

Abstract

CC chemokine receptors 2 (CCR2) and 5 (CCR5) are involved in many inflammatory diseases; however, most CCR2 and CCR5 clinical candidates have been unsuccessful. (Pre)clinical evidence suggests that dual CCR2/CCR5 inhibition might be more effective in the treatment of such multifactorial diseases. In this regard, the highly conserved intracellular binding site in chemokine receptors provides a new avenue for the design of multitarget ligands. In this study, we synthesized and evaluated the biological activity of a series of triazolopyrimidinone derivatives in CCR2 and CCR5. Radioligand binding assays first showed that they bind to the intracellular site of CCR2, and in combination with functional assays on CCR5, we explored structure–affinity/activity relationships in both receptors. Although most compounds were CCR2-selective, 39 and 43 inhibited β-arrestin recruitment in CCR5 with high potency. Moreover, these compounds displayed an insurmountable mechanism of inhibition in both receptors, which holds promise for improved efficacy in inflammatory diseases.
Introduction
CC chemokine receptors 2 (CCR2) and 5 (CCR5) are two membrane-bound G protein-coupled receptors (GPCRs), which belong to the subfamily of chemokine receptors. Chemokine receptors are widely expressed in leukocytes, and thus, they regulate different homeostatic and inflammatory leukocyte functions upon interaction with their endogenous chemokines.1,2 In general, chemokine receptors interact with multiple endogenous chemokines, such as CCL2, CCL7, and CCL8 in the case of CCR2, and CCL3, CCL4, and CCL5 in the case of CCR5.1 Furthermore, most chemokines can interact with multiple chemokine receptors, allowing for a very complex and fine-tuned system.3,4 Dysregulation of this system has been linked to the development of several pathophysiological conditions. For example, both CCR2 and CCR5 have been implicated in many inflammatory and immune diseases such as rheumatoid arthritis, multiple sclerosis, atherosclerosis, diabetes mellitus, and psoriasis,5,6 rendering these proteins attractive targets for the pharmaceutical industry. As a result, many efforts have been made to bring CCR2 and CCR5 small-molecule antagonists into the clinic although with limited success. Only maraviroc, an HIV-1 entry inhibitor selectively targeting CCR5, has been approved by the FDA and EMA,7 while all other drug candidates have failed in clinical trials.
Recently, it has been suggested that the development of multitarget drugs (designed to interact with multiple receptors) represents a more effective approach in the treatment of complex multifactorial diseases.8,9 Thus, dual targeting of CCR2 and CCR5 emerges as a potentially more efficacious strategy in diseases where both receptors are involved. Indeed, combined CCR2/CCR5 inhibition has resulted in beneficial effects in several preclinical disease models and clinical studies, further supporting the use of dual antagonists.10−12 In this regard, several antagonists with dual CCR2/CCR5 activity have been reported in the past years, including the first dual antagonist TAK-779 and the clinical candidate cenicriviroc.13 All of these antagonists bind to the extracellular region of CCR2 and CCR5, in a site overlapping with the chemokine’s binding pocket.14 Yet the crystal structures of CCR2 and CCR9 have demonstrated that chemokine receptors can also be targeted with intracellular allosteric modulators.15,16 These intracellular ligands offer a number of advantages, such as noncompetitive binding and, as a consequence, insurmountable inhibition, which is particularly important due to the high local concentration of chemokines during pathological conditions.17,18 In addition, the high conservation of this intracellular site allows for the design of multitarget antagonists.18,19 Several high-affinity intracellular ligands have been already identified for CCR220,21 but not for CCR5, although intracellular compounds developed for CCR2 or CCR4 have been reported to bind CCR5 with much lower potency.21,22
In the current study we first report that previously patented CCR2 antagonists with a triazolopyrimidinone scaffold, such as compound 8 (Figure 1),23 bind to the intracellular site of the receptor with high affinity. In addition, we show that this compound is able to inhibit CCR5 with moderate activity, suggesting a potential dual CCR2/CCR5 activity for this class of compounds. Thus, a series of novel and previously reported triazolopyrimidinone derivatives were synthesized according to published methods23 in order to obtain structure–affinity/activity relationships (SARs) in both CCR2 and CCR5. Radioligand binding assays and functional assays were used to evaluate their affinity toward CCR2 and activity toward CCR5. In addition, characterization of two selected compounds (39 and 43) in a [35S]GTPγS binding assay demonstrated that these compounds inhibit both receptors in a noncompetitive, insurmountable manner. Finally, selected compounds were docked into the CCR2 crystal structure in order to shed light on the binding mode of these derivatives, in comparison to that of the crystallized CCR2-RA-[R].15 In summary, our findings provide some insight on the CCR2/CCR5 selectivity profile of triazolopyrimidinone derivatives, as well as on the structural requirements for the design of multitarget or selective intracellular ligands for these receptors.
Figure 1.

Chemical structures of the orthosteric CCR2/CCR5 antagonist TAK-779 and the CCR2 intracellular ligands CCR2-RA-[R], SD-24, JNJ-27141491 and the triazolopyrimidinone derivative 8. [3H]-CCR2-RA-[R] was used in radioligand binding assays for CCR2.
Results and Discussion
Chemistry
Triazolo-pyrimidinone derivatives 6–43 were synthesized using a three-step synthesis approach as described by Bengtsson et al.23 (Scheme 1). First, if not commercially available, the β-keto esters 1a–n were synthesized from ethyl acetoacetate 1a and the respective bromo- or iodoalkanes 2f–h,j,k or benzyl bromide 2n. Benzylation of the β-keto esters 1a–n with the corresponding R1-substituted benzyl bromides (3a–v), at reflux, resulted in a series of benzylated β-keto esters 4aa–na, 4bb–bq, 4eq–ev in yields between 8% and 97% (Scheme 1, Table S1). Finally a cyclization reaction of the benzylated β-keto esters 4aa–na, 4bb–bq, 4eq–ev with the commercially available 3,5-diaminotriazole 5c in ionic liquid BMIM-PF6 (1-butyl-3-methylimidazolium hexafluorophosphate) at 200 °C under microwave irradiation resulted in final compounds 6, 9–43 in yields ranging from 4% to 83%. Final compound 7 (R2 = H) was synthesized using H3PO4 in ethanol conditions and 8 (R2 = Me) in p-toluenesulfonic acid monohydrate conditions.
Scheme 1. Synthesis Scheme of the Triazolopyrimidinone Derivatives 6–43.

Reagents and conditions: (i) NaH, n-BuLi, THF, overnight, 0 °C to rt (1a–e,i,l,m were commercially available); (ii) DIPEA, LiCl, THF, reflux, overnight; (iii) (8–43, R2 = NH2) BMIM-PF6, 200 °C, 1 h or (6, R2 = H) H3PO4, EtOH, 170 °C, 10 h or (7, R2 = Me) p-toluenesulfonic acid monohydrate, 180 °C, 30 min.
Biology
We have previously identified several CCR2 intracellular ligands belonging to different chemical scaffolds, such as CCR2-RA-[R], SD-24, and JNJ-27141491 (Figure 1).20,21 In contrast to CCR2 orthosteric ligands, these intracellular ligands lack a basic nitrogen and have lower molecular weights, unsaturated systems with haloarenes, and acidic groups capable of forming hydrogen bonds.18,20 Other CCR2 antagonists with similar features have been described in the literature, including the triazolo- or pyrazolopyrimidinone derivatives described in two different patents.23,24 To test whether they also bind to the intracellular site of the receptor, we synthesized “example 1” from the patent by Bengtsson et al.,23 corresponding to the triazolopyrimidinone derivative 8 in our study (Figure 1). Using a [3H]-CCR2-RA-[R] binding assay as previously described,19 we found that compound 8 fully displaced [3H]-CCR2-RA-[R] binding from U2OS cells stably expressing hCCR2b (U2OS-CCR2) with high affinity and a pseudo-Hill slope (nH) close to unity, indicating a competitive interaction with [3H]-CCR2-RA-[R] for the intracellular binding site. 8 displaced [3H]-CCR2-RA-[R] with a pKi of 8.90 ± 0.04 (Ki = 1.3 nM, Figure 2a and Table 1), consistent with its previously reported activity in a CCR2 calcium flux assay (IC50 = 16 nM).23
Figure 2.
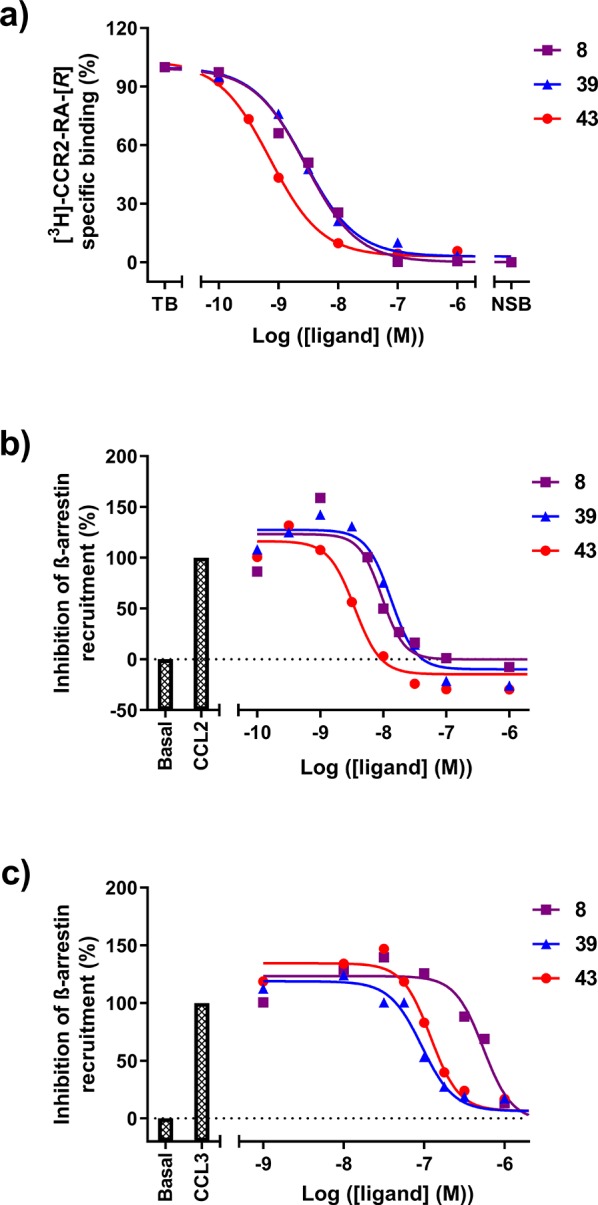
Characterization of ligands in U2OS-CCR2 and U2OS-CCR5. (a) [3H]-CCR2-RA-[R] displacement by increasing concentrations of triazolopyrimidinone derivatives 8, 39, and 43 in U2OS-CCR2 at 25 °C. Data are normalized to specific binding in the absence of compound (set as 100%). (b) Inhibition of CCL2-stimulated β-arrestin recruitment in U2OS-CCR2 by increasing concentrations of compounds 8, 39, and 43, after stimulation with an EC80 concentration of CCL2 (set as 100%). (c) Inhibition of CCL3-stimulated β-arrestin recruitment in U2OS-CCR5 by increasing concentrations of compounds 8, 39, and 43, after stimulation with an EC80 concentration of CCL3 (set as 100%). All data are from single, representative experiments performed in duplicate.
Table 1. Characterization of Compounds 6–23 in hCCR2 and hCCR5a.
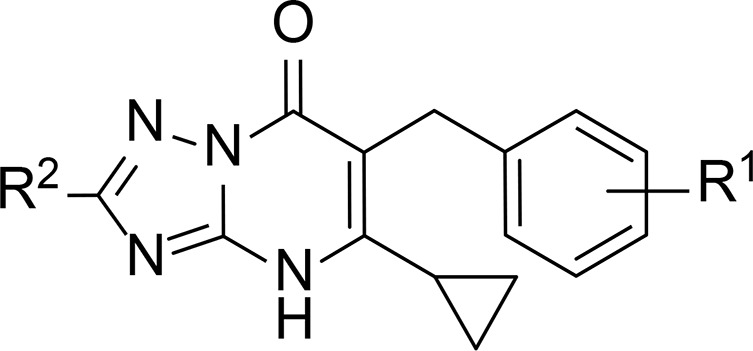
| hCCR2 | hCCR5 | |||
|---|---|---|---|---|
| compd | R1 | R2 | pKi ± SEM (Ki, nM)b | pIC50 ± SEM (IC50, nM) or inhibition at 1 μM (%)c |
| 6 | 3-Cl | H | 8.76 ± 0.01 (1.7) | 60% |
| 7 | 3-Cl | Me | 8.46 ± 0.05 (3.5) | 35% |
| 8 | 3-Cl | NH2 | 8.90 ± 0.04 (1.3) | 6.24 ± 0.004 (571) |
| 9 | H | NH2 | 8.27 ± 0.10 (5.9) | 35% |
| 10 | 2-Me | NH2 | 7.81 ± 0.05 (15.7) | 28% |
| 11 | 2-Cl | NH2 | 7.84 ± 0.03 (14.5) | 27% |
| 12 | 2-OMe | NH2 | 6.98 ± 0.04 (104.6) | –20%d |
| 13 | 3-Me | NH2 | 8.61 ± 0.03 (2.5) | 62% |
| 14 | 3-F | NH2 | 8.53 ± 0.18 (3.4) | 43% |
| 15 | 3-Br | NH2 | 9.08 ± 0.06 (0.9) | 60% |
| 16 | 3-I | NH2 | 9.06 ± 0.02 (0.9) | 66% |
| 17 | 3-OMe | NH2 | 7.89 ± 0.07 (13.0) | –27%d |
| 18 | 3-CF3 | NH2 | 8.26 ± 0.09 (5.9) | 36% |
| 19 | 4-Me | NH2 | 8.46 ± 0.03 (3.5) | –57%d |
| 20 | 4-F | NH2 | 8.39 ± 0.03 (4.1) | 31% |
| 21 | 4-Cl | NH2 | 8.74 ± 0.05 (1.8) | 31% |
| 22 | 4-Br | NH2 | 8.84 ± 0.02 (1.5) | 14% |
| 23 | 4-OMe | NH2 | 7.68 ± 0.05 (20.9) | –28% |
Data are presented as the mean pKi/pIC50 ± standard error of the mean (SEM) and mean Ki/IC50 (nM) of at least three independent experiments performed in duplicate.
pKi values from the displacement of ∼6 nM [3H]-CCR2-RA-[R] from U2OS cells stably expressing CCR2, at 25 °C.
Percent inhibition of β-arrestin recruitment in U2OS cells stably expressing CCR5 by 1 μM compound, in the presence of CCL3 (pEC80 = 7.9). pIC50 values were determined for compounds displaying more than 70% inhibition. % Inhibition values are presented as means values of at least two independent experiments, performed in duplicate.
No inhibition was observed at the concentration of 1 μM; instead some CCL3 stimulation was measured.
Previous studies have shown that some of these intracellular ligands are able to bind and inhibit multiple chemokine receptors, enabling the design of selective and multitarget inhibitors.19,21,22 In this regard, CCR5 is the closest homolog to CCR2, with >90% sequence similarity of their intracellular binding pockets. From the main interactions of CCR2-RA-[R] to CCR2, only Val2446×36 is exchanged to Leu2366×36 in CCR515 (residues named according to structure-based Ballesteros—Weinstein nomenclature25). Thus, we investigated whether compound 8 is also able to inhibit the highly homologous CCR5. However, the much lower affinity of [3H]-CCR2-RA-[R] for CCR5 compared to CCR2 hindered us from performing radioligand binding assays.21 Thus, we assessed the CCR5 activity of 8 with a functional β-arrestin recruitment assay after stimulation with CCL3, one of the endogenous agonists of CCR5. For this assay, we also included the intracellular ligands CCR2-RA-[R], SD-24 and JNJ-27141491, as well as the CCR2/CCR5 orthosteric antagonist TAK-779 as a positive control (Figure 1), since it is a potent CCR5 antagonist in a variety of functional assays.26,27
In this assay, CCL3 induced β-arrestin recruitment to U2OS cells stably expressing hCCR5 (U2OS-CCR5) with a pEC50 of 8.3 ± 0.08 (6 nM) (Figure S1a), similar to values reported in the literature.28 As expected, TAK-779 was able to completely inhibit β-arrestin recruitment induced by an EC80 concentration of CCL3 (pEC80 = 7.9 ± 0.08), when tested at a single concentration of 1 μM (Figure S1b). In contrast, none of the intracellular ligands were able to fully inhibit CCL3-induced β-arrestin recruitment to the same level as TAK-779; in fact, only compound 8 displayed more than 70% inhibition when tested at 1 μM (Figure S1b), while CCR2-RA-[R], SD-24, and JNJ-27141491 led to approximately 50% inhibition or less at the same concentration of 1 μM (Figure S1b). Consistent with this low inhibition in CCR5, it was previously shown that CCR2-RA-[R], JNJ-27141491, and SD-24 inhibited inositol phosphate (IP) formation in CCR5 with 7- to 22-fold lower potency compared to CCR2 inhibition.21 Preincubation of U2OS-CCR5 cells with increasing concentrations of TAK-779, before exposure to CCL3, resulted in an inhibitory potency (IC50) of 6 nM, consistent with previously reported values (Table S2).27 Also in agreement with a previous study,21 the reference intracellular ligand CCR2-RA-[R] inhibited CCL3-induced β-arrestin recruitment with an IC50 value of 703 nM (Table S2). Moreover, while TAK-779 inhibited CCL3-induced β-arrestin recruitment with a pseudo-Hill slope close to unity (nH = −1.1), CCR2-RA-[R] inhibition showed a significantly higher Hill slope (nH = −2.4), indicative of two different binding sites for CCL3 and CCR2-RA-[R] (Table S2).29
As compound 8 was the best CCR5 inhibitor in this assay, displaying an IC50 value of 571 nM and a Hill slope of −2.2 ± 0.3 (Figure 2c and Table 1), we then synthesized several triazolopyrimidinone derivatives to explore their structure–affinity/activity relationships (SARs) in CCR2 and CCR5. All synthesized triazolopyrimidinone derivatives were evaluated in [3H]-CCR2-RA-[R] binding assays to determine their binding affinity for CCR2 and in β-arrestin recruitment assays to determine their activity toward CCR5 (Figure 2 and Tables 1–3). In CCR5, compounds were first screened at a concentration of 1 μM, as we were only interested in dual-targeting compounds with moderate to high potencies (IC50 < 1 μM). For the same reason, only those that displayed >70% inhibition at this concentration were further evaluated in a concentration–inhibition curve to determine their potency. For better comparison, compounds 8, 39, and 43 were also tested in a CCR2 β-arrestin recruitment assay as previously described (Figure 2b).20 Finally, we determined the mechanism of inhibition of 39 and 43 in both CCR2 and CCR5 using a [35S]GTPγS binding assay (Figure 3, Table 4).
Table 3. Characterization of Compounds 37–43 in hCCR2 and hCCR5a.
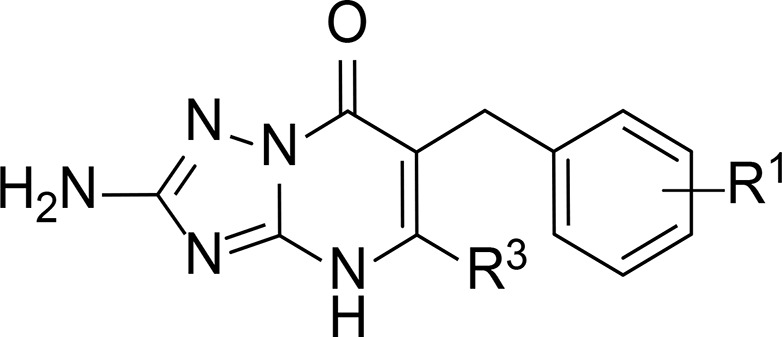
| hCCR2 | hCCR5 | |||
|---|---|---|---|---|
| compd | R1 | R3 | pKi ± SEM (Ki, nM)b | pIC50 ± SEM (IC50, nM) or inhibition at 1 μM (%)c |
| 37 | 3,4-diCl | cPr | 9.35 ± 0.05 (0.4) | 6.67 ± 0.03 (214) |
| 38 | 3,4-diCl | iPr | 9.22 ± 0.05 (0.6) | 6.91 ± 0.09 (132) |
| 39 | 2,3-diCl | iPr | 8.81 ± 0.07 (1.6) | 7.09 ± 0.07 (84) |
| 40 | 2,5-diCl | iPr | 7.65 ± 0.03 (22.5) | 20% |
| 41 | 3,5-diCl | iPr | 8.66 ± 0.05 (2.2) | 6.49 ± 0.06 (336) |
| 42 | 3,5-diBr | iPr | 8.68 ± 0.01 (2.1) | 64% |
| 43 | 3-Br, 4-Cl | iPr | 9.42 ± 0.02 (0.4) | 6.95 ± 0.04 (115) |
Data are presented as mean pKi/pIC50 ± standard error of the mean (SEM) and mean Ki/IC50 (nM) of at least three independent experiments performed in duplicate.
pKi values from the displacement of ∼6 nM [3H]-CCR2-RA-[R] from U2OS cells stably expressing CCR2, at 25 °C.
Percent inhibition of β-arrestin recruitment in U2OS cells stably expressing CCR5 by 1 μM compound, in the presence of CCL3 (pEC80 = 7.9). pIC50 values were determined for compounds displaying more than 70% inhibition. % Inhibition values are presented as mean values of at least two independent experiments, performed in duplicate.
Figure 3.

Characterization of compounds 39 and 43 as insurmountable, negative allosteric modulators using a [35S]GTPγS binding assay in hCCR2 and hCCR5. Effect of increasing concentrations of 39 and 43 in a CCL2-stimulated [35S]GTPγS binding in U2OS-CCR2 (a, b) or in a CCL3-stimulated [35S]GTPγS binding in U2OS-CCR5 (c, d), at 25 °C. Parameters obtained from the concentration–response curves (pEC50, Emax) are summarized in Table 4. Data are presented as mean ± SEM values of three experiments performed in duplicate.
Table 4. Effects of Compounds 39 and 43 in Chemokine-Stimulated [35S]GTPγS Bindinga.
| receptor | compd | pEC50 ± SEM (EC50, nM) | Emax ± SEM (%)b |
|---|---|---|---|
| hCCR2 | CCL2 | 8.10 ± 0.06 (8) | 107 ± 2 |
| CCL2 + 10 nM 39 | 7.89 ± 0.04 (13) | 91 ± 1** | |
| CCL2 + 30 nM 39 | 7.60 ± 0.07 (26)** | 75 ± 4**** | |
| CCL2 + 100 nM 39 | 7.27 ± 0.10 (56)**** | 50 ± 3**** | |
| CCL2 + 1 nM 43 | 7.91 ± 0.10 (13) | 72 ± 4**** | |
| CCL2 + 3 nM 43 | 7.53 ± 0.12 (32)*** | 51 ± 5**** | |
| CCL2 + 10 nM 43 | 6.87 ± 0.13 (148)**** | 33 ± 3**** | |
| hCCR5 | CCL3 | 8.42 ± 0.06 (4) | 108 ± 2 |
| CCL3 + 100 nM 39 | 8.35 ± 0.09 (5) | 79 ± 5**** | |
| CCL3 + 300 nM 39 | 8.14 ± 0.12 (8) | 56 ± 2**** | |
| CCL3 + 1000 nM 39 | 8.14 ± 0.17 (9) | 25 ± 4**** | |
| CCL3 + 30 nM 43 | 8.30 ± 0.05 (5) | 81 ± 3**** | |
| CCL3 + 100 nM 43 | 8.21 ± 0.05 (6) | 58 ± 1**** | |
| CCL3 + 300 nM 43 | 8.05 ± 0.06 (9)* | 35 ± 2**** |
Data represent the mean ± standard error of the mean (SEM) of three independent experiments performed in duplicate. One-way ANOVA with Dunnett’s post hoc test was used to analyze differences in pEC50 and Emax values against CCL2 or CCL3 controls.
Maximum effect (Emax) of CCL2 or CCL3 measured in the absence or presence of fixed concentrations of compound 39 and 43 in CCR2 or CCR5, respectively.
Structure–Affinity/Activity Relationships (SARs) in CCR2 and CCR5
Analysis of the triazolopyrimidinone derivatives started by modifying the amino group (R2) of the triazolo moiety (R2, Table 1). Compared to 8, removing the amino group (6) resulted in a similar affinity toward CCR2, in agreement with the similar reported IC50 values of approximately 20 nM for both compounds, when tested in a calcium flux assay.23 However, in CCR5 6 displayed a lower potency, as the inhibition of CCL3-stimulated recruitment of β-arrestin decreased to 60%, compared to 76% inhibition by 8. The introduction of a methyl group in R2 (7) was less favorable for both receptors, as both affinity for CCR2 and activity to CCR5 were reduced compared to 8. As compound 8 displayed the highest affinity/activity for both receptors, we decided to keep the amino group in R2 and explore different phenyl substituents (R1, Table 1), taking 8 as the starting point.
Compared to 8, the unsubstituted 9 showed a 5-fold decrease in affinity toward CCR2, while in CCR5 it was only able to inhibit 35% of the receptor response at 1 μM. Next, we investigated the effect of several benzyl modifications, including the influence of different substituent positions (Table 1). In the case of CCR2, meta-substituted derivatives yielded the highest affinities in this series of compounds (13–18), whereas ortho-substituted derivatives yielded the lowest (10–12). None of the ortho-substitutions led to an improvement in affinity over 8 or the unsubstituted 9. Introduction of a methyl (10) or a chloro (11) group in this position resulted in affinities lower than 10 nM, while the introduction of an electron-donating methoxy group further reduced the affinity to 105 nM (12), displaying the lowest CCR2 affinity in this series (Table 1). Moving the methyl group to meta (13) or para (19) position slightly improved the CCR2 binding affinity compared to 9, achieving the highest affinity in meta position (19, 3 nM). Similarly, moving the methoxy group to meta or para position resulted in improved affinities following the meta > para > ortho order; however, the affinities remained lower than 10 nM (17, 13 nM; 23, 21 nM), with no improvement over 9. This is consistent with functional data reported in the patent by Bengtsson et al., where similar compounds with a methoxybenzyl moiety displayed a loss of CCR2 activity compared to the unsubstituted-phenyl analogue.23 Substitution of the meta methoxy group by an electron-withdrawing CF3 group resulted in improved affinity over 17 (18, 6 nM) but no improvement over the unsubstituted 9.
The effect of introducing different halogen groups was first investigated in the meta position. Overall, an increase in size and lipophilicity from fluoro to iodo resulted in improved binding affinities toward CCR2 (F, 14 < Cl, 8 < Br, 15 ≈ I, 16). In fact, compounds 15 and 16 displayed the highest affinities in this series of derivatives (15, 0.8 nM; 16, 0.9 nM). Moving the halogen substituents to the para position resulted in a similar trend in affinity (F, 20 < Cl, 21 < Br, 22); however, their affinities were lower compared to the meta-substituted analogues. Of note, compounds with a fluorine atom in meta (14) or para (20) position displayed lower affinities than compounds with a methyl group in the equivalent position (13 and 19). To gain more insight in a potential relationship between affinity and lipophilicity as observed in the halogen series, calculated log P values (cLogP) of compounds 8–23, with R1 modifications, were plotted against their pKi values in CCR2. This analysis revealed only a slight correlation between these two parameters for this set of compounds (Figure S2a); however, this correlation was lost when all synthesized derivatives were included in this plot (Figure S2b), indicating that this is not a general trend.
In the case of CCR5, meta-substituted derivatives also outperformed their ortho- and para-substituted analogues, with some compounds displaying >60% inhibition at 1 μM; in contrast, ortho- and para-substitution resulted in compounds with low (≤31%) to marginal efficacy in CCR5, suggesting that substituents in ortho or para position are not tolerated in CCR5. Similarly as in CCR2, the introduction of a methoxy group was unfavorable, as it led to a complete loss of activity in CCR5 when tested at 1 μM (12, 17, and 23), regardless of the position, whereas electron-withdrawing groups in meta position (18, R2 = CF3) did not bring any improvement over the unsubstituted 9. Except for compound 14 bearing a meta-fluoro, which showed less than 45% inhibition, all other compounds bearing halogens in meta position led to >60% inhibition; the same was achieved when a methyl group was placed in this position (13). Overall, these data indicate that meta-substituents, especially halogens, are preferred to achieve dual CCR2/CCR5 activity, while ortho- and para-substituents lead to a lower affinity but higher selectivity toward CCR2.
As none of the other substituents in R2 led to a significant improvement in CCR5 activity over compound 8, we decided to continue with this compound and investigate the effect of replacing the cyclopropyl moiety in R3. On the basis of the chemical structure of 8 and CCR2-RA-[R] (Figure 1), we hypothesized that the cyclopropyl group in 8 interacts with Val2446×36 in CCR2 in a similar manner as the cyclohexyl group of CCR2-RA-[R].15 Thus, several triazolopyrimidinone derivatives were synthesized with different alkyl chains and aromatic groups in this position in order to investigate their SARs (Table 2). Starting with the effect of alkyl substituents, we observed that increasing the size and flexibility of the alkyl chain from n = 1 (methyl) to n = 4 (butyl) resulted in a parallel increase in CCR2 affinity (17 nM for R3 = Me (24); ∼4 nM for R3 = Et (25) and R3 = Pr (26); 2 nM for R3 = Bu (28)). However, further elongation of the chain length (n = 5–7) led to a progressive drop in affinity (7 nM for R3 = Pent (30); 22 nM for R3 = Hex (32); 178 nM for R3 = Hept (28)), indicating that linear alkyl chains longer than five carbons might not fit in this hydrophobic pocket. The same trend was observed for CCR5 activity, as only the n-propyl (26) and n-butyl (28) substituted compound led to >60% inhibition, albeit without improvement over 8 (28, 519 nM). Moreover, introduction of a hexyl or heptyl group resulted in CCL3 stimulation instead of inhibition, which was not further investigated. Increasing bulkiness via branching of alkyl groups or substitution with aliphatic rings enhanced the affinity toward CCR2, indicating that these substituents might provide a better interaction with the receptor. For instance, the introduction of both isopropyl (27) and cyclopropyl (8) groups led to an improvement in CCR2 affinity compared to the linear analogue 26. Moreover, compound 27 with an isopropyl substituent also yielded a 2-fold increase in CCR5 potency compared to the cyclic analogue 8, displaying the highest potency in this series of compounds (27, 281 nM). In line with this trend, we observed that replacing the linear pentyl group (30) with a cyclopentyl group (31) was also beneficial for CCR2, as this derivative showed a 4.5-fold increased affinity compared to 30 (31, 1.6 nM). In CCR5, 31 inhibited the CCL3-induced response with a potency of 388 nM, showing a slight improvement over compound 8. In contrast, the introduction of a 2-ethylbutyl group (29) resulted in reduced affinity/activity toward both CCR2 and CCR5. These data suggest that the isopropyl group is the preferred R3 substituent when designing CCR2/CCR5 dual antagonists, as this substituent led to the highest potency in CCR5 while maintaining a high affinity for CCR2. Next, inspired by our work on CCR1/CCR2 selectivity of pyrrolone derivatives,19 we investigated whether aromatic substituents are tolerated in this position. As expected from previous studies,19,30 the introduction of aromatic groups decreased 20-fold (34, 27 nM), 40-fold (36, 52 nM), and 122-fold (35, 159 nM) the affinity for CCR2 compared to 8. When tested in CCR5, all derivatives showed a complete loss of activity at 1 μM, indicating that aromatic groups are not favorable for selectivity or dual activity.
Table 2. Characterization of Compounds 24–36 in hCCR2 and hCCR5a.
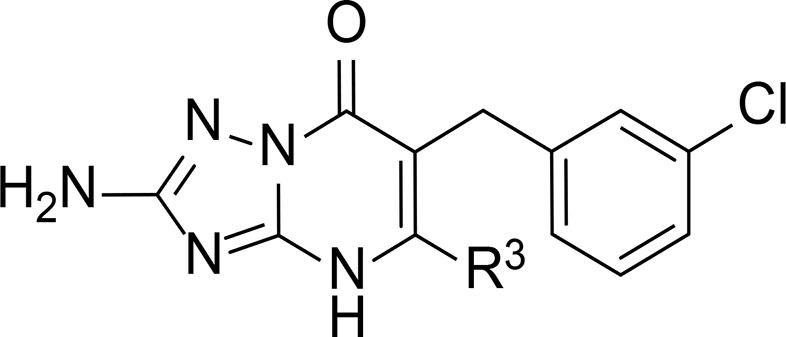
| hCCR2 | hCCR5 | ||
|---|---|---|---|
| compd | R3 | pKi ± SEM (Ki, nM)b | pIC50 ± SEM (IC50, nM) or inhibition at 1 μM (%)c |
| 8 | cPr | 8.90 ± 0.04 (1.3) | 6.24 ± 0.004 (571) |
| 24 | Me | 7.78 ± 0.07 (17.2) | –35% |
| 25 | Et | 8.40 ± 0.07 (4.0) | 29% |
| 26 | Pr | 8.46 ± 0.07 (3.6) | 64% |
| 27 | iPr | 8.72 ± 0.05 (1.9) | 6.56 ± 0.05 (281) |
| 28 | Bu | 8.64 ± 0.03 (2.3) | 6.29 ± 0.05 (519) |
| 29 | 2-EtBu | 8.20 ± 0.04 (6.4) | 29% |
| 30 | Pent | 8.14 ± 0.03 (7.2) | 38% |
| 31 | cPent | 8.81 ± 0.04 (1.6) | 6.43 ± 0.08 (388) |
| 32 | Hex | 7.66 ± 0.02 (22.0) | –63%d |
| 33 | Hept | 6.76 ± 0.05 (178.1) | –265%d |
| 34 | Ph | 7.64 ± 0.17 (26.7) | –41%d |
| 35 | 4-MePh | 6.81 ± 0.07 (158.8) | –13%d |
| 36 | CH2CH2Ph | 7.29 ± 0.05 (52.3) | –42%d |
Data are presented as the mean pKi/pIC50 ± standard error of the mean (SEM) and mean Ki/IC50 (nM) of at least three independent experiments performed in duplicate.
pKi values from the displacement of ∼6 nM [3H]-CCR2-RA-[R] from U2OS cells stably expressing CCR2, at 25 °C.
Percent inhibition of β-arrestin recruitment in U2OS cells stably expressing CCR5 by 1 μM compound, in the presence of CCL3 (pEC80 = 7.9). pIC50 values were determined for compounds displaying more than 70% inhibition. % Inhibition values are presented as mean values of at least two independent experiments, performed in duplicate.
No inhibition was observed at the concentration of 1 μM; instead some CCL3 stimulation was measured.
With the aim of finding dual CCR2/CCR5 intracellular inhibitors, we kept the isopropyl moiety in R3 and investigated the effect of having a disubstituted phenyl moiety in R1 by exploring different positions and combinations of chlorine and bromine atoms (Table 3). First and similar to 8, we kept the cyclopropyl moiety in R3 and combined it with dichlorination in meta and para positions (37). Compared to the monosubstituted analogues 8 and 21, this compound yielded an even higher affinity to CCR2 (37, 0.4 nM); moreover, its ability to inhibit CCL3-induced response in CCR5 was also improved, as the potency increased to 214 nM. By replacing the cyclopropyl of 37 with an isopropyl group (38), we retained affinity for CCR2 (0.6 nM), but the potency for CCR5 increased by almost 2-fold (132 nM), in agreement with the higher potency observed in 27 versus 8 (Table 2). Moving one chlorine atom to the ortho position, while keeping one in the adjacent meta position, yielded compound 39 with slightly lower affinity for CCR2 but even higher potency in CCR5 (39, 84 nM), indicating that although ortho substituents are not preferred in monosubstituted derivatives, they are still tolerated when placed in combination with halogens in other positions. However, placing the two halogens in the second and fifth positions was clearly detrimental for both receptors (40); in CCR2, the affinity decreased by almost 40-fold, while in CCR2, the compound was only able to inhibit 20% of the CCR5 response. Placing the two halogens in the symmetrical third and fifth positions restored the affinity/activity in both receptors (41, 2.2 nM in CCR2 and 336 nM in CCR5). Replacing the two chlorine atoms of 41 by bromine atoms yielded derivative 42, which retained affinity toward CCR2 but led to decrease in CCR5 activity, as this compound was not able to inhibit >70% of the CCL3-induced response. Finally, the combination of a bromo in meta position with a chloro in para position (42) improved both the affinity and activity to both receptors to similar levels as 37, in the case of CCR2, and 38 in the case of CCR5, indicating that halogens in adjacent positions are more favorable for activity in these receptors. Of note, compounds 37, 38, and 43 displayed the highest affinities to CCR2 in this study, while 38, 39, and 43 displayed the highest potencies to CCR5.
It is important to note that so far we are comparing data not only between two different receptors but also between two different assays: (i) a radioligand binding assay for CCR2, in the absence of agonist, which allows the determination of true affinities (pKi values); (ii) a functional assay for CCR5 in the presence of an EC80 concentration of CCL3, without further correction of their IC50 values. To better compare the activities in both receptors, we selected starting compound 8 as well as compounds 39 and 43 (with the highest potency on CCR5 and the highest affinity for CCR2, respectively) and tested these in a previously described β-arrestin recruitment assay for CCR2.20 In this assay, compound 8 inhibited CCL2-stimulated β-arrestin recruitment with a potency of 10 nM and a Hill slope of −2.7, in agreement with its allosteric binding mode. Compound 39 inhibited β-arrestin recruitment in CCR2 with a lower potency of 21 nM, while compound 43 displayed a higher potency of 4 nM, consistent with their affinities. In addition, their Hill slopes (nH = −2.5 for 39; nH = −3.4 for 43) are also indicative of a noncompetitive form of inhibition, a further confirmation of their allosteric binding site located in the intracellular region of CCR2 (Figure 2b and Table S3). Of note, the Hill slopes in CCR5 were comparable to those in CCR2 (nH = −3.7 for 39; nH = −4.4 for 43), i.e., indicating an allosteric interaction at CCR5 as well. Comparing the IC50 values obtained with the functional assays in both receptors, we observe a 4-fold difference between CCR2 and CCR5 in the case of 39, making it a potential dual-antagonist for both receptors. In contrast, the potencies in CCR2 and CCR5 differ by 29-fold in the case of 43, indicating a higher selectivity toward CCR2. Yet, network studies have suggested that partial inhibition by a low-affinity binder might be sufficient to effectively modulate cellular pathways in vivo;31 thus further studies are needed to establish the optimal activity ratio for these receptors in order to achieve in vivo efficacy.32
Mechanism of Inhibition of Selected Compounds
Selected compounds 39 and 43 were also tested in a [35S]GTPγS binding assay in both CCR2 and CCR5 in order to determine their mechanism of inhibition. In the case of CCR2, we have shown that these ligands fully displace [3H]-CCR2-RA-[R], indicating that triazolopyrimidinone derivatives bind in the same intracellular binding site. Thus, these compounds were expected to show noncompetitive, insurmountable antagonism to (orthosteric) chemokine ligands, as previously demonstrated in CCR2 with CCR2-RA-[R]20 and JNJ-27141491.33 To verify this, 39 and 43 were characterized in a previously described [35S]GTPγS binding assay on U2OS-CCR2 membranes.20 In this assay, CCL2-stimulation of [35S]GTPγS binding in CCR2 was examined in the absence or presence of fixed concentrations of 39 and 43 (Table 4 and Figure 3a,b). In the absence of antagonist, increasing concentrations of CCL2 induced [35S]GTPγS binding with an EC50 of 8 nM, in line with previously described parameters.19,20 Co-incubation of CCL2 with 39 or 43 caused a significant reduction in the maximal response of CCL2 (Emax) at all three antagonist concentrations tested. The lowest concentrations of antagonist did not affect the potency of CCL2, while higher concentrations significantly reduced the potency of CCL2 (Table 4 and Figure 3a,b). Of note, both compounds were also tested in the absence of CCL2 at a single concentration of 1 μM to determine potential inverse agonism. At this concentration they only reduced the basal [35S]GTPγS binding levels by 7–8% (Figure S3), providing too small a window to accurately determine their potencies as inverse agonists. Previously, we reported that some intracellular pyrrolone derivatives were also able to decrease the CCR2 basal activity in this assay; however, the effect seemed to be dependent on the assay conditions, such as GDP concentrations.19 Thus, more studies are needed to investigate whether the observed CCR2 constitutive activity is biologically relevant.
To confirm our hypothesis that these two compounds also bind to an allosteric site in CCR5, i.e., the intracellular binding site, we next analyzed the effect of 39 and 43 on CCL3-induced [35S]GTPγS binding in U2OS-CCR5 membranes. In agreement with previous studies, CCL3 stimulated [35S]GTPγS binding in CCR5 with a potency of 4 nM.28 Similarly as in CCR2, the two compounds were able to significantly suppress the maximal response induced by CCL3 at all concentrations tested (Table 4 and Figure 3c,d). However, in contrast to CCR2, the potency of CCL3 was only significantly reduced with the highest concentration of 43 (Table 4). Such depression of the maximal response with or without a decrease of agonist potency is typical of insurmountable antagonists,34 indicating that 39 and 43 behave as insurmountable antagonists at both CCR2 and CCR5. Of note, insurmountable antagonism can be generally achieved by two different mechanisms: allosteric binding or slow binding kinetics, i.e., slow equilibration of a competitive antagonist.34 However, insurmountable inhibition due to hemiequilibrium is only evident in preincubation experiments, where the receptor is preincubated with the antagonist before exposure to the agonist.34 In contrast, allosteric binding leads to insurmountable inhibition in co-incubation experiments, as performed in this study. These data further support our hypothesis that 39 and 43 bind to an allosteric binding site in CCR5, most probably located intracellularly.
Docking Study
To further investigate the binding mode of triazolopyrimidinone derivatives, compounds 8, 39, 40, and 43 were docked into a CCR2b model based on the crystal structure of CCR2 (PDB code 5T1A, Figures 4 and S4).15 Due to the close proximity to the intracellular binding site, several residues from the intracellular loop 3 (ICL3) had to be modeled based on the crystal structure of CCR5 (PDB code 4MBS),35 since they were mutated in the original CCR2 crystal structure to further stabilize the receptor. As seen in Figure 4a, 43 was predicted to adopt a similar binding pose as that of the previously cocrystallized ligand CCR2-RA-[R].15 The disubstituted phenyl group of 43 was constrained to overlap with the corresponding phenyl group of CCR2-RA-[R], since the disubstituted aromatic rings of JNJ-27141491 and SD-24 (Figure 1) were also predicted to overlay with the phenyl group of CCR2-RA-[R] in a previous study.21 However, the bromine group of compound 43 is predicted to form a halogen bond with the backbone of Val1×53, which might contribute to the higher affinity of 43 versus CCR2-RA-[R] (Figure 4b). The positions of the halogens in this phenyl group might also explain the results of our SAR study. For instance, compound 40 with no halogen group on position 3 is not able to form halogen bonds with the receptor; in addition, one of the chlorine groups seems to point upward toward Tyr7×53, resulting in a sterically unfavorable position and thus in the observed lower activity (Figure S4 and Table 3). Similar to 43, both 8 and 39 contain a chlorine group in position 3, promoting the formation of a halogen bond with the backbone of Val1×53, which might result in the improved activity compared to 40 (Figure S4 and Table 3). However, bromine displays a larger σ-hole and therefore a higher halogen bond strength compared to chlorine,36 which might result in the higher affinity of 43, containing a bromine group, compared to 8 or 39 containing a chlorine. As the SAR follows a similar trend in CCR5, these data suggest that the intracellular ligands share similar interactions between the aromatic group and both receptors (Table 3).
Figure 4.
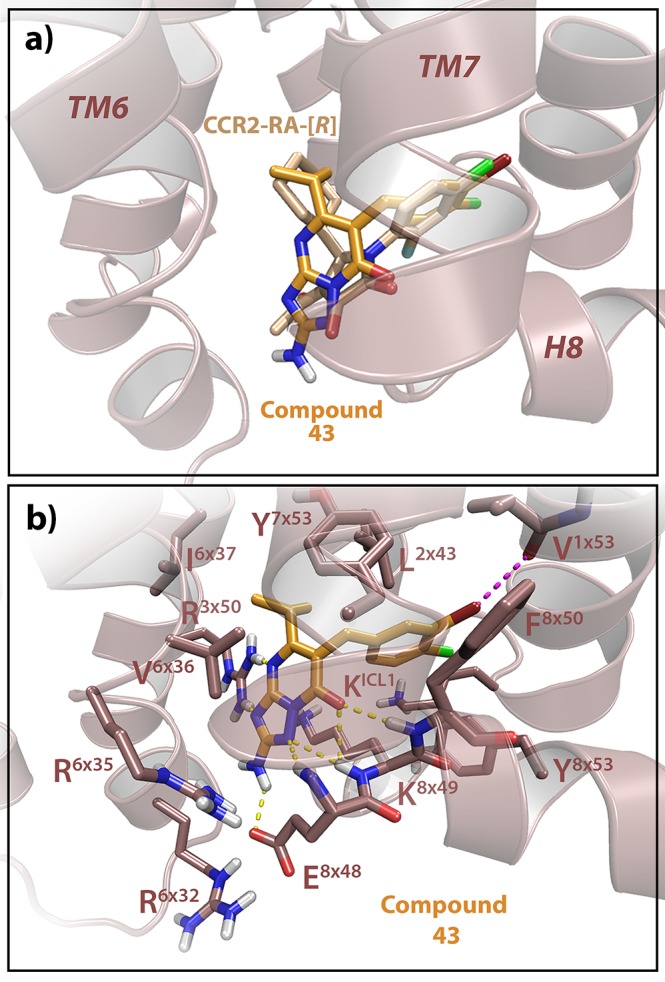
Proposed binding mode of 43 in hCCR2b. (a) Overlay of 43 with the CCR2 intracellular ligand CCR2-RA-[R], showing that 43 interacts in a similar manner as CCR2-RA-[R]. (b) Docking of 43, displaying the interactions with CCR2. The amino group in R2 makes an extra hydrogen-bond interaction with E8×48, while the bromine group in R1 makes an extra halogen bond with the backbone of V1×53, which might contribute to the improved affinity of 43 to this receptor. Model of hCCR2 is based on the crystal structure of CCR2 (PDB code 5T1A),15 and amino acid residues are labeled according to their structure-based Ballesteros–Weinstein numbers.25
In addition, the isopropyl group present in compounds 39, 40, and 43 is predicted to bind in the same position as the cyclohexyl moiety of CCR2-RA-[R], although it seems to make less interactions with Val6×36 perhaps due to the slightly different ligand orientation (Figure 4b). Previous studies have confirmed the crucial role of Val6×36 for binding affinity of some intracellular ligands in CCR2, as mutation of this residue to alanine completely abolished binding of CCR2-RA-[R] to the receptor.21 Moreover, this residue might be involved in target selectivity, as the main difference between the intracellular pockets of CCR2 and CCR5 is the single substitution of Val6×36 by Leu6×36. The steric hindrance introduced by this substitution might thus be responsible for the reduction in activity of CCR2-RA-[R] and the triazolopyrimidinone derivatives toward CCR5 compared to CCR2.21 Indeed, in the case of CCR5, only small aliphatic groups were tolerated in R3 position, such as cyclopropyl or isopropyl (Table 2), while bigger aliphatic groups resulted in improved selectivity toward CCR2. In line with the role of Val6×36 as determinant of selectivity, a previous SAR analysis of pyrrolone derivatives in CCR1, which also contains a leucine in position 6 × 36, showed that aromatic groups in the equivalent R3 position provide CCR1 selectivity versus CCR2, as aromatic groups are not tolerated in this position in CCR219 (Table 2).
The binding pose of 43 seems to be stabilized by a network of hydrogen bonds between the triazolopyrimidinone core and residues E8×48, Lys8×49, F8×50, and R3×50 (Figure 4b). Although the core of CCR2-RA-[R] and 43 binds with a different orientation, the carboxy group of both is overlaid in the same position, interacting with the backbones of Lys8×49 and F8×50. Moreover, the secondary and tertiary amino groups present in the triazolopyrimidinone core also form hydrogen bonds with the backbones of Lys8×49 and Glu8×48, as well as with the side chains of Arg3×50. Finally, the primary amino group in position R2 of compound 43 also makes an extra hydrogen bond with the side chain of E8×48. Such extended network of hydrogen bond interactions is not present with CCR2-RA-[R], and thus it might also be responsible for the higher affinity of 43 in CCR2, compared to CCR2-RA-[R]. In addition, the SAR data suggest that this interaction is also crucial in CCR5, as the removal of this amino group (compounds 6 and 7) was detrimental for CCR5 activity (Table 1). Previous studies have confirmed the importance of residues 8 × 49 and/or 8 × 50 in chemokine receptors for the binding of several intracellular ligands. For example, alanine mutations of Lys8×49 and F8×50 in CCR2 caused a 10-fold reduction or a complete loss of affinity of intracellular ligands, respectively, compared to the wild-type receptor.21 In CXCR2, alanine mutation of Lys8×49 led to a reduced affinity of three different intracellular ligands, while the mutation F8×50A only affected one of the ligands tested, indicating a different binding mode.37 Moreover, Lys8×49 has been suggested as a key residue for target selectivity between CXCR1 and CXCR2, as it is exchanged by Asn8×49 in CXCR1.38 In addition, the crystal structure of CCR9 in complex with vercirnon16 also shows a binding interaction between the ligand and Arg3238×49 and Phe3248×50.
Overall, these data suggest that although the intracellular pockets of CCR2 and CCR5 are quite conserved, the design of multitarget compounds is not quite straightforward. Moreover, several of these residues have been shown to be involved in Gαi coupling in recent cryoelectron microscopy (cryo-EM)-derived GPCR structures, including residues 3 × 50, 6 × 29, 6 × 32 to 6 × 37, 8 × 47, and 8 × 49.39−41 Similarly, homologous residues are also involved in direct interactions between rhodopsin and arrestin,42 suggesting a direct interference of these intracellular ligands with the Gαi protein and β-arrestin binding site, and the possibility of fine-tuning residue interactions for the design of biased ligands. On the basis of the SAR analysis and the docking study, these compounds could be further optimized by exploring the triazolopyrimidinone core. For instance, pyrazolopyrimidinones have also been described for CCR2,24 which might also display CCR5 activity. In addition, exploring other halogen combinations at the phenyl group in R1 or other small, bulky aliphatic groups in R3 such as cyclobutyl might lead to compounds with improved dual activity. Although out of the scope of this manuscript, ligands such as 39 with high potency in CCR5 could be investigated as potential tools to further study binding interactions in CCR5, i.e., by obtaining a radiolabeled tool compound or by obtaining a CCR5 crystal structure in complex with an intracellular ligand. Finally, these ligands can be used in future experiments designed to investigate their functional effects both in vitro and in vivo, to validate the target combination, and to establish the required level of target modulation.43
Conclusions
In this study we first confirmed that the triazolopyrimidinone derivative 8 binds to the intracellular pocket of CCR2 in a similar manner as the reference intracellular ligand CCR2-RA-[R]. Moreover, compound 8 was also able to inhibit CCR5 in a functional β-arrestin recruitment assay; thus, we took this compound as a starting point for the synthesis of a series of novel and previously described triazolopyrimidinone derivatives. Using [3H]-CCR2-RA-[R] binding assays and functional β-arrestin recruitment assays, we explored structure–affinity/activity relationships (SARs) in both receptors. Overall, these compounds were mostly selective toward CCR2; however, CCR5 activity was increased with the combination of a primary amino group in R2 position, an isopropyl moiety in R3, and two halogens placed in adjacent positions at the phenyl group in R1. Overall, these findings indicate that even though the intracellular pockets of CCR2 and CCR5 are highly conserved, selectivity of intracellular ligands can be fine-tuned, allowing the design of either selective or multitarget ligands. Evaluation of compounds 39 and 43 in a [35S]GTPγS binding assay indicates that both compounds display a noncompetitive, insurmountable mode of inhibition in CCR2 and CCR5, which might represent a therapeutic advantage in inflammatory diseases characterized by a high local concentration of endogenous chemokines, such as multiple sclerosis and rheumatoid arthritis. Thus, in diseases where selective chemokine receptor antagonists have been largely unsuccessful, the development of multitarget, intracellular ligands for CCR2 and CCR5 is warranted to further study the effects of multitarget versus selective inhibition, as these ligands may represent a novel therapeutic option in these diseases.
Experimental Section
Chemistry. General Methods
All solvents and reagents used were of analytical grade and from commercial sources. Demineralized water was used in all cases, unless stated otherwise, and is simply referred to as H2O. Microwave-based synthesis was carried out using a Biotage Initiator equipment (Biotage, Sweden). All reactions were monitored by thin-layer chromatography (TLC) using aluminum plates coated with silica gel 60 F254 (Merck), and compounds were visualized under ultraviolet light at 254 nm or via KMnO4 staining. Column chromatography for compound purification was performed using silica gel (Merck Millipore) with particle size 0.04–0.63 mm. Chemical identity of final compounds was established using 1H NMR and liquid chromatography–mass spectrometry (LC–MS). 1H NMR spectra were recorded on a Bruker AV 400 liquid spectrometer (1H NMR, 400 MHz) at room temperature. Compound 39, as one of the most soluble and one of the best CCR2/CCR5 antagonists, was fully characterized (1H NMR, 13C NMR, and attached proton test (APT)) on a Bruker AV500 spectrometer at 80 °C. The corresponding NMR spectra of compound 39 are shown in Figures S5–S7. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) in Hz. Liquid chromatography–mass spectrometry (LC–MS) of final compounds was performed using a Thermo Finnigan Surveyor LCQ Advantage Max LC–MS system and a Gemini C18 Phenomenex column (50 mm × 4.6 mm, 3 μm). Analytical purity of the compounds was determined using a Shimadzu high pressure liquid chromatography (HPLC) equipment with a Phenomenex Gemini column (3 x C18 110A column, 50 mm × 4.6 mm, 3 μm). A flow rate of 1.3 mL/min and an elution gradient of 10–90% MeCN/H2O (0.1% TFA) were used. The absorbance of the UV spectrophotometer was set at 254 nm. All compounds tested in biological assays showed a single peak at the designated retention time and were ≥95% pure. Sample preparations for HPLC and LC–MS were as follows unless stated otherwise: 0.3 mg/mL of compound was dissolved in a 1:1:1 mixture of H2O:MeOH:tBuOH. Of note, some compounds required DMSO and heat to ensure proper dissolution. None of the final compounds were identified as potential pan-assay interference compounds (PAINS) after assessment with the free ADME-Tox filtering tool (FAF-Drugs4),44,45 which uses three different PAINS filters based on Baell et al.46
General Procedure 1: Synthesis of β-Keto Esters 1f–h,j,k,n47
In a flame-dried round-bottom flask under a nitrogen atmosphere, ethyl acetoacetate (2.53 mL, 20.0 mmol, 1.00 equiv) was added dropwise to a suspension of NaH (880 mg, 22.1 mmol 1.10 equiv) in dry THF (5 mL) at 0 °C while stirring. After 20 min, n-butyllithium (20 0.0 mmol, 2.50 M solution in pentane,1.00 equiv) was added dropwise to the mixture and stirred for further 30 min. The respective alkyl halide 2f–h,j,k or benzyl bromide 2n (1.20 equiv) was subsequently added dropwise over a period of 10 min to the dianion solution after which the solution was allowed to reach rt. After 14 h, the reaction was quenched by the addition of saturated NH4Cl (aq, 80 mL). The mixture was subsequently extracted with diethyl ether (2 × 120 mL). The combined organic fractions were washed with brine (80 mL) and dried over MgSO4 followed by concentration in vacuo. The crude products were purified by flash chromatography (CH2Cl2/petroleum ether and/or EtOAc/petroleum ether as the eluent) to give the title compounds 1f–h,j,k,n as oils. Compounds 1a–e,i,l,m were commercially available.
Ethyl-3-oxoheptanoate (1f)47
Compound 1f was synthesized according to general procedure 1, using 1-bromopropane (2f, 2.00 mL, 22.0 mmol, 1.10 equiv) as starting compound. Compound was purified by silica column chromatography (1–30% EtOAc in petroleum ether). Yield: 36% (1.25 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.19 (q, J = 7.2 Hz, 2H), 3.44 (s, 2H), 2.54 (t, J = 7.4 Hz, 2H), 1.65–1.55 (m, 2H), 1.38–1.25 (m, 7H), 0.88 (t, J = 6.8 Hz, 3H) ppm.
Ethyl 5-Ethyl-3-oxoheptanoate (1g)
Compound 1g was synthesized according to general procedure 1, using 3-bromopentane (2g, 3.00 mL, 24.2 mmol, 1.21 equiv) as starting compound. Compound was purified by silica column chromatography (1–30% EtOAc in petroleum ether). Yield: 18% (630 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.17 (q, J = 7.2 Hz, 2H), 3.40 (s, 2H), 2.43 (d, J = 6.8 Hz, 2H), 1.88–1.73 (m, 1H), 1.33–1.21 (m, 7H), 0.82 (t, J = 7.4 Hz, 6H) ppm.
Ethyl 3-Oxooctanoate (1h)47
Compound 1h was synthesized according to general procedure 1, using 1-bromobutane (2h, 2.59 mL, 24.1 mmol, 1.21 equiv) as starting compound. Compound was purified by silica column chromatography (1–30% EtOAc in petroleum ether). Yield: 38% (1.41 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.19 (q, J = 7.2 Hz, 2H), 3.43 (s, 2H), 2.53 (t, J = 7.2 Hz, 2H), 1.65–1.55 (m, 2H), 1.37–1.12 (m, 7H), 0.89 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 3-Oxononanoate (1j)47
Compound 1j was synthesized according to general procedure 1, using 1-iodopentane (2j, 3.13 mL, 24.0 mmol, 1.20 equiv) as starting compound. Compound was purified by silica column chromatography (1–30% EtOAc in petroleum ether). Yield: 44% (1.76 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.19 (q, J = 7.2 Hz, 2H), 3.43 (s, 2H), 2.53 (t, J = 7.4 Hz, 2H), 1.54–1.64 (m, 2H), 1.33–1.24 (m, 9H), 0.88 (t, J = 6.8 Hz, 3H) ppm.
Ethyl 3-Oxodecanoate (1k)48
Compound 1k was synthesized according to general procedure 1 using 1-bromohexane (2k, 3.36 mL, 25.0 mmol, 1.24 equiv) as starting compound. Compound was purified by silica column chromatography (30–100% CH2Cl2 in petroleum ether). Yield: 15% (625 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.19 (q, J = 7.2 Hz, 2H), 3.43 (s, 2H), 2.53 (t, J = 7.4 Hz, 2H), 1.64–1.53 (m, 2H), 1.33–1.24 (m, 11H), 0.88 (t, J = 7.5 Hz, 3H) ppm.
Ethyl 3-Oxo-5-phenylpentanoate (1n)49
Compound 1n was synthesized according to general procedure 1, using benzyl bromide (2n, 2.90 mL, 24.4 mmol, 1.22 equiv) as starting compound. Compound was purified by silica column chromatography (1–30% EtOAc in petroleum ether). Yield: 20% (1.06 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.33–7.23 (m, 2H), 7.23–7.12 (m, 3H), 4.17 (q, J = 7.2 Hz, 2H), 3.42 (s, 2H), 2.98–2.81 (m, 4H), 1.26 (t, J = 7.2 Hz, 3H) ppm.
General Procedure 2. Benzylated β-Keto Esters 4aa–na, 4bb–bq, 4eq–ev23
LiCl (1.00 equiv) was slurried in anhydrous THF (1 mL/mmol 1a–n) in a flame-dried round-bottom flask and under an atmosphere of nitrogen. The desired β-keto ester 1a–n (1.00 equiv) was added and followed by DIPEA (2.00 equiv) and the respective benzylic halide 3a–v (1.20 equiv). The reaction mixture was refluxed for 20 h, after which the reaction was completed as indicated by TLC (5–10% EtOAc in petroleum ether). THF was removed in vacuo, the crude dissolved in EtOAc (30 mL), and this organic layer was washed with citric acid (5%, 25 mL) followed by saturated NaHCO3 (25 mL) and brine (25 mL). The organic layer was subsequently dried over MgSO4, and concentrated in vacuo to afford the crude product. The crude product was purified by flash column chromatography (5–10% EtOAc/petroleum ether) to yield the corresponding benzylated β-keto esters 4aa–na, 4bb–bq, 4eq–ev.
Ethyl 2-(3-Chlorobenzyl)-3-oxobutanoate (4aa)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxobutanoate 1a (0.37 mL, 2.92 mmol, 1.20 equiv), 3-chlorobenzyl bromide 3a (0.32 mmol, 2.43 mmol, 1.00 equiv), DIPEA (0.85 mL, 4.86 mmol, 2.00 equiv), LiCl (103 mg, 2.43 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 70% (433 mg) as a colorless oil. 1H NMR: (400 MHz, CDCl3): δ 7.21–7.14 (m, 3H), 7.09–7.04 (m, 1H), 4.16 (q, J = 7.2 Hz, 2H), 3.75 (t, J = 8.0 Hz, 1H), 3.17–3.07 (m, 2H), 2.21 (s, 3H), 1.21 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 3-cyclopropyl-2-(3-chlorobenzyl)-3-oxopropanoate (4ba).23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (7.56 mL, 51.2 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (7.06 mL, 53.8 mmol, 1.05 equiv), DIPEA (17.8 mL, 102 mmol, 2.00 equiv), LiCl (2.17 g, 51.22 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 71% (10.2 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.21–7.l6 (m, 3H), 7.10–7.05 (m, 1H), 4.17 (qd, J = 7.2, 1.6 Hz, 2H), 3.89 (t, J = 7.6 Hz, 1H), 3.15 (dd, J = 7.2 Hz, 2H), 2.08–2.02 (m, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.11–1.01 (m, 2H), 0.98–0.88 (m, 2H) ppm.
Ethyl 2-Benzyl-3-cyclopropyl-3-oxopropanoate (4bb)24
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.52 mL, 3.51 mmol, 1.20 equiv), benzyl bromide 3b (0.347 mL, 2.92 mmol, 1.00 equiv), DIPEA (1.02 mL, 5.84 mmol, 2.00 equiv), LiCl (124 mg, 2.92 mmol, 1.00 equiv), 4 mL of dry THF. Yield: 26% (105 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.29–7.25 (m, 2H), 7.22–7.18 (m, 3H), 4.20–4.12 (m, 2H), 3.91 (t, J = 7.6 Hz, 1H), 3.20 (d, J = 7.6 Hz, 2H), 2.08–2.02 (m, 1H), 1.20 (t, J = 7.2 Hz, 3H), 1.06–1.03 (m, 2H), 0.94–0.84 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(2-methylbenzyl)-3-oxopropanoate (4bc)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.63 mL, 4.27 mmol, 1.20 equiv), 2-methylbenzyl chloride 3c (0.48 mL, 3.56 mmol, 1.00 equiv), DIPEA (1.24 mL, 7.12 mmol, 2.00 equiv), LiCl (151 mg, 3.56 mmol, 1.00 equiv), 4 mL of dry THF. Yield: 80% (742 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.15–7.09 (m, 4H), 4.16 (qd, J = 7.2, 1.2 Hz, 2H), 3.91 (t, J = 7.2 Hz, 1H), 3.20 (d, J = 7.6 Hz, 2H), 2.34 (s, 3H), 2.05–2.00 (m, 1H), 1.20 (t, J = 7.2 Hz, 3H), 1.07–1.02 (m, 2H), 0.95–0.84 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(2-chlorobenzyl)-3-oxopropanoate (4bd)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.43 mL, 2.92 mmol, 1.20 equiv), 2-chlorobenzyl bromide 3d (0.32 mL, 2.43 mmol, 1.00 equiv), DIPEA (0.85 mL, 4.86 mmol, 2.00 equiv), LiCl (103 mg, 2.43 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 78% (532 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.36–7.33 (m, 1H), 7.26–7.24 (m, 1H), 7.18–7.14 (m, 2H), 4.20–4.08 (m, 3H), 3.37–3.22 (m, 2H), 2.10–2.03 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.09–1.01 (m, 2H), 0.97–0.87 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(2-methoxybenzyl)-3-oxopropanoate (4be)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.33 mL, 2.26 mmol, 1.20 equiv), 2-methoxybenzyl bromide503e (378 mg, 1.88 mmol, 1.00 equiv), DIPEA (0.66 mL, 3.76 mmol, 2.00 equiv), LiCl (79.7 mg, 1.88 mmol, 1.00 equiv), 5 mL of dry THF. Silica column chromatography in 8:1:1 petroleum ether:EtOAc:CH2Cl2. Yield: 36% (185 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.20 (td, J = 6.4, 1.6 Hz, 1H), 7.13 (dd, J = 6.0, 1.2 Hz, 1H), 6.86–6.26 (m, 2H), 4.17–4.09 (m, 2H), 4.04 (dd, J = 6.8, 1.6 Hz, 1H), 3.84 (s, 3H), 3.42–3.11 (m, 2H), 2.07–2.02 (m, 1H), 1.19 (t, J = 6.0 Hz, 3H), 1.05–1.02 (m, 2H), 0.92–0.85 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(3-methylbenzyl)-3-oxopropanoate (4bf)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.47 mL, 3.24 mmol, 1.20 equiv), 3-methylbenzyl bromide 3f (0.36 mL, 2.70 mmol, 1.00 equiv), DIPEA (0.94 mL, 5.40 mmol, 2.00 equiv), LiCl (114 mg, 2.70 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 74% (521 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.17 (t, J = 7.6 Hz, 1H), 7.02–6.96 (m, 3H), 4.19–4.10 (m, 2H), 3.90 (t, J = 7.6 Hz, 1H), 3.15 (d, J = 7.6 Hz, 2H), 2.30 (s, 3H), 2.07–2.01 (m, 1H), 1.19 (t, J = 7.2 Hz, 3H), 1.04–0.99 (m, 2H), 0.93–0.82 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(3-fluorobenzyl)-3-oxopropanoate (4bg).23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.550 mL, 3.73 mmol, 1.31 equiv), 3-fluorobenzyl bromide 3g (0.350 mL, 2.85 mmol, 1.00 equiv), DIPEA (0.940 mL, 5.39 mmol, 1.89 equiv), LiCl (0.140 g, 2.70 mmol, 0.947 equiv), 5 mL of dry THF. Yield: 100% (770 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.26–7.16 (m, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.94–6.84 (m, 2H), 4.19–4.11 (m, 2H), 3.92 (t, J = 7.6 Hz, 1H), 3.18 (dd, J = 7.6, 1.6 Hz, 2H), 2.10–2.04 (m, 1H), 1.20 (t, J = 7.2 Hz, 3H), 1.07–0.98 (m, 2H), 0.93–0.83 (m, 2H) ppm.
Ethyl 2-(3-Bromobenzyl)-3-cyclopropyl-3-oxopropanoate (4bh)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.35 mL, 2.40 mmol, 1.20 equiv), 3-bromobenzyl bromide 3h (500 mg, 2.00 mmol, 1.00 equiv), DIPEA (0.70 mL, 4.00 mmol, 2.00 equiv), LiCl (85 mg, 2.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 47% (303 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.36–7.32 (m, 2H), 7.17–7.10 (m, 2H), 4.17 (q, J = 7.2 Hz, 2H), 3.89 (t, J = 7.2 Hz, 1H), 3.15 (d, J = 8.0 Hz, 2H), 2.08–2.01 (m, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.11–1.01 (m, 2H), 0.98–0.86 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(3-iodobenzyl)-3-oxopropanoate (4bi)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.550 mL, 3.73 mmol, 1.38 equiv), 3-iodobenzyl bromide 3i (0.802 g, 2.70 mmol, 1.00 equiv), DIPEA (0.940 mL, 5.39 mmol, 2.00 equiv), LiCl (0.150 g, 2.70 mmol, 1.00 equiv), 5 mL of dry THF. Yield: quantitative (1.28 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.55–7.51 (m, 2H), 7.15 (d, J = 7.6 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 4.16 (qd, J = 7.2, 1.6 Hz, 2H), 3.87 (t, J = 7.2 Hz, 1H), 3.11 (dd, J = 8.0, 2.0 Hz, 2H), 2.06–2.00 (m, 1H), 1.20 (t, J = 6.8 Hz, 3H), 1.11–1.00 (m, 2H), 0.98–0.81 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(3-methoxybenzyl)-3-oxopropanoate (4bj)51
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.44 mL, 3.00 mmol, 1.20 equiv), 3-methoxybenzyl bromide 3j (0.35 mL, 2.50 mmol, 1.00 equiv), DIPEA (0.87 mL, 5.00 mmol, 2.00 equiv), LiCl (106 mg, 2.50 mmol, 1.00 equiv), 5 mL of dry THF. Silica column chromatography in 8:1:1 petroleum ether:EtOAc:CH2Cl2. Yield: 42% (294 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.19 (td, J = 7.2 Hz, 1.2 Hz, 1H), 6.79–6.74 (m, 3H), 4.21–4.13 (m, 2H), 3.91 (t, J = 7.6 Hz, 1H), 3.78 (s, 3H), 3.17 (d, J = 7.6 Hz, 2H), 2.08–2.02 (m, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.06–1.01 (m, 2H), 0.96–0.85 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-3-oxo-2-(3-(trifluoromethyl)benzyl)propanoate (4bk)24
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.37 mL, 2.51 mmol, 1.20 equiv), 3-(trifluoromethyl)benzyl bromide 3k (0.32 mL, 2.09 mmol, 1.00 equiv), DIPEA (0.73 mL, 4.18 mmol, 2.00 equiv), LiCl (89 mg, 2.09 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 31% (214 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 2H), 7.42–7.38 (m, 2H), 4.17 (q, J = 6.8 Hz, 2H), 3.92 (t, J = 8.0 Hz, 1H), 3.25 (d, J = 7.2 Hz, 2H), 2.10–2.02 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.12–1.01 (m, 2H), 0.99–0.86 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(4-methylbenzyl)-3-oxopropanoate (4bl)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.48 mL, 3.24 mmol, 1.20 equiv), 4-methylbenzyl bromide 3l (500 mg, 2.70 mmol, 1.00 equiv), DIPEA (0.94 mL, 5.40 mmol, 2.00 equiv), LiCl (114 mg, 2.70 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 74% (521 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.08 (s, 4H), 4.22–4.09 (m, 2H), 3.88 (t, J = 7.6 Hz, 1H), 3.15 (d, J = 7.6 Hz, 2H), 2.30 (s, 3H), 2.07–2.02 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.07–1.02 (m, 2H), 0.96–0.83 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(4-fluorobenzyl)-3-oxopropanoate (4bm)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.890 mL, 6.04 mmol, 1.14 equiv), 1-(bromomethyl)-4-fluorobenzene 3m (1.57 g, 5.29 mmol, 1.00 equiv), DIPEA (1.05 mL, 6.00 mmol, 1.13 equiv), LiCl (0.130 g, 3.00 mmol, 0.58 equiv), 5 mL of dry THF. Yield: 56% (780 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.18–7.12 (m, 2H), 6.96 (tt, J = 8.8, 2.0 Hz, 2H), 4.22–4.10 (m, 2H), 3.87 (t, J = 8.0 Hz, 1H), 3.16 (d, J = 7.6 Hz, 2H), 2.08–2.00 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.09- 0.99 (m, 2H), 0.97–0.84 (m, 2H) ppm.
Ethyl 2-(4-Chlorobenzyl)-3-cyclopropyl-3-oxopropanoate (4bn)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.43 mL, 2.92 mmol, 1.20 equiv), 4-chlorobenzyl bromide 3n (500 mg, 2.43 mmol, 1.00 equiv), DIPEA (0.85 mL, 4.86 mmol, 2.00 equiv), LiCl (103 mg, 2.43 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 66% (451 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.24 (dt J = 8.8, 2.0 Hz,, 2H), 7.13 (dt, J = 8.4, 2.0 Hz, 2H), 4.21–4.11 (m, 2H), 3.87 (dd, J = 8.0, 0.8 Hz, 1H), 3.15 (dd, J = 6.8, 1.2 Hz, 2H), 2.08–2.01 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.09–1.01 (m, 2H), 0.97–0.86 (m, 2H) ppm.
Ethyl 2-(4-Bromobenzyl)-3-cyclopropyl-3-oxopropanoate (4bo)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.880 mL, 5.97 mmol, 1.49 equiv), 1-(bromomethyl)-4-bromobenzene 3o (1.00 g, 4.00 mmol, 1.00 equiv), DIPEA (1.39 mL, 8.00 mmol, 2.00 equiv), LiCl (0.170 g, 4.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 67% (0.880 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.4 Hz, 2H), 7.07 (d, J = 8.4 Hz, 2H), 4.22–4.10 (m, 2H), 3.87 (t, J = 8.0 Hz, 1H), 3.14 (d, J = 8.2 Hz, 2H), 2.08–1.96 (m, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.10–0.99 (m, 2H), 0.99–0.85 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(4-methoxybenzyl)-3-oxopropanoate (4bp)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.57 mL, 3.83 mmol, 1.20 equiv), 4-methoxybenzyl bromide 3p (0.46 mL, 3.19 mmol, 1.00 equiv), DIPEA (1.11 mL, 6.38 mmol, 2.00 equiv), LiCl (135 mg, 3.19 mmol, 1.00 equiv), 5 mL of dry THF. Silica column chromatography in 7:1:2 petroleum ether:EtOAc:CH2Cl2. Yield: 52% (454 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.10 (dt, J = 8.8, 2.0 Hz, 2H), 6.81 (dt, J = 8.8, 2.4 Hz, 2H), 4.20–4.10 (m, 2H), 3.86 (t, J = 7.6 Hz, 1H), 3.78 (s, 3H), 3.13 (d, J = 7.6 Hz, 2H), 2.07–2.01 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.08–1.01 (m, 2H), 0.95–0.83 (m, 2H) ppm.
Ethyl 3-Cyclopropyl-2-(3,4-dichlorobenzyl)-3-oxopropanoate (4bq)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopropyl-3-oxopropanoate 1b (0.60 mL, 4.06 mmol, 1.00 equiv), 3,4-dichlorobenzyl bromide 3q (0.62 mL, 4.27 mmol, 1.05 equiv), DIPEA (1.42 mL, 8.13 mmol, 2.00 equiv), LiCl (172 mg, 4.06 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 39% (493 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 1.6 Hz, 1H), 7.03 (dd, J = 8.4, 2.0 Hz, 1H), 4.22–4.12 (m, 2H), 3.87 (t, J = 7.6 Hz, 1H), 3.18–3.09 (m, 2H), 2.09–2.02 (m, 1H), 1.23 (t, J = 7.2 Hz, 3H), 1.09–1.01 (m, 2H), 0.95–0.85 (m, 2H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxopentanoate (4ca)52
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxopentanoate 1c (0.42 mL, 2.92 mmol, 1.20 equiv), 3-chlorobenzyl bromide 3a (0.32 mL, 2.43 mmol, 1.00 equiv), NaH (117 mg, 4.86 mmol, 2.00 equiv), 5 mL of dry THF. Yield: 52% (340 mg) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.20–7.14 (m, 3H), 7.09–7.03 (m, 1H), 4.15 (qd, J = 7.5, 0.8 Hz, 2H), 3.76 (t, J = 7.5 Hz, 1H), 3.17–3.08 (m, 2H), 2.60 (dqd, J = 18.0, 7.0, 1.0 Hz, 1H), 2.37 (dqd, J = 18.5, 7.0, 0.5 Hz, 1H), 1.20 (td, J = 7.0, 1.0 Hz, 3H), 1.01 (td, J = 7.5, 1.0 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxohexanoate (4da)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxohexanoate 1d (0.46 mL, 2.92 mmol, 1.20 equiv), 2.43 mmol of 3-chlorobenzyl bromide 3a, DIPEA (0.85 mmol, 4.86 mmol, 2.00 equiv), LiCl (103 mg, 2.43 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 76% (522 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.22–7.14 (m, 3H), 7.08–7.04 (m, 1H), 4.15 (q, J = 7.2 Hz, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.18–3.07 (m, 2H), 2.54 (dt, J = 17.6, 7.2 Hz, 1H), 2.35 (dt, J = 17.2, 7.2 Hz, 1H), 1.57 (sextet, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H), 0.85 (t, J = 7.6 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-4-methyl-3-oxopentanoate (4ea)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.51 mL, 3.16 mmol, 1.20 equiv), 3-chlorobenzyl bromide 3a (0.35 mL, 2.63 mmol, 1.00 equiv), DIPEA (0.95 mL, 5.26 mmol, 2.00 equiv), LiCl (111 mg, 2.63 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 86% (640 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.20–7.14 (m, 3H), 7.07–7.04 (m, 1H), 4.14 (qd, J = 7.2, 0.8 Hz, 2H), 3.91 (t, J = 7.6 Hz, 1H), 3.17–3.07 (m, 2H), 2.66 (septet, J = 6.8 Hz, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3,4-Dichlorobenzyl)-4-methyl-3-oxopentanoate (4eq)24
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (5.00 mL, 31.0 mmol, 1.00 equiv), 3,4-dichlorobenzyl bromide 3q (5.41 mL, 37.2 mmol, 1.20 equiv), DIPEA (10.8 mL, 62.0 mmol, 2.00 equiv), LiCl (1.31 g, 30.9 mmol, 1.00 equiv), 50 mL of dry THF. Yield: 33% (3.20 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.4 Hz, 1H), 7.29–7.25 (m, 1H), 7.01 (dd, J = 8.0, 2.0 Hz, 1H), 4.15 (qd, J = 7.2, 1.6 Hz, 2H), 3.89 (t, J = 7.6 Hz, 1H), 3.16–3.04 (m, 2H), 2.69 (heptet, J = 6.8 Hz, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.07 (d, J = 6.4 Hz, 3H), 0.93 (d, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(2,3-Dichlorobenzyl)-4-methyl-3-oxopentanoate (4er)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.670 mL, 4.61 mmol, 1.00 equiv), 2,3-dichlorobenzyl bromide 3r (1.00 g, 4.17 mmol, 0.90 equiv), DIPEA (1.45 mL, 8.34 mmol, 1.81 equiv), LiCl (180 mg, 0.90 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 8% (110 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.34 (dd, J = 7.6, 1.6 Hz, 1H), 7.15 (dd, J = 7.6, 1.6 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 4.20–4.03 (m, 3H), 3.34–3.22 (m, 2H), 2.71 (heptet, J = 6.8 Hz, 1H), 1.19 (t, J = 7.2 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H) ppm.
Ethyl 2-(2,5-Dichlorobenzyl)-4-methyl-3-oxopentanoate (4es)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.204 mL, 1.39 mmol, 1.00 equiv), 2,5-dichlorobenzyl bromide 3s (500 mg, 2.08 mmol, 1.5 equiv), DIPEA (0.242 mL, 1.39 mmol, 1.00 equiv), LiCl (60 mg, 1.39 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 71% (315 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.8 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), 7.15 (dd, J = 8.4, 2.4 Hz, 1H), 4.18–4.12 (m, 2H), 4.10 (t, J = 7.2 Hz, 1H), 3.26–3.16(m, 2H), 2.73 (heptet, J = 6.8 Hz, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3,5-Dichlorobenzyl)-4-methyl-3-oxopentanoate (4et)24
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.480 mL, 3.30 mmol, 1.00 equiv), 3,5-dichlorobenzyl bromide 3t (0.440 mL, 3.12 mmol, 1.00 equiv), DIPEA (1.05 mL, 6.00 mmol, 1.82 equiv), LiCl (0.310 g, 3.00 mmol, 0.91 equiv), 5 mL of dry THF. Yield: 16% (150 mg) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.13 (t, J = 2.0 Hz, 1H), 6.99 (d, J = 2.0 Hz, 2H), 4.08 (q, J = 7.2 Hz, 2H), 3.81 (t, J = 7.6 Hz, 1H), 3.07–2.97 (m, 2H), 2.63 (heptet, J = 6.8 Hz, 1H), 1.15 (t, J = 7.2 Hz, 3H), 1.00 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H) ppm.
Ethyl 2-(3,5-Dibromobenzyl)-4-methyl-3-oxopentanoate (4eu)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.480 mL, 3.30 mmol, 1.00 equiv), 3,5-dibromobenzyl bromide 3u (1.04 g, 3.16 mmol, 0.958 equiv), DIPEA (1.05 mL, 6.00 mmol, 1.82 equiv), LiCl (0.130 g, 3.00 mmol, 0.909 equiv), 5 mL of dry. THF. Yield: 29% (0.350 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 3H), 4.15 (q, J = 7.2 Hz, 2H), 3.87 (t, J = 7.2 Hz, 1H), 3.13–3.05 (m, 2H), 2.70 (heptet, J = 6.8 Hz, 1H), 1.24 (t, J = 6.4 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H) ppm.
Ethyl 2-(3-Bromo-4-chlorobenzyl)-4-methyl-3-oxopentanoate (4ev)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 4-methyl-3-oxopentanoate 1e (0.480 mL, 3.30 mmol, 1.00 equiv), 3-bromo-4-chlorobenzyl bromide 3v (0.900 g, 3.16 mmol, 0.958 equiv), DIPEA (1.05 mL, 6.00 mmol, 1.82 equiv), LiCl (0.130 g, 3.00 mmol, 0.909 equiv), 5 mL of dry THF. Yield: 34% (0.360 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 2.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.06 (dd, J = 8.0, 2.0 Hz, 1H), 4.15 (qd, J = 7.2, 1.6 Hz, 2H), 3.88 (t, J = 7.6 Hz, 1H), 3.14–3.04 (m, 2H), 2.69 (heptet, J = 6.8 Hz, 1H), 1.22 (t, J = 7.2 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxoheptanoate (4fa)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxoheptanoate 1f (0.47 g, 2.72 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.33 mL, 2.51 mmol, 0.922 equiv), DIPEA (1.05 mL, 6.00 mmol, 2.21 equiv), LiCl (0.130 g, 3.00 mmol, 1.10 equiv), 5 mL of dry THF. Yield: 67% (0.550 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.20–7.15 (m, 3H), 7.07–7.03 (m, 1H), 7.17–4.10 (m, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.16–3.06 (m, 2H), 2.55 (dt, J = 17.2, 7.2 Hz, 1H), 2.35 (dt, J = 17.6, 7.2 Hz, 1H), 1.50 (pentet, J = 7.6 Hz, 2H), 1.28–1.19 (m, 5H), 0.85 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-5-ethyl-3-oxoheptanoate (4ga)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 5-ethyl-3-oxoheptanoate 1g (0.600 g, 3.00 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.470 mL, 3.57 mmol, 1.19 equiv), DIPEA (1.05 mL, 6.00 mmol, 2.00 equiv), LiCl (0.130 g, 3.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 31% (0.300 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.20–7.14 (m, 3H), 7.06–7.05 (m, 1H), 4.18–4.12 (m, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.17–3.06 (m, 2H), 2.45 (dd, J = 17.2, 6.8 Hz, 1H), 2.29 (dd, J = 17.2, 6.0 Hz, 1H), 1.76 (heptet, J = 6.4 Hz, 1H), 1.30–1.17 (m, 7H), 0.78 (t, J = 7.6 Hz, 6H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxooctanoate (4ha)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxooctanoate 1h (0.560 g, 3.01 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.470 mL, 3.57 mmol, 1.18 equiv), DIPEA (1.05 mL, 6.00 mmol, 1.99 equiv), LiCl (0.130 g, 3.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 21% (0.197 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.22–7.15 (m, 3H), 7.07–7.04 (m, 1H), 4.15 (q, J = 7.2 Hz, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.17–3.07 (m, 2H), 2.54 (dt, J = 17.2, 7.2 Hz, 1H), 2.36 (dt, J = 17.2, 7.2 Hz, 1H), 1.52 (pentet, J = 7.4 Hz, 2H), 1.31–1.13 (m, 7H), 0.86 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-cyclopentyl-3-oxopropanoate (4ia)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-cyclopentyl-3-oxopropanoate 1i (0.376 g, 2.04 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.240 mL, 1.83 mmol, 0.888 equiv), DIPEA (0.530 mL, 3.03 mmol, 1.47 equiv), LiCl (0.067 g, 1.57 mmol, 0.772 equiv), 5 mL of dry THF. Silica column chromatography (20–50% CH2Cl2/petroleum ether). Yield: 85% (0.483 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.20–7.15 (m, 3H), 7.08–7.02 (m, 1H), 4.13 (q, J = 7.2 Hz, 2H), 3.85 (t, J = 7.6 Hz, 1H), 3.15–3.05 (m, 2H), 2.94 (pentet, J = 8.0 Hz, 1H), 1.87–1.35 (m, 8H), 1.20 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxononanoate (4ja)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxononanoate 1j (0.601 g, 3.00 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.325 mL, 2.48 mmol, 0.825 equiv), DIPEA (1.05 mL, 6.00 mmol, 2.00 equiv), LiCl (0.130 g, 3.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 97% (0.780 g) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.20–7.15 (m, 3H), 7.07–7.01 (m, 1H), 4.14 (q, J = 6.8 Hz, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.17–3.06 (m, 2H), 2.53 (dt, J = 17.6, 7.2 Hz, 1H), 2.35 (dt, J = 17.6, 7.2 Hz, 1H), 1.50 (pentet, J = 7.2 Hz, 2H), 1.30–1.15 (m, 9H), 0.85 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxodecanoate (4ka)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxodecanoate 1k (0.650 g, 3.03 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.475 mL, 3.60 mmol, 1.20 equiv), DIPEA (1.05 mL, 6.00 mmol, 2.00 equiv), LiCl (0.128 g, 3.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 50% (0.511 g) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.21–7.16 (m, 3H), 7.07–7.04 (m, 1H), 4.15 (q, J = 7.2 Hz, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.18–3.06 (m, 2H), 2.54 (dt, J = 17.6, 7.2 Hz, 1H), 2.36 (dt, J = 17.6, 7.6 Hz, 1H), 1.51 (pentet, J = 6.8 Hz, 2H), 1.31–1.17 (m, 11H), 0.87 (t, J = 6.8 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxo-3-phenylpropanoate (4la)23
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxo-3-phenylpropanoate 1l (0.498 mL, 2.92 mmol, 1.20 equiv), 3-chlorobenzyl bromide 3a (0.320 mL, 2.43 mmol, 1.00 equiv), DIPEA (0.847 mL, 4.86, 2.00 equiv), LiCl (103 mg; 2.43 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 85% (686 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 8.0, 0.8, 2H), 7.46 (td, J = 7.6, 1.2 Hz, 1H), 7.35 (t, J = 7.6 Hz, 2H), 7.20 (s, 1H), 7.10–7.05 (m, 3H), 4.62 (t, J = 7.2 Hz, 1H), 4.05–3.95 (m, 2H), 3.31–3.20 (m, 2H), 1.01 (t, J = 7.2, 0.8 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxo-3-(p-tolyl)propanoate (4ma)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxo-3-(p-tolyl)propanoate 1m (2.30 g, 11.15 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (2.29 mL, 11.15 mmol, 1.00 equiv), DIPEA (3.88 mL, 22.3 mmol, 2.00 equiv), LiCl (473 mg; 11.15 mmol, 1.00 equiv), 30 mL of dry THF. Yield: 86% (3.18 g) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 8.0 Hz, 2H), 7.28–7.22 (m, 3H), 7.19–7.14 (m, 2H), 7.12–7.09 (m, 1H), 4.57 (t, J = 7.6 Hz, 1H), 4.13–4.05 (m, 2H), 3.33–3.23 (m, 2H), 2.41 (s, 3H), 1.12 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(3-Chlorobenzyl)-3-oxo-5-phenylpentanoate (4na)
Compound was synthesized according to general procedure 2, using the following reagents: ethyl 3-oxo-5-phenylpentanoate 1n (0.661 g, 3.00 mmol, 1.00 equiv), 3-chlorobenzyl bromide 3a (0.475 mL, 3.65 mmol, 1.20 equiv), DIPEA (1.05 mL, 6.00 mmol, 2.00 equiv), LiCl (0.128 g, 3.00 mmol, 1.00 equiv), 5 mL of dry THF. Yield: 67% (0.690 g) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.32–7.26 (m, 2H), 7.24–7.19 (m, 3H), 7.16 (t, J = 7.2 Hz, 3H), 4.13 (q, J = 7.2 Hz, 2H), 3.75 (t, J = 7.6 Hz, 1H), 3.13 (d, J = 7.6 Hz, 2H), 2.99–2.85 (m, 3H), 2.75–2.67 (m, 1H), 1.20 (t, J = 7.2 Hz, 3H) ppm.
Procedure for the Synthesis of 6-(3-Chlorobenzyl)-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (6)23,24
In a sealed microwave tube 3-amino-1,2,4-triazole 5a (66 mg, 0.78 mmol, 1.1 equiv), 4ba (200 mg, 0.71 mmol, 1.00 equiv), and H3PO4 (96 μL, 1.42 mmol, 2.00 equiv) were heated at 170 °C in 1 mL of EtOH in the microwave for 10 h. The reaction mixture was poured in water (30 mL), the pH was adjusted to pH = 12 (1 M NaOH aq), and the organics were extracted with EtOAc (3 × 30 mL). The combined extracts were dried over MgSO4 and the solvents evaporated in vacuo, resulting in 165 mg of crude mixture. The pure product was obtained by column chromatography (5% CH3OH in CH2Cl2) followed by prep HPLC gradient 10–90% CH3CN/water + 0.1%TFA yielding 4% (9 mg) as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H), 7.28–7.25 (m, 1H), 7.22–7.15 (m, 3H), 7.17–7.08 (m, 3H), 4.16 (s, 2H), 2.22–2.13 (m, 1H), 1.36–1.30 (m, 2H), 1.20–1.15 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H13ClN4O [M + H]+ 301.09, found 301.1. HPLC: 6.566 min, purity 97%.
Procedure for the Synthesis of 6-(3-Chlorobenzyl)-5-cyclopropyl-2-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (7)
In a sealed microwave tube 3-amino-5-methyl-1,2,4-triazole 5b (126 mg, 1.28 mmol, 1.2 equiv), 4ba (300 mg, 1.07 mmol, 1.0 equiv), and p-toluenesulfonic acid monohydrate (102 mg, 0.53 mmol, 0.5 equiv) were heated for 30 min at 180 °C in the microwave. As visualized by TLC, 4ba was consumed and mainly one product was formed (Rf 0.5 in 5% CH3OH in CH2Cl2). The crude product was purified by column chromatography (3% CH3OH in CH2Cl2) yielding 31% (96 mg) as a white solid. 1H NMR (400 MHz, CDCl3 + drop MeOD): δ 7.23 (s, 1H), 7.21–7.13 (m, 3H), 6.26 (br s, 2H), 4.11 (s, 2H), 2.47 (s, 2H), 2.05–1.96 (m, 1H), 1.14–1.07 (m, 2H), 1.05–1.01 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H15ClN4O [M + H]+ 315.10, found 315.1. HPLC: 6.826 min, purity 96%.
General Procedure 3. Triazolopyrimidinones 8–4323
The synthesis of compounds 8–43 was according to the following procedure: In a microwave tube a mixture of the corresponding benzylated β-keto ester 4aa–na, 4bb–bq, 4eq–ev (1.00 equiv), triazole 5c (2.00 equiv) and 1-butyl-3-methylimidazolium hexafluorophosphate (BMIM-PF6, 1 mL or 6.00 equiv) was heated at 200 °C in a microwave reactor for an hour. Afterward, the reaction was allowed to cool to room temperature and stirred in a mixture of CH2Cl2 (30 mL), H2O (10 mL), and 5–10% aqueous citric acid (1 mL) for 20–30 min. The resulting mixture was filtered over a glass filter, and the residue was washed with hot methanol. Finally, the precipitate was collected and dried in vacuo to yield the pure compounds.
2-Amino-6-(3-chlorobenzyl)-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (8)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (216 mg, 2.18 mmol, 2.00 equiv), 4ba (306 mg, 1.09 mmol, 1.00 equiv), and BMIM-PF6 (1.35 mL, 6.54 mmol, 6.00 equiv). Yield: 36% (125 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.16 (br s, 1H), 7.35–7.24 (m,2H), 7.24–7.14 (m, 2H), 6.24 (br s, 2H), 3.96 (s, 2H), 2.14–2.01 (m, 1H), 1.07–0.94 (m, 2H), 0.94–0.81 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H14ClN5O [M + H]+ 316.10, found 316.13. HPLC: 6.350 min, purity 98%.
2-Amino-6-benzyl-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (9)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (276 mg, 2.84 mmol, 2.00 equiv), 4bb (350 mg, 1.42 mmol, 1.00 equiv) and BMIM-PF6 (1.75 mL, 8.52 mmol, 6.00 equiv). Yield: 26% (105 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.11 (br s, 1H), 7.26–7.20 (m, 4H), 7.17–7.10 (m, 1H), 6.20 (br s, 2H), 3.96 (s, 2H), 2.10–2.03 (m, 1H), 1.04–0.95 (m, 2H), 0.94–0.88 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H15N5O [M + H]+ 282.14, found 282.13. HPLC: 5.655 min, purity 97%.
2-Amino-5-cyclopropyl-6-(2-methylbenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (10)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (190 mg, 1.96 mmol, 2.00 equiv), 4bc (255 mg, 0.98 mmol, 1.00 equiv), and BMIM-PF6 (1.21 mL, 5.88 mmol, 6.00 equiv). Yield: 32% (93 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.17 (br s, 1H), 7.15 (d, J = 6.8 Hz, 1H), 7.08–6.99 (m, 2H), 6.81 (d, J = 6.8 Hz, 1H), 6.21 (br s, 2H), 3.86 (s, 2H), 2.36 (s, 3H), 1.90–1.81 (m, 1H), 1.04–0.94 (m, 2H), 0.88–0.81 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O [M + H]+ 296.15, found 296.13. HPLC: 6.074 min, purity 98%.
2-Amino-6-(2-chlorobenzyl)-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (11)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (173 mg, 1.78 mmol, 2.00 equiv), 4bd (250 mg, 0.89 mmol, 1.00 equiv), and BMIM-PF6 (1.01 mL, 5.34 mmol, 6.00 equiv). Yield: 37% (104 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.14 (br s, 1H), 7.47–7.42 (m, 1H), 7.24–7.17 (m, 2H), 7.01–6.96 (m, 1H), 6.20 (br s, 2H), 4.00 (s, 2H), 1.90–1.80 (m, 1H), 0.99–0.93 (m, 2H), 0.89–0.80 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H14ClN5O [M + H]+ 316.10, found 316.13. HPLC: 6.411 min, purity 95%.
2-Amino-5-cyclopropyl-6-(2-methoxybenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (12)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (130 mg, 1.34 mmol, 2.00 equiv), 4be (185 mg, 0.67 mmol, 1.00 equiv), and BMIM-PF6 (0.83 mL, 4.02 mmol, 6.00 equiv). Yield: 43% (89 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.11 (br s, 1H), 7.15 (t, J = 6.0 Hz, 1H), 6.95 (d, J = 6.4 Hz, 1H), 6.86 (d, J = 5.6 Hz, 1H), 6.78 (t, J = 5.6 Hz, 1H), 6.18 (br s, 2H), 3.86 (s, 2H), 3.83 (s, 3H), 1.98–1.86 (m, 1H), 1.03–0.91 (m, 2H), 0.90–0.79 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O2 [M + H]+ 312.15, found 312.2. HPLC: 5.832 min, purity 98%.
2-Amino-5-cyclopropyl-6-(3-methylbenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (13)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (216 mg, 2.22 mmol, 2.00 equiv), 4bf (289 mg, 1.11 mmol, 1.00 equiv), and BMIM-PF6 (1.37 mL, 6.66 mmol, 6.00 equiv). Yield: 35% (114 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 11.98 (br s, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.04 (s, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 7.2 Hz, 1H), 6.17 (br s, 2H), 3.91 (s, 2H), 2.23 (s, 3H), 2.10–2.00 (m, 1H), 1.04–0.94 (m, 2H), 0.94–0.83 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O [M + H]+ 296.15, found 296.13. HPLC: 6.154 min, purity 97%.
2-Amino-5-cyclopropyl-6-(3-fluorobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (14)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (190 mg, 1.92 mmol, 1.45 equiv), 4bg (350 mg, 1.32 mmol, 1.00 equiv), and BMIM-PF6 (1.14 mL, 5.54 mmol, 4.19 equiv). Yield: 15% (61 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 7.40–7.20 (m, 1H), 7.16–6.85 (m, 3H), 5.81 (br s, 2H), 4.01 (s, 2H), 2.12–1.95 (m, 1H), 1.10–0.85 (m, 4H) ppm. LC–MS (ESI) m/z calcd for C15H14FN5O [M + H]+ 300.13, found 300.1. HPLC: 5.812 min, purity 97%.
2-Amino-6-(3-bromobenzyl)-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (15)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (169 mg, 1.74 mmol, 2.00 equiv), 4bh (283 mg, 0.87 mmol, 1.00 equiv), and BMIM-PF6 (1.07 mL, 5.22 mmol, 6.00 equiv). Yield: 43% (135 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.21 (br s, 1H), 7.43 (s, 1H), 7.34 (d, J = 7.2 Hz, 1H), 7.25 (d, J = 7.6 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 6.26 (br s, 2H), 3.96 (s, 2H), 2.12–2.04 (m, 1H), 1.04–0.94 (m, 2H), 0.94–0.85 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H14BrN5O [M + H]+ 360.05, found 360.2. HPLC: 6.529 min, purity 99%.
2-Amino-5-cyclopropyl-6-(3-iodobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (16)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (190 mg, 1.92 mmol, 2.32 equiv), 4bi (320 mg, 0.86 mmol, 1.00 equiv), and BMIM-PF6 (1.14 mL, 4.25 mmol, 6.00 equiv). Yield: 26% (92 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.18 (br s, 1H), 7.62 (s, 4H), 7.51 (d, J = 7.6 Hz, 1H), 7.25 (d, J = 7.6 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 6.22 (br s, 2H), 3.92 (s, 2H), 2.12–2.07 (m, 1H), 1.01–0.94 (m, 2H), 0.93–0.86 (m, 2H). LC–MS (ESI) m/z calcd for C15H14IN5O [M + H]+ 408.03, found 408.1. HPLC: 7.042 min, purity 96%.
2-Amino-5-cyclopropyl-6-(3-methoxybenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (17)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (146 mg, 1.50 mmol, 2.00 equiv), 4bj (207 mg, 0.75 mmol, 1.00 equiv), BMIM-PF6 (0.93 mL, 4.50 mmol, 6.00 equiv). Yield: 21% (48 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 11.94 (br s, 1H), 7.15 (t, J = 8.0 Hz, 1H), 6.82–6.77 (m, 2H), 6.72 (d, J = 7.6 Hz, 1H), 6.16 (br s, 2H), 3.93 (s, 2H), 3.69 (s, 3H), 2.11–2.03 (m, 1H), 1.04–0.96 (m, 2H), 0.96–0.85 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O2 [M + H]+ 312.15, found 312.13. HPLC: 5.675 min, purity 96%.
2-Amino-5-cyclopropyl-6-(3-(trifluoromethyl)benzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (18)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (150 mg, 1.54 mmol, 2.00 equiv), 4bk (242 mg, 0.77 mmol, 1.00 equiv), and BMIM-PF6 (0.95 mL, 4.62 mmol, 6.00 equiv). Yield 6% (17 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.18 (br s, 1H), 7.61 (s, 1H), 7.57–7.46 (m, 3H), 6.24 (br s, 2H), 4.06 (s, 2H), 2.17–2.08 (m, 1H), 1.03–0.95 (m, 2H), 0.94–0.86 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H14F3N5O [M + H]+ 350.13, found 350.27. HPLC: 6.727 min, purity 97%.
2-Amino-5-cyclopropyl-6-(4-methylbenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (19)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (197 mg, 2.03 mmol, 2.00 equiv), 4bl (266 mg, 1.02 mmol, 1.00 equiv), and BMIM-PF6 (1.25 mL, 6.09 mmol, 6.00 equiv). Yield: 39% (115 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.06 (br s, 1H), 7.10 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 8.0 Hz, 2H), 6.18 (br s, 2H), 3.90 (s, 2H), 2.23 (s, 3H), 2.08–2.01 (m, 1H), 1.01–0.94 (m, 2H), 0.94–0.87 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O [M + H]+ 296.15, found 296.13. HPLC: 6.279 min, purity 97%.
2-Amino-5-cyclopropyl-6-(4-fluorobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (20)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (150 mg, 1.51 mmol, 2.00 equiv), 4bm (200 mg, 0.756 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 6.39 equiv). Yield: 35% (79 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.13 (s, 1H), 7.28 (dd, J = 9.6, 2.4 Hz, 2H), 7.07 (t, J = 8.8 Hz, 2H), 6.21 (s br, 2H), 3.95 (s, 2H), 2.15–2.02 (m, 1H), 1.05–0.85 (m, 4H) ppm. LC–MS (ESI) m/z calcd for C15H14FN5O [M + H]+ 300.13, found 300.2. HPLC: 6.394 min, purity 95%.
2-Amino-6-(4-chlorobenzyl)-5-cyclopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (21)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (179 mg, 1.84 mmol, 2.00 equiv), 4bn (258 mg, 0.92 mmol, 1.00 equiv), and BMIM-PF6 (1.14 mL, 5.52 mmol, 6.00 equiv). Yield: 33% (105 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.18 (br s, 1H), 7.28 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 6.24 (br s, 2H), 3.94 (s, 2H), 2.10–2.00 (m, 1H), 1.04–0.94 (m, 2H), 0.93–0.81 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C15H14ClN5O [M + H]+ 316.10, found 316.13. HPLC: 6.393 min, purity 96%.
2-Amino-5-cyclopropyl-6-(4-bromobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (22)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (120 mg, 1.21 mmol, 2.00 equiv), 4bo (200 mg, 0.615 mmol, 1.00 equiv), and BMIM-PF6 (1,00 mL, 4.86 mmol, 7.90 equiv). Yield: 21% (47 mg) as an off-white solid. 1H NMR (400 MHz, DMSO): δ 12.15 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.24 (br s, 2H), 3.92 (s, 2H), 2.10–2.00 (m, 1H), 1.08–0.75 (m, 4H) ppm. LC–MS (ESI) m/z calcd for C15H14BrN5O [M + H]+ 360.05, found 360.1. HPLC: 6.474 min, purity 96%.
2-Amino-5-cyclopropyl-6-(4-methoxybenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (23)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (165 mg, 1.70 mmol, 2.00 equiv), 4bp (235 mg, 0.85 mmol, 1.00 equiv), and BMIM-PF6 (1.05 mL, 5.10 mmol, 6.00 equiv). Yield: 41% (109 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.07 (br s, 1H), 7.15 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 6.19 (br s, 2H), 3.88 (s, 2H), 3.69 (s, 3H), 2.12–2.01 (m, 1H), 1.04–0.95 (m, 2H), 0.94–0.86 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C16H17N5O2 [M + H]+ 312.15, found 312.07. HPLC: 5.753 min, purity 96%.
2-Amino-6-(3-chlorobenzyl)-5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (24)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (94 mg, 0.95 mmol, 2.00 equiv), 4aa (120 mg, 0.47 mmol, 1.00 equiv), and BMIM-PF6 (0.58 mL, 2.82 mmol, 6.00 equiv). Yield: 57% (77 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.56 (s, 1H), 7.30–7.21 (m, 3H), 7.17 (d, J = 7.2 Hz, 1H), 5.99 (s, 2H), 3.78 (s, 2H), 2.25 (s, 3H) ppm. LC–MS (ESI) m/z calcd for C13H12ClN5O [M + H]+ 290.08, found 290.1. HPLC: 5.709 min, purity 99%.
2-Amino-6-(3-chlorobenzyl)-5-ethyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (25)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (141 mg, 1.43 mmol, 2.00 equiv), 4ca (192 mg, 0.71 mmol, 1.00 equiv), and BMIM-PF6 (0.88 mL, 4.28 mmol, 6.00 equiv). Yield: 21% (46 mg) as a white solid. 1H NMR (500 MHz, DMSO): δ 12.57 (s, 1H) 7.29–7.51 (m, 2H), 7.22 (d, J = 8.0 Hz, 1H), 7.16 (d, J = 7.5 Hz, 1H), 6.02 (s, 2H), 3.81 (s, 2H), 2.57 (q, J = 7.5 Hz, 2H), 1.05 (t, J= 7.5 Hz, 3H) ppm. LC–MS (ESI) m/z calcd for C14H14ClN5O [M + H]+ 304.10, found 304.2. HPLC: 6.156 min, purity 95%.
2-Amino-6-(3-chlorobenzyl)-5-propyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (26)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (210 mg, 2.12 mmol, 2.00 equiv), 4da (300 mg, 1.06 mmol, 1.00 equiv), and BMIM-PF6 (1.31 mL, 6.36 mmol, 6.00 equiv). Yield: 22% (76 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.56 (s, 1H), 7.29–7.20 (m, 3H), 7.16 (d, J = 7.6 Hz, 1H), 6.01 (s, 2H), 3.81 (s, 2H), 2.53 (t, J = 7.6 Hz, 2H), 1.45 (sextet, J = 7.6 Hz, 2H), 0.85 (t, J = 7.2 Hz, 3H) ppm. LC–MS (ESI) m/z calcd for C15H16ClN5O [M + H]+ 318.11, found 318.2. HPLC: 6.660 min, purity 99%.
2-Amino-6-(3-chlorobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (27)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (284 mg, 2.87 mmol, 2.00 equiv), 1ea (407 mg, 1.44 mmol, 1.00 equiv), and BMIM-PF6 (1.76 mL, 8.63 mmol, 6.00 equiv). Yield: 21% (96 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.33 (s, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.35 (s, 1H), 7.21 (d, J = 8.0 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 6.04 (s, 2H) 3.89 (s, 2H), 3.18 (septet, J= 6.8 Hz, 1H), 1.12 (d, J= 6.8 Hz, 6H) ppm. LC–MS (ESI) m/z calcd for C15H16ClN5O [M + H]+ 318.11, found 318.2. HPLC: 6.596 min, purity 99%.
2-Amino-5-butyl-6-(3-chlorobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (28)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (250 mg, 1.51 mmol, 2.14 equiv), 1fa (210 mg, 0.708 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 6.86 equiv). Yield: 20% (47 mg) as a white solid. Purity: 98%. 1H NMR (400 MHz, DMSO) δ 12.54 (s, 1H), 7.33–7.19 (m, 3H), 7.16 (d, J = 7.3 Hz, 1H), 6.02 (s, 2H), 3.82 (s, 2H), 2.60–2.50 (m, 2H), 1.45–1.32 (m, 2H), 1.32–1.20 (m, 2H), 0.81 (t, J = 7.1 Hz, 3H) ppm. LC–MS (ESI) m/z calcd for C16H18ClN5O [M + H]+ 332.13, found 332.3. HPLC: 7.027 min, purity 98%.
2-Amino-6-(3-chlorobenzyl)-5-(2-ethylbutyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (29)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (130 mg, 1.31 mmol, 2.13 equiv), 4ga (200 mg, 0.616 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.89 equiv). Yield: 9% (20 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.49 (br s, 1H), 7.28 (t, J = 7.2 Hz, 1H), 7.24–7.18 (m, 2H), 7.14 (d, J = 7.2 Hz, 1H), 5.91 (br s, 2H), 3.82 (s, 2H), 1.58 (s, 2H), 1.30–1.15 (m, 5H), 0.77 (t, J = 6.8 Hz, 6H) ppm. LC–MS (ESI) m/z calcd for C18H22ClN5O [M + H]+ 360.16, found 360.3. HPLC: 7.640 min, purity 97%.
2-Amino-6-(3-chlorobenzyl)-5-pentyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (30)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (120 mg, 1.21 mmol, 1.88 equiv), 4ha (200 mg, 0.643 mmol; 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.56 equiv). Yield: 24% (52 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.43 (br s, 1H), 7.35–7.10 (m, 4H), 6.02 (br s, 2H), 3.82 (s, 2H), 1.45–1.15 (m, 6H), 0.79 (s, 3H) ppm. LC–MS (ESI) m/z calcd for C17H20ClN5O [M + H]+ 346.15, found 346.2. HPLC: 7.376 min, purity 98%.
2-Amino-6-(3-chlorobenzyl)-5-cyclopentyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (31)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (130 mg, 1.31 mmol, 2.00 equiv), 4ia (200 mg, 0.647 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.51 equiv). Yield: 38% (86 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.36 (br s, 1H), 7.38–7.07 (m, 4H), 6.07 (br s, 2H), 3.90 (s, 2H), 3.23 (m, 1H), 1.90–1.45 (m, 8H) ppm. LC–MS (ESI) m/z calcd for C17H18ClN5O [M + H]+ 344.13, found 344.1. HPLC: 7.049 min, purity 99%.
2-Amino-6-(3-chlorobenzyl)-5-hexyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (32)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (86 mg, 0.86 mmol, 2.00 equiv), 4ja (140 mg, 0.431 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 11.3 equiv). Yield: 83% (128 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.50 (s, 1H), 7.33–7.19 (m, 3H), 7.15 (d, J = 6.8 Hz, 1H), 6.02 (s, 2H), 3.82 (s, 2H), 2.60–2.40 (m, 2H), 1.44–1.32 (m, 2H), 1.30–1.08 (m, 6H), 0.82 (t, J = 6.5 Hz, 3H) ppm. LC–MS (ESI) m/z calcd for C18H22ClN5O [M + H]+ 360.16, found 360.3. HPLC: 7.843 min, purity 97%.
2-Amino-6-(3-chlorobenzyl)-5-heptyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (33)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (116 mg, 1.18 mmol, 2.00 equiv), 4ka (200 mg, 0.591 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 8.22 equiv). Yield: 50% (108 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.55 (s, 1H), 7.30–7.19 (m, 3H), 7.15 (d, J = 6.4 Hz, 1H), 6.01 (s, 1H), 3.82 (s, 2H), 1.46–1.32 (m, 2H), 1.27–1.10 (m, 8H), 0.83 (t, J = 6.4 Hz, 3H) ppm. LC–MS (ESI) m/z calcd for C19H24ClN5O [M + H]+ 374.18, found 374.3. HPLC: 8.200 min, purity 98%.
2-Amino-6-(3-chlorobenzyl)-5-phenyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (34)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (177 mg, 1.79 mmol, 2.00 equiv), 4la (296 mg, 0.89 mmol, 1.00 equiv), and BMIM-PF6 (1.11 mL, 5.36 mmol, 6.00 equiv). Yield: 30% (95 mg) as a white solid. 1H NMR (500 MHz, DMSO): δ 12.83 (s, 1H), 7.53–7.47 (m, 3H), 7.42 (dd, J = 8.0, 1.5 Hz, 2H), 7.20 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 7.0 Hz, 1H), 7.03 (s, 1H), 6.94 (d, J = 7.0 Hz, 1H), 6.12 (s, 2H), 3.64 (s, 2H) ppm. LC–MS (ESI) m/z calcd for C18H14ClN5O [M + H]+ 352.10, found 352.2. HPLC: 6.946 min, purity 99%.
2-Amino-6-(3-chlorobenzyl)-5-(p-tolyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (35)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (181 mg, 1.82 mmol, 2.00 equiv), 4ma (314 mg, 0.937 mmol, 1.00 equiv), and BMIM-PF6 (1.5 mL, 7.28 mmol, 7.77 equiv). Yield: 33% (108 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.80 (s, 1H), 7.33–7.28 (m, 4H), 7.26–7.14 (m, 2H), 7.05 (s, 1H), 6.96 (d, J = 7.1 Hz, 1H), 6.11 (s, 2H), 3.64 (s, 2H), 2.36 (s, 3H) ppm. LC–MS (ESI) m/z calcd for C19H16ClN5O [M + H]+ 366.20, found 366.2. HPLC: 7.342 min, purity 98%.
Procedure for the synthesis of 2-Amino-6-(3-chlorobenzyl)-5-phenethyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (36)
A mixture of 4na (213 mg, 0.57 mmol, 1.00 equiv), 3,5-diamino-4H-1,2,4-triazole 5c (116 mg, 1.16 mmol, 2.00 equiv), and ortho-phosphoric acid 85% (59.6 μL, 0.906 mmol,1.58 equiv) in EtOH (1 mL) was stirred for 1 min at 20 °C and then heated at 175 °C under microwave irradiation for 3 h. The reaction mixture was allowed to cool to room temperature, and CH2Cl2 (30 mL), water (10 mL), and aqueous citric acid (5%, 1 mL) were added. The resulting precipitate was stirred for 20 min, filtered, and the residue was washed with hot methanol, collected, and dried in vacuo to afford the title compound. Yield: 22% (49 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.67 (s, 1H), 7.32–7.18 (m, 6H), 7.17–7.10 (m, 3H), 6.06 (br s, 2H), 3.76 (s, 2H), 2.84–2.78 (m, 2H), 2.70–2.66 (m, 2H) ppm. LC–MS (ESI) m/z calcd for C20H18ClN5O [M + H]+ 380.13, found 380.2. HPLC: 7.392 min, purity 97%.
2-Amino-5-cyclopropyl-6-(3,4-dichlorobenzyl)[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (37)23
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (157 mg, 1.58 mmol, 2.00 equiv), 4bq (250 mg, 0.79 mmol, 1.00 equiv), and BMIM-PF6 (1.3 mL, 4.74 mmol, 6.00 equiv). Yield: 31% (86 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.26 (br s, 1H), 7.52–7.47 (m, 2H), 7.22 (d, J = 8.4 Hz, 2H), 6.26 (br s, 2H), 3.95 (s, 2H), 2.12–2.01 (m, 1H), 1.01–0.85 (m, 4H) ppm. LC–MS (ESI) m/z calcd for C15H13Cl2N5O [M + H]+ 350.06, found 350.1. HPLC: 6.836 min, purity 96%.
2-Amino-6-(3,4-dichlorobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (38)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (125 mg, 1.26 mmol, 2.00 equiv), 4eq (200 mg, 0.63 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.71 equiv). Yield: 34% (76 mg), as a white solid. 1H NMR (400 MHz, DMSO): δ 12.23 (br s, 1H), 7.54–7.43 (m, 2H), 7.17 (d, J = 8.0 Hz, 1H), 6.09 (s, 2H), 3.88 (s, 2H), 3.22–3.13 (m, 1H), 1.12 (d, J = 6.4 Hz, 6H) ppm. LC–MS (ESI) m/z calcd for C15H15Cl2N5O [M + H]+ 352.08, found 352.3. HPLC: 7.008 min, purity 97%.
2-Amino-6-(2,3-dichlorobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (39)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (125 mg, 1.26 mmol, 2.00 equiv), 4er (200 mg, 0.630 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.71 equiv). Yield: 48% (106 mg) as a white solid. 1H NMR (500 MHz, DMSO-d6, 80 °C): δ 7.44 (dd, J = 7.9, 1.5 Hz, 1H), 7.21 (t, J = 7.9 Hz, 1H), 6.97 (dd, J = 7.2, 1.5 Hz, 1H), 5.83 (s br, 2H), 3.98 (s, 2H), 2.96 (pentet, J = 6.9 Hz, 1H), 1.16 (d, J = 6.9 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6): δ 161.8, 156.0, 155.6, 149.7, 140.1, 131.4, 130.5, 127.7, 127.4, 126.9, 104.5, 29.4, 28.3, 19.6 ppm. LC–MS (ESI) m/z calcd for C15H15Cl2N5O [M + H]+ 352.08, found 352.2. HPLC: 7.060 min, purity 97%.
2-Amino-6-(2,5-dichlorobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (40)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (125 mg, 1.26 mmol, 2.00 equiv), 4es (200 mg, 0.630 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 7.71 equiv). Yield: 24% (54 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.41 (br s, 1H), 7.70–7.42 (m, 1H), 7.40–7.19 (m, 1H), 7.16–6.85 (m, 1H), 6.08 (br s, 2H), 3.88 (s, 2H), 3.01–2.83 (m, 1H), 1.14 (s, 6H) ppm. LC–MS (ESI) m/z calcd for C15H15Cl2N5O [M + H]+ 352.08, found 352.2. HPLC: 6.938 min, purity 96%.
2-Amino-6-(3,5-dichlorobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (41)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (96 mg, 0.97 mmol, 2 equiv), 4et (153 mg, 0.48 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 10 equiv). Yield: 18% (31 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.33 (s, 1H), 7.41 (s, 1H), 7.26 (s, 2H), 6.07 (br s, 2H), 3.89 (s, 2H), 1.13 (s, 6H) ppm. LC–MS (ESI) m/z calcd for C15H15Cl2N5O [M + H]+ 352.08, found 352.1. HPLC: 7.103 min, purity 98%.
2-Amino-6-(3,5-dibromobenzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin 7(4H)-one (42)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (97 mg, 0.98 mmol, 2.00 equiv), 4eu (200 mg, 0.49 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 9.91 equiv). Yield: 7% (14 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 12.33 (br s, 1H), 7.63 (s, 1H), 7.42 (s, 2H), 6.07 (br s, 2H), 3.88 (s, 2H), 1.13 (s, 6H) ppm. LC–MS (ESI) m/z calcd for C15H15Br2N5O [M + H]+ 439.97, found 440.1. HPLC: 7.337 min, purity 98%.
2-Amino-6-(3-bromo-4-chloro-benzyl)-5-isopropyl[1,2,4]triazolo[1,5-a]pyrimidin-7(4H)-one (43)
Compound was synthesized according to general procedure 3, using the following reagents: 3,5-diamino-4H-1,2,4-triazole 5c (109 mg, 1.10 mmol, 2.00 equiv), 4ev (200 mg, 0.553 mmol, 1.00 equiv), and BMIM-PF6 (1.00 mL, 4.86 mmol, 8.83 equiv). Yield: 24% (53 mg) as a white solid. 1H NMR (400 MHz, DMSO): δ 7.60 (d, J = 2.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.21 (dd, J = 8.0, 2.0 Hz, 1H), 6.08 (br s, 2H), 3.87 (s, 2H), 3.22–3.11 (m, 1H), 1.12 (d, J = 7.2 Hz, 6H) ppm. LC–MS (ESI) m/z calcd for C15H15BrClN5O [M + H]+ 396.02, found 396.1. HPLC: 7.080 min, purity 98%.
Chemicals and Reagents
The human recombinant chemokines CCL2 and CCL3 were purchased from PeproTech (Rocky Hill, NJ). TAK-779 was obtained from NIH AIDS reagent program (Germantown, MD, catalogue number 4983). All triazolopyrimidinone derivatives were synthesized in-house. Guanosine 5′-O-[γ-thio]triphosphate ([35S[GTPγS) (specific activity 1250 Ci/mmol) was purchased from PerkinElmer (Waltham, MA), while [3H]-CCR2-RA-[R] (specific activity 59.6 Ci mmol–1) was custom-labeled by Vitrax (Placentia, CA). Bovine serum albumin was purchased from Sigma-Aldrich (St. Louis, MO). Bicinchoninic acid (BCA) and Pierce BCA protein assay kit were purchased from Pierce Biotechnology (Thermo Scientific, Rockford, IL). Tango U2OS cells stably expressing human CCR2b (U2OS-CCR2) or human CCR5 (U2OS-CCR5) were purchased from Invitrogen (Carlsbad, CA). All other chemicals were obtained from standard commercial sources.
Cell Culture
Both U2OS-CCR2b and U2OS-CCR5 cells were cultured in McCoy’s 5A medium supplemented with 10% (v/v) fetal calf serum, 2 mM glutamine, 0.1 mM nonessential amino acids, 25 mM HEPES, 1 mM sodium pyruvate, 200 IU/mL penicillin, 200 μg/mL streptomycin, 100 μg/mL G418, 40–50 μg/mL hygromycin, and 125 μg/mL zeocin. Cells were grown until 80% confluence and cultured twice-weekly on 10 or 15 cm ⌀ plates by trypsinization. Dialyzed fetal calf serum was used when culturing cells for functional assays or as a last step before membrane preparation.
Membrane Preparation
Membranes from U2OS-CCR2 or U2OS-CCR5 cells were prepared as previously described for CCR2.20 Briefly, U2OS-CCR2 or U2OS-CCR5 cells were scraped from confluent 15 cm ⌀ plates using phosphate buffered saline (PBS) and subsequently centrifuged at 3000 rpm for 5 min. Pellets were then resuspended in ice-cold Tris buffer (50 mM Tris-HCl, 5 mM MgCl2, pH 7.4) before homogenization with an Ultra Turrax homogenizer (IKA-Werke GmbH & Co. KG, Staufen, Germany). Membranes and cytosolic contents were separated using an Optima LE-80 K ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA) at 31 000 rpm for 20 min at 4 °C. After a second cycle of homogenization and centrifugation, the final pellet was resuspended and homogenized in ice-cold Tris buffer, aliquoted, and stored at −80 °C. Finally, membrane protein concentrations were determined using a BCA protein determination assay, as described by the manufacturer (Pierce BCA protein assay kit).53
[3H]-CCR2-RA-[R] Binding Assays
For [3H]-CCR2-RA-[R] displacement assays, U2OS-CCR2b membrane homogenates (15–20 μg of total protein) were incubated with ∼6 nM [3H]-CCR2-RA-[R] and at least 6 increasing concentrations of competing ligand in a final volume of 100 μL of assay buffer (50 mM Tris-HCl, 5 mM MgCl2, 0.1% CHAPS, pH 7.4). Ligands were diluted to the desired concentration with an HP D300 digital dispenser (Tecan, Giessen, The Netherlands). Total radioligand binding did not exceed 10% of the amount added to prevent ligand depletion, and nonspecific binding was determined using 10 μM CCR2-RA-[R]. After 2 h at 25 °C, incubation was terminated by rapid filtration through a 96-well GF/B filterplate on a PerkinElmer FilterMate harvester, using ice-cold wash buffer (50 mM Tris-HCl buffer supplemented with 5 mM MgCl2 and 0.05% CHAPS, pH 7.4). Filters were washed 10 times with ice-cold wash buffer and subsequently dried at 55 °C for 30 min. After addition of 25 μL of Microscint scintillation cocktail (PerkinElmer), the filter-bound radioactivity was measured by scintillation spectrometry using the P-E 2450 Microbeta2 counter (PerkinElmer).
Tango β-Arrestin Recruitment Assay
β-Arrestin recruitment was measured using the Tango CCR2-bla or CCR5-bla U2OS cell-based assay (Invitrogen) according to the manufacturer’s protocol. Briefly, U2OS-CCR2b or U2OS-CCR5 cells were grown until approximately 80% confluence and detached by trypsinization. Cells were recovered by centrifugation at 1000 rpm for 5 min, resuspended in assay medium (FreeStyle Expression Medium, Invitrogen) to a density of 10 000 cells per well and seeded into black-wall, clear-bottom, 384-well assay plates (Corning). For agonist assays, cells were exposed to increasing concentrations of CCL2 or CCL3 for CCR2 or CCR5, respectively, for 16 h at 37 °C and 5% CO2. For antagonist assays, compounds were first diluted in assay medium containing a final DMSO concentration of 0.5% or lower. Cells were then preincubated with either 1 μM (for single-point inhibition experiments) or increasing concentrations of antagonist for 30 min at room temperature, before a 16 hour co-incubation with an EC80 concentration of CCL2 (5 nM) or CCL3 (14 nM) at 37 °C and 5% CO2. After 16 h cells were loaded in the dark with 8 μL of LiveBLAzer-FRET B/G substrate (Invitrogen) and incubated for 2 h at room temperature. Finally, fluorescence emission at 460 and 535 nm was measured in an EnVision multilabel plate reader (PerkinElmer) after excitation at 400 nm. The ratio of emission at 460 and 535 nm was calculated for each well.
[35S]GTPγS Binding Assay
To determine the mechanism of inhibition [35S]GTPγS binding assays were performed. In CCR2 the [35S]GTPγS binding assay was performed as previously described.19,20 In the case of CCR5, an amount of 10 μg of U2OS-CCR5 membranes with 0.25 mg/mL saponin was preincubated with 5 μM GDP, increasing concentrations of CCL3, and three different antagonist concentrations for 30 min at 25 °C. All dilutions were made in assay buffer containing 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 10 mM NaCl, 1 mM EDTA, and 0.05% BSA. [35S]GTPγS (0.3 nM) was added, and the mixture was co-incubated for an additional 90 min at 25 °C before harvesting. Incubation was stopped by dilution with ice-cold 50 mM Tris-HCl buffer with 5 mM MgCl2. Separation of bound and unbound radioligand was performed by rapid filtration through a 96-well GF/B filter plate as described in “[3H]-CCR2-RA-[R] Binding Assays”.
Data Analysis
All experiments were analyzed using nonlinear regression curve fitting program Prism 7 (GraphPad, San Diego, CA). EC50, EC80, Emax, and IC50 values from functional assays were obtained by nonlinear regression analysis. All values obtained are the mean ± SEM of at least three separate experiments performed in duplicate, unless stated otherwise. For radioligand binding assays, Ki values were determined using the Cheng–Prussoff equation using a KD of 6.3 nM for the radioligand.19
Computational Modeling
The inactive crystal structure of hCCR2b with BMS-681 and CCR2-RA-[R] (PDB code 5T1A)15 was used as the basis for docking compounds 8, 39, 40, and 43. Docking was performed in the Schrodinger suite,54 as previously described for the docking of CCR2-RA-[R].19 Before docking, the CCR2b crystal structure was prepared by replacing the residues between L2265×62 and R2406×32, which correspond to the M2 muscarinic acetylcholine receptor, with the CCR2b sequence using prime55−57 and CCR5 as template (PDB code 4MBS).35 Induced fit docking, with a substructure restraint on the right-hand phenyl (R1, SMARTS: “c1ccccc1”) was used to dock compound 43 in the hCCR2b model.58,59 Compounds 8, 39, and 40 were docked using regular docking (Glide-SP) on the basis of the model of compound 43. Figures 4a, 4b, and S4 were rendered using PyMOL.60
Acknowledgments
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TAK-779.
Glossary
Abbreviations Used
- CCR2
CC chemokine receptor 2
- CCR5
CC chemokine receptor 5
- GPCR
G-protein-coupled receptor
- SAR
structure–affinity/activity relationship
- U2OS
human osteosarcoma cell
- IP
inositol phosphate
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)triphosphate
- BCA
bicinchoninic acid
- cryo-EM
cryoelectron microscopy
- NEAA
nonessential amino acid
- PBS
phosphate buffered saline
- SEM
standard error of the mean
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00742.
Characterization of ligands in a U2OS-CCR5 β-arrestin-recruitment assay (Figure S1); correlation between log P (cLogP) and affinity (pKi) values in CCR2 (Figure S2); characterization of compounds 39 and 43 as potential inverse agonists in hCCR2 (Figure S3); docking of compounds 8, 39, 40, and 43 (Figure S4); list of intermediate compounds 4aa–na, 4bb–bq, 4eq–ev (Table S1); functional activity of TAK-779 and CCR2-RA-[R] in hCCR5, using a CCL3-induced β-arrestin recruitment assay (Table S2); and functional activity of compounds 8, 39, and 43 in hCCR2, using a CCL2-induced β-arrestin recruitment assay (Table S3) (PDF)
Molecular formula strings and some data (CSV)
Model of CCR2b (PDB)
Model of compound 8 (PDB)
Model of compound 39 (PDB)
Model of compound 40 (PDB)
Model of compound 43 (PDB)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bachelerie F.; Ben-Baruch A.; Burkhardt A. M.; Combadiere C.; Farber J. M.; Graham G. J.; Horuk R.; Sparre-Ulrich A. H.; Locati M.; Luster A. D.; Mantovani A.; Matsushima K.; Murphy P. M.; Nibbs R.; Nomiyama H.; Power C. A.; Proudfoot A. E.; Rosenkilde M. M.; Rot A.; Sozzani S.; Thelen M.; Yoshie O.; Zlotnik A. International union of basic and clinical pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66 (1), 1–79. 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Cotarelo P.; Gomez-Moreira C.; Criado-Garcia O.; Sanchez L.; Rodriguez-Fernandez J. L. Beyond chemoattraction: multifunctionality of chemokine receptors in leukocytes. Trends Immunol. 2017, 38 (12), 927–941. 10.1016/j.it.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Schall T. J.; Proudfoot A. E. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 2011, 11 (5), 355–363. 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- Zweemer A. J.; Toraskar J.; Heitman L. H.; IJzerman A. P. Bias in chemokine receptor signalling. Trends Immunol. 2014, 35 (6), 243–252. 10.1016/j.it.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Koelink P. J.; Overbeek S. A.; Braber S.; de Kruijf P.; Folkerts G.; Smit M. J.; Kraneveld A. D. Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol. Ther. 2012, 133 (1), 1–18. 10.1016/j.pharmthera.2011.06.008. [DOI] [PubMed] [Google Scholar]
- White G. E.; Iqbal A. J.; Greaves D. R. CC chemokine receptors and chronic inflammation—therapeutic opportunities and pharmacological challenges. Pharmacol. Rev. 2013, 65 (1), 47–89. 10.1124/pr.111.005074. [DOI] [PubMed] [Google Scholar]
- Woollard S. M.; Kanmogne G. D. Maraviroc: a review of its use in HIV infection and beyond. Drug Des., Dev. Ther. 2015, 9, 5447–5468. 10.2147/DDDT.S90580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat. Rev. Drug Discovery 2009, 8 (1), 23–33. 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- Horuk R. Promiscuous drugs as therapeutics for chemokine receptors. Expert Rev. Mol. Med. 2009, 11, e1. 10.1017/S1462399409000921. [DOI] [PubMed] [Google Scholar]
- Friedman S. L.; Ratziu V.; Harrison S. A.; Abdelmalek M. F.; Aithal G. P.; Caballeria J.; Francque S.; Farrell G.; Kowdley K. V.; Craxi A.; Simon K.; Fischer L.; Melchor-Khan L.; Vest J.; Wiens B. L.; Vig P.; Seyedkazemi S.; Goodman Z.; Wong V. W.-S.; Loomba R.; Tacke F.; Sanyal A.; Lefebvre E. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67 (5), 1754–1767. 10.1002/hep.29477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J. Leukocyte Biol. 2010, 88 (1), 41–55. 10.1189/jlb.1009671. [DOI] [PubMed] [Google Scholar]
- Fantuzzi L.; Tagliamonte M.; Gauzzi M. C.; Lopalco L.. Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. Cell. Mol. Life Sci. 2019, 10.1007/s00018-019-03255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker A.; Kokornaczyk A. K.; Strunz A. K.; Wünsch B.. Selective and dual targeting of CCR2 and CCR5 receptors: a current overview. In Chemokines: Chemokines and Their Receptors in Drug Discovery; Tschammer N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; Vol. 14, pp 187–241. [Google Scholar]
- Kothandan G.; Gadhe C. G.; Cho S. J. Structural insights from binding poses of CCR2 and CCR5 with clinically important antagonists: a combined in silico study. PLoS One 2012, 7 (3), e32864. 10.1371/journal.pone.0032864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Qin L.; Ortiz Zacarías N. V.; de Vries H.; Han G. W.; Gustavsson M.; Dabros M.; Zhao C.; Cherney R. J.; Carter P.; Stamos D.; Abagyan R.; Cherezov V.; Stevens R. C.; IJzerman A. P.; Heitman L. H.; Tebben A.; Kufareva I.; Handel T. M. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540 (7633), 458–461. 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C.; Rappas M.; Kean J.; Dore A. S.; Errey J. C.; Bennett K.; Deflorian F.; Christopher J. A.; Jazayeri A.; Mason J. S.; Congreve M.; Cooke R. M.; Marshall F. H. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540 (7633), 462–465. 10.1038/nature20606. [DOI] [PubMed] [Google Scholar]
- Turner M. D.; Nedjai B.; Hurst T.; Pennington D. J. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843 (11), 2563–2582. 10.1016/j.bbamcr.2014.05.014. [DOI] [PubMed] [Google Scholar]
- Ortiz Zacarias N. V.; Lenselink E. B.; IJzerman A. P.; Handel T. M.; Heitman L. H. Intracellular receptor modulation: novel approach to target GPCRs. Trends Pharmacol. Sci. 2018, 39 (6), 547–559. 10.1016/j.tips.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz Zacarías N. V.; van Veldhoven J. P. D.; Portner L.; van Spronsen E.; Ullo S.; Veenhuizen M.; van der Velden W. J. C.; Zweemer A. J. M.; Kreekel R. M.; Oenema K.; Lenselink E. B.; Heitman L. H.; IJzerman A. P. Pyrrolone derivatives as intracellular allosteric modulators for chemokine receptors: selective and dual-targeting inhibitors of CC chemokine receptors 1 and 2. J. Med. Chem. 2018, 61 (20), 9146–9161. 10.1021/acs.jmedchem.8b00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweemer A. J.; Nederpelt I.; Vrieling H.; Hafith S.; Doornbos M. L.; de Vries H.; Abt J.; Gross R.; Stamos D.; Saunders J.; Smit M. J.; IJzerman A. P.; Heitman L. H. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol. Pharmacol. 2013, 84 (4), 551–561. 10.1124/mol.113.086850. [DOI] [PubMed] [Google Scholar]
- Zweemer A. J.; Bunnik J.; Veenhuizen M.; Miraglia F.; Lenselink E. B.; Vilums M.; de Vries H.; Gibert A.; Thiele S.; Rosenkilde M. M.; IJzerman A. P.; Heitman L. H. Discovery and mapping of an intracellular antagonist binding site at the chemokine receptor CCR2. Mol. Pharmacol. 2014, 86 (4), 358–368. 10.1124/mol.114.093328. [DOI] [PubMed] [Google Scholar]
- Andrews G.; Jones C.; Wreggett K. A. An intracellular allosteric site for a specific class of antagonists of the CC chemokine G protein-coupled receptors CCR4 and CCR5. Mol. Pharmacol. 2008, 73 (3), 855–867. 10.1124/mol.107.039321. [DOI] [PubMed] [Google Scholar]
- Bengtsson B. A.; Blackaby W.; Cumming J.; Faull A. W.; Larsson J.; Nash I. A.; Oldham K.; Pape A.. 4H-[1, 2, 4] Triazolo [5, 1-b] Pyrimidin-7-one Derivatives As CCR2b Receptor Antagonists. Patent WO 2011/114148 A1, 2011.
- Boyd J. W.; Meo P.; Higginbottom M.; Simpson I.; Mountford D.; Savory E. D.. 7-Hydroxy-Pyrazolo [1,5-a] Pyrimidine Compounds And Their Use As CCR2 Receptor Antagonists. Patent WO 2012/041817 A1, 2012.
- Isberg V.; de Graaf C.; Bortolato A.; Cherezov V.; Katritch V.; Marshall F. H.; Mordalski S.; Pin J.-P.; Stevens R. C.; Vriend G.; Gloriam D. E. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36 (1), 22–31. 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M.; Nishimura O.; Kanzaki N.; Okamoto M.; Sawada H.; Iizawa Y.; Shiraishi M.; Aramaki Y.; Okonogi K.; Ogawa Y.; Meguro K.; Fujino M. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (10), 5698–5703. 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum S.; Kokornaczyk A. K.; Seki T.; De Maria M.; Ortiz Zacarias N. V.; de Vries H.; Weiss C.; Koch M.; Schepmann D.; Kitamura M.; Tschammer N.; Heitman L. H.; Junker A.; Wunsch B. Synthesis and biological evaluation of chemokine receptor ligands with 2-benzazepine scaffold. Eur. J. Med. Chem. 2017, 135, 401–413. 10.1016/j.ejmech.2017.04.046. [DOI] [PubMed] [Google Scholar]
- Corbisier J.; Gales C.; Huszagh A.; Parmentier M.; Springael J. Y. Biased signaling at chemokine receptors. J. Biol. Chem. 2015, 290 (15), 9542–9554. 10.1074/jbc.M114.596098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz H. Hill coefficients, dose-response curves and allosteric mechanisms. J. Chem. Biol. 2010, 3 (1), 37–44. 10.1007/s12154-009-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D.; Zhai H.-X.; Eckman J.; Higgins P.; Gillard M.; Knerr L.; Carre S.; Pasau P.; Collart P.; Grassi J. Novel, acidic CCR2 receptor antagonists: from hit to lead. Lett. Drug. Des. Discovery 2007, 4 (3), 185–191. 10.2174/157018007780077381. [DOI] [Google Scholar]
- Csermely P.; Ágoston V.; Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol. Sci. 2005, 26 (4), 178–182. 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Morphy J. R.The Challenges of Multi-Target Lead Optimization. In Designing Multi-Target Drugs; Morphy J. R., Harris C. J., Eds.; The Royal Society of Chemistry, 2012; pp 141–154. [Google Scholar]
- Buntinx M.; Hermans B.; Goossens J.; Moechars D.; Gilissen R. A.; Doyon J.; Boeckx S.; Coesemans E.; Van Lommen G.; Van Wauwe J. P. Pharmacological profile of JNJ-27141491 [(S)-3-[3, 4-difluorophenyl)-propyl]-5-isoxazol-5-yl-2-thioxo-2, 3-dihydro-1H-imidazole-4-carboxyl acid methyl ester], as a noncompetitive and orally active antagonist of the human chemokine receptor CCR2. J. Pharmacol. Exp. Ther. 2008, 327 (1), 1–9. 10.1124/jpet.108.140723. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Szczuka A. Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem. Int. 2007, 51 (5), 254–260. 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341 (6152), 1387–1390. 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56 (4), 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Salchow K.; Bond M. E.; Evans S. C.; Press N. J.; Charlton S. J.; Hunt P. A.; Bradley M. E. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharmacol. 2010, 159 (7), 1429–1439. 10.1111/j.1476-5381.2009.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. J.; Tomkinson N. P.; Wiley K. E.; Brammall A.; Bowers L.; Grahames C.; Gaw A.; Meghani P.; Shelton P.; Wright T. J.; Mallinder P. R. Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol. Pharmacol. 2008, 74 (5), 1193–1202. 10.1124/mol.107.044610. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Khoshouei M.; Thal D. M.; Liang Y.-L.; Nguyen A. T. N.; Furness S. G. B.; Venugopal H.; Baltos J.-A.; Plitzko J. M.; Danev R.; Baumeister W.; May L. T.; Wootten D.; Sexton P. M.; Glukhova A.; Christopoulos A. Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 2018, 558 (7711), 559–563. 10.1038/s41586-018-0236-6. [DOI] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; Schertler G. F. X.; Granier S.; Weis W. I.; Dror R. O.; Manglik A.; Skiniotis G.; Kobilka B. K. Structure of the μ-opioid receptor–Gi protein complex. Nature 2018, 558 (7711), 547–552. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.; Kuybeda O.; de Waal P. W.; Mukherjee S.; Van Eps N.; Dutka P.; Zhou X. E.; Bartesaghi A.; Erramilli S.; Morizumi T.; Gu X.; Yin Y.; Liu P.; Jiang Y.; Meng X.; Zhao G.; Melcher K.; Ernst O. P.; Kossiakoff A. A.; Subramaniam S.; Xu H. E. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 2018, 558 (7711), 553–558. 10.1038/s41586-018-0215-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.; Zhou X. E.; Gao X.; He Y.; Liu W.; Ishchenko A.; Barty A.; White T. A.; Yefanov O.; Han G. W.; Xu Q.; de Waal P. W.; Ke J.; Tan M. H.; Zhang C.; Moeller A.; West G. M.; Pascal B. D.; Van Eps N.; Caro L. N.; Vishnivetskiy S. A.; Lee R. J.; Suino-Powell K. M.; Gu X.; Pal K.; Ma J.; Zhi X.; Boutet S.; Williams G. J.; Messerschmidt M.; Gati C.; Zatsepin N. A.; Wang D.; James D.; Basu S.; Roy-Chowdhury S.; Conrad C. E.; Coe J.; Liu H.; Lisova S.; Kupitz C.; Grotjohann I.; Fromme R.; Jiang Y.; Tan M.; Yang H.; Li J.; Wang M.; Zheng Z.; Li D.; Howe N.; Zhao Y.; Standfuss J.; Diederichs K.; Dong Y.; Potter C. S.; Carragher B.; Caffrey M.; Jiang H.; Chapman H. N.; Spence J. C.; Fromme P.; Weierstall U.; Ernst O. P.; Katritch V.; Gurevich V. V.; Griffin P. R.; Hubbell W. L.; Stevens R. C.; Cherezov V.; Melcher K.; Xu H. E. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523 (7562), 561–567. 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay R. R.; Popovic-Nikolic M. R.; Nikolic K.; Uliassi E.; Bolognesi M. L. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl. Med. 2018, 7 (1), 3. 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce D.; Douguet D.; Miteva M. A.; Villoutreix B. O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277. 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Baell J. B.; Miteva M. A.; Villoutreix B. O. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43 (W1), W200–W207. 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Lager E.; Nilsson J.; Østergaard Nielsen E.; Nielsen M.; Liljefors T.; Sterner O. Affinity of 3-acyl substituted 4-quinolones at the benzodiazepine site of GABAA receptors. Bioorg. Med. Chem. 2008, 16 (14), 6936–6948. 10.1016/j.bmc.2008.05.049. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang Q.; Chen L. H.; Yang H.; Lu W.; Xie X.; Nan F. J. Design and synthesis of 2-alkylpyrimidine-4,6-diol and 6-alkylpyridine-2,4-diol as potent GPR84 agonists. ACS Med. Chem. Lett. 2016, 7 (6), 579–583. 10.1021/acsmedchemlett.6b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman R. K.; Johnson J. S. Nickel-catalyzed rearrangement of 1-acyl-2-vinylcyclopropanes. A mild synthesis of substituted dihydrofurans. Org. Lett. 2006, 8 (4), 573–576. 10.1021/ol052700k. [DOI] [PubMed] [Google Scholar]
- Doni E.; Mondal B.; O’Sullivan S.; Tuttle T.; Murphy J. A. Overturning established chemoselectivities: selective reduction of arenes over malonates and cyanoacetates by photoactivated organic electron donors. J. Am. Chem. Soc. 2013, 135 (30), 10934–10937. 10.1021/ja4050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amupitan J. A.; Beddoes R. L.; Mills O. S.; Sutherland J. K. 3-Methylcyclohex-2-enone derivatives as initiators of cyclisation. Part 4. Some bicyclisations. J. Chem. Soc., Perkin Trans. 1 1983, 0, 759–763. 10.1039/p19830000759. [DOI] [Google Scholar]
- Sun X.; Tymianski M.; Garman D.. Agents And Methods For Treating Ischemic And Other Diseases. Patent WO 2012/174488 A2, 2012.
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150 (1), 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Maestro, release 2017-1; Schrödinger, LLC: New York, 2017.
- Prime, release 2017-1; Schrödinger, LLC: New York, 2017.
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320 (3), 597–608. 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct., Funct., Genet. 2004, 55 (2), 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Glide Release 2017-1; Schrödinger Suite Prime 2017-1 Induced Fit Docking Protocol; Schrödinger, LLC: New York, 2017.
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49 (2), 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC: New York, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.