

Abstract
Objectives
In a multi-ethnic/racial, prospective SLE inception cohort, to determine the frequency, clinical characteristics, associations and outcomes in different types of peripheral nervous system (PNS) disease.
Methods
Patients were evaluated annually for 19 neuropsychiatric (NP) events including seven types of PNS disease. SLE disease activity, organ damage, autoantibodies, patient and physician assessment of outcome were measured. Time to event and linear regressions were used as appropriate.
Results
Of 1,827 SLE patients, 88.8% were female, 48.8% Caucasian. The mean±SD age was 35.1±13.3 years, disease duration at enrollment 5.6±4.2 months and follow-up 7.6±4.6 years. There were 161 PNS events in 139/1,827 (7.6%) patients. The predominant events were peripheral neuropathy [66/161 (41.0%)], mononeuropathy [44/161 (27.3%)] and cranial neuropathy [39/161 (24.2%)] and the majority were attributed to SLE. Multivariate Cox regressions suggested longer time to resolution in patients with prior history of neuropathy, older age at SLE diagnosis, higher SLEDAI-2K scores, and for peripheral neuropathy versus other neuropathies. Neuropathy was associated with significantly lower SF-36 physical and mental component summary scores versus patients without NP events. By physician assessment, the majority of neuropathies resolved or improved over time and this was associated with improvements in SF-36 summary scores for peripheral neuropathy and mononeuropathy.
Conclusion
PNS disease is an important component of total NPSLE and has a significant negative impact on health related quality of life. The outcome is favourable for most patients, but we noted several factors associated with longer time to resolution.
Keywords: Systemic lupus erythematosus, Neuropathy, Outcome
Involvement of the nervous system by systemic lupus erythematosus (SLE) presents clinically as a variety of neurological and psychiatric features, collectively referred to as neuropsychiatric SLE (NPSLE). Approximately one-third of NP events are directly attributable to SLE and occur in 21% of SLE patients in the first 6.6 years of their disease (1). Central nervous system (CNS) involvement accounts for over 90% of events compared to involvement of the peripheral nervous system (PNS) which accounts for most of the other events (1). Although there is a large body of work on CNS disease in SLE patients, involvement of the PNS is less well established.
Of the three current classification criteria for SLE (2-4) only the Systemic Lupus International Collaborating Clinics (SLICC) criteria include PNS events as a variable. In the American College of Rheumatology (ACR) case definitions for NPSLE (5), seven of 19 manifestations affect the PNS. The aim of the present study was to determine the frequency, characteristics, clinical and autoantibody associations and outcomes assessed by physicians and patients of these seven PNS manifestations in a large, multi-ethnic/racial, prospective, inception cohort of SLE patients.
Patients and Methods
Research study network
The study was conducted by the Systemic Lupus International Collaborating Clinics (SLICC) (6), a network of 52 investigators at 43 academic centers in 16 countries. From 1999 to 2011, a cohort of recently-diagnosed SLE patients was recruited from 31 SLICC sites in Europe, Asia, and North America. Data were collected per protocol at enrollment and annually, submitted to the coordinating centers in Toronto, Ontario and Halifax, Nova Scotia, Canada and entered into a centralized Access database. Appropriate procedures ensured data quality, management and security. The Nova Scotia Health Authority central zone Research Ethics Board, Halifax, and each of the participating centers’ institutional research ethics review boards approved the study.
Patients
Patients fulfilled the revised ACR SLE classification criteria for SLE (2), the date of which was used as the date of diagnosis, and provided written informed consent. Enrollment was permitted up to 15 months following the diagnosis. Demographic variables, education and medication history were collected. Lupus-related variables included the SLE Disease Activity Index 2000 (SLEDAI-2K) (7) and SLICC/ACR damage index (SDI) (8). Routine laboratory testing included hematological, biochemical and immunological variables required to determine SLEDAI-2K and SDI scores.
Neuropsychiatric (NP) events
An enrollment window extended from 6 months prior to the diagnosis of SLE up to the actual enrollment date. NP events were characterized within this window using the ACR case definitions for 19 NP syndromes (5). These were diagnosed by clinical evaluation supported by investigations, if clinically warranted, as per existing guidelines. Patients were seen annually with a 6-month window around the anticipated assessment date. New NP events and the status of previous NP events since the last study visit were determined at each assessment.
The ACR case definitions (5) include seven types of PNS disease: (i) peripheral neuropathy; (ii) cranial neuropathy; (iii) mononeuropathy single or multiplex; (iv) plexopathy; (v) autonomic neuropathy; (vi) acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barré syndrome) and (vii) myasthenia gravis. In view of the low frequency of the latter 4 types of PNS disease, they were not included in the detailed analyses that was restricted to peripheral neuropathy, mononeuropathy and cranial neuropathy. Recurring PNS and other NP events within the enrollment window or within each follow-up assessment period were recorded once. The date of the first such episode was taken as the onset of the event.
Attribution of NP events
In keeping with other publications on NP events within the SLICC NPSLE inception cohort, the same decision rules were used to determine the attribution of all NP events (9, 10). Factors considered in the decision rules included: (i) temporal onset of NP event(s) in relation to the diagnosis of SLE; (ii) concurrent non-SLE factor(s), such as potential causes (“exclusions”) or contributing factors (“associations”) for each NP syndrome in the glossary for the ACR case definitions of NP events(5); and (iii) “common” NP events which are frequent in normal population controls as described by Ainiala et al (11). These include isolated headaches, anxiety, mild depression (mood disorders failing to meet criteria for “major depressive-like episodes”), mild cognitive impairment (deficits in less than three of the eight specified cognitive domains) and peripheral neuropathy without electrophysiological confirmation. Two attribution decision rules of different stringency (models A and B) were used (9, 10).
Attribution Model A
NP events which had their onset within the enrollment window or subsequently and had no “exclusions” or “associations” and were not one of the NP events identified by Ainiala (11) were attributed to SLE.
Attribution Model B
NP events which had their onset within 10 years of the diagnosis of SLE and were still present within the enrollment window or onset at a later date and had no “exclusions” and were not one of the NP events identified by Ainiala (11) were attributed to SLE.
NP events that fulfilled criteria for model A (most stringent) or for model B (least stringent) were attributed to SLE. By definition, all NP events attributed to SLE using model A were included in the NP events using model B. Those events which did not fulfill these criteria were classified as a non-SLE NP event.
Outcome of PNS events
A physician generated 7-point Likert scale was completed at each follow-up assessment and compared the change in PNS events between onset and follow-up (1=patient demise, 2=much worse, 3=worse, 4=no change, 5=improved, 6=much improved, 7=resolved) (12). A patient generated SF-36 questionnaire was completed at each assessment and provided eight subscales and the mental (MCS) and physical (PCS) component summary scores (12, 13); these were not available to physicians at the time of their assessments.
Autoantibodies
Lupus anticoagulant (LAC), IgG anticardiolipin, anti-β2 glycoprotein-I, anti-ribosomal P (anti-P) and anti-NR2 glutamate receptor antibodies were measured at the Oklahoma Medical Research Foundation, USA (14-17).
Statistical analysis
The Kaplan-Meier method was used to estimate cumulative incidence for first and recurrent PNS events and the probability of not resolving neuropathy over time. We used Cox regression to examine the risk of first SLE neuropathy (either peripheral neuropathy, mononeuropathy or cranial neuropathy attributed by model B). Hazard ratios (HR) and 95% confidence intervals (CI) were calculated. Due to sparse data, logistic regression with generalized estimating equations (GEE) estimation was used to analyze grouped Likert scale outcomes (≥5 vs. ≤4) for unresolved SLE neuropathies. Cox regression was also used for analyzing the time to resolution as it examines how quickly the neuropathy events resolved while the analysis of the Likert scale outcome examines the probability of being improved (if not resolved) at a specific time point. Covariates examined included sex, race/ethnicity, SLICC sites, post-secondary education, number of ACR criteria at enrollment, age at SLE diagnosis, presence/absence of autoantibodies at baseline and, as time varying variables updated at each assessment, SDI (without NP variables), other concurrent NP events, age at SLE diagnosis, disease duration (in years), SLEDAI-2K (without NP variables, standardized by taking (x-4)/4)), presence/absence of autoantibodies at follow-up assessments and medication use since last assessment (corticosteroids, antimalarials, immunosuppressants, anticoagulants). For analyses of the physician-assessed outcomes of neuropathy, history of SLE neuropathy prior to the onset of the current event, SLE-attribution, and sub-types of neuropathies were also examined. For analyses of longitudinal SF-36 summary scores, linear regression with GEE estimation allowed for correlation of observations within patients and adjustment variables include time/visit, sex, age at SLE diagnosis, race/ethnicity/location, education, SLEDAI-2K and SDI scores (without NP variables), corticosteroids, antimalarials and immunosuppressant use since last assessment.
Results
Patients
1,827 patients were recruited between October 1999 and December 2011, from centers in the United States [n=540 (29.5%)], Europe [n=477 (26.1%)], Canada [n=418 (22.9%)], Mexico [n=223 (12.2%)] and Asia [n=169 (9.3%)] (Table 1). The number of patient assessments varied from 1 to 19 with a mean follow-up of 7.6±4.6 years and the final assessment was in September 2017.
Table 1:
Demographic and clinical features of SLE patients (n=1827) at enrolment.
| Sex (%) | Female | 1623 (88.8) |
| Male | 204 (11.2) | |
| Age (years) (mean ± SD) | 35.1 ± 13.3 | |
| Race/Ethnicity (%) | Caucasian | 891 (48.8) |
| African | 307 (16.8) | |
| Hispanic | 282 (15.4) | |
| Asian | 275 (15.1) | |
| Other | 72 (3.9) | |
| Single/Married/Other (%) | 819 (44.9)/766 (42.0)/238 (13.1) | |
| Post-secondary education (%) | 1065 (61.9) | |
| Disease duration (months) (mean ± SD) | 5.6 ± 4.2 | |
| Number of ACR criteria (mean ± SD) | 4.9 ± 1.1 | |
| ACR manifestations (%) | Malar rash | 660 (36.1) |
| Discoid rash | 227 (12.4) | |
| Photosensitivity | 653 (35.7) | |
| Oral/nasal ulcers | 678 (37.1) | |
| Serositis | 502 (27.5) | |
| Arthritis | 1368 (74.9) | |
| Renal disorder | 510 (27.9) | |
| Neurological disorder | 88 (4.8) | |
| Hematologic disorder | 1130 (61.9) | |
| Immunologic disorder | 1393 (76.2) | |
| Antinuclear antibody | 1732 (94.8) | |
| SLEDAI-2K score (mean ± SD) | 5.3 ± 5.4 | |
| *SLICC/ACR damage index score (mean ± SD) | 0.32 ± 0.74 | |
| Medications (%) | Corticosteroids | 1285 (70.3) |
| Antimalarials | 1231 (67.4) | |
| Immunosuppressants | 732 (40.1) | |
| ASA | 261 (14.3) | |
| Antidepressants | 184 (10.1) | |
| Warfarin | 99 (5.4) | |
| Anticonvulsants | 80 (4.4) | |
| Antipsychotics | 12 (0.7) | |
| Autoantibody positivity N (%) | Lupus anticoagulant | 241/1174 (20.5) |
| Anti-cardiolipin | 138/1142 (12.1) | |
| Anti-Beta2 glycoprotein-I | 163/1142 (14.3) | |
| Anti-ribosomal P Anti-NR2 |
112/1136 (9.9) 130/1064 (12.2) |
SLICC/ACR damage index not available in 1058 patients at enrollment visit when disease duration < 6 months
Neuropsychiatric (NP) manifestations
NP events (≥1) occurred in 955/1,827 (52.3%) patients and 493/1827 (27.0%) had ≥ 2 events over the study period. There were 1910 unique NP events, encompassing all 19 NP syndromes in the ACR case definitions (5). The proportion of NP events attributed to SLE varied from 17.9% (attribution model A) to 31.0% (attribution model B) and occurred in 13.5% (model A) to 21.2% (model B) of patients. Of the 1910 unique NP events, 1749 (91.6%) involved the CNS and 161 (8.4%) the PNS (5). The classification of events into diffuse and focal was 1479 (77.4%) and 431 (22.6%) respectively (10).
Peripheral nervous system manifestations
There were 161 PNS events in 139/1,827 (7.6%) patients (Table 2). Fifty-four of the 161 (33.5%) PNS events were identified at the enrollment visit (13 preceded the diagnosis of SLE by up to 4 months) and the remainder presented over the ensuing follow-up. The most frequent events were peripheral neuropathy [66/161 (41.0%)], mononeuropathy [44/161 (27.3%)] of which 17/44 (38.6%) were multiplex, and cranial neuropathy [39/161 (24.2%)]; there were few patients in the remaining categories: autonomic neuropathy [4/161 (2.5%)], myasthenia gravis [3/161 (1.9%)], acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barré syndrome) [3/161 (1.9%)] and plexopathy [2/161 (1.2%)]. For the 110 patients with peripheral neuropathy or mononeuropathy who underwent electrophysiological testing [60/110 (54.5%)] the predominant abnormality was axonal damage [25/60 (41.7%)] followed by demyelination [13/60 (21.7%)]. Of the 31 patients with peripheral neuropathy who underwent electrophysiological testing, 5/31 (16.1%) had isolated sensory neuropathy, 3/31 (9.7%) had isolated motor neuropathy and 22/31 (71%) had sensorimotor neuropathy. The most frequent cranial neuropathies were II (32.6%), VIII (27.9%), VII (9.3%), V, VI, IX (all 7%), III (4.7%), I and IV (both 2.3%). Eighty-eight of the 161 (54.7%) events in 80/139 (57.6%) patients were attributed to SLE using model A, and 118/161 (73.3%) events in 104/139 (74.8%) patients were attributed to SLE using model B attribution rules. Using model B, the majority of neuropathies were attributed to SLE, with the exception of peripheral neuropathies of which 36/66 (54.5%) were attributed to the non-SLE category. In 34/36 (94.4%) of these cases, electrophysiological studies were not done which precluded attributing the neuropathy to SLE as per the attribution rules. An alternative cause for the peripheral neuropathy was identified in only eight cases (hypothyroidism in six and vitamin deficiency in two).
Table 2:
Characteristics and attribution of peripheral nervous system (PNS) disease events in SLE patients over the duration of study.
| PNS disease | Total # PNS events |
# PNS events attributed to SLE (model A) |
# PNS events attributed to SLE (model B) |
# PNS events attributed to non- SLE causes |
|---|---|---|---|---|
| Peripheral neuropathy | 66 | 25 (37.9%) | 30 (45.5%) | 36 (54.5%) |
| Mononeuropathy single or multiplex | 44 | 24 (54.5%) | 44 (100%) | 0 |
| Cranial neuropathy | 39 | 32 (82.1%) | 35 (89.7%) | 4 (10.3%) |
| Plexopathy | 2 | 0 | 0 | 2 (100%) |
| Autonomic neuropathy | 4 | 3 (75.0%) | 3 (75.0%) | 1 (25.0%) |
| Acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barré syndrome) | 3 | 3 (100%) | 3 (100%) | 0 |
| Myasthenia gravis | 3 | 1 (33.3%) | 3 (100%) | 0 |
| Total # PNS events | 161 | 88 (54.7%) | 118 (73.3%) | 43 (26.7%) |
The estimated cumulative incidence of any PNS event regardless of attribution was 8.8% [95%CI (7.3%, 10.3%)] and for those attributed to SLE (model B attribution rule) was 6.5% [95%CI (5.2%, 7.8%)] after 10 years (see Figure 1 for rates of specific neuropathy types). In patients with a previous SLE attributed PNS event, the estimate of recurrence at 5 years after the initial PNS event was 11.7% [95%CI (4.6%, 18.4%)]. The incidence rate of first SLE PNS event was 7.4/1000 person years and the incidence of recurrence was 18.2/1000 person years.
Figure 1:
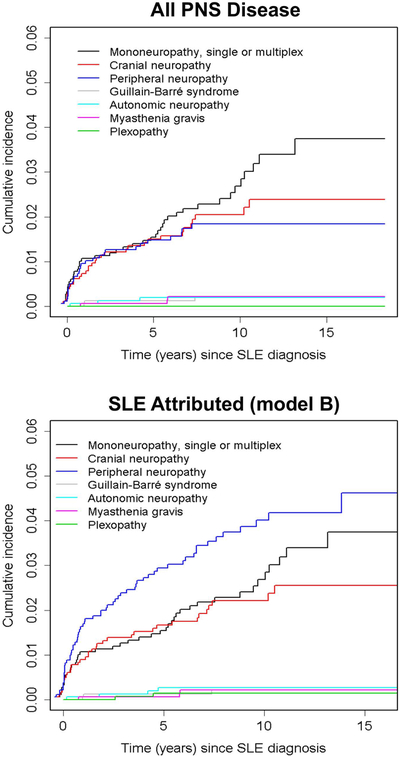
The estimated cumulative incidence of all peripheral nervous system (PNS) disease and that attributed to SLE using attribution model B.
Clinical and laboratory associations with peripheral nervous system disease attributed to SLE
Using Cox regression we looked for associations with the risk of the first episode of either peripheral neuropathy, mononeuropathy or cranial neuropathy attributed to SLE using attribution model B. There were insufficient numbers of the other neuropathies to perform this and subsequent analyses. Univariate analysis revealed a negative association [HR (95%CI)] with Asian race/ethnicity [0.40 (0.18, 0.88)] and post-secondary education [0.65, (0.43, 0.98)] and a positive association with other concurrent central [2.96 (1.66, 5.26)] or diffuse [2.58 (1.25, 5.33)] NP events (cerebrovascular disease, cognitive dysfunction, psychosis) attributed to SLE. There was no association between neuropathy and any of the autoantibodies examined. Multivariate analyses, which included these variables, indicated similar trends (Asian race/ethnicity [0.42 (0.19, 0.93)]; secondary education: [0.69 (0.45, 1.04)]; other concurrent NP events attributed to SLE [2.74 (1.49, 5.03)], but the effect of post-secondary education had a slightly wider confidence interval.
Clinical outcome of PNS events
Of 149 neuropathies (peripheral, mono and cranial) 76 (51.0%) were resolved by the end of study (27 peripheral, 23 mono and 26 cranial neuropathies). Figure 2 (upper panel) illustrates the probability of these neuropathies not resolving over time. For the total group the estimated probability at 10 years was 37.2% (27.6%, 50.0%); for peripheral neuropathy it was 42.6% [95% CI (25.4%, 71.6%)]; for mononeuropathy it was 28.6% (14.1%, 58.0%); and for cranial neuropathy it was 30.4% (18.5%, 49.8%). Although the probability of resolution was comparable for all three types of neuropathy, the time to reach resolution was most rapid for cranial neuropathy, followed by mononeuropathy and peripheral neuropathy.
Figure 2:

Physician determined change in peripheral neuropathy, mononeuropathy or cranial neuropathy (n=149) attributed to SLE and non-SLE using attribution model B. Top panel: Survival curves for resolution of all neuropathies (left) and individual neuropathies (right). Lower panel: Likert scale scores for physician assessment of outcome over the duration of followup are shifted to the right indicated improvement and this is most pronounced for cranial neuropathies (right).
In univariate Cox regression analyses, resolution times [HR (95%CI)] were negatively associated with history of neuropathy prior to the onset of the current neuropathy [0.38 (0.16, 0.88)], older age at SLE diagnosis [0.75 (0.58, 0.96), and peripheral neuropathy versus cranial neuropathy [HR: 0.44 (0.24, 0.80)] and mononeuropathy [HR 0.67 (0.41, 1.08)], 2 degree of freedom test, p=0.027. These suggest that history of neuropathy, older age at SLE diagnosis, and peripheral versus cranial neuropathy and possibly mononeuropathy were all factors indicating longer time to resolution. In multivariate analyses, we noted persistent negative associations between time to resolution and history of neuropathy [0.38 (0.16, 0.90)], older age at SLE diagnosis [0.76 (0.60, 0.98)], and for peripheral neuropathy versus cranial neuropathy [HR: 0.45 (0.25, 0.82) and versus mononeuropathy [HR: 0.74 (0.44, 1.22)], 2 degree of freedom test, p=0.034. The multivariate analyses also suggested longer time to resolution with higher SLEDAI-2K (excluding NP variables) scores [HR for an increase of 4 in SLEDAI: 0.71 (0.51, 0.99)].
Figure 2 (lower panel) summarizes the distribution of maximum and minimum Likert scale scores indicating physician assessment of outcome of neuropathies during follow-up. The Likert scale scores over the duration of follow-up are shifted to the right indicating improvement and this is most pronounced for cranial neuropathies (right). In univariate analyses, lower probabilities of improvement in unresolved neuropathies at a specific time point since onset [odds ratio (95%CI)] were associated with history of neuropathy [0.45 (0.29, 0.69)], US sites [vs. European sites 0.40 (0.84, 0.95)], longer disease duration prior to onset of neuropathy [- 0.90 (0.83, 0.99)] and presence of anti-NR2 antibodies at enrollment [0.18 (0.03, 0.91))]. The associations with geographical region (global test p-value =0.05), longer disease duration prior to onset of neuropathy (p= 0.011), and for those patients with antibody measurements available, the presence of anti-NR2 antibodies at enrollment (p=0.008) remained in the multivariate analyses.
Health-Related Quality of Life and PNS events
The association between grouped peripheral neuropathy, mononeuropathy or cranial neuropathy and SF-36 summary and sub-scale scores is illustrated in Figure 3 using data in the following three groups of patients over time: (i) any neuropathy events which occurred at or prior to the study assessment; (ii): any NP event other than a neuropathy event occurring at or prior to the study assessment; (iii) patients who never had any NP event up to the study assessment. Once assigned, each patient retained the same group membership throughout follow-up until they had a new or subsequent NP event which could trigger a change in group assignment. Utilizing scores from all assessments the lowest mean (SD) PCS score occurred in patients with neuropathies [38.9 (12.3)] compared to patients with other NP events [40.8 (11.7)] and patients without NP events [44.1 (10.9)] [overall p<0.001 after adjustment for covariates]. Similar but less marked differences in mean (SD) MCS scores were seen with the same group assignment [46.0 (12.0) vs 44.9 (12.2) vs 48.9 (10.7)] (overall p<0.0001 after adjustments). For both PCS and MCS scores there were significant differences between groups (i) and (iii) (p<0.0001 and p=0.0008, respectively) but not between groups (i) and (ii) (p>0.05) after adjustments. The group differences in PCS and MCS scores over time (Figure 3) persisted for 10 years of follow-up (global p-values for group effects <0.0001 after adjustments). Utilizing scores from all assessments the mean group differences in individual SF-36 subscale scores in the same three groups of patients (Figure 3), indicated that at least six of eight self-reported health domains were lower in patients with SLE neuropathy compared to the other two groups.
Figure 3:

Association of SF-36 summary and subscale scores with neuropathy (peripheral neuropathy, mononeuropathy, cranial neuropathy) attributed to SLE and non-SLE using attribution model B for the following 3 patient groups: (i) neuropathy events which occurred at or prior to the study assessment; (ii) any NP event other than neuropathy event occurring at or prior to the study assessment;(iii) patients who never had any NP event up to the study assessment. Upper two panels: SF-36 physical component summary (PCS) and mental component summary (MCS) scores with neuropathy over time. Bottom panel: comparison of individual subscale scores in the 3 patient groups. The SF-36 subscales are VT = Vitality, SF = Social function, RE = Role emotion, MH = Mental health, PF = Physical function, RP = Role physical, BP = Bodily pain, GH= General health.
The change in patient self-report HRQoL following physician determined resolution of peripheral neuropathy, mononeuropathy or cranial neuropathy was examined by comparing SF-36 scores in the following groups of patients (Figure 4): (i) active peripheral neuropathy; (ii) resolved peripheral neuropathy; (iii) active mononeuropathy; (iv) resolved mononeuropathy; (v) active cranial neuropathy; (vi) resolved cranial neuropathy; (vii) any active NP event other than peripheral neuropathy, mononeuropathy or cranial neuropathy; (viii) any resolved NP event other than peripheral neuropathy, mononeuropathy or cranial neuropathy; (ix) patients who never had any NP event. Due to the small number of unique patients for some groups, adjustments for other variables were not performed in the linear regression with GEE estimation. In parallel with physician determined resolution, there was a clinically and statistically significant improvement in PCS scores for peripheral and mononeuropathies and a similar improvement in MCS scores for peripheral neuropathies. These changes were similar to that seen in patients with other non-neuropathy NP events and the final PCS and MCS scores were similar to those reported by patients who never had an NP event.
Figure 4:

The change in SF-36 physical component summary (PCS) and mental component summary (MCS) scores following resolution of neuropathy (peripheral neuropathy, mononeuropathy, cranial neuropathy) attributed to SLE and non-SLE using attribution model B for the following patient groups: (i) peripheral neuropathy events (n=235) which occurred at or prior to the study assessment up to its resolution; (ii) resolved peripheral neuropathy (n=130) up to their last follow-up or recurrence of peripheral neuropathy; (iii) mononeuropathy events (n=135) which occurred at or prior to the study assessment up to its resolution; (iv) resolved mononeuropathy (n=120) up to their last follow-up or recurrence of mononeuropathy; (v) cranial neuropathy events (n=89) which occurred at or prior to the study assessment up to its resolution; (vi) resolved cranial neuropathy events (n=130) up to their last follow-up or recurrence of cranial neuropathy; (vii) any NP event (n=2718) other than peripheral neuropathy, mononeuropathy or cranial neuropathy events occurring at or prior to the study assessment; (viii) resolved any NP event (n=2307)other than peripheral neuropathy, mononeuropathy or cranial neuropathy up to their last follow-up or recurrence; (ix) patients who never had any NP event (n=6064) up to the study assessment.
Discussion
The focus of the current study was PNS disease in a large international inception cohort of patients in the first decade following the diagnosis of SLE. PNS manifestations in SLE were confirmed to be uncommon (7.6% of patients) and of the seven ACR case definitions for PNS disease in NPSLE, only peripheral neuropathy (41.0% of PNS events), mononeuropathy (27.3% of PNS events) and cranial neuropathy (24.2% of PNS events) occurred with notable frequency. Although peripheral neuropathy was frequently attributed to non-SLE causes, this was because 28/66 (42.4%) of these patients did not undergo electrophysiological testing, which precluded attributing the neuropathy to SLE. In only a minority of cases was an alternative cause for peripheral neuropathy identified and thus we cannot exclude the possibility that many more of the peripheral neuropathies could have been due to SLE. Physician determined outcomes were generally favourable although the speed of resolution differed between the three types of neuropathy and was most rapid for cranial neuropathy. The occurrence of PNS disease was associated with a reduction in patient self-report HRQoL. Following resolution this improved significantly for peripheral and mononeuropathy but not for cranial neuropathy.
Although PNS manifestations are well recognized in SLE patients, the literature consists largely of individual case reports and small case series. There have been three large, single center, prevalent cohort studies of SLE patients (18-20) with longitudinal follow-up in one (18). Oomatia et al (19) reported peripheral neuropathy in 123 (5.9%) of 2,097 patients that was attributed to SLE in 66.7% of cases and associated with lower SLE disease activity and cumulative organ damage. A cross-sectional study by Toledano et al (20) utilized the ACR case definitions to characterize PNS disease. Overall, 93 of 524 (17.7%) patients had disease attributed to SLE. The most frequent manifestation was peripheral neuropathy (36.6%), followed by mononeuropathy (23.7%), cranial neuropathy, myasthenia gravis (7.5%, each) and Guillain-Barré syndrome (1.1%). In the most comprehensive study to date by Florica et al (18), 207 (14%) of 1533 patients had PNS disease that was attributed to SLE in 60% of cases. Peripheral neuropathy was diagnosed in 56%, cranial neuropathy in 13%, mononeuropathy in 11% and mononeuritis multiplex in 9% of patients with PNS disease. Electrophysiological studies were available in 126 (60.8%) of 207 patients and indicated axonal neuropathy in 70% and demyelination in 20% of patients, regardless of attribution to SLE and non-SLE causes. Using a nested case-control design, those with PNS events had significantly more CNS involvement, higher SLE disease activity and lower patient self-report HRQoL compared with patients without PNS events.
The current study supports and expands the findings of previous work (18-21). The overall frequency of PNS events (7.6%) in our study was higher than that reported by Oomatia et al (19) (5.9%) but lower than that in the other two large cohort studies (18, 20) (14% and 17.7%. This is to be expected in view of the differences between inception and prevalent disease cohorts. Our findings on the relative frequency of different types of PNS events, as defined by the ACR case definitions, are in alignment with the findings of Florica et al (18) and Toledano et al (20), as is the proportion of PNS events attributed to SLE (18, 19). The current study demonstrates that PNS disease increases over time, at least over the first 10 years. This is in contrast to some other NP manifestations [e.g. seizures (22), cerebrovascular events (23)] and non-NP manifestations of SLE [e.g. nephritis (24)] which have a strong predilection to present early in the disease course and frequently as part of the initial manifestation of SLE. The outcome of the different PNS manifestations, as determine by physician assessment, indicated a similar degree of improvement and resolution across neuropathy type, although the rate of improvement was most rapid for cranial neuropathies. Factors associated with a slower improvement were older age at SLE diagnosis, longer disease duration at onset of neuropathy, active SLE outside of the nervous system and recurrent PNS events. There was no association between the onset of PNS events and any of the selected panel of autoantibodies, including previously reported associations with anti-ribosomal P (25) and anticardiolipin antibodies (26). The presence of anti-NR2 antibodies was associated with a slower rate of improvement of PNS events which has not previously been reported and requires further study to demonstrate reproducibility and/or plausibility of this result.
One of the goals of our study was to determine the impact of PNS events on patient reported HRQoL, as reflected by SF-36 summary and subscale scores, as this has only been examined in one previous study (18). In comparison to patients without NP events, the occurrence of any of the three most frequent neuropathies was associated with a significant reduction in HRQoL which was comparable to that seen with other NP events. As expected, the negative effect on HRQoL was most profound on patient reported physical function although mental function was also impacted. The study by Florica et al (18) reported similar findings. We also examined the potential reversibility of low HRQoL by analyzing the change in SF-36 summary scores in patients who had a physician determined resolution of neuropathy. There were statistically significant and clinically meaningful improvements in HRQoL scores reported by patients who had resolution of peripheral neuropathy and mononeuropathy but not for cranial neuropathy. Baseline PCS scores, generated at the first annual assessment following the onset of the NP event, were better for patients with cranial neuropathy than for the other neuropathies and thus had less potential to improve. Due to the rapid improvement in cranial neuropathies (figure 2), the first SF-36 summary scores following their onset were not adversely affected as occurred in patients with peripheral neuropathy and mononeuropathy, both of which had a slower recovery (figure 2). Discrepancy between physician and patient reported outcomes has been seen in other SLE manifestations (23, 27). This emphasizes the importance of capturing both physician and patient perspectives on the potential benefit of an intervention, be it in the treatment of individual patients or in the context of clinical trials.
There are some limitations to the current study. First, specialized investigations such as nerve conduction studies and test batteries for autonomic dysfunction (28) were not routinely performed on all patients but left to the discretion of individual investigators. Likely, the universal application of such investigations would have detected additional PNS abnormalities. However, our research protocol more accurately reflects what is done in clinical practice which was a deliberate strategy of our study. Furthermore, these investigations would not have helped to determine causal attribution for neuropathies (18) Second, the unavailability of autoantibody data for some patients and restriction to a panel of autoantibodies more suited to CNS disease may have limited our ability to fully assess the role of autoantibodies in the pathogenesis of PNS events. For example, some studies have reported associations with anti-Sm (29) and anti-Ro (30) and with the more specialized anti-ganglioside antibodies (31). Third, as this was an observational cohort study, any association between immunosuppressive therapies and outcome of neuropathies was difficult to determine and we could not reliably identify symptomatic neurotropic therapies or the specific indications for their use. Similarly, SLE disease activity and autoantibodies were measured at annual assessments which usually did not coincide precisely with the onset of neuropathies. Finally, although the ACR classification of NPSLE is quite detailed and extensive, the reports of PNS events were not reviewed centrally by a neurologist and there are PNS disease manifestations described in SLE that are not captured. These include small fiber neuropathy (19, 32) and chronic inflammatory demyelinating polyradiculoneuropathy (33). The former could account for some of the peripheral sensory neuropathies in patients with normal electrophysiological testing and both entities should be considered in any revision of the ACR case definitions.
There are also many strengths to our study. These include a large disease inception cohort of SLE patients, the long-term prospective study design using a standardized protocol for data collection and the identification of all PNS events with application of decision rules for determination of attribution. Overall, the results of our study provide a comprehensive overview of the frequency and characteristics of PNS disease in SLE patients, the impact on HRQoL and the outcome with current treatment modalities for SLE. The findings provide a benchmark for the assessment of future treatment modalities.
Acknowledgments
Financial support
John G. Hanly (Canadian Institutes of Health Research grant MOP-88526)
Dr. Sang-Cheol Bae’s work was supported in part by NRF-2017M3A9B4050335, Republic of Korea.
Dr Caroline Gordon is supported by Lupus UK, Sandwell and West Birmingham Hospitals NHS Trust and the National Institute for Health Research (NIHR)/Wellcome Trust Birmingham Clinical Research Facility. The views expressed are those of the authors(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
The Hopkins Lupus Cohort is supported by the NIH (grant AR43727 and 69572).
The Montreal General Hospital Lupus Clinic is partially supported by the Singer Family Fund for Lupus Research.
Dr. Clarke holds The Arthritis Society Chair in Rheumatic Diseases at the University of Calgary.
Dr. Paul R. Fortin holds a tier 1 Canada Research Chair on Systemic Autoimmune Rheumatic Diseases at Université Laval.
Dr. Bruce is a National Institute for Health Research (NIHR) Senior Investigator and is supported by Arthritis Research UK, the NIHR Manchester Biomedical Centre and the NIHR/Wellcome Trust Manchester Clinical Research Facility. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Dr. Soren Jacobsen is supported by the Danish Rheumatism Association (A3865) and the Novo Nordisk Foundation (A05990).
Dr. Ramsey-Goldman’s work was supported by the NIH (grants 5UL1TR001422–02, formerly 8UL1TR000150 and UL-1RR-025741, K24-AR-02318, and P60AR064464 formerly P60-AR-48098).
Dr. Mary Anne Dooley’s work was supported by the NIH grant RR00046.
Dr. Ruiz-Irastorza is supported by the Department of Education, Universities and Research of the Basque Government.
Dr Isenberg and Dr Rahman are supported by and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Center.
References
- 1.Hanly JG, Li Q, Su L, Urowitz MB, Gordon C, Bae SC, et al. Cerebrovascular Events in Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken). 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. [DOI] [PubMed] [Google Scholar]
- 3.Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmajuk G, Hoyer BF, Aringer M, Johnson SR, Daikh DI, Dorner T, et al. Multicenter Delphi Exercise to Identify Important Key Items for Classifying Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken). 2018;70(10):1488–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599–608. [DOI] [PubMed] [Google Scholar]
- 6.Isenberg D, Ramsey-Goldman R. Systemic Lupus International Collaborating Group--onwards and upwards? Lupus. 2006;15(9):606–7. [DOI] [PubMed] [Google Scholar]
- 7.Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–91. [PubMed] [Google Scholar]
- 8.Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39(3):363–9. [DOI] [PubMed] [Google Scholar]
- 9.Hanly JG, Urowitz MB, Sanchez-Guerrero J, Bae SC, Gordon C, Wallace DJ, et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis Rheum. 2007;56(1):265–73. [DOI] [PubMed] [Google Scholar]
- 10.Hanly JG, Urowitz MB, Su L, Sanchez-Guerrero J, Bae SC, Gordon C, et al. Short-term outcome of neuropsychiatric events in systemic lupus erythematosus upon enrollment into an international inception cohort study. Arthritis Rheum. 2008;59(5):721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ainiala H, Hietaharju A, Loukkola J, Peltola J, Korpela M, Metsanoja R, et al. Validity of the new American College of Rheumatology criteria for neuropsychiatric lupus syndromes: a population-based evaluation. Arthritis Rheum. 2001;45(5):419–23. [DOI] [PubMed] [Google Scholar]
- 12.Hanly JG, Urowitz MB, Jackson D, Bae SC, Gordon C, Wallace DJ, et al. SF-36 summary and subscale scores are reliable outcomes of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(6):961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thumboo J, Fong KY, Ng TP, Leong KH, Feng PH, Thio ST, et al. Validation of the MOS SF-36 for quality of life assessment of patients with systemic lupus erythematosus in Singapore. J Rheumatol. 1999;26(1):97–102. [PubMed] [Google Scholar]
- 14.Merrill JT, Zhang HW, Shen C, Butman BT, Jeffries EP, Lahita RG, et al. Enhancement of protein S anticoagulant function by beta2-glycoprotein I, a major target antigen of antiphospholipid antibodies: beta2-glycoprotein I interferes with binding of protein S to its plasma inhibitor, C4b-binding protein. Thromb Haemost. 1999;81(5):748–57. [PubMed] [Google Scholar]
- 15.Merrill JT, Shen C, Gugnani M, Lahita RG, Mongey AB. High prevalence of antiphospholipid antibodies in patients taking procainamide. J Rheumatol. 1997;24(6):1083–8. [PubMed] [Google Scholar]
- 16.Erkan D, Zhang HW, Shriky RC, Merrill JT. Dual antibody reactivity to beta2-glycoprotein I and protein S: increased association with thrombotic events in the antiphospholipid syndrome. Lupus. 2002;11(4):215–20. [DOI] [PubMed] [Google Scholar]
- 17.Hanly JG, Urowitz MB, Siannis F, Farewell V, Gordon C, Bae SC, et al. Autoantibodies and neuropsychiatric events at the time of systemic lupus erythematosus diagnosis: results from an international inception cohort study. Arthritis Rheum. 2008;58(3):843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Florica B, Aghdassi E, Su J, Gladman DD, Urowitz MB, Fortin PR. Peripheral neuropathy in patients with systemic lupus erythematosus. Semin Arthritis Rheum. 2011;41(2):203–11. [DOI] [PubMed] [Google Scholar]
- 19.Oomatia A, Fang H, Petri M, Birnbaum J. Peripheral neuropathies in systemic lupus erythematosus: clinical features, disease associations, and immunologic characteristics evaluated over a twenty-five-year study period. Arthritis Rheumatol. 2014;66(4):1000–9. [DOI] [PubMed] [Google Scholar]
- 20.Toledano P, Orueta R, Rodriguez-Pinto I, Valls-Sole J, Cervera R, Espinosa G. Peripheral nervous system involvement in systemic lupus erythematosus: Prevalence, clinical and immunological characteristics, treatment and outcome of a large cohort from a single centre. Autoimmun Rev. 2017;16(7):750–5. [DOI] [PubMed] [Google Scholar]
- 21.Bortoluzzi A, Piga M, Silvagni E, Chessa E, Mathieu A, Govoni M. Peripheral nervous system involvement in systemic lupus erythematosus: a retrospective study on prevalence, associated factors and outcome. Lupus. 2019;28(4):465–74. [DOI] [PubMed] [Google Scholar]
- 22.Hanly JG, Urowitz MB, Su L, Gordon C, Bae SC, Sanchez-Guerrero J, et al. Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Ann Rheum Dis. 2012;71(9):1502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanly JG, Li Q, Su L, Urowitz MB, Gordon C, Bae SC, et al. Cerebrovascular Events in Systemic Lupus Erythematosus: Results From an International Inception Cohort Study. Arthritis Care Res (Hoboken). 2018;70(10):1478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanly JG, O’Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, et al. The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatology (Oxford). 2016;55(2):252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaber W, Ezzat Y, El Fayoumy NM, Helmy H, Mohey AM. Detection of asymptomatic cranial neuropathies in patients with systemic lupus erythematosus and their relation to antiribosomal P antibody levels and disease activity. Clin Rheumatol. 2014;33(10):1459–66. [DOI] [PubMed] [Google Scholar]
- 26.Ubogu EE, Zaidat OO, Suarez JI. Acute motor-sensory axonal neuropathy associated with active systemic lupus erythematosus and anticardiolipin antibodies. J Clin Rheumatol. 2001;7(5):326–31. [DOI] [PubMed] [Google Scholar]
- 27.Golder V, Ooi JJY, Antony AS, Ko T, Morton S, Kandane-Rathnayake R, et al. Discordance of patient and physician health status concerns in systemic lupus erythematosus. Lupus. 2018;27(3):501–6. [DOI] [PubMed] [Google Scholar]
- 28.Straub RH, Zeuner M, Lock G, Rath H, Hein R, Scholmerich J, et al. Autonomic and sensorimotor neuropathy in patients with systemic lupus erythematosus and systemic sclerosis. J Rheumatol. 1996;23(1):87–92. [PubMed] [Google Scholar]
- 29.Huynh C, Ho SL, Fong KY, Cheung RT, Mok CC, Lau CS. Peripheral neuropathy in systemic lupus erythematosus. J Clin Neurophysiol. 1999;16(2):164–8. [DOI] [PubMed] [Google Scholar]
- 30.Su YJ, Huang CR, Chang WN, Tsai NW, Kung CT, Lin WC, et al. The association between autoantibodies and peripheral neuropathy in lupus nephritis. Biomed Res Int. 2014;2014:524940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Labrador-Horrillo M, Martinez-Valle F, Gallardo E, Rojas-Garcia R, Ordi-Ros J, Vilardell M. Anti-ganglioside antibodies in patients with systemic lupus erythematosus and neurological manifestations. Lupus. 2012;21(6):611–5. [DOI] [PubMed] [Google Scholar]
- 32.Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol. 2006;63(3):401–4. [DOI] [PubMed] [Google Scholar]
- 33.Vina ER, Fang AJ, Wallace DJ, Weisman MH. Chronic inflammatory demyelinating polyneuropathy in patients with systemic lupus erythematosus: prognosis and outcome. Semin Arthritis Rheum. 2005;35(3):175–84. [DOI] [PubMed] [Google Scholar]