

Variation in GBA confers a spectrum of increased risk for Parkinson’s disease. Blauwendraat et al. report that an individual’s overall Parkinson’s disease genetic risk score modifies the effect of GBA variants on disease risk and age at onset. Genes implicated in lysosomal function exert the most influence on GBA-associated risk.
Keywords: GBA, SNCA, CTSB, Parkinson’s disease, modifiers
Abstract
Parkinson’s disease is a genetically complex disorder. Multiple genes have been shown to contribute to the risk of Parkinson’s disease, and currently 90 independent risk variants have been identified by genome-wide association studies. Thus far, a number of genes (including SNCA, LRRK2, and GBA) have been shown to contain variability across a spectrum of frequency and effect, from rare, highly penetrant variants to common risk alleles with small effect sizes. Variants in GBA, encoding the enzyme glucocerebrosidase, are associated with Lewy body diseases such as Parkinson’s disease and Lewy body dementia. These variants, which reduce or abolish enzymatic activity, confer a spectrum of disease risk, from 1.4- to >10-fold. An outstanding question in the field is what other genetic factors that influence GBA-associated risk for disease, and whether these overlap with known Parkinson’s disease risk variants. Using multiple, large case-control datasets, totalling 217 165 individuals (22 757 Parkinson’s disease cases, 13 431 Parkinson’s disease proxy cases, 622 Lewy body dementia cases and 180 355 controls), we identified 1691 Parkinson’s disease cases, 81 Lewy body dementia cases, 711 proxy cases and 7624 controls with a GBA variant (p.E326K, p.T369M or p.N370S). We performed a genome-wide association study and analysed the most recent Parkinson’s disease-associated genetic risk score to detect genetic influences on GBA risk and age at onset. We attempted to replicate our findings in two independent datasets, including the personal genetics company 23andMe, Inc. and whole-genome sequencing data. Our analysis showed that the overall Parkinson’s disease genetic risk score modifies risk for disease and decreases age at onset in carriers of GBA variants. Notably, this effect was consistent across all tested GBA risk variants. Dissecting this signal demonstrated that variants in close proximity to SNCA and CTSB (encoding cathepsin B) are the most significant contributors. Risk variants in the CTSB locus were identified to decrease mRNA expression of CTSB. Additional analyses suggest a possible genetic interaction between GBA and CTSB and GBA p.N370S induced pluripotent cell-derived neurons were shown to have decreased cathepsin B expression compared to controls. These data provide a genetic basis for modification of GBA-associated Parkinson’s disease risk and age at onset, although the total contribution of common genetics variants is not large. We further demonstrate that common variability at genes implicated in lysosomal function exerts the largest effect on GBA associated risk for disease. Further, these results have implications for selection of GBA carriers for therapeutic interventions.
Introduction
Heterozygous functional GBA variants are one of the most common genetic risk factors for Parkinson’s disease and Lewy body dementia (LBD), found in 3–20% of patients in different populations (Lesage et al., 2011; Gan-Or et al., 2015b; Blauwendraat et al., 2018a; Guerreiro et al., 2018; Rivas et al., 2018). In a homozygous or compound heterozygous state, GBA variants may cause Gaucher disease, an autosomal recessive lysosomal storage disorder. GBA encodes the lysosomal enzyme glucocerebrosidase (GCase), and it is hypothesized that loss of GCase activity leads to a reduced ability to degrade α-synuclein, encoded by SNCA. Aggregated, misfolded α-synuclein protein deposits in the form of Lewy bodies and Lewy neurites are the pathological hallmarks of Parkinson’s disease and LBD.
Notably, GBA variants that do not cause Gaucher disease but do confer increased risk for Parkinson’s disease and LBD have been identified. It is hypothesized that while these variants result in reduced GCase activity, the activity is not low enough to cause Gaucher disease. Multiple rare GBA variants have been described in Parkinson’s disease in different populations. More common variants include p.E326K, p.T369M, p.N370S and p.L444P, whose frequencies vary with ethnicity and are each found on different haplotypes (Blauwendraat et al., 2018a; Leija-Salazar et al., 2019). In some ethnicities, such as the Ashkenazi Jewish population, certain GBA variants are found in about 5% of unaffected individuals, and 17–20% of Parkinson’s disease patients (Gan-Or et al., 2008; Sidransky et al., 2009; Ruskey et al., 2019).
In European populations, the p.E326K risk variant is found in ∼2% of the general population (https://gnomad.broadinstitute.org/). However, the majority of GBA variant carriers will not develop Parkinson’s disease, implying that there are other genetic and/or environmental factors that affect the penetrance of these variants. Studies that have examined the penetrance of GBA variants in carriers suggest it is age-related and is typically between 10% and 30% (Anheim et al., 2012; Rana et al., 2013). Furthermore, penetrance differs across GBA variants, with high-risk variants leading to earlier disease onset compared to lower risk variants (Gan-Or et al., 2015a). Interestingly, it was demonstrated that various GBA variants, including p.E326K, p.T369M and p.N370S have similar effects on GCase activity in humans, reducing it by 18–46% on average (Alcalay et al., 2015). Additionally, genotype-phenotype studies have shown that Parkinson’s disease patients with GBA variants have an earlier age at onset, faster disease progression, and higher rates of non-motor symptoms, such as rapid eye movement (REM) sleep behaviour disorder (RBD), autonomic dysfunction, hallucinations and cognitive decline, compared to those with non-GBA associated Parkinson’s disease (Gan-Or et al., 2018).
In recent years, GBA has become a prominent target for therapeutic development, and the first gene-specific phase 2 clinical trial in Parkinson’s disease is currently ongoing for GBA-positive Parkinson’s disease patients (ClinicalTrials.gov Identifier: NCT02906020). One of the concerns in performing such a trial is that, despite the randomization for treatment and placebo groups, these groups will remain unbalanced in terms of factors that affect their progression, which can be a significant confounder of trial outcome (Leonard et al., 2018). In addition, it is likely that future preventive clinical trials will also target populations that are at high risk for Lewy body diseases, such as GBA risk variant carriers with prodromal symptoms. Therefore, identifying factors that can affect the penetrance and clinical presentation of GBA-associated Parkinson’s disease will be crucial for trial design. Furthermore, modifiers of penetrance age at onset may also become drug targets.
In the current study, we gathered data from multiple sources, including the International Parkinson’s Disease Genomics Consortium (IPDGC) and the UK Biobank (UKB), to identify GBA variants in both cases and controls and used 23andMe and whole-genome sequencing data for further validation. Subsequently, we used genome-wide association studies (GWAS) and genetic risk scoring to identify genetic variants that modify the penetrance and age at onset of GBA-associated Parkinson’s disease. We then examined protein levels of the top hits in forebrain neurons differentiated from induced pluripotent stem cells of individuals with and without GBA variants.
Materials and methods
Genotyping data
International Parkinson Disease Genomics Consortium genotyping data
Genotyping data (all Illumina platform based) was obtained from IPDGC members, collaborators, and public resources (Supplementary Tables 1 and 2). All datasets underwent quality control separately, both on individual-level data and variant-level data before imputation as previously described (Nalls et al., 2018; Blauwendraat et al., 2019). Refer to the online Supplementary material for a detailed description of data processing.
For GWAS analyses, variants passing the following post-imputation criteria were included: R2 > 0.8, minor allele frequency > 0.05, and genotype missingness < 0.15 across subsets. Case-control GWAS were performed using RVTESTS with default settings (Zhan et al., 2016) using logistic regression with sex, age (if not available the mean age of all data were used), principal components (PC) 1–5, and dataset origin as covariates. age at onset GWAS was performed in GBA carriers only using RVTESTS linear regression with age at onset as a continuous phenotype and sex, PCs 1–5 and dataset origin as covariates. Cases without age information were excluded from the age at onset GWAS, and individuals with two GBA variants were excluded from all analyses to prevent bias (Supplementary Table 2).
Lewy body dementia genotyping data
LBD cases and controls were genotyped for ongoing projects at the Neurodegenerative Diseases Research Unit (NDRU) using the NeuroChip genotyping array (Illumina). Genotyping was performed as previously described (Blauwendraat et al., 2017). All cases met the McKeith criteria for probable or definite disease (McKeith, 2006). For more details on this dataset see Supplementary Table 2. Genotype quality control, processing, and analysis was performed similar to the above described IPDGC genotype data. For analyses, variants that passed the following post-imputation criteria were included: imputation R2 > 0.8 and minor allele frequency > 0.05. Case-control analyses were performed using RVTESTS using logistic regression on genotype dosages with sex, and PCs 1–5 as covariates. Age was not used since the majority of the individuals had only age of death information available.
McGill GBA genotyping data
Ashkenazi Jewish Parkinson’s disease cases were genotyped at McGill University using the Illumina Human OmniExpress Array and custom SNPs of the NeuroX array (Nalls et al., 2015a). Sanger sequencing or targeted next-generation sequencing confirmed GBA p.N370S status. For more details on this dataset, see Supplementary Table 2. GBA variant carriers from the McGill dataset included samples from Columbia University, NY, USA (Alcalay et al., 2015) and Sheba Medical Center, Tel Hashomer, Israel (Ruskey et al., 2019). Genotype quality control, processing, and analysis was performed similar to the above described genotyping data.
UK Biobank data
Imputed UKB genotype data (v3) were downloaded (April 2018) under application number 32663 (Sudlow et al., 2015; Bycroft et al., 2018). All European ancestry individuals (based on code 22006) were screened for the common GBA coding variants p.E326K, p.T369M (both directly genotyped) and p.N370S (imputed R2 = 0.81). Parkinson’s disease cases were identified using ICD10 code ‘G20’ from data fields 41202 (Diagnoses - main ICD10), 41204 (Diagnoses - secondary ICD10) and 40002 [Contributory (secondary) causes of death: ICD10]. Additionally, self-report Parkinson’s disease cases were included from data field 20002 with code ‘1262’. ‘Proxy’ Parkinson’s disease cases were included by using individuals who reported a parent with Parkinson’s disease (paternal Parkinson’s disease, data field 20107 and maternal Parkinson’s disease, data field 20110), which have been previously shown to share genetic risk with Parkinson’s disease cases (Nalls et al., 2018). Controls were set as subjects with no report of Parkinson’s disease, no parent with Parkinson’s disease and no sibling with Parkinson’s disease (data field 20111). Covariates were obtained from the data fields: genetic sex (22001) batch (22000), age of recruitment (21022), and Townsend index (189). Individuals were filtered for relatedness (PIHAT>0.125) based on the pre-imputed data. Imputed genotypes were converted to PLINK2 .pgen files using PLINK2 (version v2.00) and filtered for missingness (<0.05) and minor allele frequency (>0.05), Hardy-Weinberg equilibrium of P ≥ 1 × 10−6 in controls and imputation quality (R2 > 0.8). GWAS was performed using PLINK2 logistic regression with covariates including batch, age of recruitment, Townsend index, and five PCs created using FlashPCA (Abraham et al., 2017). All UKB analyses were performed using population controls with age of recruitment ≥60 and for GBA carrier case-control analyses, the case-control ratio was set to 1:9 by randomly selected controls.
Additional datasets
Additional results for variants of interest were obtained from 23andMe and whole-genome sequencing data obtained from various resources. Analyses were performed using similar and identical methods as described above. Associations in 23andMe were performed using logistic regression correcting for age, sex, genotyping platform (v4 or v5) and five PCs. Genome-sequencing data were obtained from multiple ongoing projects at the Laboratory of Neurogenetics, see Supplementary material for a detailed description. Sample sizes can be found in Supplementary Table 3.
Genetic risk score analyses
To assess the influence of the genetic risk score (GRS), we calculated the GRS using the effect sizes from the most recent Parkinson’s disease GWAS variants (Nalls et al., 2018) (see Supplementary Table 4). Note that most of the data are included in the latest Parkinson’s disease GWAS; however, for the calculation of the GRS in the UKB data we excluded the UKB summary statistics. Overall, at least 65 of these variants of interest were imputed with high quality (R2 > 0.8) in the included datasets, allowing for a maximum of 15% variant missingness across any constituent data subset due to differing genotyping backbones. For the IPDGC dataset, rs356182 was replaced by rs356219, due to a higher imputation quality (R2 > 0.82 and D' 0.959 between variants in the IPDGC reference panel). Variants from the GBA locus and LRRK2 p.G2019 were excluded from the GRS in analyses including GBA carriers only. The GRS was calculated and processed using PLINK (v1.90) for each individual as described previously (Nalls et al., 2015b). To make the scale of analyses relating to the Parkinson’s disease GRS more easily interpretable, Parkinson’s disease GRSs were converted to Z-scores. GRS associations were performed with a logistic regression in R (v3.5.1) using case-control status or linear regression with age at onset as the outcome phenotype. As covariates, we included: sex, age (in case-control status comparison only), the first 10 PCs, and the dataset origin (due to the fact that the IPDGC data are derived from multiple sources see Supplementary Table 2). Area under the curve risk predictions based on the GRS were performed using the pROC package (Robin et al., 2011).
Additional analyses
GBA variants were annotated using ANNOVAR (Wang et al., 2010). Nucleotide positions in GBA were based on build hg19 and amino acid positions were based on NM_001005742. Note that historically these GBA variants are also known as p.E365K → p.E326K (rs2230288), p.T408M → p.T369M (rs75548401), and p.N409S → p.N370S (rs76763715). Where possible, association results were meta-analysed with METAL (v.2011-03-25) using default settings (Willer et al., 2010) or using the metafor R package (2.0) using fixed effects models (Viechtbauer, 2010). We excluded SNPs with a heterogeneity I2 statistic > 80% and variants that were present in only one GWAS. Limited population stratification was seen despite low sample numbers in some GWAS (Supplementary Table 5). Differences in age at onset between GBA variants was tested using linear regression in R using age at onset as the dependent variable, correcting for sex, PCs 1–5, and the dataset origin as covariates. Conditional GCTA-cojo analyses (Yang et al., 2011, 2012) were performed to identify whether there were multiple independent signals in loci of interest, using the previously described IPDGC reference panel (Nalls et al., 2018; Blauwendraat et al., 2019). Genetic interaction analyses were performed using R (version 3.5) using an interaction term created using SNP1 + 1 × SNP2 + 1, followed by a regression using both variants and covariates (PCs 1–5, sex and age if available).
Exploratory gene expression values and functional annotation of identified risk variants were obtained by mining datasets including: the largest brain expression quantitative trait loci (eQTL) study to date (Qi et al., 2018), the GTEx QTL portal V7 (https://gtexportal.org/) (Consortium, 2013) and the NABEC dataset [phs001300.v1.p1 (Gibbs et al., 2010)]. LocusZoom (Pruim et al., 2010) and LocusCompare plots (Liu et al., 2019) were generated for the regions of interest and were compared to the latest published Parkinson’s disease GWAS. Single-nuclei RNAseq expression of three frozen prefrontal cortex samples and four frozen hippocampal samples were obtained from dbGaP accession phs000424.v8.p1 (Habib et al., 2017). Data were processed using Seurat and genes of interest were highlighted to explore expression patterns (Butler et al., 2018).
GBA variant validation
To validate the genotyping and imputation of GBA variants, we obtained next-generation sequencing (targeted resequencing and genome sequencing) data from two parallel IPDGC projects. GBA carrier genotypes were merged using PLINK (v1.90) with both the resequencing and whole genome sequencing data. Overlapping individuals were identified using > 0.9 PIHAT values. Additionally, the majority of the PROBAND dataset individuals were Sanger sequenced for the full GBA gene as described elsewhere (Malek et al., 2018). In the McGill dataset, GBA variants were confirmed using Sanger sequencing or targeted next generation sequencing as previously described (Ruskey et al., 2019). Overall, the concordance rate between the imputed/genotype data and the sequence data for the presence of at least one p.N370S and p.T369M allele was 100% and for p.E326K was 98% and 94% in the next-generation and Sanger sequencing data, respectively (see Supplementary material for more details).
Induced pluripotent stem cells and neuron analysis
Forebrain neuron differentiation
Induced pluripotent stem cells (iPSCs) from the Parkinson’s Progression Marker Initiative (PPMI) were selected based on their genotype at GBA and rs1293298 then differentiated to forebrain type neurons. IPSCs were grown in E8 media (ThermoFisher) on Matrigel® until 90% confluent. When cells had grown to confluence they were transitioned to N3 media (50% DMEM/F12, 50% Neurobasal™ with 1× penicillin-streptomycin, 0.5× B-27™ minus vitamin A, 0.5× N2 supplement, 1× GlutaMAX™, 1× NEAA, 0.055 mM 2-mercaptoethanol and 1 μg/ml Insulin) plus 1.5 μM dorsomorphin (Tocris Bioscience) and 10 μM SB431542 (Stemgent) as previously reported (Burkhardt et al., 2013). Media was replaced every day for 11 days. On Day 12 dorsomorphin and SB431542 were removed and cells continued to be fed each day with N3. N3 was supplemented with 0.05 μM retinoic acid from Days 16 to 20. The differentiating cells were split 1:2 with Accutase® and seeded with ROCK inhibitor onto poly-l-ornithine (Sigma), fibronectin (2 μg/ml) and laminin (0.2 μg/ml) coated plates on Day 20. Cells were then fed daily with N4 media (same as N3 plus 0.05 μM retinoic acid, 2 ng/ml BDNF and 2 ng/ml GDNF) until Day 26 when they were frozen in Synth-a-Freeze™. Vials were thawed as needed in N4 media. Differentiation state was confirmed by immunocytochemistry for neuronal markers β-III tubulin (Novus Biologicals, NB100-1612) and MAP2 (Santa Cruz Biotechnology, sc-20172) with nuclei counterstained for Hoechst 33342 (ThermoFisher Scientific, H3570).
Protein analysis
Protein lysates were collected in buffer with 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.3% Triton™ 10% glycerol, 1× protease inhibitors, and 1× phosphatase inhibitors and run as previously reported (Blauwendraat et al., 2018b) on 4–20% Criterion™ TGX™ gels (Bio-Rad) in Tris-glycine running buffer and transferred to nitrocellulose membranes (Bio-Rad) using the Trans-blot® Turbo system (Bio-Rad). Membranes were blocked in Odyssey® Blocking buffer (LI-COR) diluted by 50% in phosphate-buffered saline. Primary antibodies for cathepsin B (Abcam, ab92955) and α-synuclein (BD Biosciences, 610787) were incubated overnight at 4°C. Primary antibody to β-III tubulin (Novus Biologicals, NB100-1612) was incubated for 1 h at room temperature. LI-COR secondary IRDye® antibodies were incubated 1 h at room temperature. Proteins were visualized on the Odyssey® CLx Imaging System (LI-COR) and quantified using Image Studio Lite (LI-COR).
Data availability
Summary statistics from the GWAS are available at https://pdgenetics.org/resources.
Results
Initial data overview
We included 21 478 Parkinson’s disease cases and 24 388 controls from IPDGC datasets; 1176 Parkinson’s disease cases, 13 431 proxy Parkinson’s disease cases, and 155 325 population controls from the UKB dataset; 103 GBA p.N370S Parkinson’s disease cases from the McGill dataset; and 622 LBD cases and 782 controls from the NDRU dataset (Supplementary Table 2). In the Haplotype Reference Consortium (HRC) reference panel, 10 coding GBA variants are present (Supplementary Table 6), with varying imputation qualities as common variants are more easily imputed than variants with lower frequencies. Additionally, some GBA variants were directly genotyped in some datasets (Supplementary Table 1). The three most common variants (p.E326K, p.T369M and p.N370S) had the highest quality, we therefore selected these three variants for all subsequent analyses. This resulted in a total of 1691 GBA carriers with Parkinson’s disease, 81 with LBD, 711 Parkinson’s disease proxy cases, and 7624 control GBA carriers. We excluded individuals carrying two GBA variants to ensure conservative estimates and reduce bias due to the association with Gaucher disease (Supplementary Table 2). All included GBA variants (p.E326K, p.T369M and p.N370S) have previously been shown to be enriched in cases in all datasets.
Parkinson’s disease genetic risk score modifies disease risk in GBA carriers
Exploring the potential effect of Parkinson’s disease case-control GWAS loci, we calculated the Parkinson’s disease GRS, both including and excluding GBA variants and LRRK2 p.G2019S for all three datasets. To compare the effect size associated with the GRS, we examined four groups within each dataset: controls without a GBA risk variant, Parkinson’s disease cases without a GBA risk variant, controls with a GBA risk variant, and Parkinson’s disease cases with a GBA risk variant. Additionally, for the UKB dataset, we included proxy-Parkinson’s disease cases (having a parent with Parkinson’s disease) (Fig. 1). As expected, the GRS is significantly associated with risk in GBA-negative Parkinson’s disease cases compared to controls and interestingly, also in the NDRU-LBD dataset (Fig. 1 and Table 1). This effect is highly similar between GBA carrier controls and GBA carrier cases, when excluding GBA and LRRK2 variants from the GRS (Fig. 2 and Table 1). This effect is consistent in subsets based on all three GBA variants (Supplementary Fig. 1). Although this was not significant in the (smaller) p.N370S group, the direction and effect size were highly similar to the other GBA variants [Supplementary Fig. 1, odds ratios (ORs) ranging from 1.31 to 1.55]. Notably, we do not observe a consistent difference in genetic risk between Parkinson’s disease or LBD cases with and without GBA variants when excluding GBA and LRRK2 in the genetic risk (Table 1).
Figure 1.

Parkinson disease GRS box plots divided by disease status and GBA carrier status. GRS differences between GBA negative controls, GBA negative Parkinson’s disease or LBD cases, GBA positive controls and GBA positive cases. In each dataset, a highly similar effect is observed clearly separating the subgroups. PD = Parkinson’s disease.
Table 1.
Associations between the Parkinson disease GRS and case-control status
| Outcome | Case-control | GBA+, Case-controla | GBA+ case versus GBA− casea | ||||||
|---|---|---|---|---|---|---|---|---|---|
| P | OR (95%CI) | Cases, n; controls, n | P | OR (95%CI) | Cases, n; controls, n | P | OR (95%CI) | GBA+ cases, n; GBA− cases, n | |
| IPDGC | 1.877 × 10−267 | 1.531 (1.51–1.56) | 21 478; 24 388 | 2.75 × 10−12 | 1.464 (1.36–1.57) | 1506; 905 | 0.00747 | 0.926 (0.87–0.98) | 1392; 18 062 |
| UKB | 9.156 × 10−55 | 1.557 (1.5–1.61) | 1176; 155 185 | 1.96 × 10−4 | 1.578 (1.34–1.82) | 82; 6679 | 0.674 | 1.050 (0.82–1.28) | 82; 1094 |
| UKB proxy-case | 1.284 × 10−35 | 1.138 (1.12–1.16) | 13 291; 155 185 | 2.12 × 10−5 | 1.217 (1.13–1.31) | 711; 6679 | NA | NA | NA |
| NDRU-LBD | 6.88 × 10−8 | 1.370 (1.26–1.48) | 622; 782 | 0.0798 | 1.565 (1.06–2.07) | 81; 40 | 0.892 | 1.017 (0.78–1.26) | 81; 541 |
GRS associations performed per group. Odds ratios are based on a Z-score scale as described in the methods. NA = not applicable; SE = standard error; UKB = UK Biobank.
For these associations, the GBA region and LRRK2 p.G2019S were excluded when calculating the GRS.
Figure 2.

Parkinson disease GRS box plots divided by disease status and GBA carrier status excluding GBA in the GRS calculation. GRS differences between GBA negative controls, GBA negative Parkinson’s disease or Lewy body dementia cases, GBA positive controls and GBA positive cases when excluding the GBA region and LRRK2 p.G2019S from the calculations. In each dataset, a highly similar effect is observed clearly separating the cases and controls. However, there is no clear separation between GBA cases and GBA negative cases or GBA negative controls and GBA positive controls. PD = Parkinson’s disease.
When assessing the predictive power of the GRS in GBA positive cases versus GBA-positive controls (excluding GBA and LRRK2 variants in GRS calculations), these values were slightly lower compared to the reported predictive power of the GRS in Parkinson’s disease cases as previously described by Nalls et al. (2018) [area under the curve (AUC) = 0.651, 95% confidence interval (CI) 0.617–0.684], with AUC of 0.614 (95% CI 0.591–0.637) in the IPDGC dataset and 0.622 (95% CI 0.561–0.684) in the UKB dataset (Supplementary Table 7 and Supplementary Figs 2–4). As expected, the NDRU-LBD dataset performed poorer than the Parkinson’s disease cases with an AUC of 0.570 (95% CI 0.540–0.600), supporting the notion of partial overlapping and shared genetic risk between Parkinson’s disease and LBD.
SNCA and CTSB loci as genetic modifiers of disease risk in GBA carriers
To identify potential genetic modifiers of GBA-associated Parkinson’s disease penetrance, we performed a GWAS comparing Parkinson’s disease patients with GBA variants and controls with GBA variants. We first examined the association using all three GBA variants together (p.E326K, p.T369M and p.N370S) in the IPDGC and the UKB dataset, followed by a meta-analysis of both. After meta-analysing the GBA data in both datasets, no genome-wide significant variants were identified (Supplemental Fig. 5).
When solely examining the 90 Parkinson’s disease variants previously identified in a Parkinson’s disease case-control GWAS (Nalls et al., 2018), variants in two Parkinson’s disease loci passed Bonferroni multiple test correction for association with Parkinson’s disease, SNCA and CTSB. The strongest association was observed with rs356219 in the SNCA locus (meta-P = 9.26 × 10−7, OR = 1.375, 95% CI = 1.211–1.563, Table 2 and Supplementary Fig. 6). In addition to SNCA, the CTSB locus rs1293298 (meta-P = 9.74 × 10−6, OR = 0.725, 95% CI = 0.629–0.836, Table 2 and Supplementary Fig. 7) was also significantly associated with GBA penetrance after correction for multiple testing (Supplementary Table 8). Additional investigation of these two variants was performed in the 23andMe Parkinson’s disease case-control dataset and in a WGS dataset (Table 2). Meta-analyses of these results showed an increase of significance level for the CTSB variant (P = 6.34 × 10−6) and a genome-wide significant P-value (P = 2.23 × 10−11) for the SNCA variant (Table 2 and Supplementary Figs 9 and 10). The majority of these effects are mostly driven by p.E326K carriers likely due to the larger sample size, but the effect size was consistent for each of the three variants when tested individually in the IPDGC dataset (Supplementary Table 9 and Supplementary Figs 6 and 7).
Table 2.
Meta-analyses of significant GWAS signals and interaction effects
| IPDGC | UKB | Meta | 23andMe | LNG Genomes | Meta_all | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P | OR (CI) | P | OR (CI) | P | OR (CI) | P | OR (CI) | P | OR (CI) | P | OR (CI) | |
| Association | ||||||||||||
| rs356219 (SNCA) | 6.37 × 10−5 | 1.315 (1.150–1.504) | 3.99 × 10−4 | 2.085 (1.388–3.132) | 9.26 × 10−7 | 1.376 (1.211–1.563) | 6.89 × 10−7 | 1.273 (1.157–1.400) | 0.6645 | 0.921 (0.635–1.336) | 2.23 × 10−11 | 1.290 (1.198–1.391) |
| rs1293298 (CTSB) | 8.27 × 10−5 | 0.740 (0.637–0.860) | 0.0304478 | 0.614 (0.394–0.955) | 9.74 × 10−6 | 0.726 (0.630–0.837) | 0.0564 | 0.898 (0.805–1.003) | 0.05908 | 0.6616 (0.431–1.016) | 6.34 × 10−6 | 0.822 (0.755–0.895) |
| Interaction | ||||||||||||
| rs356219 × GBA | 0.7025 | 1.029 (0.889–1.190) | 0.00639 | 1.600 (1.141–2.244) | 0.152 | 1.103 (0.965–1.261) | 0.773 | 0.985 (0.891–1.089) | 0.3697 | 0.823 (0.537–1.261) | 0.653 | 1.018 (0.941–1.102) |
| rs1293298 × GBA | 0.00647 | 0.796 (0.676–0.938) | 0.28833 | 0.802 (0.534–1.205) | 0.00347 | 0.797 (0.685–0.928) | 0.554 | 0.966 (0.860–1.084) | 0.0663 | 0.625 (0.379–1.032) | 0.0113 | 0.890 (0.813–0.974) |
Two variants were significant after correction of multiple testing: rs356219 (SNCA) and rs1293298 (CTSB) in the initial used datasets. Additional datasets were assessed and overall both P-values became more significant. The interaction effects between the two variants and GBA were not replicated in the external datasets.
Evidence for a potential genetic interaction between CTSB and GBA
Interestingly, the OR of the CTSB variant rs1293298 association in the GBA GWAS is lower compared to the Parkinson’s disease case-control GWAS [Supplementary Table 4 (Nalls et al., 2018) OR = 0.911, 95% CI = 0.891–0.932 (Supplementary Fig. 8)], while for the SNCA variant this was not the case. This suggests a potential interaction between CTSB and GBA. Examining this possibility further resulted in a significant interaction term after meta-analysing effects of the IPDGC and UKB data (Table 2). As expected, no consistent significant interaction term was identified between SNCA and GBA (Table 2 and Supplementary Fig. 11). Replication of these results was investigated in the 23andMe and LNG Genomes data for both interactions. In both datasets the interaction terms were not significant, although the direction of effect was the same for CTSB × GBA (Table 2 and Supplementary Fig. 12).
Genetic dissection of the CTSB GWAS locus
To evaluate the potential molecular mechanism for increased risk of Parkinson’s disease in GBA carriers, we explored gene expression and eQTL datasets for the two significant associated loci, SNCA (rs356219) and CTSB (rs1293298). For the SNCA locus, several previous reports showed that risk variants in this locus likely increase SNCA expression (Soldner et al., 2016; Pihlstrøm et al., 2018). For the CTSB, no comprehensive assessment has yet been performed. In both the Parkinson’s disease GWAS and the GBA GWAS, rs1293298 appears to be the index variant and both GWAS signals appears to be highly similar (Fig. 3A–C).
Figure 3.

Genetic dissection of the CTSB locus. (A) LocusZoom plot of the CTSB region from the latest Parkinson’s disease GWAS (Nalls et al., 2018). (B) LocusZoom plot of the CTSB region from the latest current GBA carrier GWAS. (C) LocusCompare plot of the CTSB region showing high correlation between the Parkinson’s disease GWAS (x-axis) and the GBA carrier GWAS (y-axis). (D) LocusCompare plot of the CTSB region showing high correlation between the Parkinson’s disease GWAS (x-axis) and the brain QTL data (Qi et al., 2018) (y-axis).
The protective allele (A) of the CTSB variant rs1293298 increases CTSB expression in the brain in both GTEx and NABEC datasets and was significant in the latest meta-analyses of brain eQTL data (Supplementary Table 10). Results from the NABEC dataset suggest that specific isoforms of CTSB are differentially regulated and the QTL effect is mainly for the most highly expressed transcript (isoform: uc003wuq.3, Supplementary Fig. 13). Interestingly the QTL signal shows high correlation with the Parkinson’s disease GWAS signal (Fig. 3D).
Exploring post-mortem brain-derived single nuclei RNAseq data showed that CTSB is mainly expressed in neurons and microglia in a pattern that is similar to SNCA expression (Supplementary Fig. 14). This suggests that the biological effects of these variants possibly take place in these cell types, which is in line with pathology and previous literature. We attempted to examine GBA expression levels, but they were too low to assess.
Given the function of cathepsin B (encoded by CTSB) as lysosomal protease, which is of interest for Parkinson’s disease pathogenesis in general (McGlinchey and Lee, 2015), we explored other members of the cathepsin family for: (i) genetic association in the most recent Parkinson’s disease GWAS; (ii) genetic association in GBA GWAS; and (iii) the presence of an eQTL at each locus. In addition to the peak at CTSB, only CTSE has a Parkinson’s disease GWAS peak in close proximity; however, this locus is already nominated as the NUCKS1/RAB29 locus (Supplementary Table 11). Interestingly, for almost all cathepsins a brain eQTL has been identified. However, with the exception of CTSB, none of the eQTLs were associated with Parkinson’s disease (Supplementary Table 11). These results suggest that expression differences in other cathepsins, including CTSD and CTSL, which were previously suggested to degrade α-synuclein (McGlinchey and Lee, 2015), do not modulate risk for disease.
Cathepsin B and α-synuclein levels in iPSC-derived neurons from GBA carriers and non-carriers
We examined the potential relationship between GBA and CTSB using PPMI iPSCs differentiated to forebrain neurons. For this analysis we used iPSCs from individuals with and without the GBA p.N370S variant to interrogate cathepsin B and α-synuclein protein levels in the context of GBA variants. All lines used were heterozygous for rs1293298 at the CTSB locus and neuronal differentiation was confirmed by immunocytochemistry for β-III tubulin and MAP2 (Supplementary Fig. 15). Previous work has shown that cathepsin B protein is highly cleaved and processed before becoming active (Ritonja et al., 1985; Chan et al., 1986). The protein can be cleaved to a final heavy (24 kDa) and light chain (5 kDa) or they can remain uncleaved and run at 29 kDa. Processing of cathepsin B is cell type-dependent (Chen et al., 2019) and in our cells we see a double band pattern corresponding to the heavy plus light chain and the heavy chain alone. When quantifying both bands we observed a significantly lower (P = 0.0291; unpaired t-test, n = 6) level of active cathepsin B protein in GBA variant carriers compared to cells without GBA variants (GBA wild-type = 0.528 ± 0.0930; GBA p.N370S = 0.263 ± 0.0467) (Fig. 4). These results show that the GBA variant carriers have reduced levels of active cathepsin B, which may result in even lower lysosomal protease activity and increased accumulation of protein aggregates in neurons. Although the forebrain neurons carrying the GBA variant did not have increased levels of α-synuclein (Supplementary Fig. 16) it is possible that the overall reduction in lysosomal proteases allows for a faster accumulation of α-synuclein aggregates as neurons age.
Figure 4.

Cathepsin B protein expression is decreased in iPSC derived neurons from GBA p.N370S carriers. Forebrain neurons differentiated from patient iPSCs with GBA mutations have decreased cathepsin B protein expression. (A) Representative western blot showing protein levels of active cathepsin B (CTSB, 24 and 29 kDa) and neuron associated β-III tubulin (50 kDa) in forebrain neurons differentiated from individuals with and without Parkinson’s disease-associated GBA variants (n = 2 lines from each genotype; all lines tested are heterozygous at rs1293298). (B) Quantification of the mean active cathepsin B protein levels relative to β-III tubulin shows a significant decrease (*P < 0.05; unpaired t-test, n = 2 lines of each genotype with three technical replicates each) in cathepsin B levels in forebrain neurons differentiated from GBA p.N370S carriers compared to neurons from wild-type (WT) GBA carriers. M = protein marker; error bars represent standard error of the mean. Note that both the heavy chain and heavy plus light chain bands were quantified in this analysis.
Genetic modifiers of age at onset in GBA associated Parkinson’s disease cases
Information on age at onset was available for the majority of the IPDGC GBA Parkinson’s disease cases [89.9% n = 1353 cases, average = 60.54 years, standard deviation (SD) = 11.42]. Cases with two GBA variants (either homozygous or compound heterozygous, n = 38) had a lower age at onset compared to single variant carriers; however, no statistically significant difference was detected [P = 0.107; linear regression, Beta = −2.93 years, standard error (SE) = 1.82, two GBA variant case average = 56.81, SD = 10.50 versus single GBA variant case average = 60.54, SD = 11.42]. The age at onset of GBA Parkinson’s disease cases was significantly lower compared to non-GBA Parkinson’s disease cases [linear regression, P = 9.70 × 10−6, Beta = −1.43, SE = 0.324, GBA Parkinson’s disease case average (one GBA mutation) = 60.54, SD = 11.42 versus non-GBA case average = 62.06, SD = 12.01]. On average p.N370S cases had the earliest age at onset followed by p.E326K and p.T369M. Of note, the LRRK2 p.G2019S variant was also identified in 14 GBA carriers (nine p.N370S and five p.E326K carriers) and did not seem to influence the age at onset P = 0.44 (linear regression, Beta = 2.41, SE = 3.15, GBA Parkinson’s disease LRRK2 p.G2019S carriers average = 63.86, SD = 10.87 versus GBA Parkinson’s disease non-LRRK2 p.G2019S average = 60.29, SD = 11.80), which is consistent with previous findings, although power was limited given the small number of included individuals (Yahalom et al., 2019).
To identify potential GBA age at onset modifiers, we performed a GWAS using age at onset as phenotype in all GBA positive Parkinson’s disease cases (n = 1353). No genome-wide hits were identified, most likely because of the relatively low sample size. When investigating previously identified Parkinson’s disease loci, no variants passed Bonferroni correction. However, nominally significant signals were identified for several Parkinson’s disease loci, including SNCA and TMEM175 (Supplementary Table 12). Similar to Parkinson’s disease in general, the Parkinson’s disease GRS is significantly associated with a lower age at onset in GBA carriers (Fig. 5) (Blauwendraat et al., 2019). This effect was mainly driven by p.E326K carriers. However, both p.T369M and p.N370S carriers also show the same trend towards lower age at onset with a similar effect size that is close to significance likely due to lower sample numbers (Fig. 5). This association was similar in an independent p.N370S Ashkenazi Jewish ancestry dataset, again demonstrating the same general trend and similar effect size. A meta-analysis of the three tested GBA variants resulted in a significant P-value of 0.0050 (Fig. 5 and Supplementary Fig. 17).
Figure 5.
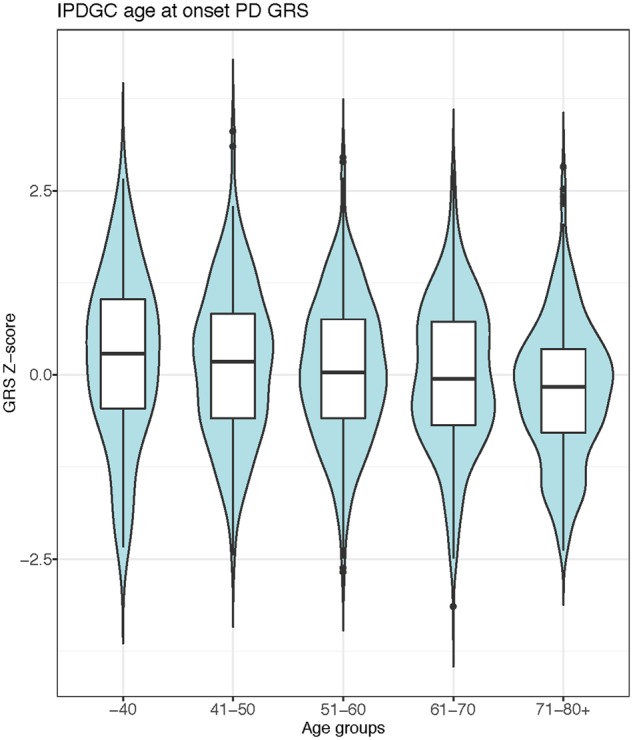
Parkinson’s disease GRS significantly influences the age at onset of GBA positive cases. Parkinson’s disease cases carrying a GBA variant were grouped by ages and a clear and significant decline was seen for the GRS. PD = Parkinson’s disease.
Discussion
In the current study, we analysed multiple large datasets to examine whether genetic variants affect the penetrance and age at onset of GBA-associated neurodegeneration. Our data show that at least 7% of the European Parkinson’s disease population carries a common protein coding GBA risk variant versus ∼4.5% of the general population. It is noteworthy that this number is an underestimation because in some datasets p.T369M and p.N370S were not genotyped or reliably imputed, and rarer disease-linked GBA variants were not captured. For LBD, these numbers are likely to be higher as seen in the NDRU-LBD dataset with ∼13% of the LBD cases carrying a GBA variant.
One of the major findings of this study is that Parkinson’s disease and LBD cases with GBA variants often also carry a substantial number of other Parkinson’s disease associated risk variants, which modify disease risk and age at onset. The GRS is significantly higher in cases with a GBA variant compared to controls with a GBA variant (Table 1) with effect sizes that are very similar to general Parkinson’s disease compared to controls. These findings suggest that GRS could be considered when designing clinical trials for GBA Parkinson’s disease/LBD. Pre-trial genetic analysis could assist in stratifying patients according to Parkinson’s disease-related genetic variants, on top of GBA. Therefore, future clinical trials involving GBA carriers should include a comprehensive genetic assessment prior to enrolment, as previously suggested (Leonard et al., 2018). Disease predictions based on current genetic knowledge should be combined with non-genetic data, such as family history and pre-symptomatic phenotypes including RBD and smell test to maximize disease prediction (Nalls et al., 2015c).
The link between Parkinson’s disease and lysosomal pathways has already been established by both genetic and functional studies (Gan-Or et al., 2015c; Robak et al., 2017; Ysselstein et al., 2019). Moreover, in the latest Parkinson’s disease GWAS (Nalls et al., 2018) numerous lysosomal and autophagy-related genes were associated with Parkinson’s disease, emphasizing the central role of the lysosomal-autophagy pathway in disease. Interestingly, the two main loci identified that influence disease risk in GBA carriers are variants in close proximity to SNCA and CTSB (rs356219 and rs1293298), both implicated in the lysosomal autophagy pathway. The SNCA locus is well-studied and the current hypothesis is that variants in this locus increase SNCA expression of either total α-synuclein or specific mRNA isoforms, resulting in an increased risk of Parkinson’s disease (Soldner et al., 2016; Pihlstrøm et al., 2018).
For the CTSB locus, no functional studies have been performed to dissect this locus in Parkinson’s disease to date, although several genetic studies have nominated CTSB as causal gene in this region via either QTL analyses or Mendelian randomization (Nalls et al., 2018; Li et al., 2019). Therefore, it remains unclear whether CTSB is the causal gene in this locus. However, there is evidence linking CTSB to Parkinson’s disease pathways. In this study, we see a decrease in active cathepsin B protein levels in iPSC-derived neurons from GBA variant carriers compared to non-carriers, suggesting a further reduction in lysosomal protease function in these cases. Depletion of TMEM175, another lysosomal gene associated with Parkinson’s disease, significantly decreases protein expression and enzyme activity of cathepsin B in rat hippocampal neurons (Jinn et al., 2017, 2019). Cathepsin B has also been suggested to be involved in α-synuclein lysosomal degradation (McGlinchey and Lee, 2015). Moreover, ceramide, which is one of the lipids produced by GCase, has been shown to be an activator of cathepsin B-related pathways (Liu et al., 2016). Overall, these results fit with the general model that GBA risk variant carriers have impaired lysosomal function due to reduced or abolished GCase activity (Alcalay (Alcalay et al., 2015). The independent associations of the SNCA and CTSB loci with GBA associated Parkinson’s disease suggest that increased SNCA expression and/or decreased CTSB mRNA expression may further impair lysosomal function, resulting in an increased risk of Parkinson’s disease. Additionally, we identified that cathepsin B protein levels are reduced in p.N370S neurons compared to wild-type. Overall, these findings suggest that cathepsin B might play a larger role in Parkinson’s disease than previously thought and that increasing CTSB/cathepsin B levels could be a potential therapeutic strategy. However, extensive functional in vitro and in vivo work is needed to validate these effects.
Despite the large datasets included in this study, it has several limitations. First, only one genome-wide significant locus was identified (SNCA) after meta-analysing with the replication datasets, most likely because of the lack of power caused by the relatively low number of GBA variant carriers available. However, when we sought to reduce the burden of genome-wide correction by examining previously implicated Parkinson’s disease risk loci, a significant association with the CTSB locus was identified. Second, the majority of the analyses were based on aggregated genotype data derived from several different genotyping arrays and centres/countries. As such, some datasets did not contain data on all three of the GBA variants of interest. It is also important to consider that different centres/countries might have used different measures of age at onset or age at diagnosis and that included controls may develop Parkinson’s disease at a later age. However, using the current data, we are still able to identify large effects, such as those driven by the GRS. Larger future initiatives specifically focused on GBA carriers will resolve these limitations and are needed to confirm these associations. Third, because the majority of the data included here were also used in the most recent Parkinson’s disease GWAS meta-analysis (Nalls et al., 2018); there could be a bias towards the identification of known Parkinson’s disease risk signals in this GBA dataset. We addressed this potential bias by looking for replication in several datasets that have not been included in the most recent Parkinson’s disease GWAS meta-analysis, such as the UKB Parkinson’s disease cases, the LBD dataset and the McGill GBA dataset. Unfortunately, we did not have a replication dataset for the GBA age at onset results, so these results require replication in an independent dataset. Fourth, some datasets might have excluded known GBA carriers prior to genotyping, which would result in an underestimation of total GBA carriers and the associated risk estimates. Of note, another relatively common GBA variant p.L444P (rs421016) is currently not imputable, as it is not included in the HRC reference panel and is in a complex region of the genome with a highly similar pseudogene. Finally, the functional data presented in Fig. 4 suggests a functional interaction between GBA and CTSB; however, it does not functionally validate the expression changes associated with the rs1293298 variant. Additional, larger experiments are needed to explore these findings further.
Despite these limitations, several conclusions can be drawn from our results. First, the risk of Parkinson’s disease in GBA carriers is influenced by specific variants at loci that are known to be associated more generally with Parkinson’s disease risk, including SNCA and CTSB, the latter possibly through interaction with GBA. In addition, GBA associated risk is significantly affected by the GRS; however, the total contribution of the GRS is not large. In other words, common genetic factors do not fully explain the partial penetrance of GBA variants in Parkinson’s disease, hence rare variants, environmental and ageing-related factors probably contribute to GBA penetrance. Furthermore, variants in known Parkinson’s disease loci, such as SNCA and TMEM175, as well as the GRS, might affect the age at onset of GBA-associated parkinsonism at a similar scale compared to the effect seen in typical Parkinson’s disease. Overall, these results confirm the importance of the lysosomal pathway in Parkinson’s disease and further supports CTSB and SNCA as interesting candidate genes for functional analysis in both GBA-associated and general Parkinson’s disease.
Supplementary Material
Acknowledgements
We would like to thank all of the subjects who donated their time and biological samples to be part of this study. We would also like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). The authors thank Ole Andreassen and the DemGene consortium for genotyping of the Oslo cohort. Samples used in the analyses presented in this article were obtained from the Golub Capital iPSC Parkinson's Progression Markers Initiative (PPMI) Sub-study (www.ppmi-info.org/cell-lines). As such, the investigators within PPMI contributed to the design and implementation of PPMI and/or provided data and collected samples but did not participate in the analysis or writing of this report. For up-to-date information on the study, visit www.ppmiinfo.org. Data used in the preparation of this article were obtained from the PPMI database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. Data and biospecimens used in preparation of this manuscript were obtained from the Parkinson’s Disease Biomarkers Program (PDBP) Consortium, part of the National Institute of Neurological Disorders and Stroke at the National Institutes of Health. Investigators include: Roger Albin, Roy Alcalay, Alberto Ascherio, DuBois Bowman, Alice Chen-Plotkin, Ted Dawson, Richard Dewey, Dwight German, Xuemei Huang, Rachel Saunders-Pullman, Liana Rosenthal, Clemens Scherzer, David Vaillancourt, Vladislav Petyuk, Andy West and Jing Zhang. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript. DNA panels from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds) were used in this study, as well as clinical data. The Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council and the Brains for Dementia research, a joint venture between Alzheimer's Society and Alzheimer's Research UK. The research was also partly funded by the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle Hospitals NHS Foundation Trust and Newcastle University. We thank the following brain banks for providing brain tissues: Banner Sun Health Research Institute, New York Brain Bank, Newcastle Brain Tissue Resource, University of Michigan Brain Bank, University of California San Diego Brain Bank, Duke University Brain Bank, Virginia Commonwealth University Brain Bank, and the Georgetown University Brain Bank. We would like to thank the NIA Baltimore Longitudinal Study of Aging for contributing tissue samples to the Johns Hopkins Alzheimer’s Disease Research Center. We are grateful to members of the North American Brain Expression Consortium (NABEC) for contributing DNA samples. This research has been conducted using the UK Biobank Resource under Application Number 33601. We would like to thank the NIH Neuro Brain Bank (https://neurobiobank.nih.gov) for contributing tissue samples. Tissue samples for genotyping were provided by the Johns Hopkins Morris K. Udall Center of Excellence for Parkinson’s Disease Research (NIH P50 NS38377) and the Johns Hopkins Alzheimer’s Disease Research Center. Members of the 23andMe Research Team are provided in the Supplementary material.
Funding
For a complete overview of members, acknowledgements and funding, please see the Supplemental material and/or http://pdgenetics.org/partners. This work was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services; project numbers 1ZIA-NS003154, Z01-AG000949-02 and Z01-ES101986. This work was also financially supported by the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program. The Columbia University cohort is supported by the Parkinson’s Foundation, the National Institutes of Health [K02NS080915, and UL1 TR000040], and the Brookdale Foundation. PPMI, a public-private partnership, is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, UCB, and Golub Capital. This study was supported in part by grants from the National Institutes of Health: U19-AG03365, P50 NS38377, and P50-AG005146. We are grateful for the support of the entire BIOCARD study team at Johns Hopkins University. Additionally, we acknowledge the contributions of the Geriatric Psychiatry Branch (GPB) in the intramural program of NIMH who initiated the BIOCARD study.
Competing interests
M.A.N. reported receiving support from a consulting contract between Data Tecnica International and the National Institute on Aging (NIA), National Institutes of Health (NIH), and consulting for the Michael J. Fox Foundation, Vivid Genomics, Lysosomal Therapeutics Inc., and Neuron23, Inc, among others. No other disclosures were reported. Z.G-O. has received consultancy fees from Lysosomal Therapeutics Inc., Idorsia, Denali, Prevail Therapeutics and Inception Sciences. K.H., P.C., and members of the 23andMe Research Team are employees of 23andMe, Inc. and hold stock or stock options in 23andMe.
Glossary
Abbreviation
- GRS =
genetic risk score
- GWAS =
genome-wide association studies
- IPDGC =
International Parkinson’s Disease Genomics Consortium
- IPSC =
induced pluripotent stem cell
- LBD =
Lewy body dementia
- PC =
principal component
- PPMI =
Parkinson’s Progression Marker Initiative
References
- Abraham G, Qiu Y, Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017; 33: 2776–8. [DOI] [PubMed] [Google Scholar]
- Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo S-H, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 2015; 138: 2648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012; 78: 417–20. [DOI] [PubMed] [Google Scholar]
- Blauwendraat C, Bras JM, Nalls MA, Lewis PA, Hernandez DG, Singleton AB, et al. Coding variation in GBA explains the majority of the SYT11-GBA Parkinson's disease GWAS locus. Mov Disord 2018a; 33: 1821–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Faghri F, Pihlstrom L, Geiger JT, Elbaz A, Lesage S, et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 2017; 57: 247.e9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrøm L, et al. Parkinson's disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 2019; 34: 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Reed X, Kia DA, Gan-Or Z, Lesage S, Pihlstrom L, et al. Frequency of Loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol 2018b; 75: 1416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 2013; 56: 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018; 36: 411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018; 562: 203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SJ, San Segundo B, McCormick MB, Steiner DF. Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc Natl Acad Sci U S A 1986; 83: 7721–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou C, Yu H, Tao L, An Y, Zhang X, et al. 27-Hydroxycholesterol contributes to lysosomal membrane permeabilization-mediated pyroptosis in co-cultured SH-SY5Y cells and C6 cells. Front Mol Neurosci 2019; 12: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet 2013; 45: 580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z, Amshalom I, Bar-Shira A, Gana-Weisz M, Mirelman A, Marder K, et al. The Alzheimer disease BIN1 locus as a modifier of GBA-associated Parkinson disease. J Neurol 2015a; 262: 2443–7. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015b; 84: 880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015c; 11: 1443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008; 70: 2277–83. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson's disease and other synucleinopathies. Curr Neurol Neurosci Rep 2018; 18: 44. [DOI] [PubMed] [Google Scholar]
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 2010; 6: e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 2018; 17: 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 2017; 14: 955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Blauwendraat C, Toolan D, Gretzula CA, Drolet RE, Smith S, et al. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 2019. doi: 10.1093/hmg/ddz136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Drolet RE, Cramer PE, Wong AH-K, Toolan DM, Gretzula CA, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 2017; 114: 2389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med 2019; 7: e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H, Blauwendraat C, Krohn L, Faghri F, Iwaki H, Furgeson G, et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson's disease. bioRxiv 2018. doi: 10.1101/427385. [DOI] [PMC free article] [PubMed]
- Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet 2011; 20: 202–10. [DOI] [PubMed] [Google Scholar]
- Li YI, Wong G, Humphrey J, Raj T. Prioritizing Parkinson's disease genes using population-scale transcriptomic data. Nat Commun 2019; 10: 994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet 2019; 51: 768–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget 2016; 7: 83907–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of -associated Parkinson's disease at presentation in the UK study. J Neurol Neurosurg Psychiatry 2018; 89: 702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci U S A 2015; 112: 9322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis 2006; 9: 417–23. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Parkinson's disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable risk. biorXiv 2018. doi: 10.1101/388165.
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, et al. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 2015a; 36: 1605.e7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Escott-Price V, Williams NM, Lubbe S, Keller MF, Morris HR, et al. Genetic risk and age in Parkinson's disease: continuum not stratum. Mov Disord 2015b; 30: 850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, McLean CY, Rick J, Eberly S, Hutten SJ, Gwinn K, et al. Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol 2015c; 14: 1002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrøm L, Blauwendraat C, Cappelletti C, Berge-Seidl V, Langmyhr M, Henriksen SP, et al. A comprehensive analysis of SNCA -related genetic risk in sporadic Parkinson disease. Ann Neurol 2018; 84: 117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP,, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 2018; 9: 2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013; 15: 146–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritonja A, Popovic T, Turk V, Wiedenmann K, Machleidt W. Amino acid sequence of human liver cathepsin B. FEBS Lett 1985; 181: 169–72. [DOI] [PubMed] [Google Scholar]
- Rivas MA, Avila BE, Koskela J, Huang H, Stevens C, Pirinen M, et al. Insights into the genetic epidemiology of Crohn's and rare diseases in the Ashkenazi Jewish population. PLoS Genet 2018; 14: e1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 2017; 140: 3191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S to analyze and compare ROC curves. BMC Bioinform 2011; 12: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskey JA, Greenbaum L, Roncière L, Alam A, Spiegelman D, Liong C, et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson's disease. Eur J Med Genet 2019; 62: 65–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009; 361: 1651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016; 533: 95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015; 12: e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw 2010; 36: 1–48. [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahalom G, Greenbaum L, Israeli-Korn S, Fay-Karmon T, Livneh V, Ruskey JA, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson's disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord 2019; 62: 179–84. [DOI] [PubMed] [Google Scholar]
- Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2012; 44: 369–75, S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysselstein D, Shulman JM, Krainc D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov Disord 2019; 34: 614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 2016; 32: 1423–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics from the GWAS are available at https://pdgenetics.org/resources.