

Ciliates, a eukaryotic clade that is over 1 billion years old, are defined by division of genome function between transcriptionally inactive germline micronuclei and functional somatic macronuclei. To date, most analyses of gene family evolution have been limited to cultivable model lineages (e.g., Tetrahymena, Paramecium, Oxytricha, and Stylonychia). Here, we focus on the uncultivable Karyorelictea and its understudied sister class Heterotrichea, which represent two extremes in genome architecture.
KEYWORDS: transcriptomics, gene family evolution, genetic code evolution, phylogenomics, Ciliophora, uncultivable microbes
ABSTRACT
Ciliates, a eukaryotic clade that is over 1 billion years old, are defined by division of genome function between transcriptionally inactive germline micronuclei and functional somatic macronuclei. To date, most analyses of gene family evolution have been limited to cultivable model lineages (e.g., Tetrahymena, Paramecium, Oxytricha, and Stylonychia). Here, we focus on the uncultivable Karyorelictea and its understudied sister class Heterotrichea, which represent two extremes in genome architecture. Somatic macronuclei within the Karyorelictea are described as nearly diploid, while the Heterotrichea have hyperpolyploid somatic genomes. Previous analyses indicate that genome architecture impacts ciliate gene family evolution as the most diverse and largest gene families are found in lineages with extensively processed somatic genomes (i.e., possessing thousands of gene-sized chromosomes). To further assess ciliate gene family evolution, we analyzed 43 single-cell transcriptomes from 33 ciliate species representing 10 classes. Focusing on conserved eukaryotic genes, we use estimates of transcript diversity as a proxy for the number of paralogs in gene families among four focal clades: Karyorelictea, Heterotrichea, extensive fragmenters (with gene-size somatic chromosomes), and non-extensive fragmenters (with more traditional somatic chromosomes), the latter two within the subphylum Intramacronucleata. Our results show that (i) the Karyorelictea have the lowest average transcript diversity, while Heterotrichea are highest among the four groups; (ii) proteins in Karyorelictea are under the highest functional constraints, and the patterns of selection in ciliates may reflect genome architecture; and (iii) stop codon reassignments vary among members of the Heterotrichea and Spirotrichea but are conserved in other classes.
INTRODUCTION
Most work on genome evolution in ciliates has focused on a few cultivable model lineages (e.g., Tetrahymena and Paramecium) that represent only a small proportion of biodiversity within this ancient (∼1 billion-year-old) clade. In the present study, we analyze data from a diverse sample of uncultivable ciliates, particularly the class Karyorelictea, which has very few published molecular data, that we isolated by hand from diverse environments.
Single-cell transcriptomics (SCT) have yielded insights in diverse fields, including microbial ecology, neurobiology, stem cell research, and cancer research (1–3). Developed in 2009 for analyses of blastomere transcriptomes in mice, SCT has since been used in a large number of studies focusing on microbes, primarily on bacteria (4, 5). Single-cell transcriptome techniques were first applied to ciliates, the focus of the present study, by Kolisko et al. (6), who reported that data generated from single-cell transcriptomics recovered approximately 90% of transcripts found from traditional total RNA extraction of stable cultures. Unsurprisingly, the number of assembled transcriptomes in SCT experiments varies with cell size and among individuals within species, the latter likely due to differences in life history stages (6, 7). Nevertheless, a major strength of single-cell transcriptomics is the recovery of gene sequences from uncultivable lineages, which constitute the majority of microbial eukaryotes.
Ciliates are a group of microbial eukaryotes that have somatic and germline genomes in separate nuclei sharing a common cytoplasm. In ciliates, somatic macronuclei are transcriptionally active and possess an atypical genome architecture: somatic chromosomes are often gene-dense, lack centromeres, exist at high copy number (∼45 N in Tetrahymena thermophila and ∼15,000 N in Stylonychia lemnae [8–10]), and in some lineages, are extensively fragmented to generate gene-sized somatic chromosomes (e.g., ∼2.2 kbp on average in S. lemnae [11]). In all but one class of ciliates, the class Karyorelictea, these processed somatic nuclei divide by amitosis, a noncanonical form of nuclear division (i.e., lacks clear spindles and without clear chromosome condensation) that divides the polyploid somatic macronuclei (12). The germline genome remains quiescent throughout asexual cycles, becoming transcriptionally active only during the sexual phases. Unlike the somatic genome, the germline chromosomes are genomically conventional (i.e., they possess centromeres and are several megabases long [10, 13]).
A challenge for interpreting microbial transcriptome data is the use of alternative genetic codes (14–16) since many ciliates, and an increasing number of other eukaryotic lineages, have been shown to reassign one or more canonical stop codons to various amino acids (17–20). In ciliates, genetic codes tend to fall into one of three classes: (i) standard (UAA, UAG, and UGA) stop codons are used for translation termination (i.e., canonical “universal” genetic code; e.g., Dileptus [21] [Cl: Litostomatea], Nyctotherus [22] [Cl: Armophorea], and Stentor [23] [Cl: Heterotrichea]); (ii) UAG and UAA are recognized as translation termination signals, with UGA coding for cysteine or tryptophan, e.g., Euplotes (24) (Cl: Spirotrichea) and Blepharisma japonicum (15) (Cl: Heterotrichea), respectively; and (iii) UGA is the sole functional stop codon, whereas UAA and UAG are translated into glutamine (e.g., Tetrahymena [25], Paramecium [26] [Cl: Oligohymenophorea], Oxytricha and Stylonychia [14] [Cl: Spirotrichea]), tyrosine (Mesodinium rubrum [15]), or glutamic acid (Campanella umbellaria [15] [Cl: Oligohymenophorea]). Even more unusual, Condylostoma magnum (15, 16) (Cl: Heterotrichea) follows none of the three strategies and reassigns all three standard stop codons to sense codons; in this lineage, interpreting the function of stop codons depends on their context in the mRNA (i.e., proximity to the 3′ untranslated region (UTR) and poly(A) tail [16]).
In addition, the majority of the species from which we isolated and collected transcriptome data lack reference genomes/transcriptomes even from closely related taxa. Thus, contamination removal, open reading frame (ORF) prediction, and gene family assignment from de novo transcriptome assembly is another major challenge. In the present study, we rely on PhyloToL (27), a taxon- and gene-rich bioinformatic pipeline that has been successfully used to analyze high-throughput sequencing (HTS) data from diverse eukaryotes (27). PhyloToL allows for a conservative approach addressing bioinformatic bleeding, contamination, and sequencing/assembly errors associated with HTS data.
Previous work has linked genome architecture to gene family evolution in ciliates (28). First, protein coding genes in ciliates tend to evolve faster than in other eukaryotes (29, 30). Second, ciliates with extensively fragmented somatic genomes (i.e., gene-sized somatic chromosomes) have more of and more diverse paralogs compared to ciliates without extensively fragmented somatic chromosomes (31, 32). However, these observations are mostly limited to taxa from the large Intramacronucleata clade (referred to here as the Im-clade), particularly the model ciliates Tetrahymena, Paramecium, and Oxytricha. Moreover, many analyses of gene family evolution have focused on only a few conserved genes, such as actin, α-tubulin, HSP90, dynein heavy chain family (32–36), and lineage specific genes, such as pheromones in Euplotes (37). Gene family evolution in other genes and across the other major ciliate subphylum Postciliodesmatophora (referred to as the Po-clade), containing the classes Karyorelictea and Heterotrichea, remains poorly understood.
To expand our knowledge of ciliate genome evolution, we sampled uncultivable ciliates, focusing on the understudied classes Karyorelictea and Heterotrichea (Po-clade) that have distinct genome features. The somatic nuclei in all Karyorelictea are described as paradiploid (i.e., have similar DNA content to the germline nuclei), lack the ability to divide by amitosis, and are differentiated from germline nuclei during both asexual and sexual cycles (38). In contrast, Heterotrichea, the sister clade to the Karyorelictea, contain highly amplified somatic genomes (e.g., ∼1,000 to ∼13,000 times more DNA compared to germline nuclei [39]) that are often housed in large nuclei resembling a chain of beads, and the somatic macronucleus is capable of amitosis, dividing with extramacronuclear microtubules (versus intramacronuclear microtubules for members in Im-clade) (10). Thus, even though Karyorelictea and Heterotrichea group together as sister clades, they represent strikingly different nuclear characteristics.
Here, we use single-cell transcriptome analyses of uncultivable ciliates to investigate the impact of these variable somatic genome structures on patterns of gene family evolution. We characterize transcripts from 43 individuals representing 33 species and 10 classes, including 11 species of Karyorelictea and 6 species of Heterotrichea focusing on transcript diversity, transcript divergence, and stop codon reassignments across the ciliate phylogeny. Using bioinformatics tools such as PhyloToL (27) and RELAX (40), we analyze 509 genes to assess the relationship between patterns of molecular evolution and genome architecture.
RESULTS
Single-cell transcriptomes.
We collected single-cell transcriptomic data from 43 individuals representing 33 species from ten ciliate classes, focusing on the poorly studied classes Karyorelictea and Heterotrichea (Table 1), and we combined these data with 13 transcriptomes available from public databases (see Table S3 in the supplemental material). After using PhyloToL (27; https://github.com/Katzlab/PhyloTOL) to remove rRNA sequences and potential prokaryotic contaminants, our single-cell transcriptomes yielded an average of 3,278 (range, 213 to 12,012) transcripts falling into 1,665 (range, 159 to 3,894) distinct gene families (GF; Table S1). For the newly generated data, we combined transcriptomes from individuals belonging to the same species (as determined by shared small subunit-rDNA sequences) for analyses of GF evolution (see Materials and Methods for details).
TABLE 1.
Summary of ciliate single-cell transcriptomes included in the present work, focusing on diverse species from ten classes of ciliatesa
| Focal clade | Class | No. of genera | No. of species | WTA | Species |
|---|---|---|---|---|---|
| Karyorelictea | Karyorelictea | 7 | 11 | 14 | Cryptopharynx sp., Geleia acuta, Geleia sinica, Geleia sp., Kentrophoros sp., Loxodes sp. 1, Loxodes sp. 2, Loxodes striatus, Remanella sp., Trachelocercidae sp. 1, Trachelocercidae sp. 2 |
| Heterotrichea | Heterotrichea | 5 | 6 | 8 | Anigsteinia sp., Blepharisma americanum, Climacostomum sp., Spirostomum ambiguum, Spirostomum minus, Stentor roeselii |
| Extensive fragmenters | Armophorea | 2 | 2 | 2 | Brachonella spiralis, Metopus sp. |
| Phyllopharyngea | 1 | 1 | 2 | Chilodonella uncinata | |
| Non-extensive fragmenters | Colpodea | 1 | 1 | 2 | Bursaria truncatella |
| Litostomatea | 4 | 4 | 6 | Didinium nasutum, Rimaleptus mucronatus, Litonotus sp., Spathidium sp. | |
| Nassophorea | 1 | 1 | 2 | Zosterodasys sp. | |
| Oligohymenophorea | 3 | 3 | 3 | Frontonia sp., Lembadion sp., Vorticella sp. | |
| Plagiopylea | 2 | 2 | 2 | Parasonderia sp., Sonderia sp. | |
| Prostomatea | 2 | 2 | 2 | Prorodon ovum, Nolandia orientalis |
Detailed information of the ciliate single-cell transcriptomes included in the present work. GF, gene family. Average and median K-mer coverage per transcript are assessed after combining individuals from same species. Related to sampling in Materials and Methods. Download Table S1, XLSX file, 0.02 MB (18.9KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ciliate transcriptomes that are obtained from public databases. Related to transcript diversity analyses in Materials and Methods. Download Table S3, XLSX file, 0.01 MB (9.2KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
For comparison, we separate the Im-clade into two non-monophyletic groups, those with extensively fragmented somatic genomes (EF; Armophorea, Spirotrichea, and Phyllopharyngea; 12 species, Table 1 and Fig. 1) and those with putative non-extensively fragmented genomes (NEF; 16 species, Table 1 and Fig. 1). This allows us to evaluate the impact of extensively fragmented somatic genomes, which are known to contribute to GF expansion (31, 41). For all subsequent analyses, we focus on the 509 conserved eukaryotic GFs present in at least 1 member of all four focal clades: Karyorelictea, Heterotrichea, EF, and NEF.
FIG 1.

Rank curve and boxplot showing lower average transcript diversity of Karyorelictea (blue), compared to extensive fragmenters (EF, light gray), non-extensive fragmenters (NEF, dark gray), and Heterotrichea (yellow), in analyses of 509 gene families. All pairwise comparisons are significant (P < 0.05, Kruskal-Wallis nonparametric test). GF, gene family.
Transcript diversity in ciliates.
To evaluate patterns of GF evolution across ciliates, we estimated the transcript diversity per GF for each taxon in our data set and then assessed the patterns within four groups: Karyorelictea, Heterotrichea, EF, and NEF. Among the 509 gene families included in the analyses, the average transcript diversity in Karyorelictea is the lowest among the four focal clades (Fig. 1). Among the four groups, we observe the following order of average transcript diversity per GF per taxon: Heterotrichea > EF > NEF > Karyorelictea (1.25, 0.92, 0.67, and 0.18, respectively). In over half of all GFs (319/509; 62.67%), each karyorelictid species included in our analyses possessed a single transcript. Similarly, the median number of transcripts per GF (i.e., paralogs) among the Karyorelictea is significantly lower than those of Heterotrichea, EF, and NEF (P < 0.001, Kruskal-Wallis two-sided test). In Karyorelictea, the variability in average number of transcripts per GF is also lower compared to the other three groups (interquartile range/median: Karyorelictea = 0.36, Heterotrichea = 1.25, EF = 0.84; NEF = 0.73; Fig. 1). To our surprise, the average transcript diversity of Heterotrichea is the greatest (P << 0.001, Kruskal-Wallis two-sided test) and most variable among the four groups, indicating greater paralog diversity of highly expressed genes.
Selection analysis.
We also investigated the strength of selection acting on gene families in our four focal groups using RELAX (42), assessing intensification versus relaxation based on selection intensity parameter (K) with the NEF chosen as the reference group. In nearly half of the alignments tested (236/503 or 46.9%, Table 1; see also Table S5 in the supplemental material) we were able to detect differences (P < 0.05) in selective strengths between the four focal groups with the RELAX test (Table 1; see also Table S5). The direction of change (intensification versus relaxation relative to NEF) was evenly split among alignments for EF branches (120/236, 116/236). However, for both Heterotrichea (179/236, 57/236) and Karyorelictea (148/236, 88/236; Table 1; see also Table S5), branches evolved under stronger selection (compared to NEF) more frequently than under weaker selection. Because intensification of selection could be a consequence of either stronger negative selection (here we use ω to represent the dN/dS ratio [i.e., the ratio of nonsynonymous to synonymous evolutionary changes (or substitutions)]; lower ω for ω < 1) and/or stronger positive selection (higher ω for ω > 1), we further examined patterns of ω variation across groups. Specifically, we computed means of ω values conditioned on ω < 1 (i.e., the negatively selected component of the distribution) based on the fits from the partitioned exploratory RELAX models (Fig. S1). Based on previous observations on the relationship between genome architecture and patterns of molecular evolution (31, 32, 43), we tested the a priori ordering of groups: Karyorelictea < NEF < Heterotrichea < EF and found a statistically significant trend (P < 0.001, Jonckheere-Terpstra test). In other words, our inferences are consistent with stronger functional constraints in the Karyorelictea compared to either reduced constraints and/or positive selection operating on lineages in the non-monophyletic EF group.
Maximum-likelihood estimates for alignmentwide mean of ω in the negative selection regime (ω ≤ 1) obtained by partitioned exploratory RELAX models. Box plots and individual points are shown side by side for each group. EF, extensive fragmenters; H, Heterotrichea; K, Karyorelictea; NEF, non-extensive fragmenters. Download FIG S1, DOCX file, 0.1 MB (153KB, docx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relaxation/intensification parameters (K) for each branch group relative to NEF are shown (sorted by P value). Related to Table 2. Download Table S5, XLSX file, 0.03 MB (36.8KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To compare the extent to which paralogs and orthologs were subject to episodic diversifying selection, we computed, for each alignment that contained both orthologs and paralogs from the same taxonomic group, the ratio of paralog branches subject to episodic selection to the total number of paralog branches and the analogous ratio for orthologs. Comparing these fractions within a specific alignment ensures that the power to detect selection is comparable between ortholog and paralog branches. For all four taxonomic groups, a significantly higher fraction of branches was selected among paralogs than orthologs in three groups, except Karyorelictea (see Fig. 2).
FIG 2.

The significant difference in diversifying positive selection between paralogs and orthologs in 3 of our four focal groups estimated by aBSREL suggests that diversifying selection may occur following duplications, with Karyorelictea as the exception. The numbers above each group are group means, and P values are from two-sided Wilcoxon test for differences in medians. *, Significant test results.
Stop codon usage and reassignment.
We assessed stop codon usage by calculating in-frame stop codon frequency, and determined amino acid reassignments by evaluating conserved sites within alignments among the diverse ciliates (see Materials and Methods for more details). We inferred a complex pattern and a considerable diversity of stop codon usage and reassignment across the ciliate phylogeny.
Our data from 943 GFs shared among at least eight of the ten ciliate classes are consistent with the three major patterns previously described (15, 16) (Fig. 3): ciliates using the “universal” genetic code (UAR and UGA being stop; 11/46), those reassigning UAR to amino acids (26/46), and those that have reassigned UGA (9/46). The “ciliate” (UGA as the sole stop codon) codon table is the most common alternative genetic code in our taxonomic sampling (26/46), with the standard universal codon also being prevalent among ciliates (11/47; Fig. 3). The remaining types, the Blepharisma and Euplotes codes (UAR as stop codon; UGA coding for tryptophan and cysteine, respectively), Chilodonella code (UAA as stop codon; UGA and UAG coding glutamine), Mesodinium code (UGA as stop codon; UAR reassigned to tyrosine), and the context-dependent Condylostoma code, together only represent a small proportion of ciliates (9/46).
FIG 3.
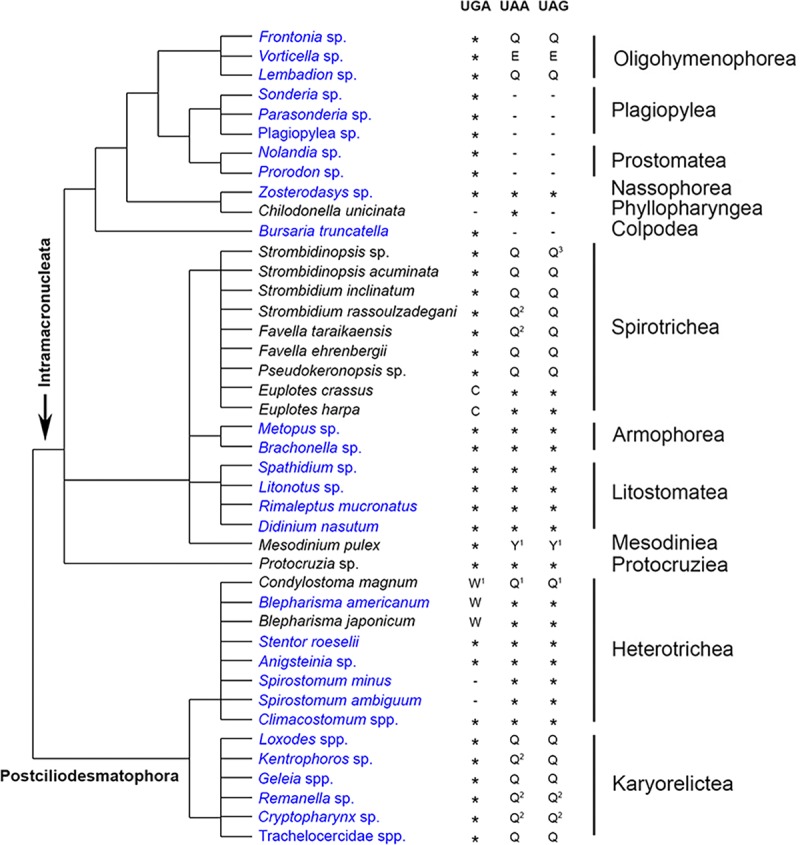
Putative stop codon usage and reassignment from single-cell transcriptomes in the present work shows variable patterns among classes. Data collected from this work are marked in blue. Phylogenetic tree topology is according to Adl et al. (61), with relationships within classes represented as polytomies; since the ciliate phylogeny is still under active debate, please see the alternative topology in Gao et al. (62). *, Serves as stop codon; –, not predominantly used as stop codon. Superscript numbers: 1, uncertainty of reassignment; 2, reassignment inferred from Swart et al. (16); 3, reassignment inferred from members from the same taxonomic group. For Strombidinopsis sp., we also found small and equal numbers of cases where K was as a reassignment for UAG.
DISCUSSION
The three main insights from this study are that (i) single-cell transcriptomics provides numerous insights into genome evolution (e.g., gene family sizes and stop codon reassignments) in uncultivable ciliates, (ii) features of genome architecture in ciliates (i.e., extensive chromosome fragmentation, high polyploidy, and paradiploidy) influence patterns of gene family evolution, and (iii) expanded taxonomic sampling reveals conservation of genetic code usage within some classes (e.g., Armophorea, Litostomatea, and Karyorelictea) and variability in others (e.g., Oligohymenophorea, Spirotrichea, and Heterotrichea). As a resource for the community, we generated single-cell transcriptomic analyses of uncultivable ciliates (Table 1) and deposited the data in the NCBI database (BioProject no. PRJNA573114; BioSample no. SAMN12807523 to SAMN12807565). These data substantially expand on the analyses of molecular evolution in ciliates by shifting the focus from cultivable model ciliates, e.g., Tetrahymena and Paramecium (44, 45), to a more comprehensive sampling of ciliate lineages.
Among the four focal clades (i.e., Karyorelictea and Heterotrichea in the Po-clade and EF and NEF in the Im-clade), the Karyorelictea possess the lowest transcript diversity (Fig. 1). This suggests that the inability of Karyorelictea to divide their somatic macronuclei (i.e., the lack of amitosis) limits GF evolution (i.e., paralog diversity); unlike other ciliates, Karyorelictea must develop a new somatic genome from the germline with every division, thus exposing any mutations accumulated in the germline in each new somatic nucleus. We speculate that this process changes fitness landscapes compared to ciliates that divide somatic nuclei by amitosis, enabling the removal of deleterious mutations through phenotypic assortment (32, 46), (i.e., stochastic distribution of alleles during somatic nuclear division). In fact, for many ciliates numerous asexual generations are necessary before sexual “maturity” (e.g., 80 to 100 generations in certain clones of Tetrahymena pyriformis [47] and 15 to 46 generations in Euplotes crassus [48]), during which time there is a greater opportunity to acquire potentially compensatory mutations in the germline nuclei while removing potentially deleterious mutations from their somatic nuclei (32). These compensatory mutations then appear in newly developed somatic nuclei following conjugation, allowing ciliates with amitosis to explore adaptive landscapes. Hence, the inability to undergo somatic macronuclear division (i.e., amitosis) in the Karyorelictea may explain the maintenance of fewer and less-divergent paralogs per gene family observed in this study (Fig. 1).
In contrast, the Heterotrichea, the sister class of Karyorelictea, has the greatest transcript diversity among the ciliates in this study. The average number of paralogs per gene in Heterotrichea are even greater than that of ciliates with extensively fragmented somatic genomes, which are known to have large gene families composed of divergent paralogs (32, 49) (Fig. 1). All heterotrichs studied to date have extremely high somatic ploidy levels (∼1,000 to ∼13,000 N), indicating a massive amplification process during somatic macronuclear differentiation (38, 39, 50). Maurer-Alcalá et al. (51) have previously shown relatively high copy numbers of protein coding genes in Blepharisma americanum. If this is true for the majority of protein coding genes in heterotrichs, “errors” generated during the differentiation and amplification of a new somatic genome might contribute to the observed high transcript diversity. Intriguingly, many members in the Heterotrichea have a somatic macronucleus organized as “beads on a chain” (also observed in some other ciliate clades, e.g., Litostomatea and Spirotrichea; Fig. 4), and with only one or two beads from the somatic nucleus, Stentor is able to recover and regenerate itself from a partial piece (as little as 1/100th of the cell [52, 53]). At the same time, they have many germline micronuclei, e.g., 12 to 30 in Climacostomum virens (54) and up to 49 in Fabrea salina (54) (Fig. 4), and the accumulation of mutations in each individual germline nucleus might also contribute to the high transcript diversity in Heterotrichea. Further research on the physical distribution of gene copies in the nucleus is needed to assess whether there is any spatial variation in the distribution of paralogs within asexually dividing Heterotrichea somatic macronuclei.
FIG 4.

Summary of features of the four focal ciliate groups, including the ability of somatic division, somatic ploidy, the structure of somatic genome, the average transcript diversity, and patterns of selection estimated by average dN/dS ratio, and, based on their nuclear/genome features, how likely was it that compensatory mutations would occur in each group when mildly deleterious mutations are present. Unknown features are indicated by a question mark (“?”). Diagrams of representative members of each group are drawn with somatic nuclei in empty circles and germline nuclei in filled circles (black). Oral structures are shown in light gray.
Consistent with previous studies (32), we also detected a higher average transcript diversity in ciliates with extensively fragmented somatic genomes (the EF group) compared to the non-extensive fragmenters (the NEF group) (31, 49) (Fig. 1; P = 0.00153, Kruskal-Wallis non-parametric test). Our results further support the hypothesis that gene-size chromosomes enhance the rates of gene family evolution (32, 41). By breaking up gene-linkage during amitotic divisions of the somatic macronucleus, stochastic assortment of gene-sized chromosomes during amitosis is likely more efficient at purging deleterious mutations rapidly and maintaining a higher fitness that is further influenced by epigenetics (31, 55). Meanwhile, periods of sexual immaturity allow the possibility for compensatory mutations to appear in the germline, generating greater paralog diversity (32, 41) (Fig. 4).
We also estimate the selection strength among the four focal groups using the selection intensity parameter K and the group average dN/dS value. The null hypothesis is that there should be no significant difference among the four groups if genome architecture does not impact patterns of gene family evolution. To our surprise, the Karyorelictea and Heterotrichea have more gene families under intensified selection compared with NEF reference group (Table 2). Similarly, there is a trend of selection strength (measured by group average dN/dS values) among the four focal groups (Karyorelictea > NEF > Heterotrichea > EF; Fig. S1). The intensification and lowest dN/dS values suggest that Karyorelictea is under the most selective constraint, whereas the EF group is under the most relaxed selection, which could be either relaxed purifying selection or weak positive selection. Our analyses are at odds with the null hypothesis (i.e., that genome architecture and patterns of sequence evolution are not correlated) and further emphasize the impact of different genome architectures, including programmed genome rearrangements, on gene family evolution. We also tested for episodic diversifying selection between paralogs and orthologs for each group (Fig. 2). Here, a significantly higher proportion of paralogous branches under episodic selection is detected in the Heterotrichea, EF, and NEF groups, which indicates that paralogs are more likely to undergo more functional differentiation after duplication events compared to orthologs; in contrast, the generally limited number of paralogs in Karyorelictea do not show significant selective difference compared to orthologs (Fig. 2).
TABLE 2.
Summary of 236 significant (P ≤ 0.05) RELAX group results among the 503 alignments tested
| Selection strength | RELAX result (median K)a
|
||
|---|---|---|---|
| Heterotrichea | Extensive fragmenters | Karyorelictea | |
| Intensification | 179 (1.51) | 120 (1.36) | 148 (1.53) |
| Relaxation | 57 (0.803) | 116 (0.750) | 88 (0.660) |
Intensification/relaxation values for the selection for Karyorelictea, Heterotrichea, and extensive fragmenters were measured relative to non-extensive fragmenter branches. The median values of intensification/relaxation coefficients (K) are shown in parentheses.
We further demonstrate the diversity of patterns of stop codon usage in ciliates, and the increase in sampling shows contrasting patterns among ciliate classes. Stop codon usage appears to be conserved in some classes (e.g., in Armophorea, Litostomatea, and Karyorelictea), whereas stop codon reassignments are variable in other classes (e.g., in Heterotrichea and Spirotrichea; Fig. 3). This is consistent with previous hypotheses that mutations in the eukaryotic release factor 1 (eRF1), altering its ability to recognize certain stop codons, has evolved independently in different ciliate lineages (14–17, 56). We estimate stop codon usage in the class Karyorelictea and find all species use UGA as a stop codon, while UAR is reassigned to glutamine in Loxodes spp., Geleia spp., Trachelocercidae spp., and Kentrophoros sp. (the reassignment of UAA in Kentrophoros sp. is unclear in our data; thus, the reassignment as glutamine is inferred from other karyorelictid members). This is one of the most common types of stop codon usage patterns in ciliates, which is also found in the classes Oligohymenophorea, Colpodea, Plagiopylea, Prostomatea, Nassophorea, and Spirotrichea (UAR in Vorticella sp. (Oligohymenophorea) is reassigned to glutamic acid instead of glutamine, and we were unable to extract reassignments for several species due to insufficient data based on our criteria; Fig. 3). Heterotrichea remains the clade hosting the greatest diversity of genetic codes, including the extreme case, Condylostoma magnum, which has reassigned all three conventional stop codons and where translation termination is context dependent (15, 16). Together, these data indicate that rates of changes in stop codons are variable among ciliates, though certainly faster than other well-sampled eukaryotic clades (57).
Synthesis.
Our analyses demonstrate the relationship between somatic macronuclear genome architecture and patterns of gene family evolution in ciliates: paralog diversity is lowest in the “paradiploid” class Karyorelictea, greater in ciliates with extensively processed genomes, and highest in the highly polyploid Heterotrichea. Similarly, there is a distinct difference in patterns of gene family evolution among those ciliates able to divide their somatic nuclei and those that cannot (i.e., Karyorelictea), which suggests that life history intersects with genome architecture in driving evolutionary patterns in ciliates. At the broadest level, our data suggest a macroevolutionary pattern, i.e., that genome architecture must be considered when developing models of molecular evolution, at least in ciliates.
MATERIALS AND METHODS
Sampling.
Chilodonella uncinata, Blepharisma americanum, Rimaleptus mucronatus, Didinium nasutum, and Bursaria truncatella were obtained from cultures, and all other taxa were collected from diverse sample sites, including a marine sandy beach, a freshwater tank, and a fen (see details in Table S1). Freshwater samples were directly poured into 5-cm petri dishes for ciliate isolation, while marine samples with sand grains were filtered through 35-μm-pore size mesh then kept in 5-cm petri dishes before single-cell transcriptome amplification.
Single-cell transcriptomes.
Individual cells were isolated by hand using glass pipettes and washed in filtered (0.2 μm) in situ water three to five times prior to being placed in a minimal volume of nuclease-free water (<1 μl) in a microcentrifuge tube. Transcriptomes of the individual ciliates were generated using the SMART-Seq v4 Ultra Low Input RNA kit for sequencing (Clontech, catalog numbers 634895 and 634896) according to the manufacturer’s instructions, adjusting all measurements to a quarter reaction volume. After transcriptome amplification, we used a Nextera XT DNA library preparation kit (96 samples; Illumina, catalog no. FC-131-1096) and a Nextera XT Index kit v2 set A (96 indexes, 384 samples; Illumina, catalog no. FC-131-2001) to construct libraries for HTS. The resulting libraries were sequenced on a HiSeq 4000 (Illumina) lane at the genome sequencing center (University of California at San Diego) or at the Institute for Genome Sciences, University of Maryland, Baltimore, MD.
Taxonomy assignment.
We collected all available small subunit (SSU) rRNA gene sequences of ciliates from the National Center for Biotechnology Information (NCBI) database and then performed BLAST searches of rRNA gene sequences isolated from each transcriptome. We inspected the top matching contigs for each cell to determine taxonomy based on identity and overlap (see the rRNA report in Table S2 and in the supplemental material).
rRNA report of each transcriptome for taxonomy identification. Related to taxonomy assignment in Materials and Methods. Download Table S2, XLSX file, 0.01 MB (11.5KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transcriptome assembly and analyses.
The output paired-end reads were trimmed with an individual quality trimming score and a minimum length of 100 bp with BBTools (58) and assembled with rnaSPAdes (part of the SPAdes v3.10.1 package [59]). After assembly, the output transcriptome was processed through a suite of custom Python scripts (part of the PhyloToL pipeline [27; https://github.com/Katzlab/PhyloTOL]). We applied PhyloToL using default settings, a relatively conservative approach, which has been successfully benchmarked in analyses of ancient eukaryotic gene families (27). The processing includes (i) the removal of contaminating rRNAs, potential mitochondrial sequences, and apparent prokaryotic transcripts; (ii) the assignment of transcripts to homologous gene families (based on the OrthoMCL database); (iii) the identification of putative ORFs from the transcripts; and (iv) the removal of transcripts of >98% nucleotide identity across ≥70% of their length to larger transcripts, which likely represent a pool of alleles, recent paralogs, and sequencing/assembly errors. The removal of potential eukaryotic contaminants was performed using outputs from the PhyloToL pipeline. For the 10 species with two (or more) available transcriptomes, we combined all transcriptomes for each species by removing partial transcripts (>98% nucleotide identity across ≥70% of length to a larger transcript) in the pool of transcriptomes (Table S1). We report average and median K-mer coverage for each data set (Table S7).
Number of transcripts in each studied species in the selected 509 gene families. Related to transcript diversity analyses in Materials and Methods. EF, extensive fragmenters; NEF, non-extensive fragmenters. *, Individual cells that are combined for the analyses, respectively. Download Table S7, XLSX file, 0.2 MB (165KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
SSU rRNA gene sequences from focal individuals in the present study. Download Dataset S1, TXT file, 0.1 MB (62.4KB, txt) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transcript diversity.
Together with 13 transcriptome data sets obtained from public databases (see the details in Table S3), we assessed transcript diversity in 509 GFs that contain at least one transcript in each focal clade, Karyorelictea (11 species) and Heterotrichea (8 species) in the Po-clade and extensive fragmenters (12 species) and non-extensive fragmenters (15 species) in the Im-clade (Tables S1 and S7). We counted the number of unique transcripts present in each GF for each species, and then we calculated the average transcript diversity for each clade in two ways, both including and excluding the “0” values representing the absence of transcripts in a given gene family. Here, we only show the results including the “0” values, since both analyses (with and without “0” values) are consistent (Fig. 1). To evaluate the patterns of transcript diversity, we performed boxplot analyses and Kruskal-Wallis and Mann-Whitney/Wilcoxon tests to visualize the variance among the four clades in R (42).
Evaluating selection profiles by a phylogenetic test of selection.
We compared selection strengths between the four defined taxonomic groups (see transcript diversity) in an alignment using a group-level extension of the RELAX test (40). The test operates on a tree where branches are partitioned into N+1 nonoverlapping sets, where N sets comprise the groups of interest and the remaining set contains the “nuisance” (or unlabeled) branches. In our case, branch groups were computed as follows. Each leaf is assigned to one of the Karyorelictea (K), non-extensive fragmenters (NEF), Heterotrichea (H), and extensive fragmenters (EF) groups based on the ciliate species that it belongs to. Internal branches are labeled bottom-up (post-order tree traversal). A branch is designated as a member of K, NEF, H, or EF if and only if all of its descendant branches have the same label; otherwise, it receives no label.
RELAX models variation in selection strengths, via the ω (dN/dS) ratio, among sites and branches, and between groups by fitting discrete distributions to the data via maximum likelihood. Sites and branches in NEF group, which is designated as the reference group (the choice of reference should not influence test results, and NEF was chosen since it is generally the largest group, and this property facilitates numerical convergence and stability), are modeled with a 3-bin ω distribution, 0 ≤ ω1 ≤ ω2 ≤ 1 ≤ ω3. A p1 proportion of branch/site combinations evolve with ω1, p2 with ω2, and p3 with ω3 (p1 + p2 + p3 = 1). Proportions are shared among all branch groups, and ω values are scaled using the group-specific relaxation/intensification coefficient K[g], so that ωg = ωK[g]. When K[g] > 1, all of the ω values move further away from 1 (neutrality), encoding intensification of both negative and positive selection, and when K[g] < 1, all of the ω values move closer to 1 (neutrality), representing relaxation of both negative and positive selection. Branches in the nuisance group are modeled with their own distribution of ω values and proportions.
The RELAX test compares the model where three values of K[g] are estimated from the data (one per branch group) with the model where K[g] = 1 (selection strength does not vary between groups). Significance is assessed via a likelihood ratio test with the χ2 asymptotic distribution with 3 degrees of freedom for computing P values. As with all group tests, this test does not identify for differences between any specified pair of groups, but rather for differences between any groups. We also fitted models where all parameters of the ω distributions were estimated separately for each branch group in order to better characterize the nature of selective processes (partitioned exploratory models [40]).
aBSREL.
To derive branch level estimates of selective regimes, we ran the aBSREL (60) procedure on gene family alignments. This method estimates the suitable number of ω regimes for each branch, fits ω and proportion parameters, and tests, for every branch, whether or not it has evidence of ω > 1 using a likelihood ratio test.
Assessment of stop codon usage and reassignment.
We developed custom Python scripts (https://github.com/yyan823/SCT_ciliates) to predict the in-frame stop codon usage and stop codon reassignment from each transcriptome. In brief, we collected transcripts with homologous gene family annotations and forced translation using TAA, TAG, or TGA as the only stop codon, respectively. We then calculated the frequencies of the other two traditional stop codons being in-frame. The stop codon(s) with substantial lower in-frame frequency(ies) were considered the most likely stop codon(s) for translation termination and used for further analyses. Those stop codons with heightened in-frame frequencies were then evaluated to determine their likely reassignment. For estimates of stop codon reassignments, we collected transcriptomic data from all 33 ciliate species we sampled, as well as 13 ciliate transcriptomes from the NCBI online database (https://www.ncbi.nlm.nih.gov/; Table S2), and selected 943 homologous gene families from seven well-annotated ciliate genomes (Table S4) as a reference and built alignments for each transcriptome. Conserved columns (across > 50% of the column) where stop codons were present in the taxon of question, were collected by a custom Python script (available upon request) and checked manually to calculate the frequency of the reassigned sense amino acid (Table S6).
Ciliate genomes used for stop codon analyses that are obtained from public databases. Related to assessment of stop codon usage and reassignment in Materials and Methods. Download Table S4, XLSX file, 0.01 MB (9.1KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Counts for assessments of stop codon reassignments in ciliates that are not using conventional stop codon usage (UAR and UGA). Related to Fig. 3. Download Table S6, XLSX file, 0.01 MB (14.6KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data availability.
Data for single-cell transcriptomic analyses of uncultivable ciliates were deposited in the NCBI database under BioProject accession number PRJNA573114 and BioSample accession numbers SAMN12807523 to SAMN12807565.
ACKNOWLEDGMENTS
This study is supported by two National Institutes of Health (NIH) awards (R15GM113177 and R15HG010409) and a National Science Foundation Go-LIFE award (DEB-1541511) to L.A.K. and two NIH awards (R01 GM093939 and R01 AI134384) to S.L.K.P.
We thank members in the Katz Lab for insightful discussions and helpful suggestions on the manuscript. We also thank James Gaffney of the Knight Lab for technical help.
Y.Y. led the identification and isolation of ciliates. Y.Y. and X.X.M.-A. designed and carried out experiments, analyzed data, and wrote the paper. S.L.K.P. developed analytical tools, analyzed data, and contributed to the paper. R.K. provided advice on methods and supported the first round of data collection. L.A.K. supervised the project and contributed to the experimental design, analyses, and writing of the manuscript.
The authors declare no competing interests.
Footnotes
This article is a direct contribution from Laura A. Katz, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Peter Vd'ačný, Comenius University in Bratislava; Sujal Phadke, J. Craig Venter Institute; Mireille Betermier, Université Paris Sud; Claude Thermes, Institute for Integrative Biology of the Cell; and Zhenzhen Yi, South China Normal University.
Citation Yan Y, Maurer-Alcalá XX, Knight R, Kosakovsky Pond SL, Katz LA. 2019. Single-cell transcriptomics reveal a correlation between genome architecture and gene family evolution in ciliates. mBio 10:e02524-19. https://doi.org/10.1128/mBio.02524-19.
REFERENCES
- 1.Eberwine J, Lovatt D, Buckley P, Dueck H, Francis C, Kim TK, Lee J, Lee M, Miyashiro K, Morris J, Peritz T, Schochet T, Spaethling J, Sul J-Y, Kim J. 2012. Quantitative biology of single neurons. J R Soc Interface 9:3165–3183. doi: 10.1098/rsif.2012.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saliba AE, Westermann AJ, Gorski SA, Vogel J. 2014. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu S, Qing T, Zheng Y, Jin L, Shi L. 2017. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget 8:53763–53779. doi: 10.18632/oncotarget.17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taniguchi Y, Choi PJ, Li G-W, Chen H, Babu M, Hearn J, Emili A, Xie XS. 2010. Quantifying Escherichia coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Z, Chen L, Zhang W. 2017. Tools for genomic and transcriptomic analysis of microbes at single-cell level. Front Microbiol 8:1831. doi: 10.3389/fmicb.2017.01831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolisko M, Boscaro V, Burki F, Lynn DH, Keeling PJ. 2014. Single-cell transcriptomics for microbial eukaryotes. Curr Biol 24:R1081–R1082. doi: 10.1016/j.cub.2014.10.026. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Hu SK, Campbell V, Tatters AO, Heidelberg KB, Caron DA. 2017. Single-cell transcriptomics of small microbial eukaryotes: limitations and potential. ISME J 11:1282–1285. doi: 10.1038/ismej.2016.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doerder FP, Deak JC, Lief JH. 1992. Rate of phenotypic assortment in Tetrahymena thermophila. Dev Genet 13:126–132. doi: 10.1002/dvg.1020130206. [DOI] [PubMed] [Google Scholar]
- 9.Eisen JA, Coyne RS, Wu M, Wu D, Thiagarajan M, Wortman JR, Badger JH, Ren Q, Amedeo P, Jones KM, Tallon LJ, Delcher AL, Salzberg SL, Silva JC, Haas BJ, Majoros WH, Farzad M, Carlton JM, Smith RK, Garg J, Pearlman RE, Karrer KM, Sun L, Manning G, Elde NC, Turkewitz AP, Asai DJ, Wilkes DE, Wang Y, Cai H, Collins K, Stewart BA, Lee SR, Wilamowska K, Weinberg Z, Ruzzo WL, Wloga D, Gaertig J, Frankel J, Tsao C-C, Gorovsky MA, Keeling PJ, Waller RF, Patron NJ, Cherry JM, Stover NA, Krieger CJ, del Toro C, Ryder HF, Williamson SC, Barbeau RA, Hamilton EP, Orias E. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol 4:e286. doi: 10.1371/journal.pbio.0040286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prescott DM. 1994. The DNA of ciliated protozoa. Microbiol Rev 58:233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aeschlimann SH, Jönsson F, Postberg J, Stover NA, Petera RL, Lipps H-J, Nowacki M, Swart EC. 2014. The draft assembly of the radically organized Stylonychia lemnae macronuclear genome. Genome Biol Evol 6:1707–1723. doi: 10.1093/gbe/evu139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujiu K, Numata O. 2000. Reorganization of microtubules in the amitotically dividing macronucleus of Tetrahymena. Cell Motil Cytoskeleton 46:17–27. [DOI] [PubMed] [Google Scholar]
- 13.Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, Russ C, Zeng Q, Fan L, Levin JZ, Shea T, Young SK, Hegarty R, Daza R, Gujja S, Wortman JR, Birren BW, Nusbaum C, Thomas J, Carey CM, Pritham EJ, Feschotte C, Noto T, Mochizuki K, Papazyan R, Taverna SD, Dear PH, Cassidy-Hanley DM, Xiong J, Miao W, Orias E, Coyne RS. 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife 5:e19090. doi: 10.7554/eLife.19090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lozupone CA, Knight RD, Landweber LF. 2001. The molecular basis of nuclear genetic code change in ciliates. Curr Biol 11:65–74. doi: 10.1016/s0960-9822(01)00028-8. [DOI] [PubMed] [Google Scholar]
- 15.Heaphy SM, Mariotti M, Gladyshev VN, Atkins JF, Baranov PV. 2016. Novel ciliate genetic code variants, including the reassignment of all three stop codons to sense codons in Condylostoma magnum. Mol Biol Evol 33:2885–2889. doi: 10.1093/molbev/msw166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swart EC, Serra V, Petroni G, Nowacki M. 2016. Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166:691–702. doi: 10.1016/j.cell.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alkalaeva E, Mikhailova T. 2017. Reassigning stop codons via translation termination: how a few eukaryotes broke the dogma. Bioessays 39:1600213. doi: 10.1002/bies.201600213. [DOI] [PubMed] [Google Scholar]
- 18.Lekomtsev SA. 2007. Non-standard genetic codes and translation termination. Mol Biol (Mosk) 41:964–972. [PubMed] [Google Scholar]
- 19.Santos J, Monteagudo A. 2011. Simulated evolution applied to study the genetic code optimality using a model of codon reassignments. BMC Bioinformatics 12:56. doi: 10.1186/1471-2105-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cocquyt E, Gile GH, Leliaert F, Verbruggen H, Keeling PJ, De Clerck O. 2010. Complex phylogenetic distribution of a non-canonical genetic code in green algae. BMC Evol Biol 10:327. doi: 10.1186/1471-2148-10-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricard G, de Graaf RM, Dutilh BE, Duarte I, van Alen TA, van Hoek AH, Boxma B, van der Staay GWM, Moon-van der Staay SY, Chang W-J, Landweber LF, Hackstein JHP, Huynen MA. 2008. Macronuclear genome structure of the ciliate Nyctotherus ovalis: single-gene chromosomes and tiny introns. BMC Genomics 9:587. doi: 10.1186/1471-2164-9-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slabodnick MM, Ruby JG, Reiff SB, Swart EC, Gosai S, Prabakaran S, Witkowska E, Larue GE, Fisher S, Freeman RM, Gunawardena J, Chu W, Stover NA, Gregory BD, Nowacki M, Derisi J, Roy SW, Marshall WF, Sood P. 2017. The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr Biol 27:569–575. doi: 10.1016/j.cub.2016.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turanov AA, Lobanov AV, Fomenko DE, Morrison HG, Sogin ML, Klobutcher LA, Hatfield DL, Gladyshev VN. 2009. Genetic code supports targeted insertion of two amino acids by one codon. Science 323:259–261. doi: 10.1126/science.1164748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caron F, Meyer E. 1985. Does Paramecium primaurelia use a different genetic code in its macronucleus? Nature 314:185–188. doi: 10.1038/314185a0. [DOI] [PubMed] [Google Scholar]
- 26.Horowitz S, Gorovsky MA. 1985. An unusual genetic code in nuclear genes of Tetrahymena. Proc Natl Acad Sci U S A 82:2452–2455. doi: 10.1073/pnas.82.8.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerón-Romero MA, Maurer-Alcalá XX, Grattepanche JD, Yan Y, Fonseca MM, Katz LA. 2019. PhyloToL: a taxon/gene-rich phylogenomic pipeline to explore genome evolution of diverse eukaryotes. Mol Biol Evol 36:1831–1842. doi: 10.1093/molbev/msz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zufall RA, Katz LA. 2007. Micronuclear and macronuclear forms of beta-tubulin genes in the ciliate Chilodonella uncinata reveal insights into genome processing and protein evolution. J Eukaryot Microbiol 54:275–282. doi: 10.1111/j.1550-7408.2007.00267.x. [DOI] [PubMed] [Google Scholar]
- 29.Katz LA, Bornstein J, Lasek-Nesselquist E, Muse SV. 2004. Dramatic diversity of ciliate histone H4 genes revealed by comparisons of patterns of substitutions and paralog divergences among eukaryotes. Mol Biol Evol 21:555–562. doi: 10.1093/molbev/msh048. [DOI] [PubMed] [Google Scholar]
- 30.Grant JR, Katz LA. 2014. Building a phylogenomic pipeline for the eukaryotic tree of life: addressing deep phylogenies with genome-scale data. PLoS Curr 6:ecurrents.tol.c24b6054aebf3602748ac042ccc8f2e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao F, Song WB, Katz LA. 2014. Genome structure drives patterns of gene family evolution in ciliates, a case study using Chilodonella uncinata (Protista, Ciliophora, Phyllopharyngea). Evolution 68:2287–2295. doi: 10.1111/evo.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zufall RA, McGrath CL, Muse SV, Katz LA. 2006. Genome architecture drives protein evolution in ciliates. Mol Biol Evol 23:1681–1687. doi: 10.1093/molbev/msl032. [DOI] [PubMed] [Google Scholar]
- 33.Israel RL, Kosakovsky Pond SL, Muse SV, Katz LA. 2002. Evolution of duplicated alpha-tubulin genes in ciliates. Evolution 56:1110–1122. doi: 10.1111/j.0014-3820.2002.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 34.Kim OT, Yura K, Go N, Harumoto T. 2004. Highly divergent actins from karyorelictean, heterotrich, and litostome ciliates. J Eukaryot Microbiol 51:227–233. doi: 10.1111/j.1550-7408.2004.tb00551.x. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan V, Wilkes DE. 2016. Evolution of the dynein heavy chain family in ciliates. J Eukaryot Microbiol 63:138–141. doi: 10.1111/jeu.12245. [DOI] [PubMed] [Google Scholar]
- 36.Yi Z, Huang L, Yang R, Lin X, Song W. 2016. Actin evolution in ciliates (Protist, Alveolata) is characterized by high diversity and three duplication events. Mol Phylogenet Evol 96:45–54. doi: 10.1016/j.ympev.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 37.Pedrini B, Suter-Stahel T, Vallesi A, Alimenti C, Luporini P. 2017. Molecular structures and coding genes of the water-borne protein pheromones of Euplotes petzi, an early diverging polar species of Euplotes. J Eukaryot Microbiol 64:164–172. doi: 10.1111/jeu.12348. [DOI] [PubMed] [Google Scholar]
- 38.Raikov IB. 1982. The protozoan nucleus: morphology and evolution. Springer-Verlag, New York, NY. [Google Scholar]
- 39.Wancura MM, Yan Y, Katz LA, Maurer-Alcalá XX. 2018. Nuclear features of the heterotrich ciliate Blepharisma americanum: genomic amplification, life cycle, and nuclear inclusion. J Eukaryot Microbiol 65:4–11. doi: 10.1111/jeu.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. 2015. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 32:820–832. doi: 10.1093/molbev/msu400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katz LA, Kovner AM. 2010. Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata. J Exp Zool B Mol Dev Evol 314:480–488. doi: 10.1002/jez.b.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R Foundation for Statistical Computing. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 43.Maurer-Alcalá XX, Katz LA. 2016. Nuclear architecture and patterns of molecular evolution are correlated in the ciliate Chilodonella uncinata. Genome Biol Evol 8:1634–1642. doi: 10.1093/gbe/evw099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chalker DL. 2008. Dynamic nuclear reorganization during genome remodeling of Tetrahymena. Biochim Biophys Acta 1783:2130–2136. doi: 10.1016/j.bbamcr.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Betermier M, Duharcourt S. 2014. Programmed rearrangement in ciliates: Paramecium. Microbiol Spectr 2:6. doi: 10.1128/microbiolspec.MDNA3-0035-2014. [DOI] [PubMed] [Google Scholar]
- 46.Merriam EV, Bruns PJ. 1988. Phenotypic assortment in Tetrahymena thermophila: assortment kinetics of antibiotic-resistance markers, tsA, death, and the highly amplified rDNA locus. Genetics 120:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nanney DL. 1974. Aging and long-term temporal regulation in ciliated protozoa: a critical review. Mech Ageing Dev 3:81–105. doi: 10.1016/0047-6374(74)90008-6. [DOI] [PubMed] [Google Scholar]
- 48.Dini F, Nyberg D. 1992. Development of sexual maturity in the ciliate Euplotes crassus: sources of variation in the timing of maturity. Dev Genet 13:41–46. doi: 10.1002/dvg.1020130107. [DOI] [PubMed] [Google Scholar]
- 49.Maurer-Alcalá XX, Knight R, Katz LA. 2018. Exploration of the germline genome of the ciliate Chilodonella uncinata through single-cell omics (transcriptomics and genomics). mBio 9:01836-17. doi: 10.1128/mBio.01836-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ovchinnikova L, Cheissin E, Selivanova G. 1965. Photometric study of the DNA content in the nuclei of Spirostomum ambiguum (Ciliata, Heterotricha). Acta Protozool 3:69–78. [Google Scholar]
- 51.Maurer-Alcalá XX, Yan Y, Pilling O, Knight R, Katz LA. 2018. Twisted tales: insights into genome diversity of ciliates using single-cell ‘omics. Genome Biol Evol 10:1927–1938. doi: 10.1093/gbe/evy133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lillie FR. 1896. On the smallest parts of Stentor capable of regeneration; a contribution on the limits of divisibility of living matter. J Morphol 12:239–249. doi: 10.1002/jmor.1050120105. [DOI] [Google Scholar]
- 53.Tartar V. 1961. The biology of Stentor. Pergamon Press, New York, NY. [Google Scholar]
- 54.Kim JH, Shin MK. 2015. Novel discovery of two heterotrichid ciliates, Climacostomum virens and Fabrea salina (Ciliophora: Heterotrichea: Heterotrichida) in Korea. Anim Syst Evol Divers 31:182–190. doi: 10.5635/ASED.2015.31.3.182. [DOI] [Google Scholar]
- 55.Yerlici VT, Landweber LF. 2014. Programmed genome rearrangements in the ciliate Oxytricha, p 389–407. In Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer S. (ed), Mobile DNA III. ASM Press, Washington, DC. doi: 10.1128/microbiolspec.MDNA3-0025-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tourancheau AB, Tsao N, Klobutcher LA, Pearlman RE, Adoutte A. 1995. Genetic code deviations in the ciliates: evidence for multiple and independent events. EMBO J 14:3262–3267. doi: 10.1002/j.1460-2075.1995.tb07329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Osawa S, Muto A, Jukes TH, Ohama T. 1990. Evolutionary changes in the genetic code. Proc Biol Sci 241:19–28. doi: 10.1098/rspb.1990.0060. [DOI] [PubMed] [Google Scholar]
- 58.Brushnell B. 2016. BBMap short read aligner. University of California, Berkeley, CA: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/. [Google Scholar]
- 59.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 32:1342–1353. doi: 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adl SM, Bass D, Lane CE, Lukeš J, Schoch CL, Smirnov A, Agatha S, Berney C, Brown MW, Burki F, Cárdenas P, Čepička I, Chistyakova L, Del Campo J, Dunthorn M, Edvardsen B, Eglit Y, Guillou L, Hampl V, Heiss AA, Hoppenrath M, James TY, Karnkowska A, Karpov S, Kim E, Kolisko M, Kudryavtsev A, Lahr DJG, Lara E, Le Gall L, Lynn DH, Mann DG, Massana R, Mitchell EAD, Morrow C, Park JS, Pawlowski JW, Powell MJ, Richter DJ, Rueckert S, Shadwick L, Shimano S, Spiegel FW, Torruella G, Youssef N, Zlatogursky V, Zhang Q. 2019. Revisions to the classification, nomenclature, and diversity of eukaryotes. J Eukaryot Microbiol 66:4–119. doi: 10.1111/jeu.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao F, Warren A, Zhang Q, Gong J, Miao M, Sun P, Xu D, Huang J, Yi Z, Song W. 2016. The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci Rep 6:24874. doi: 10.1038/srep24874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed information of the ciliate single-cell transcriptomes included in the present work. GF, gene family. Average and median K-mer coverage per transcript are assessed after combining individuals from same species. Related to sampling in Materials and Methods. Download Table S1, XLSX file, 0.02 MB (18.9KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ciliate transcriptomes that are obtained from public databases. Related to transcript diversity analyses in Materials and Methods. Download Table S3, XLSX file, 0.01 MB (9.2KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Maximum-likelihood estimates for alignmentwide mean of ω in the negative selection regime (ω ≤ 1) obtained by partitioned exploratory RELAX models. Box plots and individual points are shown side by side for each group. EF, extensive fragmenters; H, Heterotrichea; K, Karyorelictea; NEF, non-extensive fragmenters. Download FIG S1, DOCX file, 0.1 MB (153KB, docx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relaxation/intensification parameters (K) for each branch group relative to NEF are shown (sorted by P value). Related to Table 2. Download Table S5, XLSX file, 0.03 MB (36.8KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
rRNA report of each transcriptome for taxonomy identification. Related to taxonomy assignment in Materials and Methods. Download Table S2, XLSX file, 0.01 MB (11.5KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Number of transcripts in each studied species in the selected 509 gene families. Related to transcript diversity analyses in Materials and Methods. EF, extensive fragmenters; NEF, non-extensive fragmenters. *, Individual cells that are combined for the analyses, respectively. Download Table S7, XLSX file, 0.2 MB (165KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
SSU rRNA gene sequences from focal individuals in the present study. Download Dataset S1, TXT file, 0.1 MB (62.4KB, txt) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ciliate genomes used for stop codon analyses that are obtained from public databases. Related to assessment of stop codon usage and reassignment in Materials and Methods. Download Table S4, XLSX file, 0.01 MB (9.1KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Counts for assessments of stop codon reassignments in ciliates that are not using conventional stop codon usage (UAR and UGA). Related to Fig. 3. Download Table S6, XLSX file, 0.01 MB (14.6KB, xlsx) .
Copyright © 2019 Yan et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Data for single-cell transcriptomic analyses of uncultivable ciliates were deposited in the NCBI database under BioProject accession number PRJNA573114 and BioSample accession numbers SAMN12807523 to SAMN12807565.