, Sobel E, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2, 6, 10, 14 tetramethylpentadecane (pristane). Am J Pathol. 2006;168:1227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ronnblom LE, Alm GV, Oberg KE. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann Intern Med. 1991;115(3):178–83. [DOI] [PubMed] [Google Scholar]
- 8.Wilson LE, Widman D, Dikman SH, Gorevic PD. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum. 2002;32(3):163–73. [DOI] [PubMed] [Google Scholar]
- 9.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197(6):777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Sobel E, Satoh M, et al. Deficiency of the Type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56:3770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69(2):376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. 2016;213(12):2527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12(12):1159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017;38(8):542–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–86. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Nan J, Willard B, Wang X, Yang J, Stark GR. Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. The EMBO journal. 2017;36(2):202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han S, Zhuang H, Shumyak S, Wu J, Li H, Yang LJ, et al. A Novel Subset of Anti-Inflammatory CD138(+) Macrophages Is Deficient in Mice with Experimental Lupus. J Immunol. 2017;199(4):1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, et al. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity. 2017;47(5):890–902 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S, Zhuang H, Shumyak S, Wu J, Xie C, Li H, et al. Liver X Receptor Agonist Therapy Prevents Diffuse Alveolar Hemorrhage in Murine Lupus by Repolarizing Macrophages. Front Immunol. 2018;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee PY, Sykes DB, Ameri S, Kalaitzidis D, Charles JF, Nelson-Maney N, et al. The metabolic regulator mTORC1 controls terminal myeloid differentiation. Sci Immunol. 2017;2:eaam6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–8. [DOI] [PubMed] [Google Scholar]
- 23.Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol. 2014;107:4 22, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–8. [DOI] [PubMed] [Google Scholar]
- 28.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. [DOI] [PubMed] [Google Scholar]
- 29.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. [DOI] [PubMed] [Google Scholar]
- 30.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172(7):4410–7. [DOI] [PubMed] [Google Scholar]
- 31.Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, et al. Type-I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J Immunol. 1998;161(11):6250–7. [PubMed] [Google Scholar]
- 33.Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007;204(10):2259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic acids research. 2013;41(Database issue):D1040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–80. [DOI] [PubMed] [Google Scholar]
- 36.Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate and nonclassical human monocyte subsets. Blood. 2011;118:e16–e31. [DOI] [PubMed] [Google Scholar]
- 37.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211(6–8):609–18. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Lee PY, Kellner E, Paulus M, Switanek J, Xu Y, et al. Monocyte surface expression of Fcgamma receptor RI (CD64), a biomarker reflecting Type-I interferon levels in systemic lupus erythematosus. Arthritis ResTher. 2010;12(3):R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose T, Grutzkau A, Hirseland H, Huscher D, Dahnrich C, Dzionek A, et al. IFNalpha and its response proteins, IP-10 and SIGLEC-1, are biomarkers of disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2013;72(10):1639–45. [DOI] [PubMed] [Google Scholar]
- 40.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–82. [DOI] [PubMed] [Google Scholar]
- 41.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benoit P, Maguire D, Plavec I, Kocher H, Tovey M, Meyer F. A monoclonal antibody to recombinant human IFN-alpha receptor inhibits biologic activity of several species of human IFN-alpha, IFN-beta, and IFN-omega. Detection of heterogeneity of the cellular type I IFN receptor. J Immunol. 1993;150(3):707–16. [PubMed] [Google Scholar]
- 43.Peng L, Oganesyan V, Wu H, Dall’Acqua WF, Damschroder MM. Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs. 2015;7(2):428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurtado-Guerrero I, Pinto-Medel MJ, Urbaneja P, Rodriguez-Bada JL, Leon A, Guerrero M, et al. Activation of the JAK-STAT Signaling Pathway after In Vitro Stimulation with IFNss in Multiple Sclerosis Patients According to the Therapeutic Response to IFNss. PloS one. 2017;12(1):e0170031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Araya RE, Goldszmid RS. IFNAR1 Degradation: A New Mechanism for Tumor Immune Evasion? Cancer Cell. 2017;31(2):161–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia C, Vijayan M, Pritzl CJ, Fuchs SY, McDermott AB, Hahm B. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J Virol. 2015;90(5):2403–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, et al. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol. 2011;186(8):4994–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol. 2012. [DOI] [PubMed] [Google Scholar]
- 50.Steinbach F, Henke F, Krause B, Thiele B, Burmester GR, Hiepe F. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann Rheum Dis. 2000;59(4):283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis. 2015;74(7):1441–9. [DOI] [PubMed] [Google Scholar]
- 52.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203(12):2613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.