

Abstract
Combination therapy has emerged as an efficient way to deliver chemotherapeutics for treatment of glioblastoma. It provides collaborative approach of targeting cancer cells by acting via multiple mechanisms, thereby reducing drug resistance. However, the presence of impermeable blood brain barrier (BBB) restricts the delivery of chemotherapeutic drugs into the brain. To overcome this limitation, we designed a dual functionalized liposomes by modifying their surface with transferrin (Tf) and a cell penetrating peptide (CPP) for receptor and adsorptive mediated transcytosis, respectively. In this study, we used two different CPPs (based on physicochemical properties) and investigated the influence of insertion of CPP to Tf-liposomes on biocompatibility, cellular uptake, and transport across the BBB both in vitro and in vivo. The biodistribution profile of Tf-CPP liposomes showed more than 10 and 2.7 fold increase in doxorubicin and erlotinib accumulation in mice brain, respectively as compared to free drugs with no signs of toxicity.
Keywords: biodistribution, glioblastoma, combination therapy, in vitro brain tumor model, dual functionalized liposomes
Graphical Abstract
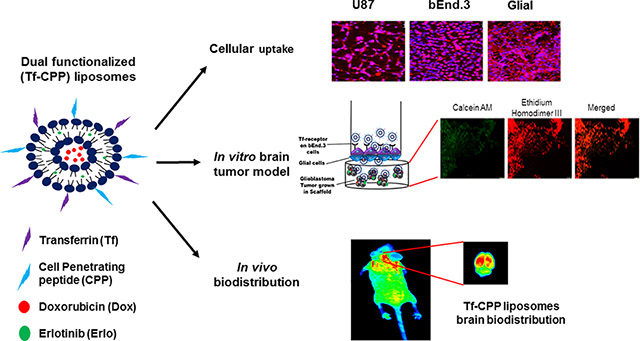
Blood brain barrier prevents the delivery of chemotherapeutics into brain. To overcome this issue, we designed a doxorubicin and erlotinib loaded dual functionalized liposomal delivery system, surface modified with transferrin (Tf) and a cell penetrating peptide to enhance their translocation across the BBB into glioblastoma tumor in brain via receptor mediated transcytosis and enhanced cell penetration, both in vitro and in vivo. Our results demonstrated several fold increase in the concentration of anticancer drugs across the co-culture endothelial barrier as well as in mice brain. Thus, we believe that this study would have high impact for treating patients with glioblastoma.
Background
The selective and impermeable nature of the blood brain barrier (BBB) prevent the delivery of anticancer therapeutics into the brain and therefore annul the systemic administration of such drugs for the treatment of glioblastoma multiforme (GBM). Glioblastoma is a brain tumor arises from astrocytes with poor prognosis. Due to its ability of rapid reproduction and infiltration into different parts of brain, which make GBM highly life threatening. The median survival time for patients with GBM is 12–15 months after diagnosis [1,2]. The conventional treatment options for the treatment of GBM includes surgical resections, radiotherapy or chemotherapy. However, these treatment options are not enough to treat GBM effectively. Chemotherapy is the most common treatment option for GBM. However, these chemotherapeutic drugs have undesirable toxic effects due to off targeting to healthy brain cells [3,4]. Therefore, there is a need to develop a non-invasive glioblastoma targeting delivery system which can efficiently translocate across the BBB and successfully deliver anticancer chemotherapeutics to the glioblastoma such as nanoparticles, exosomes, and focused ultrasound. Exosomes have been studied to deliver anticancer chemotherapeutics into brain, but possess some limitations including exosomes aggregation with cellular proteins, long time consuming procedure as well as require specialized equipment [5]. The focus ultrasound technique involves the disruption of the BBB, thereby allowing anticancer chemotherapeutics to cross the BBB. However, this technique is associated with the potential risk of skull heating, thermal coagulation, and formation of gas bubbles which can lead to unpredictable effects, from the BBB disruption to gross hemorrhage [6–9]. Considering the limitations of these non-invasive techniques we developed a dual functionalized nanoparticulate system without causing physiological disruption of the BBB.
There are many active targeting strategies associated with nanoparticles for the efficient delivery of chemotherapeutics across the BBB. The most common approach involves the receptor mediated transcytosis [10]. There are several receptors expressed on the surface of brain endothelial to facilitate the translocation of amino acid, glucose, or nucleic acid [11,12]. Therefore, this non-invasive active targeting can be exploited to deliver chemotherapeutics into the brain. Transferrin receptors (TfR) are expressed on brain endothelial cells as well as glioblastoma cells, which can used for active targeting to delivery chemotherapeutics to the glioblastoma [13–16]. However, receptor mediated active targeting is shown to be restricted due to the receptor saturation [17,18]. In addition, the endosomal entrapment of such delivery system by endosomes is another limitation in the effective delivery of nanocarriers [19,20]. Therefore, to overcome these limitations, another ligand such as cell penetrating peptide (CPP) needs to be incorporated to improve the delivery of the nanocarriers [19–23]. CPPs are short cationic peptide used to improve internalization of nanocarriers. The rationale of using two ligands is to overcome the abovementioned limitations by synergistically increase the translocation of nanocarriers across the BBB into the brain via dual mechanisms. In this study, we used two CPPs (TAT and QLPVM) based on their physicochemical properties. TAT is a cationic peptide with more hydrophilic amino acids residues and derived from the trans-activating protein of the HIV type-1 [24,25]. This peptide has been shown efficient delivery of cargoes from nanoparticles to proteins, peptides and nucleic acids [26]. The penta peptide QLPVM has more hydrophobic amino acid residues and derived from Bax-binding domain of Ku70 protein [27,28]. It has been studied for enhanced cell penetration and cell death inhibition properties [29]. The impact of this study was to design and develop a dual modified liposomal system, surface modified with transferrin (Tf) and a CPP (TAT or QLPVM) to efficient delivery of anticancer chemotherapeutics, doxorubicin (Dox) and erlotinib (Erlo) for the effective treatment of GBM.
The combination therapy is an effective treatment option for cancer which combines two or more chemotherapeutic agents. This therapy provides several advantages by targeting different mechanisms, reduce drug resistance and offer therapeutic benefits, thereby decreasing tumor growth and metastatic potential of cancer cells. Combination therapy shows synergistic approach by acting in a dual mechanism, thereby decreasing therapeutic dosage of individual drug. The co-delivery of Dox along with Erlo, an epidermal growth factor receptor inhibitor synergizes the anticancer effect by inducing the cytotoxicity in tumor cells through apoptosis with DNA damaging ability of Dox.
Due to complex pathology of brain tumor makes in vitro brain tumor model extremely difficult. Apart from the complexities associated, we designed a robust in vitro brain tumor model. The model comprised of 3-dimensionally grown tumor inside the porous scaffold which simulates in vivo like conditions where the liposomes are required to first translocate across the endothelial barrier before reaching and delivering drugs to the tumor site. In the present study, we determined the transport ability of CPP coupled Tf liposomes (Tf-TAT and Tf-QLPVM) across the in vitro brain tumor model as well as in vivo in mice. Based on the physiochemical properties of CPPs, we evaluated the ability of Tf-CPP to co-deliver chemotherapeutics in glioblastoma (U87), brain endothelial (bEnd.3) and glial cells. We performed the cytotoxicity and hemolysis studies to determine the biocompatibility of Tf-CPP liposomes for in vivo administration. We also assessed the biodistribution potential of Tf-CPP liposomes to co-deliver anticancer chemotherapeutics into mice brain.
Methods
The methods are explained in the supplementary materials in detail.
Animal experiments in mice
All animals were procured from the Jackson laboratory (Bar Harbor, ME). The experiments were performed using Male/female nude mice (nu/J; stock #002019) in accordance with the animal protocol # A17074 approved by the Institutional Animal Care and Use Committee (IACUC) at North Dakota State University. The animals were housed under controlled temperature conditions with 12 h dark/ 12 h light cycles with free access of water and food. The experiments were started after 7 days acclimation period.
In vivo biodistribution and biocompatibility of liposomes
To determine the biodistribution of liposomes, mice were randomly divided into groups. Each group was consisted of 6 mice including 3 males and 3 females. Each group of mice were intravenously injected via tail with either PBS, free Dox, Free Erlo, Dox and Erlo loaded plain, Tf, TAT, QLPVM, Tf-TAT, and Tf-QLPVM at a dose 15.2 μmoles/ kg of body weight. At predetermined interval of 24 h, mice were sacrificed and various organs (brain, lungs, heart, liver, kidney, and spleen) were harvested and blood samples were collected. The harvested organs were washed with PBS and stored at −80°C until assayed. Group injected with PBS was considered as control. The various organs were homogenized, followed by extraction of drugs in acetonitrile: methanol (9:1). Then, the drugs extracted sample was centrifuged at 10,000 rpm for 15 min at 4°C. Then, the supernatant was evaporated using vacuum evaporator. The residual drugs exacted sample was further reconstituted in Methanol: PBS pH 5.5 (1:1) and vortexed. The unwanted proteins were separated from the reconstituted sample was again centrifuged at 4°C for 15 min at 10,000. The quantitative estimation of drugs was performed using HPLC. The Dox analysis was done as per described in Dox loading and the quantitative estimation of Erlo distribution was performed with some modifications as per Erlo loading. The mobile was comprised of 0.2 M potassium phosphate buffer pH 3.0: acetonitrile (52:48) at a room temperature with a flow rate 0.6 ml/min [30]. All the data was normalized and represented as the percent injected dose per gram of the tissue (%ID/g). To determine the qualitative distribution, mice in each group were intravenously injected lissamine-rhodamine labeled liposomes at a dose of 15.2 μmoles/ kg of body weight through tail vein. Mice were sacrificed 24h post-injection and the whole body as well as ex-vivo fluorescent images were taken by Kodak in vivo imaging system FX (Carestream Health Inc., Rochester, NY). The images were acquired by setting rhodamine channel.
Various organs including brain, heart, lungs, liver, spleen, and kidneys were histologically evaluated to determine biocompatibility of liposomes post injection. After sacrificing mice, the organs were harvested and fixed in 10% neutralized buffer formalin. Paraffin sectioning of organs were done and stained with hematoxylin and eosin (H&E) staining. The tissue slides were observed for histopathological evaluation.
Results
Synthesis and characterization of Liposomes
The coupling of Tf and CPPs were performed to the terminal end of the DSPE-PEG(2000)-NHS via nucleophilic substitution reaction. The primary amine groups in Tf and CPPs formed stable amide bonds when reacted with the activated NHS ester group of PEG(2000)–DSPE in slightly alkaline pH 8– 9. More than 75 % of Tf and CPPs were coupled to PEG(2000)–DSPE. As depicted in Table 1, the mean particle size and zeta potential of all liposomal formulations were less than 200 nm and in near neutral range, respectively. The results demonstrated that the surface modification of liposomes with Tf and CPPs (TAT and QLPVM) did not significantly (p > 0.05) change in their particle sizes. The entrapment efficiencies of Dox and Erlo for all the liposomes were approximately 66% and 53%, respectively. As shown in Table 1, there is no significant (p > 0.05) difference in the entrapment efficiencies of both the drugs in all liposomal formulation regardless of surface modification.
Table 1.
Particle size distribution, polydispersity index, zeta potential and entrapment efficiency of various liposomal formulations
| Liposomes | Particle size (nm) | PDIa | Zeta Potential (mV) | Dox EEb (%) | Erlo EEb (%) |
|---|---|---|---|---|---|
| Plain | 174.45 ± 10.24 | 0.173 ± 0.02 | 5.85 ± 2.37 | 64.80 ± 1.98 | 53.79 ± 1.48 |
| Tf | 177.45 ± 3.69 | 0.212 ± 0.02 | −6.50 ± 1.08 | 65.32 ± 2.67 | 53.84 ± 1.10 |
| TAT | 173.60 ± 1.57 | 0.259 ± 0.02 | 20.35 ± 2.06 | 66.91 ± 2.54 | 52.67 ± 2.28 |
| Tf-TAT | 174.90 ± 4.45 | 0.254 ± 0.03 | 15.03 ± 3.94 | 65.72 ± 3.57 | 54.61 ± 1.18 |
| QLPVM | 171.90 ± 2.45 | 0.197 ± 0.04 | 19.27 ± 4.66 | 65.06 ± 1.20 | 51.82 ± 1.46 |
| Tf-QLPVM | 175.57 ± 4.57 | 0.246 ± 0.02 | 14.87 ± 0.53 | 66.47 ± 1.87 | 53.37 ± 1.10 |
Polydispersity index (PDI).
Entrapment efficiency (EE). The data represented as mean ± SD, (n=4).
The data represented as mean ± SD, (n=4).
In vitro biocompatibility of liposomes
The determination of liposomes compatibility was accessed in all three cell lines (U87, bEnd.3 and glial cells) which observed to be non-toxic. The results showed the cell viability of more than 85% up to a phospholipid concentration of 200 nMoles, relative to the untreated control group (Figure 1). In case of all three cell lines, the viability decreased as the concentration of phospholipid increased regardless of cell types. The viability at 600 nMoles were observed to be less than 69% for U87, bEnd.3 and glial cells, respectively. At higher phospholipid concentration, the cell viabilities were observed to be lowered with Tf-QLPVM liposomes in all cell lines. This can be attributed in relation to the hydrophobic nature of QLPVM, which increases the interaction of this peptide with plasma membrane, resulting in increased membrane destabilization and permeabilization through binding to intracellular targets [31,32]. The cell viabilities of CPP liposomes were lower compared to Tf coupled, plain and Tf-CPP liposomes in all three cell lines. This is can be due to their higher cationic charge.
Figure 1.

In vitro cell viability plots of different phospholipid concentrations of various liposomes on (A) U87, (B) bEnd.3 and (C) Glial cells. Data represented as mean ± SD, (n=3).
Cellular uptake of liposomes
The cellular uptake of different liposomal formulations was evaluated quantitatively as well as qualitatively in three different cell lines following 2 h of incubation. The fluorescence images in Figure 2A, B &C show the uptake of lissamine rhodamine labeled liposomes in all three cell lines. Tf-CPP liposomes showed strong fluorescence pattern all through cytoplasm and nucleus in comparison to single ligand or plain liposomes. Moreover, Tf liposomes displayed higher uptake than CPP liposomes and plain liposomes. As shown in Figure 2D and E, both Tf-TAT and Tf-QLPVM liposomes showed more than 70 % of cellular uptake of Dox and Erlo in U87, bEnd.3 and glial cells. As compared to the cellular uptake of dual functionalized liposomes, the plain liposomes had only ~ 30 % of uptake, which demonstrated statistically significant (p < 0.05) difference. In addition, the dual functionalized liposomes exhibited significantly (p < 0.05) higher uptake as compared to single ligand liposomes. Thus, the results demonstrated the importance of dual mechanisms of uptake over single ligand.
Figure 2.

Cellular uptake of various liposomes. Fluorescence images (10X magnification) showed uptake of lissamine rhodamine labeled liposomes after 2 h incubation in (A) U87, (B) bEnd.3 and (C) Glial cells (red; excitation/emission wavelengths: 560/583 nm). Nuclei were stained with Hoechst 33342 (blue; excitation/emission wavelengths: 350/461 nm). The graph plots represent the cellular uptake plots of (D) Dox and (E) Erlo encapsulated various liposomes in U87, bEnd.3 and Glial cells after 2 h incubation. Data represented as mean ± SD, (n=4). Statistically significant (p < 0.05) differences are shown as (*) with plain liposomes, with (†) Dox, (#) Erlo, (‡) Tf-liposomes, (§) TAT-liposomes, and (¥) QLPVM liposomes.
Hemocompatibility assessment
The liposomes were prepared to be injected intravenously into mice, thus it is important to determine the hemolytic potential of the liposomes prior to in vivo administration. The hemolysis study evaluates the hemoglobin release after the nonspecific interactions between positively charged liposomes with negatively charged erythrocytes membrane upon damage. Up to 10% of hemolysis is considered as non-toxic and biocompatible. As shown in Figure 3A, the percent hemolysis increased with increasing in the phospholipid concentrations. Plain, Tf and Tf-CPP liposomes demonstrated less than 9% hemolysis up to a concentration of 600 nMoles of phospholipids. However, Tf-QLPVM liposomes showed significantly (p < 0.05) higher hemolysis at a concentration of 800 nMoles of phospholipids as compared to Tf-TAT liposomes. Therefore, Tf-CPP liposomes are non-toxic and biocompatible for in vivo administration.
Figure 3.

(A) The graph represnts the percent hemolytic activity of various liposomes on RBCs after 1 h incubation. Significant (p < 0.05) differences are shown as with ( *) plain liposomes, (†) TAT-liposomes, and (#) QLPVM-liposomes. (B) The histological sections of PLGA-chitosan scaffold show tumor cell proliferation at different time points (10X magnification). (C) The graphical plot demonstrates the TEER value for the co-culture (glial and endothelial cells) and endothelial monolayer model only. Statistically significant (p < 0.05) difference is shown as (*) with endothelial monolayer. Data represented as mean ± SD, (n=4). (D) The plot represents the percent transport of various liposomes loaded with doxorubicin across in vitro brain tumor model, over a period of 24 h. Statistically significant (p < 0.05) higher are demonstrated in comparison to to (*) free drug. Data represented as mean ± SD, (n=3).
3-dimensional tumor growth and co-culture endothelial barrier integrity
As depicted in Figure 3B, The PLGA-chitosan scaffold demonstrated excellent 3-dimensional U87 tumor growth with time, thereby displayed biocompatible and porous nature of the scaffold. The H&E staining images showed the presence of the pores, which make the scaffold compatible for attachment of U87 cells and leading the growth of tumor in a 3-dimensional microenvironment. The images showed the dense growth of tumor cells on day 21 of tumor inoculation. The 3-dimensional tumor culture scaffold was combined with the co-culture endothelial barrier insert carrying brain endothelial cells (bEnd.3) on the luminal side and glial cells on the abluminal side of the polyethylene terephthalate (PET) membrane of insert. The permeability and integrity of the co-culture endothelial barrier was evaluated by measuring the transendothelial electrical resistance (TEER). As compared to the co-culture endothelial barrier model, the endothelial monolayer model had TEER value of 121.44 ± 6.13 Ω cm2, which demonstrated significantly (p < 0.05) lower as compared to the co-culture endothelial barrier model (189.58 ± 7.47 Ω cm2) (Figure 3C). Thus, the co-culture endothelial model with high TEER value was used to quantify the transcytosis as well as in vitro anti-tumor efficacy of various liposomes.
Transport across the endothelial barrier
Liposomal transport across the co-culture endothelial barrier was evaluated using in vitro brain tumor model. The liposomal transport was studied in the presence of 10% FBS to mimic in vivo like conditions. The percent liposomal transport increased from 8.5% for TAT to 12.72% for Tf-TAT liposomes and from 7.72% for QLPVM to 11.23% for Tf-QLPVM liposomes (Figure 3D). Tf-TAT liposomes showed non-significantly higher transport as compared to Tf-QLPVM liposomes. Thus, the dual functionalized liposomes demonstrated significantly (p < 0.05) higher transport across the co-culture endothelial barrier in comparison to Tf or CPP liposomes which shows the significance of dual mechanisms of transport through receptor targeting and increased cell penetration.
In vitro anti-tumor efficacy of liposomes using in vitro brain tumor model
An in vitro brain tumor model was used to evaluate the anti-tumor efficacy of Tf-CPP liposomes. The tumor regression was determined by quantifying the percent tumor cells viability housed in the scaffold using MTT assay and further confirmed by performing live/dead cell staining. The in vitro brain tumor model was treated with various Dox and Erlo loaded liposomal formulations for 24 h on day 21 of tumor inoculation. On day 28th of tumor inoculation, the treated scaffolds were quantified to evaluate the percent tumor cell viability. The percent tumor cell viability was decreased from 75.64 ± 1.71 for Dox and Erlo loaded plain liposomes to 44.39 ± 1.49 and 44.87 ± 1.86 for Tf-TAT and Tf-QLPVM liposomes, respectively (Figure 4A). The co-delivery of Dox and Erlo to the tumor cells demonstrated the potential of combination therapy by increasing the anti-cancer effect from the delivered drugs, thereby significant decrease (p < 0.05) in the percent tumor cell viability. In addition, Tf-CPP liposomes revealed the excellent antitumor efficacy as compared to single ligand or plain liposomes. The Tf-TAT and Tf-QLPVM liposomes exhibited no significant (p > 0.05) difference in the percent tumor cell viability. The results revealed the efficient co-delivery of Dox and Erlo from Tf-CPP liposomes across the co-culture endothelial barrier resulted in tumor regression. The efficacy of Dox and Erlo loaded Tf-CPP liposomes was further confirmed by performing live/dead cell fluorescence staining on treated tumor housed scaffold. The treated scaffold fluorescence images revealed higher number of dead tumor cells (Figure 4B).
Figure 4.

In vitro anti-tumor efficacy of various liposomes. (A) The Graphical plot shows the percent tumor cell viability after treatment with various Dox and Erlo loaded liposomes using an in vitro brain tumor model for 24 h. Significant (p < 0.05) differences with (*) plain liposomes, (#) free Dox-Erlo, (‡) Tf-liposomes, (†) TAT-liposomes, and (¥) QLPVM liposomes were observed. Data represented as mean ± SD, (n=4). (B) The fluorescence images show tumor cell death in scaffold after treatment.
Biodistribution of liposomes
The biodistribution of various liposomes was performed after 24 h of intravenous administration in mice. For qualitative determination, mice were injected with lissamine rhodamine labeled liposomes and post 24 h of injection the whole mice body and various organs were imaged by an in vivo imaging system. As depicted in Figure 5A and B, the images revealed the strong fluorescent intensity in the mice brain injected with Tf-CPP liposomes as compared to single ligand or plain liposomes, thereby showed the accumulation of liposomes in the mice brain. The ex vivo images of brain further confirmed the accumulation of liposomes. However, a higher fluorescent intensity was observed also in liver and spleen. HPLC analysis was performed to determine the quantitative estimation of liposomes in various organs by homogenization of organs followed by extraction of drugs. The biodistribution of Tf-CPP liposomes demonstrated more than 10 and 2.7 fold increase in the accumulation of Dox and Erlo in mice brain, respectively which exhibit significantly (p < 0.05) higher as compared to free drugs administration (Figure 6A and B). However, Tf-TAT liposomes showed non-significant (p > 0.05) rapid accumulation in liver and spleen as compared to Tf and Tf-QLPVM liposomes.
Figure 5.

(A) In vivo fluorescence imaging of mice intravenously injected via tail with various lissamine rhodamine labeled liposomes at 24 h time point. (B) Ex-vivo fluorescence imaging of various organs isolated from mice after 24 h invtravenous injection.
Figure 6.

The plots demonstrating the biodistrubition of various Dox and Erlo loaded liposomes at 24 h time point after intravenous injection. (A) The biodistribution of Dox and (B) the biodistribution of Erlo. The data are expressed as percent injected dose (% ID)/gram of tissue; (mean ± SD; n = 6). Statistically significant (p < 0.05) differences with (#) plain liposomes and (*) free drugs were observed.
In vivo biocompatibility study
The biocompatibility study of liposomes was performed by histological examination of various tissue sections of mice organs (Figure 7). The cationic charge and possibility of non-specific interactions by CPPs have shown organ toxicities in previously published report [33]. The mice group administered PBS was considered as a control. From the histological images of tissue sections from mice after liposomal administration showed no signs of change in morphology. In addition, the tissue sections were observed for any evidence of necrosis, nuclei enlargement or inflammation as compared to control group. Mice were injected with a dose of 15.2 μmoles of phospholipid/kg of body weight showed no signs of toxicity or inflammation in any of the tissues. Thus, histological evaluation showed that Tf-CPP liposomes are non-toxic and biocompatible.
Figure 7.

Histological evaluation of various liposomes in different organ sections post injection. Mice injected with PBS were used as controls.
Discussion
The liposomes were prepared using thin film hydration method while dual functionalized liposomes were formulated via post-insertion method. The post-insertion method is a spontaneous process which helps in insertion of an active ligand into preformed liposomes [34]. In addition, in this process the PEG derivative interacts with the hydrophobic part of lipid membrane. The advantage of using this method of insertion of ligand is to ensure appropriate targeting efficiency, prevent the nonspecific interactions of ligands with liposomes, eliminate the possibility of degradation of encapsulated moiety and stabilize the conformation of large targeting proteins [35–38]. The PEGylation of liposomes not only improves their stability but also increases their mean residence time in the body including in the brain. The addition of PEG act as a steric stabilizer, thereby reducing aggregation of liposomes and improving their stability. The PEG forms a hydrophilic layer on liposomes and reduces their protein absorption as well as nonspecific interactions with macrophages which prevents their removal from the body by macrophage system [39,40]. The coupling of ligand to the terminal end of PEG chains prevents the interference in the binding and internalization of liposomes to tumor cells, thereby maintaining the properties [41,42]. The surface charge of CPP coupled liposomes was positive, while the incorporation of transferrin to the CPP coupled liposomes counter balanced the positive charge of CPPs and imparted near neutral characteristics to Tf-CPP liposomes. The overall surface charge of the Tf-coupled liposomes was negative, which is attributed to the presence of negative charge of transferrin protein.
Due to strong hydrophobic nature, erlotinib is encapsulated in the phospholipid bilayer. However, the hydrophilic doxorubicin was encapsulated into liposomes using pH gradient method. The dried lipid film was hydrated with 300 mM citric acid pH 5.0. The external pH of liposomes was changed using 300 mM sodium carbonate to create pH gradient. The inner acidic buffer protonates Dox intra-liposomally, thereby helping diffusion of unionized Dox to the core of the liposomes from outside.
Tf-CPP liposomes demonstrated excellent biocompatibility upto 200 nMoles of phospholipid concentration. Tf-TAT liposomes showed higher cell viability (non-significantly) as compared to Tf-QLPVM liposomes. This can be explained by the greater interaction of hydrophobic amino acid residues with plasma membrane present in QLPVM which led to membrane destabilization and permeabilization via binding to intracellular targets [31,32]. Due to higher cationic charge of CPP, the cell viabilities of CPP liposomes were lower compared to Plain, Tf and Tf-CPP liposomes.
The higher uptake of Tf-CPP liposomes is attributed by dual mechanisms of receptor targeting and improved cell penetration. In addition, the positively charged CPP liposomes interacted electrostatically with negatively charged membrane and facilitated the uptake of liposomes through adsorptive mediated transcytosis [43]. Therefore, the rapid and higher uptake of Tf-CPP liposomes demonstrated synergistic effect of interacting with cell membrane followed by binding of Tf to Tf receptor, thereby leading to increase in cellular uptake in all three cell lines.
The cationic macromolecules and cell penetrating peptides may induce nonspecific interactions which can interfere with the erythrocytes’ membrane integrity and cause hemolysis [22,44]. Also, such nonspecific interactions may affect the targeting ability, half-life and reproducibility of delivery system [44,45]. In addition, cationic macromolecules may induce various adverse effects including thrombosis and embolization. Up to 600 nMoles of phospholipids concentration, Tf-CPP liposomes showed excellent hemocompatibility. However, at higher phospholipid concentration, Tf-QLPVM showed slight hemolysis. This can be attributed due to the presence of more hydrophobic amino acid residues in QLPVM, thereby increasing its interaction with lipophilic erythrocytes’ membrane [46]. At high phospholipid concentration, Tf liposomes demonstrated higher percent hemolysis as compared at low phospholipid concentration. This is due to the Tf protein aggregation at high concentration, thereby leading to nonspecific interactions of Tf aggregates with erythrocytes and resulting in destabilization of membrane as compared to decreased interactions of Tf and erythrocytes’ membrane [12].
The high TEER value of the co-culture endothelial barrier (bEnd.3 and glial cells) attribute to the formation of tight barrier and this can be explained due to the localization and upregulation of junctional proteins around cell borders as well as the physical strength by the glial cells [22,47,48]. Moreover, the absence of glial cells, in the endothelial monolayer attributed to the formation of loose barrier as compared to the co-culture endothelial barrier because of lack of initiation and maintenance of barrier properties. Therefore, the co-culture endothelial barrier was use to study transport as well as anti-tumor efficacy of Tf-CPP liposomes.
The in vitro brain tumor model was designed by placing the co-culture endothelial barrier on tumor grown scaffold. This model mimics in vivo like tumor environment. The advantage of this model is that the liposomal formulation before reaching to the tumor cells inside scaffold, first has to translocate through the co-culture endothelial barrier. The liposomal transport across the co-culture endothelial barrier was evaluated using in vitro brain tumor model in the presence of 10% FBS to mimic in vivo like conditions. Tf-CPP liposomes demonstrated significantly (p < 0.05) higher transport (more than 11%) across the co-culture endothelial barrier as compared to single ligand or plain liposomes. Tf-TAT liposomes showed non-significantly higher transport as compared to Tf-QLPVM liposomes. This can be explained by the presence of higher positive charge of TAT which demonstrated strong electrostatic interaction with the cell membrane, thereby increasing the endothelial transport of Tf-TAT liposomes. CPP liposomes showed the transport through adsorptive mediated transcytosis while Tf liposomes were transported via receptor mediated transcytosis. The initial binding of CPP liposomes can be interfered by the presence of serum protein. However, Tf liposomes demonstrated specific binding to its receptor, thereby eliminating nonspecific interactions with serum protein. Moreover, the lower transport of plain and CPP liposomes is attributed due to the entrapment in endothelial cell layer and absence of dual mechanisms. Therefore, Tf-CPP liposomes showed higher transport and highlighting the importance of dual mechanisms of receptor and adsorptive mediated transcytosis across the co-culture endothelial barrier. In vitro anti-tumor efficacy of liposomes were evaluated using in vitro brain tumor model by quantifying the percent tumor cell viability of U87 cells housed in the porous PLGA-chitosan scaffold. The co-delivery of Dox and Erlo to the tumor cells demonstrated the potential of combination therapy by increasing the anti-tumor effect from the delivered drugs, thereby significant decrease (p < 0.05) in the percent tumor cell viability. Tf-CPP efficiently translocated across the co-culture endothelial barrier, followed by endocytosis into the U87 cells in the scaffold and co-delivered Dox and Erlo to tumor cells which resulted in tumor regression.
Due to complexity of in vivo environment, we studied the biodistribution of liposomes in mice and determined the ability of Tf-CPP liposomes in translocation across the BBB into the brain. The fluorescent images showed the strong fluorescent intensity in the mice brain injected with Tf-CPP liposomes, thereby showing the accumulation of liposomes in the mice brain after 24 h. The ex vivo images of brain further confirmed the accumulation of liposomes. However, a higher fluorescent intensity was observed also in liver and spleen. The incorporation of CPP to the Tf-liposomes significantly increased the accumulation of Dox and Erlo into the brain as compared to single ligand or plain liposomes. However, Tf-TAT liposomes showed non-significant (p > 0.05) rapid accumulation in liver and spleen as compared to Tf-QLPVM liposomes. This is due to higher cationic charge on the liposomes which could have triggered non-specific interactions and rapidly captured by the phagocytic cells, thereby resulting in accumulation in the major macrophage organs. Tf-liposomes penetration into brain was significantly (p < 0.05) lesser than Tf-CPP liposomes. This could be due to Tf receptor saturation which probably restricted the further translocation of liposomes into the brain. In addition, the presence of Tf receptors in liver, spleen and heart resulted in higher uptake of Tf-liposomes in these organs [49,50]. Plain liposomes majorly transported to liver, spleen and lungs, thereby showing significantly (p < 0.05) lesser uptake as compared to Tf-CPP liposomes across the BBB. Tf-QLPVM liposomes showed greater transport to the lungs as compared to Tf-TAT liposomes. This can be explained due to the greater interaction of these liposomes with erythrocytes. Pharmacokinetic characteristics of both the drugs might cause differences in blood distribution. Several studies have been published demonstrating the pharmacokinetic profiles of erlotinib and doxorubicin [51–53]. The elimination half-life of erlotinib (5.2 h) is shorter than doxorubicin (10 h) [52,53]. In addition, He et al. reported lower AUC (0.6 μg*h/ml) and Cmax (0.4 μg/ml) for erlotinib as compared to AUC (6 μg*h/ml) and Cmax (2.3 μg/ml) for doxorubicin [51]. However, higher clearance (3 ml/h) for erlotinib as compared to clearance (0.7 ml/h) for doxorubicin [51]. Therefore, the shorter half-life and higher clearance resulted in rapid elimination of erlotinib from the system. The accumulation of TAT and QLPVM liposomes was majorly seen in liver and spleen due to the cationic charge and non-specific interactions. Therefore, the incorporation of CPP to the Tf-liposomes demonstrated higher translocation across the BBB and higher accumulation into brain via dual mechanisms of receptor and adsorptive mediated transcytosis.
Mice were injected with liposomal dose of 15.2 μmoles of phospholipid/kg of body weight which was based on the results obtained from the in vitro cytotoxicity and hemolysis studies. The tissue sections were observed for any evidence of necrosis, nuclei enlargement or inflammation as compared to control group. The liver section was carefully observed for any signs of ballooning of hepatocytes or enlargement of nuclei and spleen was examined for signs of necrosis, which confirmed no evidence of necrosis in the tissue section. Heart was also examined for any evidence of myofibrillar loss or diffuse fibrosis of myocardium. Tissue sections from brain, kidneys and lungs were also carefully examined and showed no evidence of any type of toxicities. Therefore, Tf-CPP liposomes showed no signs of toxicity or inflammation in any of the tissues. In summary, Tf-TAT and Tf-QLPVM liposomes are non-toxic and biocompatible formulations resulting in high translocation of chemotherapeutics into brain. Therefore, we believe these dual functionalized liposomes exhibit great potential in delivering chemotherapeutics across the BBB into brain in a safe and efficient way, thereby benefiting the patients with glioblastoma.
Supplementary Material
Acknowledgements
The work was supported by National Institutes of Health (NIH) grant RO1 AG051574 and National Science Foundation (NSF) under North Dakota Established Program to Stimulate Competitive Research (ND EPSCoR) Track 1 Cooperative OIA-1355466.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS, Breast Cancer Metastasis to the Central Nervous System, Am. J. Pathol 167 (2005) 913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Johnson DR, O’Neill BP, Glioblastoma survival in the United States before and during the temozolomide era., J. Neurooncol 107 (2012) 359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- [3].Lichter AS, Lawrence TS, Recent Advances in Radiation Oncology, N. Engl. J. Med 332 (1995) 371–379. doi: 10.1056/NEJM199502093320607. [DOI] [PubMed] [Google Scholar]
- [4].Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R, Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol 2 (2007) 751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [5].Gomari H, Forouzandeh Moghadam M, Soleimani M, Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle, Onco. Targets. Ther 11 (2018) 5753–5762. doi: 10.2147/OTT.S173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vykhodtseva NI, Hynynen K, Damianou C, Histologic effects of high intensity pulsed ultrasound exposure with subharmonic emission in rabbit brain in vivo, Ultrasound Med. Biol 21 (1995) 969–979. doi: 10.1016/0301-5629(95)00038-S. [DOI] [PubMed] [Google Scholar]
- [7].Vykhodtseva N, McDannold N, Hynynen K, Progress and problems in the application of focused ultrasound for blood-brain barrier disruption, Ultrasonics. 48 (2008) 279–296. doi: 10.1016/j.ultras.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lin Y-J, Chen K-T, Huang C-Y, Wei K-C, Non-invasive focused ultrasound-based synergistic treatment of brain tumors, J. Cancer Res. Pract. 3 (2016) 63–68. doi: 10.1016/j.jcrpr.2016.05.001. [DOI] [Google Scholar]
- [9].Dong X, Current Strategies for Brain Drug Delivery, Theranostics. 8 (2018) 1481–1493. doi: 10.7150/thno.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wong HL, Wu XY, Bendayan R, Nanotechnological advances for the delivery of CNS therapeutics., Adv. Drug Deliv. Rev 64 (2012) 686–700. doi: 10.1016/j.addr.2011.10.007. [DOI] [PubMed] [Google Scholar]
- [11].Qian ZM, Li H, Sun H, Ho K, Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway., Pharmacol. Rev 54 (2002) 561–587. [DOI] [PubMed] [Google Scholar]
- [12].Sharma G, Modgil A, Layek B, Arora K, Sun C, Law B, Singh J, Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: Biodistribution and transfection, J. Control. Release 167 (2013) 1–10. doi: 10.1016/j.jconrel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- [13].Sharma G, Lakkadwala S, Modgil A, Singh J, The Role of Cell-Penetrating Peptide and Transferrin on Enhanced Delivery of Drug to Brain, Int. J. Mol. Sci 17 (2016) 806. doi: 10.3390/ijms17060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Prabhakar K, Afzal SM, Kumar PU, Rajanna A, Kishan V, Brain delivery of transferrin coupled indinavir submicron lipid emulsions-Pharmacokinetics and tissue distribution, Colloids Surfaces B Biointerfaces. 86 (2011) 305–313. doi: 10.1016/j.colsurfb.2011.04.013. [DOI] [PubMed] [Google Scholar]
- [15].Skarlatos S, Yoshikawa T, Pardridge WM, Transport of [125I]transferrin through the rat blood-brain barrier., Brain Res. 683 (1995) 164–171. [DOI] [PubMed] [Google Scholar]
- [16].Zhu X, Zhou H, Liu Y, Wen Y, Wei C, Yu Q, Liu J, Transferrin/aptamer conjugated mesoporous ruthenium nanosystem for redox-controlled and targeted chemo-photodynamic therapy of glioma, Acta Biomater. 82 (2018) 143–157. doi: 10.1016/j.actbio.2018.10.012. [DOI] [PubMed] [Google Scholar]
- [17].Sharma G, Modgil A, Sun C, Singh J, Grafting of cell-penetrating peptide to receptor-targeted liposomes improves their transfection efficiency and transport across blood-brain barrier model, J. Pharm. Sci 101 (2012) 2468–2478. doi: 10.1002/jps.23152. [DOI] [PubMed] [Google Scholar]
- [18].Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H, Dual-ligand modification of PEGylated liposomes shows better cell selectivity and efficient gene delivery, J. Control. Release 153 (2011) 141–148. doi: 10.1016/j.jconrel.2011.03.012. [DOI] [PubMed] [Google Scholar]
- [19].Zong T, Mei L, Gao H, Cai W, Zhu P, Shi K, Chen J, Wang Y, Gao F, He Q, Synergistic dual-ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals, Mol. Pharm 11 (2014) 2346–2357. doi: 10.1021/mp500057n. [DOI] [PubMed] [Google Scholar]
- [20].Bolhassani A, Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer, Biochim. Biophys. Acta - Rev. Cancer 1816 (2011) 232–246. doi: 10.1016/j.bbcan.2011.07.006. [DOI] [PubMed] [Google Scholar]
- [21].Lakkadwala S, Singh J, Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model, Colloids Surfaces B Biointerfaces. 173 (2019) 27–35. doi: 10.1016/j.colsurfb.2018.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lakkadwala S, Singh J, Dual Functionalized 5-Fluorouracil Liposomes as Highly Efficient Nanomedicine for Glioblastoma Treatment as Assessed in an In Vitro Brain Tumor Model, J. Pharm. Sci 107 (2018) 2902–2913. doi: 10.1016/j.xphs.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tian Y, Mi G, Chen Q, Chaurasiya B, Li Y, Shi D, Zhang Y, Webster TJ, Sun C, Shen Y, Acid-Induced Activated Cell-Penetrating Peptide-Modified Cholesterol-Conjugated Polyoxyethylene Sorbitol Oleate Mixed Micelles for pH-Triggered Drug Release and Efficient Brain Tumor Targeting Based on a Charge Reversal Mechanism, ACS Appl. Mater. Interfaces 10 (2018) 43411–43428. doi: 10.1021/acsami.8b15147. [DOI] [PubMed] [Google Scholar]
- [24].Frankel AD, Pabo CO, Cellular uptake of the tat protein from human immunodeficiency virus., Cell. 55 (1988) 1189–1193. [DOI] [PubMed] [Google Scholar]
- [25].Green M, Loewenstein PM, Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein., Cell. 55 (1988) 1179–1188. [DOI] [PubMed] [Google Scholar]
- [26].Zorko M, Langel U, Cell-penetrating peptides: mechanism and kinetics of cargo delivery., Adv. Drug Deliv. Rev 57 (2005) 529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- [27].Gomez JA, Gama V, Yoshida T, Sun W, Hayes P, Leskov K, Boothman D, Matsuyama S, Bax-inhibiting peptides derived from Ku70 and cell-penetrating pentapeptides., Biochem. Soc. Trans 35 (2007) 797–801. doi: 10.1042/BST0350797. [DOI] [PubMed] [Google Scholar]
- [28].Yoshida T, Tomioka I, Nagahara T, Holyst T, Sawada M, Hayes P, Gama V, Okuno M, Chen Y, Abe Y, Kanouchi T, Sasada H, Wang D, Yokota T, Sato E, Matsuyama S, Bax-inhibiting peptide derived from mouse and rat Ku70., Biochem. Biophys. Res. Commun 321 (2004) 961–966. doi: 10.1016/j.bbrc.2004.07.054. [DOI] [PubMed] [Google Scholar]
- [29].Gomez JA, Chen J, Ngo J, Hajkova D, Yeh I-J, Gama V, Miyagi M, Matsuyama S, Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity., Pharmaceuticals (Basel) 3 (2010) 3594–3613. doi: 10.3390/ph3123594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lakkadwala S, dos Santos Rodrigues B, Sun C, Singh J, Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma, J. Control. Release 307 (2019) 247–260. doi: 10.1016/j.jconrel.2019.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].El-Andaloussi S, Jarver P, Johansson HJ, Langel U, Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study., Biochem. J 407 (2007) 285–292. doi: 10.1042/BJ20070507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saar K, Lindgren M, Hansen M, Eiríksdóttir E, Jiang Y, Rosenthal-Aizman K, Sassian M, Langel Ü, Cell-penetrating peptides: A comparative membrane toxicity study, Anal. Biochem 345 (2005) 55–65. doi: 10.1016/j.ab.2005.07.033. [DOI] [PubMed] [Google Scholar]
- [33].Ramana LN, Sethuraman S, Ranga U, Krishnan UM, Development of a liposomal nanodelivery system for nevirapine., J. Biomed. Sci 17 (2010) 57. doi: 10.1186/1423-0127-17-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nag O, Awasthi V, Surface Engineering of Liposomes for Stealth Behavior, Pharmaceutics. 5 (2013) 542–569. doi: 10.3390/pharmaceutics5040542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Iden DL, Allen TM, In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach., Biochim. Biophys. Acta 1513 (2001) 207–216. [DOI] [PubMed] [Google Scholar]
- [36].Moreira JN, Ishida T, Gaspar R, Allen TM, Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity., Pharm. Res. 19 (2002) 265–269. [DOI] [PubMed] [Google Scholar]
- [37].Torchilin VP, Khaw BA, Smirnov VN, Haber E, Preservation of antimyosin antibody activity after covalent coupling to liposomes, Biochem. Biophys. Res. Commun 89 (1979) 1114–1119. doi: 10.1016/0006-291X(79)92123-5. [DOI] [PubMed] [Google Scholar]
- [38].Marqués-Gallego P, De Kroon AIPM, Ligation strategies for targeting liposomal nanocarriers, Biomed Res. Int 2014 (2014) 12. doi: 10.1155/2014/129458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].(Chezy) Barenholz Y, Doxil® — The first FDA-approved nano-drug: Lessons learned, J. Control. Release 160 (2012) 117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- [40].Chen Y, Liu L, Modern methods for delivery of drugs across the blood-brain barrier, Adv. Drug Deliv. Rev 64 (2012) 640–665. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- [41].Zhao B-X, Zhao Y, Huang Y, Luo L-M, Song P, Wang X, Chen S, Yu K-F, Zhang X, Zhang Q, The efficiency of tumor-specific pH-responsive peptide-modified polymeric micelles containing paclitaxel., Biomaterials. 33 (2012) 2508–2520. doi: 10.1016/j.biomaterials.2011.11.078. [DOI] [PubMed] [Google Scholar]
- [42].Wang Z, Yu Y, Dai W, Lu J, Cui J, Wu H, Yuan L, Zhang H, Wang X, Wang J, Zhang X, Zhang Q, The use of a tumor metastasis targeting peptide to deliver doxorubicin-containing liposomes to highly metastatic cancer, Biomaterials. 33 (2012) 8451–8460. doi: 10.1016/j.biomaterials.2012.08.031. [DOI] [PubMed] [Google Scholar]
- [43].Trabulo S, Cardoso AL, Mano M, De Lima MCP, Cell-Penetrating Peptides-Mechanisms of Cellular Uptake and Generation of Delivery Systems, Pharmaceuticals (Basel). 3 (2010) 961–993. doi: 10.3390/ph3040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Antohi S, Brumfeld V, Polycation-cell surface interactions and plasma membrane compartments in mammals. Interference of oligocation with polycationic condensation., Zeitschrift Fur Naturforschung. Sect. C, Biosci 39 (1984) 767–775. [DOI] [PubMed] [Google Scholar]
- [45].Zhu S, Qian F, Zhang Y, Tang C, Yin C, Synthesis and characterization of PEG modified N-trimethylaminoethylmethacrylate chitosan nanoparticles, Eur. Polym. J 43 (2007) 2244–2253. doi: 10.1016/j.eurpolymj.2007.03.042. [DOI] [Google Scholar]
- [46].Sharma G, Modgil A, Zhong T, Sun C, Singh J, Influence of short-chain cell-penetrating peptides on transport of doxorubicin encapsulating receptor-targeted liposomes across brain endothelial barrier, Pharm. Res 31 (2014) 1194–1209. doi: 10.1007/s11095-013-1242-x. [DOI] [PubMed] [Google Scholar]
- [47].Janzer RC, Raff MC, Astrocytes induce blood–brain barrier properties in endothelial cells, Nature. 325 (1987) 253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- [48].Arthur FE, Shivers RR, Bowman PD, Astrocyte-mediated induction of tight junctions in brain capillary endothelium: an efficient in vitro model, Dev. Brain Res. 36 (1987) 155–159. doi: 10.1016/0165-3806(87)90075-7. [DOI] [PubMed] [Google Scholar]
- [49].Deaglio S, Capobianco A, Cali A, Bellora F, Alberti F, Righi L, Sapino A, Camaschella C, Malavasi F, Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and a polyclonal antiserum., Blood. 100 (2002) 3782–3789. doi: 10.1182/blood-2002-01-0076. [DOI] [PubMed] [Google Scholar]
- [50].Jefferies WA, Brandon MR, V Hunt S, Williams AF, Gatter KC, Mason DY, Transferrin receptor on endothelium of brain capillaries., Nature. 312 (1984) 162–163. [DOI] [PubMed] [Google Scholar]
- [51].He Y, Su Z, Xue L, Xu H, Zhang C, Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy, J. Control. Release 229 (2016) 80–92. doi: 10.1016/j.jconrel.2016.03.001. [DOI] [PubMed] [Google Scholar]
- [52].Gao Y, Shen JK, Choy E, Zhang Z, Mankin HJ, Hornicek FJ, Duan Z, Pharmacokinetics and tolerability of NSC23925b, a novel P-glycoprotein inhibitor: preclinical study in mice and rats, Sci. Rep 6 (2016) 25659. doi: 10.1038/srep25659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Meany HJ, Fox E, McCully C, Tucker C, Balis FM, The plasma and cerebrospinal fluid pharmacokinetics of erlotinib and its active metabolite (OSI-420) after intravenous administration of erlotinib in non-human primates., Cancer Chemother. Pharmacol. 62 (2008) 387–392. doi: 10.1007/s00280-007-0616-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.