

Abstract
Background:
In recent years, tRFs(transfer RNA-Derived Fragments) and transfer RNADerived Stress-induced RNAs (or tRNA halves) have been shown to have vital roles in cancer biology.We aimed to reveal the expression profile of tRNA-derived fragments in breast cancer tissues in the study, and to explore their potential as biomarkers of breast cancer.
Methods:
We characterized the tRNA-derived fragments expression profile from 6 paired clinical breast cancer tissues and adjacent normal samples. Then we selected 6 significantly expressed tRNAderived fragments and screened the genes for validation by using Quantitative Real-time PCR. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes biological pathway were finally analyzed.
Results:
We found 30 differentially expressed tRNA-derived fragments across our dataset, out of which 17 were up-regulated, and 13 were down-regulated. Compared with 16 clinical breast cancer tissues and adjacent normal tissues by qPCR, the results demonstrated that tRF-32-Q99P9P9NH57SJ(FC = -2.6476, p = 0.0189), tRF-17-79MP9PP (FC = -4.8984, p = 0.0276) and tRF-32-XSXMSL73VL4YK (FC = 6.5781, p = 0.0226) were significantly expressed in breast cancer tissues(p < 0.001). tRF-32-XSXMSL73VL4YK was significantly up-regulated, and tRF-32-Q99P9P9NH57SJ and tRF-17-79MP9PP were significantly down-regulated in which the expressionpatterns were similar to the sequencing results. The top ten significant results of GO and KEGG pathways enrichment analysis were presented.
Conclusion:
Our studies have demonstrated that there were significantly expressed tRNA-derived fragments in breast cancer tissues. They are hopefully to become biomarkers and would be valuable researches in this area.
Keywords: Breast cancer, TRNA-derived fragments, Expression profile, Biomarker, Genomes, Gene ontology
1. INTRODUCTION
Breast cancer has now become the most common cancer all over the world as a major health problem [1]. It has both a higher incidence and mortality rate in women malignant tumours [2]. Effective treatment of breast cancer depends on early diagnosis and therapy [3]. However, it is difficult to achieve this goal because the diagnosis of breast cancer is lack of sensitive and effective biomarkers [4]. In recent years, studies have shown that tRNA-derived fragments are related to various pathological conditions, even pathogenic factors, such as respiratory syncytial virus infection [5],
cancer [6-8], neurodegenerative diseases [9], inherited metabolic diseases [10], etc. The tRNA-derived fragments are roughly divided into two categories: tRFs (transfer RNA-Derived Fragments) and transfer RNA-Derived Stress-induced RNAs (or tRNA halves) with specific molecular size, nucleotide composition, physiological function and biogenesis [11]. Several studies have revealed that the expression of tRNA-derived fragments is linked with proliferation, especially in tumor cells [12], but the generation of tRNA-derived fragments may only be produced because of the rampant transcription of cancer cells [13]. In addition, increased studies also show that the expression level of tRNA halves increases under stress conditions, although stress induces tRNA halves accumulation, the concentration of full-length tRNAs does not change [14]. The composition and quantity of tRNA-derived fragments are highly dependent on cell type and disease state [15]. Telonis, et al. independently validated several tRFs from the database in clinical samples from different breast cancer subtypes which demonstrated that the heterogeneity and stability of tRNA-derived fragments make them possible to be biomarkers for cancer diagnosis [15, 16]. The recent increase in rapid and inexpensive RNA sequencing may allow us to accurately identify tRNA-derived fragments from sRNA(small RNA) deep-sequencing data and evaluate their expression in multiple cancers [17, 18]. In particular, the Mintbase and the Mintmap software based on this database can help us better identify the tRNA-derived fragments subfamily and unknown tRNA-derived fragments [19, 20]. With the help of abundant data and information, those differentially expressed tRNA-derived fragments would be further studied.
Therefore, in our study, we explored the expression level of tRNA-derived fragments in the initial diagnosis of breast cancer. We constructed a database of tRNA-derived fragments expressed in clinical breast cancer samples and identified the most differentially expressed tRNA-derived fragments. We conducted qPCR (Quantitative Real-time PCR) analysis in the clinical samples of two paired groups, to verify the difference of expression of the selected tRNA-derived fragments, and explored the biological pathway analysis of GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes biological pathway).
2. METHODS
2.1. Tissue Specimens
In this study, six patients with breast cancer were selected for gene screening. A further ten cases were reported then and finally, there were sixteen cases of breast cancer tissue used for independent cohort validation. The tissue samples were resected in Jiangsu Cancer Hospital (Nanjing, China), which were also paired with normal tissues adjacent to cancerous tissues. All specimens were confirmed by histology and no treatment was administered to patients prior to diagnosis. The samples were frozen freshly in liquid nitrogen and stored in -80 degrees celsius until the specimens were used. The study was approved and supervised by the Clinical Research Ethics Committee of Nanjing Medical University. Written informed consent was obtained from all the patients.
2.2. Pretreatment of tRNA-derived Fragments
Total RNA was qualified by agarose gel electrophoresis and quantified using NanoDrop ND-1000 (Shanghai, China). Total RNA was first pretreated as follows to remove some RNA modifications that interfere with small RNA-seq library construction by rtStar™ tRF&tiRNA Pretreatment Kit (Cat# AS-FS-005, Arraystar, MD, USA). The rtStar™ tRF&tiRNA Pretreatment Kit is designed to remove the modifications that interfere with small RNA cDNA library construction of qPCR. These modifications include terminal modifications that block adaptor ligation to the RNA ends and internal methylations that hinder reverse transcription for cDNA synthesis. As described in the manufacturer's instructions, 3’-aminoacyl (charged) deacylation to 3’-OH for 3’ adaptor ligation, 3’-cP (2’,3’-cyclic phosphate) removal to 3’-OH for 3’ adaptor ligation, 5’-OH (hydroxyl group) phosphorylation to 5’-P for 5’-adaptor ligation, m1A and m3C were demethylated for efficient reverse transcription.
2.3. Sequencing Library Preparation
The pretreated total RNA was used to prepare the sequencing library by rtStar™ First-Strand cDNA Synthesis Kit (3’ and 5’ adaptors) (Cat#: AS-FS-003, Arraystar, MD, USA) in the following steps: 1) 3'-adapter ligation; 2) 5'-adapter ligation; 3) cDNA (complementary DNA) synthesis; 4) PCR amplification; 5) size selection of 135~160bp PCR amplified fragments (corresponding to 15~40nt small RNA size range). The prepared tRF&tiRNA-seq libraries were finally quantified using BioAnalyzer2100(Agilent, California, USA) by Agilent DNA 1000 chip kit (Agilent, part # 5067-1504) to quantified.
2.4. Sequencing and Expression Analysis
The libraries were denatured with 0.1M NaOH to generate single-stranded DNA molecules and diluted to a loading volume of 1.3ml and loading concentration of 1.8pM. Diluted libraries were loaded onto reagent cartridge and forwarded to sequencing run on the Illumina NextSeq 500 system using NextSeq 500 V2 kit (#FC-404-2005, Illumina, California, USA), according to the manufacturer’s instructions. Raw data files in FASTQ format were generated by the Illumina sequencer which were examined by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) software (v0.11.3). Subsequently, the 3’ adapter sequence was trimmed by Cutadapt (https://doi.org/10.14806/ej.17.1. 200) from the clean reads and the reads with lengths shorter than 15 nt were discarded. As the 5’-adaptor was also used as the sequencing primer site, the 5’-adaptor sequence is not present in the sequencing reads. The trimmed reads were recorded in the FASTA format and then were aligned to mature-tRNA and pre-tRNA sequences from GtRNAdb (Genomic tRNA Database) using NovoAlign software (v2.07.11).
2.5. Different Expression of tRNA-derived Fragments for Validation
The tRNA-derived fragments expression levels were measured and normalized as tag counts per million of total aligned tRNA reads (TPM, Trans Per Million). When comparing two groups of profile differences, we used the normalized tag number of tRNAs annotated in GtRNAdb, including the tag count of each sample. The “fold change” (i.e. the ratio of the group averages) between the groups for each tRNA-derived fragment was computed. tRNA-derived fragments having fold changes ≥2 and with FDR (false discovery rate) modified p-value<0.05 were selected as the significantly, differentially expressed tRNA-derived fragments. Image analysis and base calling were performed using Solexa pipeline v1.8 (Off-Line Base Caller software, v1.8). TPM values of all tRNA-derived fragments were plotted in the Scatter plot. The Volcano plot provides a visualization method to perform a quick visual identification of the tRNA-derived fragments displaying large-magnitude changes which are also statistically significant. Outputs analysis could be filtered by deleting the duplicate sequence and ranking the differentially expressed genes according to fold change and p-value by using Microsoft Excel’s Data/Sort & Filter functionalities. Studies have shown that using GtRNAdb database alone will generate incorrect result [15, 20]. Hence, complete matching sequences were selected, and each sequence was checked using the MINTbase v2.0 framework (https://cm.jefferson.edu/MINTbase/) to make sure that each sequence has a MINTbase unique ID. The repeated annotation of the same tRNA-derived fragments was deleted. The tRNA-derived fragments with the potential of coding protein and low expression were removed. By cluster analysis, the eligible indexes were obtained and the Heatmap was drawn. The Scatter plot, Volcano plot and Heatmap were all performed in R environment for statistical computing and graphics.
2.6. Primer Design and Quantitative Real-time PCR
Six of the high different degrees and the qualified requirements for signal values were selected. Total RNA was extracted from 16 clinical samples with TRIZOL (Invitrogen life technologies). cDNA was synthesized with the rtStar™ First-Strand cDNA Synthesis Kit (3’ and 5’ adaptor) (Cat# AS-FS-003, Arraystar, MD, USA). The primers were designed for the tRNA-derived fragments particularly by Primer 5.0 and were synthesized with 2X PCR master mix (Arraystar, MD, USA). U6 was utilized as an internal control. Quantitative Real-time PCR was performed on ViiA 7 Real-time PCR System (Applied Biosystems, MA, USA). The relative expression level of each tRNA-derived fragments was calculated with the 2-△Ct method.
2.7. GO and KEGG Biological Pathway Analysis
The Gene Ontology project provides a controlled vocabulary to describe gene and gene product attributes in any organism (http://www.geneontology.org). The ontology covers three domains: Biological Process, Cellular Component and Molecular Function. Fisher’s exact test is used to detect whether there is more overlap between the DE list and the GO annotation list than would be expected by chance. The p-value denotes the significance of GO terms enrichment in the DE genes. The lower the p-value, the more significant the GO Term. Pathway analysis is functional analysis of mapping genes to KEGG pathways (http://www.genome.jp/kegg/). We regarded the -log10 (p) as the enrichment score that indicated the significance of correlation.
2.8. Statistical Analysis
Statistical analyses were performed with SPSS 23.0 (SPSS Inc, Chicago, IL, USA) and GraphPad prism 7.0 (Graphpad Software Inc, CA, USA). Wilcoxon test was used to examine significant differences of expression of tRNA-derived fragments in cancerous and adjacent normal tissues. The association between tRNA-derived fragments expression and clinicopathological characteristics of breast cancer was analyzed by Pearson’s chi-squared test and Fisher’s exact test. The value of p <0.05 was declared statistically significant.
3. RESULTS
3.1. Expression Profiles of tRNA-derived Fragments
The expression levels of tRNA-derived fragments in six tissue specimens of breast cancer patients and adjacent normal tissues were analyzed. The expression signatures of tRNA-derived fragments were reviewed using scatter plot, volcano plot and hierarchical clustering analyses. The scatter plots revealed that tRNA-derived fragments were differentially expressed between breast cancer and adjacent normal tissues (Fig. 1a). TPM values of all tRNA-derived fragments showed a more than 2.0fold change (Default fold change value is 2.0). In the scatter plot, 496 of the 1910 total genes were up-regulated and 639 down-regulated expressed in the cancer tissue compared to the normal tissue adjacent to the cancerous tissues. The Volcano plot provides a visualization method to perform quick visual identification of the tRNA-derived fragments displaying large-magnitude changes. The plot is constructed by plotting -log10 (p) on the y-axis, and tRF&tiRNA expression log2-scaled fold change (FC ≥ 2 and p<0.05) between the two experimental groups on the x-axis (Fig. 1b). In the difference results list, each detected tRF&tiRNA had an ID number. In comparison to the genome, one molecule might be compared to the precursor of several tRNAs. The duplicate, mismatched and imperfect sequences were deleted, and we finally found 30 significantly differentially expressed genes. A total of 17 tRNA-derived fragments were up-regulated, and 13 tRNA-derived fragments were down-regulated (Table 1). Heatmap of gene expression data obtained from the control group and the experimental group, includes the genes that were selected by the Coefficient of Variation (CV) based on TPM counts (Fig. 1c). Each row represents one gene and each column represents one sample. The color in the panel represents the relative expression level (log2-transformed). The sequencing raw data of all tRNA-derived fragments genes have been stored in GEO (Gene Expression Omnibus). The assigned GEO accession numbers are GSE123967 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123967).
Fig. (1).
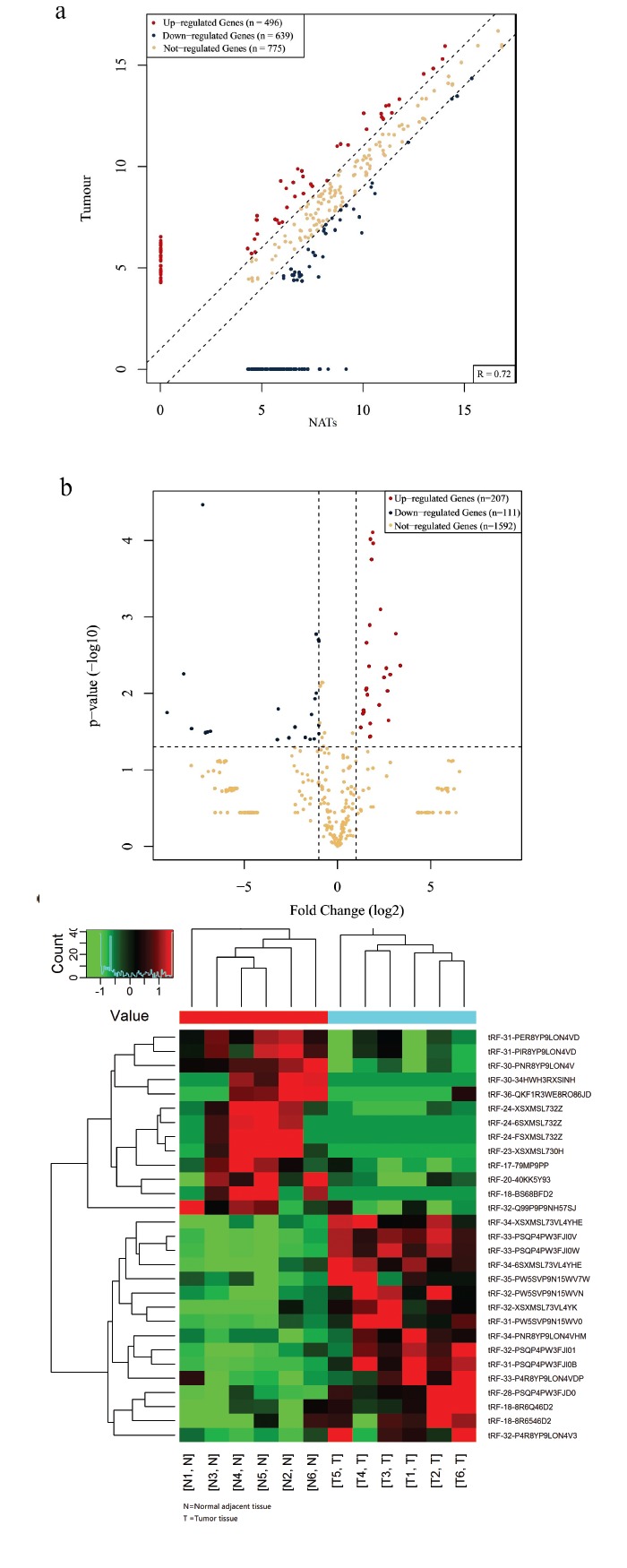
tRNA-derived fragments sequencing analysis of breast cancer tissues Scatter plots (a) of tRNA-derived fragments signal values showed genes above the top line (red dots, up-regulation) or below the bottom line (green dots, down-regulation) between two compared groups. Gray dots indicate tRNA-derived fragments without differential expression. Volcano plot (b) suggested 2.0fold change differentially expressed tRNA-derived fragments with statistical significance (Red: up-regulated; Green: down-regulated). Gray circles indicated non-differentially expressed tRNA-derived fragments, whether fold-change or p-value is not satisfied. Heatmap (c) of gene expression data showed hierarchical clustering of tRNA-derived fragments with expression changes (FC ≥ 2, p<0.05). Red and green colors represent up-regulated and down-regulated genes respectively. (The color version of the figure is available in the electronic copy of the article).
Table 1. Thirty significantly differential expressed tRNA-derived fragments.
|
MINTbase Unique ID
(sequence derived) |
Sequence
(5’-3’) |
tDF Type | Anticodon(s) | Fold Change | p value | FDR |
|---|---|---|---|---|---|---|
| Up-regulated Genes | ||||||
| tRF-32-XSXMSL73VL4YK | TGCCGTGATCGTATAGTGGTTAGTACTCTGCG | 5‘-tRF | HisGTG (n) | 6.5781 | 0.0226 | 0.17708 |
| tRF-33-PSQP4PW3FJI0W | GCCCGGCTAGCTCAGTCGGTAGAGCATGAGACT | 5‘-half | LysCTT (n) | 3.7331 | 0.0001 | 0.01100 |
| tRF-33-PSQP4PW3FJI0V | GCCCGGCTAGCTCAGTCGGTAGAGCATGAGACC | 5‘-half | LysCTT (n) | 3.6596 | 0.0001 | 0.01100 |
| tRF-18-8R6546D2 | TCCCCGGCACCTCCACCA | 3'-tRF | AlaAGC (n) AlaTGC (n) |
3.3616 | 0.0364 | 0.19936 |
| tRF-18-8R6Q46D2 | TCCCCGGCATCTCCACCA | 3'-tRF | AlaTGC (n) AlaCGC (n) |
3.3406 | 0.0248 | 0.18437 |
| tRF-35-PW5SVP9N15WV7W | GCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGT | 5‘-half | HisGTG (n) | 3.281 | 0.0371 | 0.19936 |
| tRF-34-6SXMSL73VL4YHE | GGCCGTGATCGTATAGTGGTTAGTACTCTGCGTT | 5'-half | HisGTG (n) | 3.1888 | 0.0044 | 0.08534 |
| tRF-32-PSQP4PW3FJI01 | GCCCGGCTAGCTCAGTCGGTAGAGCATGAGAC | 5‘-half | LysCTT (n) | 2.9049 | 0.0022 | 0.05282 |
| tRF-28-PSQP4PW3FJD0 | GCCCGGCTAGCTCAGTCGGTAGAGCATG | 5'-tRF | LysCTT (n) | 2.9021 | 0.0086 | 0.09614 |
| tRF-33-P4R8YP9LON4VDP | GCATGGGTGGTTCAGTGGTAGAATTCTCGCCTG | 5‘-half | GlyGCC (n) | 2.8576 | 0.009 | 0.09614 |
| tRF-32-P4R8YP9LON4V3 | GCATGGGTGGTTCAGTGGTAGAATTCTCGCCT | 5‘-half | GlyGCC (n) | 2.6216 | 0.0175 | 0.14423 |
| tRF-34-PNR8YP9LON4VHM | GCATTGGTGGTTCAGTGGTAGAATTCTCGCCTGC | 5‘-half | GlyGCC (n) | 2.6003 | 0.0167 | 0.14278 |
| tRF-32-PW5SVP9N15WVN | GCCGTGATCGTATAGTGGTTAGTACTCTGCGT | 5'-half | HisGTG (n) | 5.5853 | 0.0062 | 0.08971 |
| tRF-31-PSQP4PW3FJI0B | GCCCGGCTAGCTCAGTCGGTAGAGCATGAGA | 5'-tRF | LysCTT (n) | 4.89 | 0.0008 | 0.05255 |
| tRF-31-PW5SVP9N15WV0 | GCCGTGATCGTATAGTGGTTAGTACTCTGCG | 5'-tRF | HisGTG (n) | 4.6602 | 0.0142 | 0.12892 |
| tRF-33-PW5SVP9N15WV0E | GCCGTGATCGTATAGTGGTTAGTACTCTGCGTT | 5‘-half | HisGTG (n) | 6.0763 | 0.0047 | 0.08533 |
| tRF-34-XSXMSL73VL4YHE | TGCCGTGATCGTATAGTGGTTAGTACTCTGCGTT | 5'-half | HisGTG (n) | 8.6323 | 0.0017 | 0.05281 |
| Down-regulated Genes | ||||||
| tRF-31-PIR8YP9LON4VD | GCACTGGTGGTTCAGTGGTAGAATTCTCGCC | 5‘-half | ValCAC (n) GlyCCC (n) |
-2.0206 | 0.0337 | 0.19601 |
| tRF-30-PNR8YP9LON4V | GCATTGGTGGTTCAGTGGTAGAATTCTCGC | 5'-tRF | GlyGCC (n) GlyCCC (n) |
-2.2326 | 0.0017 | 0.05281 |
| tRF-31-PER8YP9LON4VD | GCAATGGTGGTTCAGTGGTAGAATTCTCGCC | 5‘-half | GluTTC (n) | -2.3354 | 0.0118 | 0.11362 |
| tRF-32-Q99P9P9NH57SJ | GCTTCTGTAGTGTAGTGGTTATCACGTTCGCC | 5‘-half | ValCAC (n) | -2.6476 | 0.0189 | 0.14836 |
| tRF-20-40KK5Y93 | CTAAGCCAGGGATTGTGGGT | i-tRF | ArgCCT (n) | -3.3527 | 0.0378 | 0.19936 |
| tRF-17-79MP9PP | GTTTCCGTAGTGTAGTG | 5'-tRF | ValCAC (n) ValAAC (n) |
-4.8984 | 0.0276 | 0.18437 |
| tRF-36-QKF1R3WE8RO86JD | GCGGGAGACCGGGGTTCGATTCCCCGACGGGGAGCC | 3'-tRF | AspGTC (n) | -6.1712 | 0.0382 | 0.19936 |
| tRF-24-XSXMSL732Z | TGCCGTGATCGTATAGTGGTTAGT | 5'-tRF | HisGTG (n) | -9.1749 | 0.016 | 0.14278 |
| tRF-18-BS68BFD2 | AACCGGGCGGAAACACCA | 3'-tRF | ValCAC (n) ValAAC (n) |
-114.2616 | 0.0314 | 0.19318 |
| tRF-24-6SXMSL732Z | GGCCGTGATCGTATAGTGGTTAGT | 5'-tRF | HisGTG (n) | -127.0568 | 0.0321 | 0.19318 |
| tRF-30-34HWH3RXSINH | CCGTGATCGTATAGTGGTTAGTACTCTGCG | i-tRF | HisGTG (n) | -137.1639 | 0.0327 | 0.19318 |
| tRF-24-FSXMSL732Z | AGCCGTGATCGTATAGTGGTTAGT | 5'-tRF | HisGTG (n) | -229.9317 | 0.029 | 0.18908 |
| tRF-23-XSXMSL730H | TGCCGTGATCGTATAGTGGTTAG | 5'-tRF | HisGTG (n) | -570.2959 | 0.0179 | 0.14556 |
3.2. Validation of Quantitative Real-time PCR
The 6 selected tRNA-derived fragments were up-regulated tRF-32-XSXMSL73VL4YK, tRF-28-PSQP4PW3FJD0, tRF-33-PSQP4PW3FJI0V, tRF-31-PSQP4PW3FJI0B and down-regulated tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, respectively. Table 2 enumerated the primer sequences and the optimal annealing temperature. U6 was used for tRNA-derived fragments template normalization. The results demonstrated that tRF-32-XSXMSL73VL4YK, tRF-32-Q99P9P9NH57SJ and tRF-17-79MP9PP were significantly expressed in 16 breast cancer tissues, compared with adjacent samples (Fig. 2a-c). The up and down-regulated expression patterns were similar to the results which were observed in the sequencing analyzed in heatmap as said above (Fig. 2d). Expression of tRNA-derived fragments was quantified by qPCR in breast cancer tissues and adjacent samples in which expressions were normalized by U6 RNA in each sample (Fig. 3). Compared to the adjacent tissues, relative expression of tRF-32-XSXMSL73VL4YK, tRF-32-Q99P9P9NH57SJ and tRF-17-79MP9PP was differential significantly (p<0.0001).
Table 2. The primer sequences and the optimal annealing temperature in quantitative real-time PCR.
| Genes | Two-way Primer Sequence | Optimal Annealing Temperature (°C) | Length(bp) |
|---|---|---|---|
| U6 | F:5’GCTTCGGCAGCACATATACTAAAAT3’ R:5’CGCTTCACGAATTTGCGTGTCAT3’ |
60 | 89 |
| tRF-31-PSQP4PW3FJI0B | F:5’ AGTTCTACAGTCCGACGATCGC 3' R:5’ GATCTTCTCATGCTCTACCGACTG 3’ |
60 | 56 |
| tRF-32-XSXMSL73VL4YK | F:5’ ATCTGCCGTGATCGTATAGTGGTT 3' R:5’ ACGTGTGCTCTTCCGATCTCG 3’ |
60 | 54 |
| tRF-28-PSQP4PW3FJD0 | F:5’ CGGCTAGCTCAGTCGGTAGA 3' R:5’ CGTGTGCTCTTCCGATCTCAT 3’ |
60 | 43 |
| tRF-17-79MP9PP | F:5’ TCTACAGTCCGACGATCGTTTC 3' R:5’ TGCTCTTCCGATCTCACTACACTA 3’ |
60 | 48 |
| tRF-33-PSQP4PW3FJI0V | F:5’ ATCGCCCGGCTAGCTCAGT 3' R:5’ TGTGCTCTTCCGATCTGGTCTC 3’ |
60 | 52 |
| tRF-32-Q99P9P9NH57SJ | F:5’ TTCTACAGTCCGACGATCGCT 3' R:5’ GGCGAACGTGATAACCACTACA 3’ |
60 | 50 |
Fig. (2).
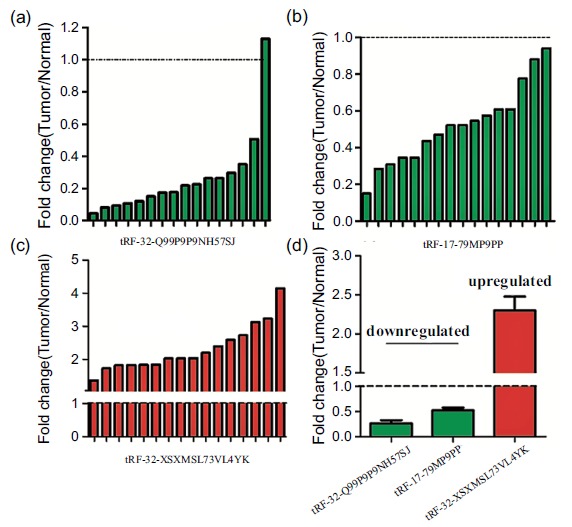
The tRNA-derived fragments expression in breast cancer tissues. Data relative to the paired samples were presented. (a): Fold changes of tRF-32-Q99P9P9NH57SJ in each individual paired sample. (b): Fold changes of tRF-17-79MP9PP in each individual paired sample. (c): Fold changes of tRF-32-XSXMSL73VL4YK in each individual paired sample. (d): Bar Chart presented that compared with adjacent tissues, tRF-32-XSXMSL73VL4YK were up-regulated genes, while tRF-17-79MP9PP and tRF-32-Q99P9P9NH57SJ were down-regulated genes in breast cancer tissues.
Fig. (3).
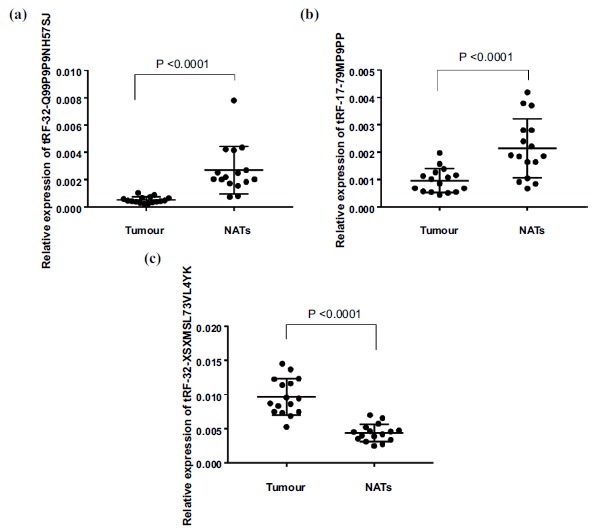
The expression level of tRNA-derived fragments in breast cancer patients and adjacent subjects were identified by qPCR. (a): tRF-32-Q99P9P9NH57SJ (p<0.0001). (b): tRF-17-79MP9PP (p<0.0001). (c): tRF-32-XSXMSL73VL4YK (p<0.0001).
3.3. tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL73VL4YK in MINTbase and Correlations with Clinical Characteristics
We checked the information of tRF-32-Q99P9P 9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL73 VL4YK
in version 2.0 of MINTbase and also listed the number and location of all genomic loci (Table 3). From MINTbase, we have found that tRF-32-XSXMSL73VL4YK has 9 genomic locations and tRF-32-Q99P9P9NH57SJ has only one. Both of them have exclusive tRNA space. tRF-17-79MP9PP has 18 genomic locations but it has no exclusive tRNA space. Fig. 4 also showed the column row links to their parental tRNA isodecoders and pointed out their anticodons. The TCGA module of MINTbase provides the information separately for each of 32 TCGA cancer types (https://doi.org/10.1101/135517). tRF-32-XSXMSL 73VL4YK and tRF-32-Q99P9P9NH57SJ have no reported database in TCGA currently but tRF-17-79MP9PP has. The correlation of tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL 73VL4YK expression levels with clinicopathological factors of breast cancer patients was also analyzed (Table 4). tRF-32-Q99P9P9NH57SJ expression was significantly correlated with TNM stage (p = 0.012) and lymph node metastasis (p = 0.013), but there were no obvious changes between tRF-17-79MP9PP (p = 0.614 and 0.091 respectively) and tRF-32-XSXMSL73VL4YK (p = 0.317 and 0.302 respectively). Furthermore, we examined the relationship of tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP and tRF-32-XSXMSL 73VL4YK between breast cancer subtypes and expression levels, but none of them were statistically significant (p =0.375,0.375 and 0.522 respectively).
Table 3. tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL73VL4YK in MINTbase.
|
MINTbase Unique ID
(sequence derived) |
Genomic Locations | Fragment Length |
MINTbase Alternative IDs
(GRCh37 assembly-derived) |
Anticodon(s) | Exclusive to tRNA space? | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Up-regulated Genes | ||||||||||
| tRF-32-XSXMSL73VL4YK | 9 | 32 | trna111_HisGTG_1_-_147774845_147774916@-1T.31.32, trna118_HisGTG_1_-_145396881_145396952@-1T.31.32, trna16_HisGTG_1_+_146544773_146544844@-1T.31.32, trna21_HisGTG_1_+_147753471_147753542@-1T.31.32, trna1_HisGTG_15_+_45493349_45493420@-1T.31.32, trna8_HisGTG_15_-_45492611_45492682@-1T.31.32, trna9_HisGTG_15_-_45490804_45490875@-1T.31.32, trna33_HisGTG_6_+_27125906_27125977@-1T.31.32, trna7_HisGTG_9_-_14433938_14434009@-1T.31.32 |
HisGTG (n) | yes | |||||
| Down-regulated Genes | ||||||||||
| tRF-32-Q99P9P9NH57SJ | 1 | 32 | trna152_ValCAC_6_-_27248049_27248121@1.32.32 | ValCAC (n) | yes | |||||
| tRF-17-79MP9PP | 18 | 17 | trna37_ValAAC_6_+_27203288_27203360@1.17.17 trna136_ValAAC_6_-_27648885_27648957@1.17.17 trna139_ValAAC_6_-_27618707_27618779@1.17.17 trna15_ValAAC_5_-_180615416_180615488@1.17.17 trna2_ValAAC_3_+_169490018_169490090@1.17.17 trna12_ValAAC_5_-_180645270_180645342@1.17.17 trna4_ValAAC_5_+_180591154_180591226@1.17.17 trna5_ValAAC_5_+_180596610_180596682@1.17.17 trna132_ValAAC_6_-_27721179_27721251@1.17.17 trna133_ValCAC_6_-_27696327_27696399@1.17.17 trna90_ValCAC_1_-_149684088_149684161@1.17.17 trna98_ValCAC_1_-_149298555_149298627@1.17.17 trna85_ValCAC_1_-_161369490_161369562@1.17.17 trna10_ValCAC_5_-_180649395_180649467@1.17.17 trna18_ValCAC_5_-_180529253_180529325@1.17.17 trna2_ValCAC_5_+_180524070_180524142@1.17.17 trna6_ValCAC_5_+_180600650_180600722@1.17.17 trna9_ValCAC_6_+_26538282_26538354@1.17.17 |
ValCAC (n) ValAAC (n) |
no | |||||
Fig. (4).
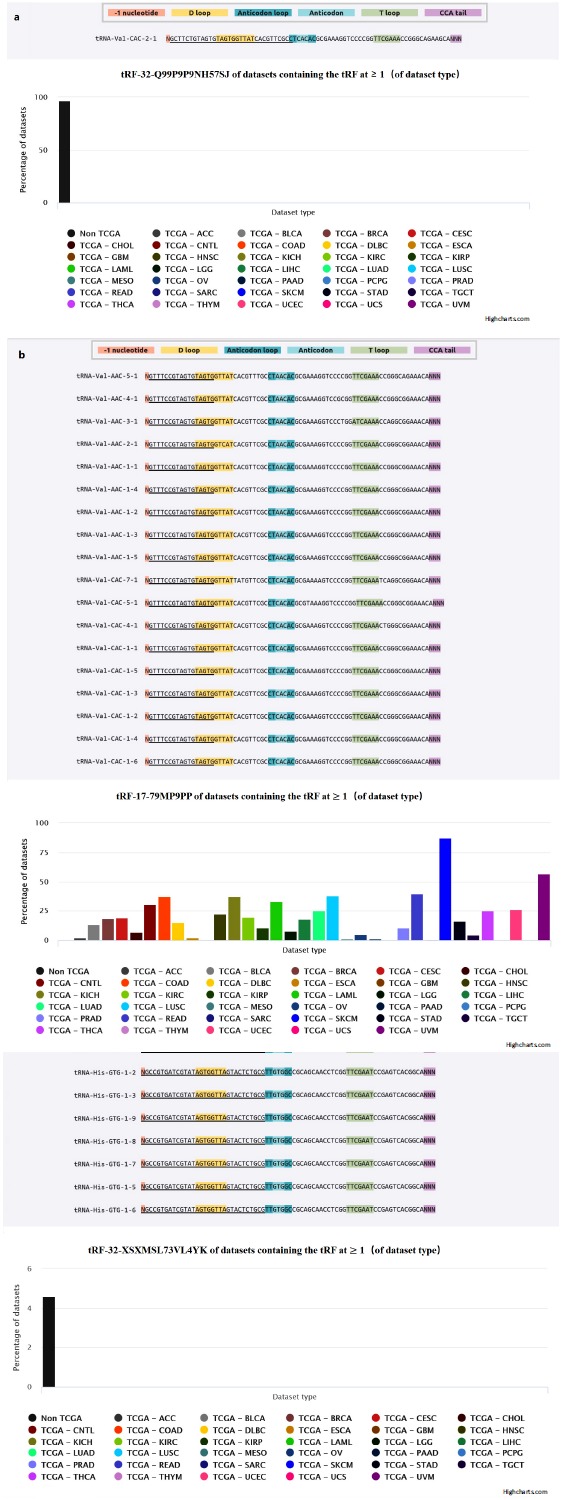
MINTbase (https://cm.jefferson.edu/MINTbase/) summarizes the known information of the corresponding tRNA-derived fragments in the current public database. (a): The candidate mature tRNA sources and datasets with RPM ≥ 1.0) from TCGA of tRF-32-Q99P9P9NH57SJ. (b): The candidate mature tRNA sources and datasets with RPM ≥ 1.0) from TCGA of tRF-17-79MP9PP. (c): The candidate mature tRNA sources and datasets with RPM ≥ 1.0) from TCGA of tRF-32-XSXMSL73VL4YK.
Table 4. Correlation between significantly expressed tRNA-derived fragments and their clinicopathological characteristics.
| Characteristics | N |
tRF-32-Q99P9P9NH57SJ
Low Expression (≤ median) |
tRF-32-Q99P9P9NH57SJ
High Expression (> median) |
p |
tRF-17-79MP9PP
Low Expression (≤ Median) |
tRF-17-79MP9PP
High Expression (> Median) |
p |
tRF-32-XSXMSL73VL4YK
Low Expression (≤ Median) |
tRF-32-XSXMSL73VL4YK
High Expression (> Median) |
p |
|---|---|---|---|---|---|---|---|---|---|---|
| Number | 16 | 7 | 9 | - | 7 | 9 | - | 8 | 8 | - |
| Age (years) | - | - | - | - | - | - | - | - | - | - |
| ≤ 50 | 7 | 2 | 5 | 0.280 | 1 | 6 | 0.036* | 3 | 4 | 0.614 |
| >50 | 9 | 5 | 4 | - | 6 | 3 | - | 5 | 4 | - |
| Tumor stage | - | - | - | - | - | - | - | - | - | - |
| I-II | 8 | 1 | 7 | 0.012* | 3 | 5 | 0.614 | 5 | 3 | 0.317 |
| III- IV | 8 | 6 | 2 | - | 4 | 4 | - | 3 | 5 | - |
| Lymph node metastasis | - | - | - | - | - | - | - | - | - | - |
| No | 10 | 2 | 8 | 0.013* | 6 | 4 | 0.091 | 4 | 6 | 0.302 |
| Yes | 6 | 5 | 1 | - | 1 | 5 | - | 4 | 2 | - |
| Subtype | - | - | - | - | - | - | - | - | - | - |
| Triple negative | 3 | 2 | 1 | 0.375 | 2 | 1 | 0.375 | 1 | 2 | 0.522 |
| Non-tri-negative | 13 | 5 | 8 | - | 5 | 8 | - | 7 | 6 | - |
* Statistical significance (p<0.05).
3.4. GO and KEGG Pathway Analysis of tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL 73VL4YK
For subsequent research, we predicted the target gene of tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXM SL73VL4YK through GO and KEGG pathways. The top ten significantly results of GO and KEGG pathways enrichment analysis are presented in Fig. 5. The GO analysis covered three domains: BP (Biological Process), CC (Cellular Component) and MF (Molecular Function). The p-value denoted the significance of GO terms enrichment in the differential genes (p<0.05) and the enrichment score was regarded as the -log(p). In the BP analysis, the most enriched tRF-32-Q99P9P9NH57SJ was in the regulation of nucleobase-containing compound metabolic process (GO: 0019219). In the CC, the analysis showed that enrichment mainly occurred at intracellular (GO:0005622). For MF, the analysis revealed that the most significant enrichment was transcription regulator activity (GO: 0140110). In the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, tRF-32-Q99P9P9NH57SJ was meaningfully enriched in the regulation of lipolysis in adipocytes (hsa04923) (Fig. 5a). The most significant results in BP, CC, MF and KEGG of tRF-17-79MP9PP (Fig. 5b) were respectively Golgi vesicle transport (GO:0048193), protein complex (GO:0043234), DNA binding transcription factor activity (GO:0003700) and AMPK signaling pathway (hsa04152). For tRF-32-XSXMSL 73VL4YK (Fig. 5c), the positive regulation of myelination (GO:0031643), Golgi apparatus (GO:0005794), transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding (GO:0001228) and drug metabolism-other enzymes (hsa00983) were the most significantly enriched term, separately.
Fig. (5).
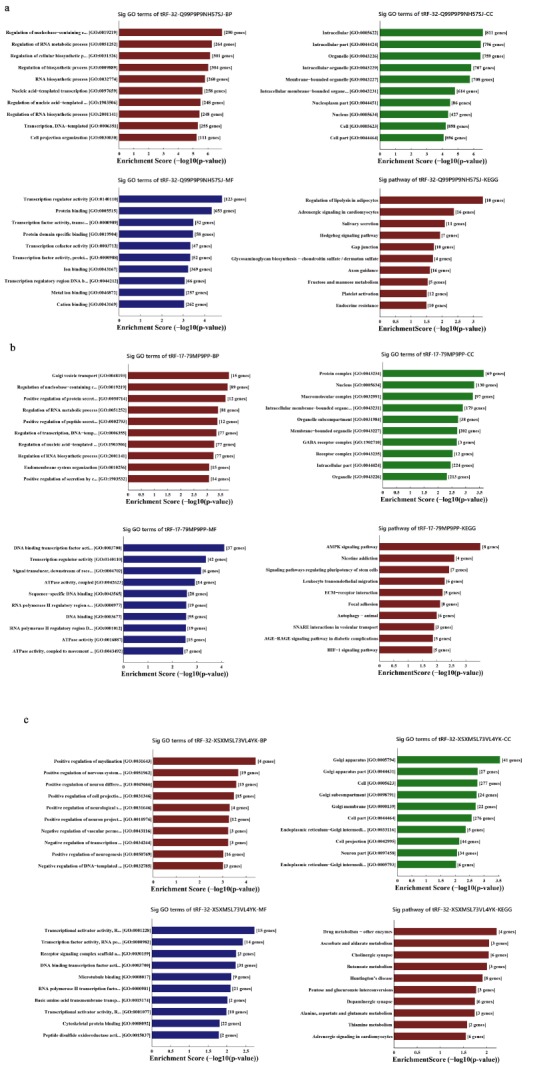
GO and KEGG pathway analysis of the tRNA-derived fragments: (a): The bar plot showed the top ten Enrichment Score value of the significant enrichment terms with tRF-32-Q99P9P9NH57SJ. (b): The bar plot showed the top ten Enrichment Score value of the significant enrichment terms with tRF-17-79MP9PP. (c): The bar plot showed the top ten Enrichment Score value of the significant enrichment terms with tRF-32-XSXMSL73VL4YK.
4. DISCUSSION
In the early studies, it was confirmed that tRNA-derived fragments were not a random by-product of tRNA degeneration or synthesis, but rich and novel classes of short RNA with an exact sequence structure, specific expression patterns and biological roles [21]. With the rapid development of sequencing technology in recent years, the regulatory mechanisms of tRNA-derived fragments have been further studied. In addition to the MINTbase and MINTmap [19, 20] mentioned in the introduction section, Ling-Ling Zheng et al. developed an integrated web-based computing system to identify tRNA-derived fragments from sRNA deep-sequen- cing data and evaluate their expression in multiple cancers [22]. Goodarzi et al. investigated tRNA-derived fragments suppressed growth under serum-starvation, cancer cell invasion, and metastasis by breast cancer cells. Tumor suppressive pathway by attenuating the induction of tRFs might generalize to other tRNA fragments [23]. Sobala et al. presented that 5'-tRFs could inhibit the process of protein translation without the need for complementary target sites in the mRNA [24]. Olvedy, et al. have established some tRFs as novel candidate biomarkers for the early detection of recurrent aggressive in prostate cancer [25].
Telonis, et al. have screened the tRNA-derived fragments from the data of the BRCA repository of The Cancer Genome Atlas (TCGA) at the National Institutes of Health (NIH). They have reported that the specific type of i-tRFs were identified first and verified in tested breast tumor and adjacent normal samples [15]. Our study was based on the extraction of fresh breast cancer tissues, sequencing to obtain a list of different expressed tRNA-derived fragments. Ebhardt, et al. also found that there were diversities in the expression of tRNA-derived fragments in different tissues and they were heavily decorated by RNA modifications that interfere with small RNA-seq library construction [26]. Hence, before the experiment started, we preprocessed total RNA samples using the specialized kits in accordance with the manufacturer’s instructions to ensure the endpoints. Then we sequenced tRNA-derived fragments expression profiles on Illumina NextSeq 500 system and finally identified 30 significantly expressed tRNA-derived fragments. A total of 17 tRNA-derived fragments were up-regulated, and 13 tRNA-derived fragments were down-regulated. After comparing with the Mintbase, we found that the 30 tRNA-derived fragments contained various types.
We did not select i-tRF for validation because of its special type, which required special analysis [15, 20, 27]. We selected 6 tRNA-derived fragments from other types by using qPCR in 16 paired breast cancer samples. The results suggested that tRF-32-XSXMSL73VL4YK was significantly up-regulated, tRF-32-Q99P9P9NH57SJ and tRF-17-79MP9 PP were significantly down-regulated, which were consistent with Dhahbi, et al. They found that there were significant changes in up- and down-regulated of specific 5′-tRNA halves in the serum samples of breast cancer [28].
When we investigated the relationship between tRNA-derived fragments expression and clinical characteristics, there are connections in tumour stage and lymph node metastasis of tRF-32-Q99P9P9NH57SJ with no obvious correlation in tRF-17-79MP9PP and tRF-32-XSXMSL73VL4YK. However, significant literatures has emphasized that the different abundance and expression differences of tRNA-derived fragments depend on tissue type and tissue state. Both their studies were conducted under the subtype of three-negative breast cancer [29, 30]. We are currently doing an elementary exploration of the different expressions in tRNA-derived fragments between normal tissue and clinical cancer tissue specimens. Therefore, there was no special subdivision of subtype in breast cancer when the specimens were selected. The subtypes of triple-negative and non-triple negative breast cancer were both tested in the tissues we used to sequence and validate sequencing results. No more sequencing of tissue specimens was possible due to funding constraints. More information is required to be collected and explored the implications. In the following studies, we will expand the sample size for a certain subtype of breast cancer, and conduct further analysis.
In GO and KEGG pathway analyses, for subsequent research reference, we conducted target gene prediction analysis. We have found the biological functions and potential pathways of tRF-32-Q99P9P9NH57SJ, tRF-17-79MP9PP, tRF-32-XSXMSL73VL4YK. The top ten results of GO enrichment analysis were presented in results showed above. The results of the pathway analysis needed to be supported by the later protein functional experiments, which have laid a foundation for the follow-up mechanism research. Further studies are needed to investigate the molecular mechanism and biological function of tRNA-derived fragments.
The above studies have suggested that these genes were likely to be biomarkers in the diagnosis of breast cancer. The tRNA-derived fragments are highly enriched in biological tissues and fluids [31, 32]. Although the current screening of humoral biomarkers is mainly concentrated on miRNA, it opens up a broad prospect for us to develop less invasive and sensitive biomarkers for tRNA-derived fragments. They have high equivalent and stability in a variety of body fluids, extensive involvement in pathological processes [33-35], differently expressed in solid tumours and hematological malignancies [36], and also have a strong resolution between cancer patients and normal controls [37].
CONCLUSION
Taken together, the detection of tRNA-derived fragments expression profile provides a new diagnosis process for breast cancer. We forecast that tRNA-derived fragments would become promising biomarkers for cancer diagnosis.
ACKNOWLEDGEMENTS
Feng Yan conceived the experiments. Xiaoming Wang and Yining Yang performed the experiments and bioinformatic analyses. Da Wei, Pan Jiang and Yufeng Yao reviewed the pathological specimen. Yining Yang wrote the manuscript. Xuyan Tan, Xuelian Mao, Dongping Mo and Ting Wang analyzed the results. All authors reviewed the manuscript. The authors thank Dr. Feng Yan for expert assistance with manuscript preparation.
LIST OF ABBREVIATIONS
- tRFs
tRNA-derived Fragments
- ncRNA
Non-coding RNA
- sRNA
Small RNA
- cDNA
Complementary DNA
- NATs
Normal Adjacent Tissues
- FC
Fold Change
- qPCR
Quantitative real-time Polymerase Chain Reaction
- TPM
Trans Per Million
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- BP
Biological Process
- CC
Cellular Component
- MF
Molecular Function
- FDR
False Discovery Rate
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved and supervised by the Clinical Research Ethics Committee of Nanjing Medical University, China.
HUMAN AND ANIMAL RIGHTS
No animals were used for the study. All the reported experiments on humans were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Written informed consent was obtained from all the patients for the study.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in the [GEO (Gene Expression Omnibus)] at [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123967], reference number [GSE123967], public release date [17-12-2021].
FUNDING
The study was supported by the Funding from the National Nature Science Foundation of China (Funding number: 81871718), and the Funding of Medicine innovation teams and Leading Talents of Jiangsu Health Department (Funding number: CXTDA2017017) and Project of Jiangsu provincial Six Talent Peaks (Funding number: WSN-057).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Miller K.D., Siegel R.L., Lin C.C., Mariotto A.B., Kramer J.L., Rowland J.H., Stein K.D., Alteri R., Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016;66(4):271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 2.Zeng H., Chen W., Zheng R., Zhang S., Ji J.S., Zou X., Xia C., Sun K., Yang Z., Li H., Wang N., Han R., Liu S., Li H., Mu H., He Y., Xu Y., Fu Z., Zhou Y., Jiang J., Yang Y., Chen J., Wei K., Fan D., Wang J., Fu F., Zhao D., Song G., Chen J., Jiang C., Zhou X., Gu X., Jin F., Li Q., Li Y., Wu T., Yan C., Dong J., Hua Z., Baade P., Bray F., Jemal A., Yu X.Q., He J. Changing cancer survival in China during 2003-15: a pooled analysis of 17 population-based cancer registries. Lancet Glob. Health. 2018;6(5):e555–e567. doi: 10.1016/S2214-109X(18)30127-X. [DOI] [PubMed] [Google Scholar]
- 3.Wu X., Zeng R., Wu S., Zhong J., Yang L., Xu J. Comprehensive expression analysis of miRNA in breast cancer at the miRNA and isomiR levels. Gene. 2015;557(2):195–200. doi: 10.1016/j.gene.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 4.Hamam R., Hamam D., Alsaleh K.A., Kassem M., Zaher W., Alfayez M., Aldahmash A., Alajez N.M. Circulating microRNAs in breast cancer: novel diagnostic and prognostic biomarkers. Cell Death Dis. 2017;8(9):e3045. doi: 10.1038/cddis.2017.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J., Liu S., Chen Y., Fu Y., Silver A.J., Hill M.S., Lee I., Lee Y.S., Bao X. Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J. Gen. Virol. 2017;98(7):1600–1610. doi: 10.1099/jgv.0.000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magee R.G., Telonis A.G., Loher P., Londin E., Rigoutsos I. Profiles of miRNA isoforms and tRNA fragments in prostate cancer. Sci. Rep. 2018;8(1):5314. doi: 10.1038/s41598-018-22488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pekarsky Y., Balatti V., Palamarchuk A., Rizzotto L., Veneziano D., Nigita G., Rassenti L.Z., Pass H.I., Kipps T.J., Liu C.G., Croce C.M. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc. Natl. Acad. Sci. USA. 2016;113(18):5071–5076. doi: 10.1073/pnas.1604266113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honda S., Kirino Y. SHOT-RNAs: A novel class of tRNA-derived functional RNAs expressed in hormone-dependent cancers. Mol. Cell. Oncol. 2016;3(2):e1079672. doi: 10.1080/23723556.2015.1079672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco S., Dietmann S., Flores J.V., Hussain S., Kutter C., Humphreys P., Lukk M., Lombard P., Treps L., Popis M., Kellner S., Holter S.M., Garrett L., Wurst W., Becker L., Klopstock T., Fuchs H., Gailus-Durner V., Hrabe de Angelis M., Karadottir R.T., Helm M., Ule J., Gleeson J.G., Odom D.T., Frye M. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014;33(18):2020–2039. doi: 10.15252/embj.201489282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkatesh T., Suresh P.S., Tsutsumi R. tRFs: miRNAs in disguise. Gene. 2016;579(2):133–138. doi: 10.1016/j.gene.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 11.Shen Y., Yu X., Zhu L., Li T., Yan Z., Guo J. Transfer RNA-derived fragments and tRNA halves: biogenesis, biological functions and their roles in diseases. J. Mol. Med. (Berl.) 2018;96(11):1167–1176. doi: 10.1007/s00109-018-1693-y. [DOI] [PubMed] [Google Scholar]
- 12.Green D., Fraser W.D., Dalmay T. Transfer RNA-derived small RNAs in the cancer transcriptome. Pflugers Arch. 2016;468(6):1041–1047. doi: 10.1007/s00424-016-1822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun C., Fu Z., Wang S., Li J., Li Y., Zhang Y., Yang F., Chu J., Wu H., Huang X., Li W., Yin Y. Roles of tRNA-derived fragments in human cancers. Cancer Lett. 2018;414:16–25. doi: 10.1016/j.canlet.2017.10.031. [DOI] [PubMed] [Google Scholar]
- 14.Saikia M., Hatzoglou M. The many virtues of tRNA-derived stress-induced RNAs (tiRNAs): discovering novel mechanisms of stress response and effect on human health. J. Biol. Chem. 2015;290(50):29761–29768. doi: 10.1074/jbc.R115.694661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Telonis A.G., Loher P., Honda S., Jing Y., Palazzo J., Kirino Y., Rigoutsos I. Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget. 2015;6(28):24797–24822. doi: 10.18632/oncotarget.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keam S.P., Hutvagner G. tRNA-derived Fragments (tRFs): emerging new roles for an ancient RNA in the regulation of gene expression. Life (Basel) 2015;5(4):1638–1651. doi: 10.3390/life5041638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P., Mudunuri S.B., Anaya J., Dutta A. tRFdb: a database for transfer RNA fragments. Nucleic Acids Res. 2015;43:D141–D145. doi: 10.1093/nar/gku1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoogstrate Y., Jenster G., Martens-Uzunova E.S. FlaiMapper: computational annotation of small ncRNA-derived fragments using RNA-seq high-throughput data. Bioinformatics. 2015;31(5):665–673. doi: 10.1093/bioinformatics/btu696. [DOI] [PubMed] [Google Scholar]
- 19.Pliatsika V., Loher P., Magee R., Telonis A.G., Londin E., Shigematsu M., Kirino Y., Rigoutsos I. MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projects. Nucleic Acids Res. 2018;46:D152–D159. doi: 10.1093/nar/gkx1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loher P., Telonis A.G., Rigoutsos I. MINTmap: fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci. Rep. 2017;7:41184. doi: 10.1038/srep41184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y.S., Shibata Y., Malhotra A., Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009;23(22):2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng L.L., Xu W.L., Liu S., Sun W.J., Li J.H., Wu J., Yang J.H., Qu L.H. tRF2Cancer: a web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acids Res. 2016;44(W1):W185–W193. doi: 10.1093/nar/gkw414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodarzi H., Liu X., Nguyen H.C., Zhang S., Fish L., Tavazoie S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161(4):790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sobala A., Hutvagner G. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10(4):553–563. doi: 10.4161/rna.24285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olvedy M., Scaravilli M., Hoogstrate Y., Visakorpi T., Jenster G., Martens-Uzunova E.S. A comprehensive repertoire of tRNA-derived fragments in prostate cancer. Oncotarget. 2016;7(17):24766–24777. doi: 10.18632/oncotarget.8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ebhardt H.A., Tsang H.H., Dai D.C., Liu Y., Bostan B., Fahlman R.P. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009;37(8):2461–2470. doi: 10.1093/nar/gkp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magee R., Telonis A.G., Cherlin T., Rigoutsos I., Londin E. Assessment of isomiR discrimination using commercial qPCR methods. Noncoding RNA. 2017;3(2):1–12. doi: 10.3390/ncrna3020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhahbi J.M., Spindler S.R., Atamna H., Boffelli D., Martin D.I. Deep sequencing of serum small RNAs identifies patterns of 5′ tRNA half and YRNA fragment expression associated with breast cancer. Biomark. Cancer. 2014;6:37–47. doi: 10.4137/BIC.S20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Telonis A.G., Rigoutsos I. Race disparities in the contribution of miRNA isoforms and tRNA-derived fragments to triple-negative breast cancer. Cancer Res. 2018;78:1140–1154. doi: 10.1158/0008-5472.CAN-17-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui Y., Huang Y., Wu X., Zheng M., Xia Y., Fu Z., Ge H., Wang S., Xie H. Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J. Cell. Physiol. 2019;234(6):8740–8751. doi: 10.1002/jcp.27533. [DOI] [PubMed] [Google Scholar]
- 31.Balatti V., Nigita G., Veneziano D., Drusco A., Stein G.S., Messier T.L., Farina N.H., Lian J.B., Tomasello L., Liu C.G., Palamarchuk A., Hart J.R., Bell C., Carosi M., Pescarmona E., Perracchio L., Diodoro M., Russo A., Antenucci A., Visca P., Ciardi A., Harris C.C., Vogt P.K., Pekarsky Y., Croce C.M. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA. 2017;114(30):8071–8076. doi: 10.1073/pnas.1706908114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soares A.R., Santos M. Discovery and function of transfer RNA-derived fragments and their role in disease. Wiley Interdiscip. Rev. RNA. 2017;8(6):e1423. doi: 10.1002/wrna.1423. [DOI] [PubMed] [Google Scholar]
- 33.Cech T.R., Steitz J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Keam S.P., Sobala A., Ten Have S., Hutvagner G. tRNA-derived rna fragments associate with human Multisynthetase Complex (MSC) and modulate ribosomal protein translation. J. Proteome Res. 2017;16(2):413–420. doi: 10.1021/acs.jproteome.6b00267. [DOI] [PubMed] [Google Scholar]
- 35.Li Q., Hu B., Hu G.W., Chen C.Y., Niu X., Liu J., Zhou S.M., Zhang C.Q., Wang Y., Deng Z.F. tRNA-derived small non-coding RNAs in response to ischemia inhibit angiogenesis. Sci. Rep. 2016;6:20850. doi: 10.1038/srep20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao C., Tolkach Y., Schmidt D., Kristiansen G., Muller S.C., Ellinger J. 5′-tRNA halves are dysregulated in clear cell renal cell carcinoma. J. Urol. 2018;199(2):378–383. doi: 10.1016/j.juro.2017.07.082. [DOI] [PubMed] [Google Scholar]
- 37.Nientiedt M., Deng M., Schmidt D., Perner S., Muller S.C., Ellinger J. Identification of aberrant tRNA-halves expression patterns in clear cell renal cell carcinoma. Sci. Rep. 2016;6:37158. doi: 10.1038/srep37158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of the article is available in the [GEO (Gene Expression Omnibus)] at [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123967], reference number [GSE123967], public release date [17-12-2021].